

Abstract
The receptor tyrosine kinase HER2 is an oncogene amplified in invasive breast cancer and its overexpression in mammary epithelial cell lines is a strong determinant of a tumorigenic phenotype. Accordingly, HER2-overexpressing mammary tumors are commonly indicative of a poor prognosis in patients. Several quantitative proteomic studies have employed two-dimensional gel electrophoresis in combination with tandem mass spectrometry, which provides only limited information about the molecular mechanisms underlying HER2/neu signaling. In the present study, we used a SILAC-based approach to compare the proteomic profile of normal breast epithelial cells with that of Her2/neu-overexpressing mammary epithelial cells, isolated from primary mammary tumors arising in MMTV-Her2/neu transgenic mice. We identified 23 proteins with relevant annotated functions in breast cancer, showing a substantial differential expression. This included overexpression of creatine kinase, retinol-binding protein 1, thymosin beta 4 and tumor protein D52, which correlated with the tumorigenic phenotype of Her2-overexpressing cells. The differential expression pattern of two genes, gelsolin and retinol binding protein 1, was further validated in normal and tumor tissues. Finally, an in silico analysis of published cancer microarray datasets revealed a 23-gene signature which can be used to predict the probability of metastasis-free survival in breast cancer patients.
Keywords: Cancer biomarker, Her2, quantitative proteomics and SILAC
Introduction
Overexpression of growth factor receptors leads to alterations of downstream signal transduction pathways. These signaling alterations cause aberrant changes in cell proliferation rate and cell-cell signaling that lead to genetic instability and transformation of normal cells into tumor cells [1, 2]. The human epidermal growth factor receptor 2 (HER2) is a ligand-independent receptor tyrosine kinase (RTK) used by cells to amplify the signaling cascades from other growth factor receptors [2]. This RTK is highly expressed in about 30% of breast cancer cases that are referred to as HER2-positive; and its overexpression is indicative of low survival rates in breast cancers [3, 4].
HER2 is homologous to the epidermal growth factor receptor (EGFR), also known as HER1. In humans, there are four HER receptor tyrosine kinase homologues (HER1-4) [2]. They form homo or hetero dimers and autophosphorylate a conserved set of tyrosine residues that, in turn, recruit various adaptor proteins. As a result, growth promoting signals are relayed to the nucleus, where immediate-early genes are induced transcriptionally [2]. HER2 is a potent oncogenic kinase, in part because of its ability to dimerize with other members of the HER family of receptors, EGFR, HER3 and HER4, leading to its activation even in the absence of an extracellular ligand [5, 6]. Its overproduction is sufficient to confer a tumorigenic phenotype to an immortalized cell line [7, 8].
In addition to gene expression profiling studies [9-12], various published proteomic studies have been published that have investigated the proteomic profile of either HER2-positive mammary tissues [13-19] or HER2-expressing cell lines [20-26]. Here, we report a quantitative proteomics investigation of a Her2-overexpressing epithelial cell line (H6O5), isolated from a primary mammary tumor of MMTV-Her2/neu transgenic mice. We compared the proteome of H6O5 cells to that of a non-transformed mouse mammary cell line, C127. Our results show upregulation of Tumor protein D52, Creatine kinase, Retinol-binding protein 1 and Thymosin beta 4. Gelsolin 1 and thrombospondin 1 were among the proteins found to be downregulated. Based on a statistical analysis of published microarray data, we show that these proteins may be novel potential biomarkers to predict clinical outcomes of breast cancer patients.
Materials and Methods
Establishment of cell lines from tumors of Her2/neu-transgenic mice
Primary mammary tumors from 6-month old transgenic mice were removed and rinsed 3 times in PBS with 100U/ml penicillin and 100 mg/ml streptomycin. Each tumor was then minced into small pieces (0.5 mm each) with a sterile scalpel, and digested in Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium containing, 10μg/ml human insulin, 1% penicillin/streptomycin and 0.25g/80 ml of collagenase A in a 37°C shaker rocking at 100 rpm for 2-3 hours. The cell suspension was then centrifuged at about 1200 g for 5 min, and the pellet containing the cells was washed with 20ml of DMEM/F12 medium containing 20% FBS twice and cultured in DMEM/F12 medium supplemented with 10% FBS, 10 μg/ml human insulin, 1% penicillin/streptomycin until the cells formed a subconfluent monolayer. A few cell lines derived from independent tumors were obtained. In particular, clone H605 was maintained in culture for 30 passages and well characterized.
Cell growth assays
Cellular growth curves were obtained by plating 1× 105 cells/well on 6-well plates (Falcon) with DMEM/F12 medium supplemented with 10% FBS, 10 μg/ml human insulin, 1% penicillin/streptomycin. The cells were counted on days 1, 3, 5 and 7. The experiment was repeated three times in duplicate. The doubling time was calculated based on the following formula: Doubling time= ln2 / ((ln (A/Ao) / t) Where A = cell numbers at time t; Ao = initial cell number.
Cell cycle analysis
H605 cells (2× 105 cells in 2ml) were seeded into each well of 6-well plates. After 24 h, the cells were trypsinized, washed, resuspended in PBS and fixed in 70% ethanol. Fixed cells were treated with RNAase (0.1 mg/ml) and stained with propidium iodide (40 mg/ml) on ice for one hour and analyzed on a Becton-Dickson FACScan flow cytometer. The data was analyzed using CellQuest software.
In vivo transplantation analysis of tumorigenicity of H6O5 tumor cells
Tumorigenicity in vivo was assessed by injection of 5 × 105 H6O5 cells into number 4 mammary glands of mouse mammary tumor virus (MMTV)-neu transgenic female mice at 4~6 weeks of age. Mammary tumor growth was monitored and measured weekly by a calibrator. The tumor sizes were calculated using the following formula: Volume=1/2 length × height2. Mammary tumors and lung tissue were harvested from mice bearing tumors for about 60 days. Histological examination was performed as described previously [27].
Cell culture
We used a two-state SILAC strategy [28] to compare the whole-cell proteome of Her2/neu-overexpressing mammary epithelial cells with that of a normal mammary epithelial cell line C127 (from ATCC) that does not express Her2/neu. In our experimental design, H6O5 cells were cultured in media containing heavy isotope labeled 13C6 Arg and 13C6 Lys, whereas C127 cells were cultured in medium containing normal light amino acids. A detailed explanation of how the samples were processed is provided as supplementary information [29].
Both cell lines, H6O5 and C127, cells were maintained at 37°C and 5% CO2, in Dulbeccos Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. For large scale proteomics experiments, 15 cm tissue culture-treated plates were used. Five plates were cultured for each cell line, making a total of 10 dishes.
Liquid chromatography and tandem mass spectrometry (LC-MS/MS)
Tandem mass spectrometry analysis of SILAC labeled peptides was carried out on a quadrupole time-of-flight (QSTAR) or an LTQ-Orbitrap XL mass spectrometer. The methods used for each instrument and the strategy for data analysis and interpretation are provided in detail as supplementary information [28, 29].
Quantitative real-time PCR analysis
Quantitative real time RT-PCR analysis was performed to verify the proteomic results. RNA samples were extracted from C127 and H605 cells, normal mouse mammary tissues and primary tumors of MMTV-Her2/neu transgenic mice. Reverse transcription reaction was performed as follows: 1 μg of DNase-treated total RNA, 0.5 μg of anchored oligo(dT)15 primer, and 500 μM dNTPs (New England Biolabs) were heated for 5 min at 65 °C; 1× first strand buffer (Invitrogen), 0.01 M dithiothreitol, and 200 units of Superscript II (Invitrogen) were added, and reverse transcription was carried out, in a 20-μl reaction, for 50 min at 42 °C and terminated by heating for 15 min at 70 °C. To assess for potential contamination of solutions, a control containing all reagents, but devoid of RNA, was included. In addition, a control containing all reagents, except the Superscript II, was included for each sample in order to monitor for possible residual genomic DNA in the RNA preparations.
The quantitative RT-PCR was performed using the fluorescent dye SYBR Green Master Mix following standard protocols on an ABI PRISM 7300 sequence detection system (Applied Biosystems, CA). The data were first analyzed using the Sequence Detector Software SDS 2.0 (Applied Biosystems). Results were calculated and normalized relative to the GAPDH control by using the Microsoft Excel program. The relative expression values were calculated relative to GAPDH by using the 2-ΔCT method [29]. The data shown here represent the average of three independent experiments. T-test was performed to show that there are significant differences in the expression of these tested genes among samples.
In silico clinical data analysis
To access the clinical relevance of identified specific genes, we used the list of 23 proteins from Table 1 to perform a statistical analysis of published microarray data. After translating the signature genes into UnigeneIDs, we extracted the gene expression information from published data sets of breast cancer patients [30, 31] and normalized it by samples. We applied the supervised principle component method [32] for testing the performance of the proposed gene set. The principal component of this data set was extracted and used to derive a regression model to predict the survival time from microarray expressions of our biomarker genes. The samples were then divided into highrisk and low-risk groups by comparing to their median survival time. The p-values were calculated using the log-rank test, and differences were considered statistically significant at p<0.05.
Table 1.
| Gene identification | Protein name | SILAC H/L ratio
|
Real- Time PCR Ratioa | ||
|---|---|---|---|---|---|
| QSTAR | LTQ-Oritrap | ||||
| a | |||||
| gi∣11230802 | Actinin A4 | 3.8 | 3.3 | 1.5 | |
| gi∣7304887 | Annexin A3 | 4.4 | 5.2 | 15.8 | |
| gi∣6174396 | AHNAK | 0.5 | 0.2 | 0.5 | |
| gi∣47059073 | Thrombospondin 1 | 0.3 | 0.2 | 0.8 | |
| gi∣28916693 | Gelsolin 1 | 0.2 | 0.6 | 0.02 | |
| gi∣7305295 | Myosin heavy polypeptide 9 | 0.8 | 0.1 | 0.8 | |
| gi∣50355692 | Lamin A isoform A | 0.9 | 0.2 | 0.6 | |
| gi∣33859624 | S100 A4 | 0.5 | 0.1 | 0.3 | |
| gi∣6755809 | Talin 1 | 0.8 | 0.1 | 0.4 | |
| gi∣10946578 | Thymosin beta 4 X | 6.3 | 9.3 | 2.5 | |
|
|
|||||
| b | |||||
| gi∣31981515 | Ribosomal protein L7 | 0.4 | 0.1 | 0.8 | |
| gi∣6755358 | Ribosomal protein L8 | 0.9 | 0.1 | 0.6 | |
| gi∣33186863 | Ribosomal protein L13a | NDb | 0.1 | 0.9 | |
| gi∣13385044 | Ribosomal protein L35 | 0.8 | 0.2 | 1.8 | |
| gi∣51873060 | Eukaryotic translation elongation factor 1 alpha 1 | 0.7 | 0.2 | 0.4 | |
| gi∣33859482 | Eukaryotic translation elongation factor 2 | 0.8 | 0.2 | 0.2 | |
|
|
|||||
| c | |||||
| gi∣31981562 | Pyruvate kinase | 0.5 | 0.2 | 2.3 | |
| gi∣6753428 | Creatine kinase | ND | 15.8 | 8.9 | |
| gi∣6753036 | Aldehyde dehydrogenase 2 | 0.6 | 0.3 | 0.5 | |
| gi∣6755300 | Retinol-binding protein | >99 | 23.3 | 1112.2 | |
|
|
|||||
| d | |||||
| gi∣6678682 | Galectin 1/3 | 0.3 | 0.3 | 0.9 | |
| gi∣31543113 | Plastin 2 (lymphocyte cytosolic protein 1) | ND | 1.7 | 20.4 | |
| gi∣19526912 | Suppression of tumorigenicity 13 | 1.3 | 0.8 | 0.4 | |
The PCR was performed as described under “Materials and Methods.” The expression level of each gene in the C127 cells was normalized to GAPDH control for each sample. The ratio represents the relative mRNA expression level of each gene in H605 cells compared to C127 cells.
ND, not detected.
Results and Discussion
Cellular characterization of H6O5 cells
The use of a conditionally-induced Her2/neu transgenic mouse model has been used previously in two proteomics-based, biomarker discovery projects [17, 20]. The proteomics strategy in these papers is based on label-free quantitation of protein/peptide ratios [17, 20]. In this study, we used SILAC proteomics to characterize a cell line derived from primary tumors arising in MMTV-Her2/neu transgenic mice. When maintained in culture, H6O5 cells are polygonal and exhibit an epithelial-like morphology (Figure 1a). When cultured in DMEM/F12 medium supplemented with 10% BCS and 10μg/ml human insulin, these cells have a population doubling time of 23 hours (Figure 1b). Cell cycle analysis under these conditions shows that 65.1% of the cells were in G1 phase, 10.4% in the S phase, and 24.1% in G2/M phase (Figure 1c).
Figure 1. In vitro characterization of H605 cells.
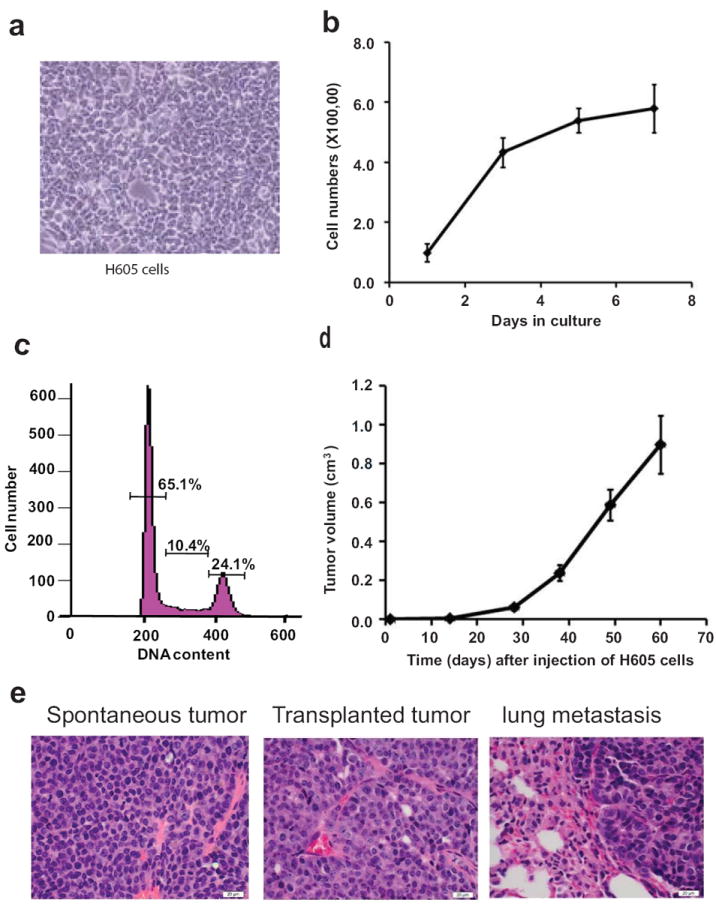
a) The morphology of H605 cells was observed with an inverted microscope (400× magnification). b) To establish a cell growth curve in DMEM/F12 medium, 1× 105 cells were seeded into each well of 6-well plates for culture and were counted at day 1, 3, 5 and 7. c) Cell cycle analysis was performed by flow cytometry in order to determine the percentage of cells at different stages of cell cycle. d) The tumor growth kinetics was observed in syngeneic transplanted animals. H605 cells (5 × 105) were injected into number 4 mammary glands of MMTV-neu 4-6 weeks old transgenic female mice (n=8). Mammary tumors were measured weekly by calibrator. The average tumor sizes are shown in the figure. e) Histopathological analysis of paraffin embedded sections of tissues. Hematoxylin and eosin (H&E) staining was performed on three different samples: spontaneously arising tumors of MMTV-neu transgenic mice; primary tumors arising in the transplanted animals; and metastatic lesion in the lung of transplanted mice.
To investigate whether the malignant potential was still maintained in the H6O5 cell line, 5 × 105 cells were injected into mammary glands of MMTV-Her2/neu transgenic mice. The tumors arose as solid masses that became palpable after about 10 days post-inoculation and grew rapidly (Figure 1d). At 60 days post inoculation, the animals were sacrificed and tumors were harvested. A histopathological analysis revealed that the morphology of the grafted tumors was comparable to that of the primary mammary adenocarcinomas arising in MMTV-neu transgenic mice (Figure 1e). In addition, we found that 6 out of 8 mice developed lung metastases. However, no metastatic lesions were detected in other organs of these transplanted mice including brain, liver or bone. These results are in agreement with previous studies indicating that MMTV-Her2/neu transgenic mice only develop lung metastases [33].
SILAC-based quantitative proteomics of H6O5 cells
We performed a two-state SILAC experiment [28] in which H6O5 cells were labeled by growing in heavy amino acid containing medium and their proteomic profile was compared with that of C127 cells grown in light amino acid containing medium (Figure 2a). A total of 501 unique proteins were identified and quantitated from duplicate experiments. The first sample set was analyzed on a quadruple time-of-flight mass spectrometer (QSTAR) and resulted in identification of 220 proteins. The other sample set was analyzed on an LTQ-Orbitrap XL mass spectrometer and resulted in 415 proteins. By comparing the results obtained from the two instruments we found an overlap of 115 proteins (Figure 2b). Additionally, the common proteins detected in both experiments exhibited significant correlation between the two SILAC ratios (r > 0.7) (Figure 2c). Two supplementary files contain the list of peptides and the corresponding proteins identified with the QSTAR and the LTQ-Orbitrap instruments are provided as supplementary data. Data is also provided about the relative protein levels obtained from their SILAC-derived values. Supporting the notion that equal amounts of C127 and H6O5 cells were mixed proportionately for proteomic analysis, we found that the large majority of proteins had H/L ratios ranging from 0.5 to 2.0 (Figure 2c and supplementary combined list of proteins and ratios).
Figure 2. Experimental strategy for SILAC-basedproteomics.
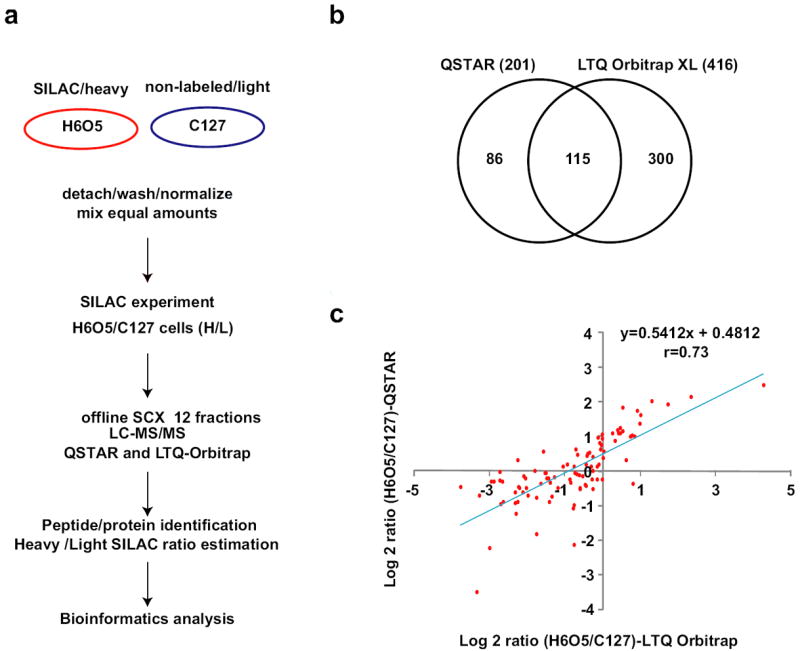
a) Proteomics workflow. This scheme summarizes the experimental strategy described in the Results and Discussion section; b) Total amount of proteins identified. This diagram shows the number of proteins identified by each MS/MS instrument used, and the corresponding overlapping results; c) SILAC ratios obtained by different MS/MS instruments. The peptide/protein ratios obtained by both instruments correlated significantly (r > 0.7).
Functional classification of the proteins identified
We next classified the identified proteins based on available gene ontology and protein function databases. For this we used the PubMed database from the National Center for Biotechnology Information (NCBI) and several bioinformatics and proteomics resources, including the Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov/), the Universal Protein Resource (UniProt) (http://www.uniprot.org), the Expert Protein Analysis System (EXPASY) Proteomics Server (http://ca.expasy.org), and the Gene Ontology project (http://www.geneontology.org/index.shtml). The proteins identified by proteomic analysis were classified into 8 categories (supplementary protein classification file): 1) membrane and cytoskeleton; 2) metabolism; 3) gene expression; 4) ribosome; 5) ATP hydrolysis and chaperone; 6) redox homeostasis; 7) proteasome and proteases; 8) signaling cascades; and 9) undetermined function (protein classification file and supplementary figure 1).
To illustrate the quality of the protein identifications reported, we present the MS and MS/MS spectra of 4 selected proteins from the data obtained from the LTQ-Orbitrap mass spectrometer (Figure 3). We used a 3-fold change in heavy/light (H/L) SILAC ratios as a cut-off to define that a protein had a significant SILAC ratio. From the annotated function classification and the SILAC ratios obtained, we selected a list of relevant proteins with respect to tumor biology, most of which had significant changes in SILAC ratios on at least one of the instruments (Table 1). The protein profile obtained aftermanual curation of the data (Supplementary Figure 1), and in particular Table 1, explains the ability of H6O5 cells to form solid tumors when grafted onto mice (Figure 1d). In brief, H6O5 cells were found to overexpress Thymosin beta 4, an actin-binding protein previously described as having a role in angiogenesis [34, 35], while having low protein levels of thrombospondin 1, an angiogenesis inhibitor, that binds to proteins on the cell surface, thereby modulating cell motility and cell adhesion events [36, 37].
Figure 3. Representative peptide MS/MS fragmentation pattern and H/L precursor ion ratio spectrum.
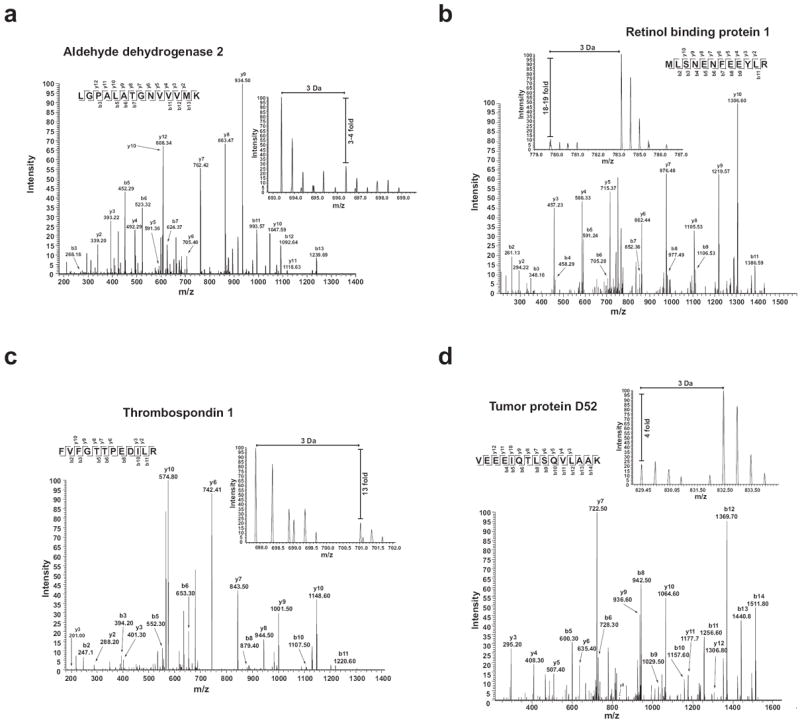
The representative MS/MS and MS spectra from 6 proteins are shown. a) Aldehyde dehydrogenase 2; b) Retinol binding protein 1; c) Thrombospondin 1 and d) Tumor protein D52. In each panel, the inset shows the relative ratio of Heavy to Light versions of each precursor ion.
We analyzed the SILAC sample on a QTOF and an LTQ-Orbitrap mass spectrometer in order to obtain a pair of technical replicates obtained. Despite the overall good correlation in the SILAC ratios derived from the 2 different instruments (r > 0.7), we found some variability in the values obtained for the proteins that showed a significant SILAC-ratio change (Table 1). Such variability was more pronounced in those ratios with pronounced SILAC changes, such as retinol-binding protein 1 (Table 1) and was minimal in those proteins with a 1:1 ratio, explaining why the overall correlation in SILAC values obtained from two different instruments was good.
An important aspect of our results is that the patterns of down-regulation or up-regulation in the proteins identified by the two instruments remained constant, despite the differences in the relative SILAC ratios (Table 1). Furthermore, the variability observed is not unusual, and rather highlights the importance validating the relative quantitative values obtained in discovery-based shot-gun proteomics experiments, such as the ones reported here.
Identification of novel biomarkers
To corroborate our quantitative proteomic results, we confirmed all proteins listed in Table 1 at the mRNA level by quantitative RT-PCR in H6O5 and C127 cells. The relative mRNA expression levels in H605 cells versus C127 cells are listed in Table 1 and several PCR products were run on a 1% agarose gel as shown in Figure 4a. Estimating mRNA levels by RT-PCR is a straightforward strategy in comparison to the Western blot-based validation of proteins for which in many cases, a specific antibody is not available. Our results show that > 90% of the mRNA values (21 out of 23 genes) correlated with the SILAC-based protein ratios.
Figure 4. RT-PCR analyses of selected genes in H6O5 and C127 cell lines and primary tumors.
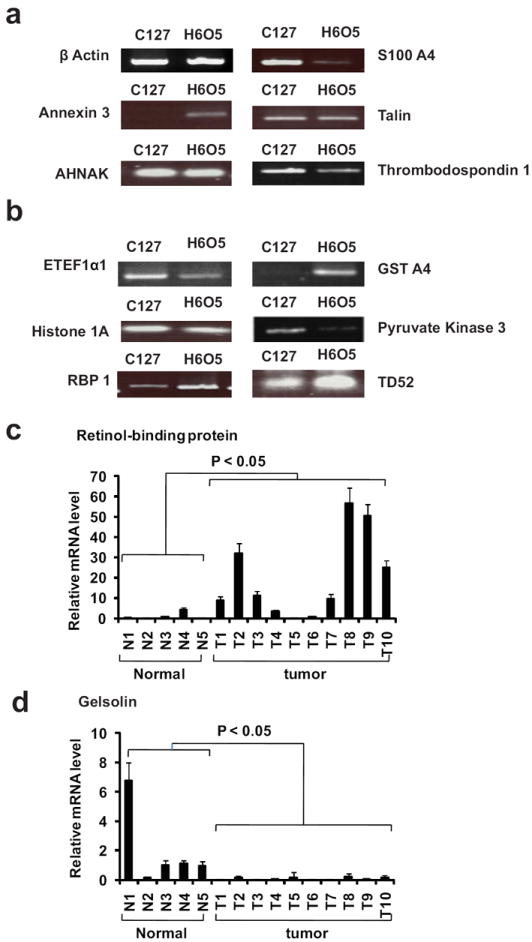
The RT-PCR results for mRNA levels of a selected list of proteins are presented. The RT-PCR analysis was performed on RNA samples isolated from H6O5 and C127 cells. a) Genes coding for membrane and cytoskeleton-associated proteins. b) Genes coding for intracellular proteins. c) Relative mRNA expression levels of Retinol-binding protein (RBP) in normal mammary gland and Her2/neu-induced primary tumors. Quantitative real-time RT-PCR analysis was performed on RNA samples from a panel of normal mammary glands (N1-N6) and Her2/neu-induced primary mammary tumors (T1-T10).. d) Relative mRNA expression levels of Gelsolin in normal mammary gland and Her2/neu-induced primary tumors as described in above.
An exception was in the case of Pyruvate kinase and Ribosomal protein L35 that were found to be down-regulated in our SILAC experiments, but upregulated at mRNA level in H605 cells by quantitative PCR analysis. A simple explanation to this discrepancy in our results would be incorrect assignment of peptides to these two proteins. At least in the case of pyruvate kinase, this is highly unlikely, because its identification score and the number of peptides observed were amongst the highest. Another possibility is that protein levels are indeed lower due to degradation via proteases or ubiquitin-mediated protein turnover. Thus, it is possible that HER2 overexpressed in H6O5 cells induces the degradation of certain proteins such as L35 and pyruvate kinase. Regardless of the explanation to this discrepancy, the occurrence of disparate relative values in mRNA and protein levels is not surprising, but rather reflects the intricate post-transcriptional regulation path followed by an mRNA transcript before it gets translated to produce a protein.
Next, we selected two functionally relevant candidate genes, gelsolin 1, which was downregulated and retinol-binding protein 1, which was upregulated, to further validate their expression patterns in primary tumors. Gelsolin 1, an actin-binding protein regulated by calcium, is commonly downregulated in invasive breast carcinoma and is thus a negative prognostic factor in HER-2-positive epidermal growth factor receptor-positive breast cancers [38, 39]. Furthermore, an extracellular form of this protein has been identified as downregulated in conditioned medium of pancreatic cancer cell cultures [40]. Cellular retinol-binding protein 1, on the other hand, regulates the bioavailability of retinol/vitamin A by preventing the action of retinol degrading enzymes[41]. A study in endometrial carcinoma has shown that the levels of retinolbinding protein 1 inversely correlated with tumor grade progression[41].
We used quantitative real-time RT-PCR assay to determine the relative expression levels of gelsolin 1 and retinol-binding protein 1 in normal mammary gland tissues and Her2/neu-induced primary tumors (Figure 4c and 4d). Consistent with the quantitative proteomic profile, gelsolin expression was significantly downregulated in tumors while retinol-binding protein 1 expression was upregualted (student’s t test, p value < 0.05).
To further evaluate the clinical relevance of the identified proteins, we used the list of 23 proteins from Table 1 to perform a statistical analysis of published microarray data [30, 31]. Using UnigeneIDs of the corresponding genes, we extracted the gene expression information from published data sets of breast cancer patients [30, 31]. We applied a supervised principle component method to test the performance of this set of genes [26]. The principal component of this data set was extracted and used to derive a regression model to predict the survival time from expression levels of this set of genes in the microarray dataset. The samples were then divided into high-risk and low-risk groups by comparing their median survival time. Our analysis revealed that the 23-gene mRNA expression signature could be used to predict the probability of metastasis-free survival in breast cancer patients with statistical confidence (Figure 5). The statistical analysis using two microarray datasets from two independent groups showed very similar results, indicating that the quantitative protein profiling that we present here has clinical relevance and could be used to develop a novel biomarker signature in HER2/neu-positive breast cancer.
Figure 5. Prediction of clinical outcome based on a 23-gene signature.
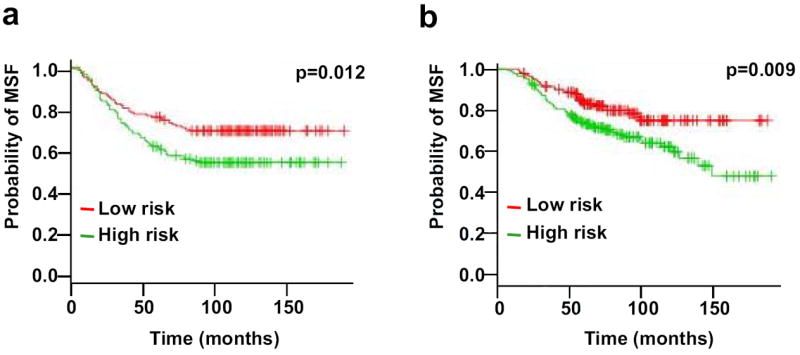
Kaplan-Meier analysis was performed to predict the probability of metastasis-free survival using published microarray datasets. a) Microarray dataset from Wang group [30]. This data set includes 286 lymph node-negative breast cancer patients who received no adjuvant treatment when clinical samples were collected. b) Microarray dataset from van de Vijver group [31]. A total of 295 patients had stage I or II breast cancer and were younger than 53 years old; 151 had lymph-node-negative disease, and 144 had lymphnode- positive disease. X axis represents time of survival. Y axis represents the probability of metastasis-free survival. MFS, metastasis-free survival
In this study, we have used an integrative proteomic and genomic analysis to discover novel biomarkers in HER2/neu-positive breast cancer. HER2/neu-positive breast cancer is generally associated with particular aggressiveness, tumor recurrence, enhanced metastasis, induced chemotherapy resistance and worse prognosis [42]. Both genomic and proteomic approaches have been used to profile the HER2-positive breast cancers and cell lines with the common goal to identify novel biomarkers. The use of a conditionally-induced Her2/neu transgenic mouse model has been used previously in two proteomics-based, biomarker discovery projects [13, 17]. The proteomics strategy, while different in these reports, is based on label-free quantitation of protein/peptide ratios [18]. In the first case a novel statistical algorithm was applied to identify proteins with a relevant protein ratio that were further validated by multiple reaction monitoring (MRM) targeted-mass spectrometry [17]. The most recent of these reports follows up on the aforementioned publication, but uses a custom-built database to identify novel splicing isoforms with a potential as tumor biomarkers [18]. Overall, these and other proteomic strategies offer unique and rather complimentary avenues for biomarker discovery in mammary tumors [13, 17-19].
Several studies have reported the mRNA expression profiles of Her2-positive tumors from microarray analysis [43, 44]. However, validation of the long lists of biomarker candidates identified using either proteomic or genomic approaches is challenging. Conventional validation assays such as RT-PCR, ELISA and Western blots are prohibitively resource and time-intensive [45]. To relieve the bottleneck between the discovery and validation phrase, efforts are being made to integrate both proteomic and genomic platforms to discover biomarkers [24, 46]. In line with these efforts, we first used a SILAC proteomic approach to identify differentially expressed proteins in Her2/neu-positive cells, followed by validation at the mRNA level and in silico analysis of published microarray data. This integrative strategy allowed us to identify a 23-gene signature for prediction of clinical outcome of breast cancers. This study demonstrates how the integration of proteomic and genomic gene expression data may prove useful in accelerating cancer biomarker discovery.
Conclusions remarks
In this report, we describe the cellular and proteomic characterization of H6O5, which is a Her2/neu positive cell line derived from a spontaneous tumor arising in Her2/neu transgenic mice. Upon confirming that H6O5 cells are tumorigenic when grafted onto mice, we used SILAC quantitative proteomics to establish a protein signature of these cells by manual curation of the literature and gene ontology databases. We found that the protein profile obtained was consistent with the tumorigenic phenotype of H6O5 cells. We next validated mRNA expression patterns of a short list of functionally relevant proteins from Table 1. We performed this analysis in the two cell lines, H6O5 and C127; and additionally in primary Her2/neu mammary tumors. Further statistical analysis of published cancer microarray datasets indicated that the mRNA expression pattern of a 23-gene signature correlates with clinical outcome in breast cancer patients. We therefore conclude that these proteins may serve as novel biomarkers in breast cancer patients.
Supplementary Material
Acknowledgments
This work was in part supported by the Elsa U. Pardee Cancer Foundation grant (B94AFFAA), the American Cancer Society Research Award (RSG-10-067-01-TBE) and NIH grant (3P20RR017698-08) to HC. This project was funded in part by a grant from the NIH Roadmap initiative U54 RR020839 (AP), a contract N01-HV-28180 from the National Heart Lung and Blood Institute (AP) and a Department of Defense Era of Hope Scholar award (W81XWH-06-1-0428) to AP.
Non-standard abbreviations
- 2D
two-dimensional
- CSC
class-specific correlations
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- FBS
fetal bovine serum
- H
heavy
- H and E
hematoxylin and eosin
- L
light
- LC
liquid chromatography
- MFS
metastasis-free survival
- MMTV
mouse mammary tumor virus
- MS/MS
tandem mass-spectrometry
- PCR
polymerase chain reaction
- QTOF
quadrupole time of flight
- RP
reverse phase
- rpm
revolutions per minute
- RT
reverse transcription
- RTK
receptor tyrosine kinase
- SCX
strong cation exchange
- SILAC
stable isotope labeling of amino acids in cell culture
Footnotes
The authors have declared no conflict interests.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 3.Hondermarck H. Breast cancer: when proteomics challenges biological complexity. Mol Cell Proteomics. 2003;2:281–291. doi: 10.1074/mcp.R300003-MCP200. [DOI] [PubMed] [Google Scholar]
- 4.Sorlie T, Perou CM, Tibshirani R, Aas T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho HS, Mason K, Ramyar KX, Stanley AM, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 6.Garrett TP, McKern NM, Lou M, Elleman TC, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 7.Kim IY, Yong HY, Kang KW, Moon A. Overexpression of ErbB2 induces invasion of MCF10A human breast epithelial cells via MMP-9. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–6130. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertucci F, Finetti P, Cervera N, Esterni B, et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123:236–240. doi: 10.1002/ijc.23518. [DOI] [PubMed] [Google Scholar]
- 10.Hegde PS, Rusnak D, Bertiaux M, Alligood K, et al. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther. 2007;6:1629–1640. doi: 10.1158/1535-7163.MCT-05-0399. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Groth J, Sossey-Alaoui K, Hawthorn L, et al. Aberrant expression of novel and previously described cell membrane markers in human breast cancer cell lines and tumors. Clin Cancer Res. 2005;11:4357–4364. doi: 10.1158/1078-0432.CCR-04-2107. [DOI] [PubMed] [Google Scholar]
- 12.Wilson KS, Roberts H, Leek R, Harris AL, Geradts J. Differential gene expression patterns in HER2/neu-positive and -negative breast cancer cell lines and tissues. The American journal of pathology. 2002;161:1171–1185. doi: 10.1016/S0002-9440(10)64394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rauser S, Marquardt C, Balluff B, Deininger SO, et al. Classification of HER2 receptor status in breast cancer tissues by MALDI imaging mass spectrometry. Journal of proteome research. 9:1854–1863. doi: 10.1021/pr901008d. [DOI] [PubMed] [Google Scholar]
- 14.Somiari RI, Sullivan A, Russell S, Somiari S, et al. High-throughput proteomic analysis of human infiltrating ductal carcinoma of the breast. Proteomics. 2003;3:1863–1873. doi: 10.1002/pmic.200300560. [DOI] [PubMed] [Google Scholar]
- 15.Huang HL, Stasyk T, Morandell S, Dieplinger H, et al. Biomarker discovery in breast cancer serum using 2-D differential gel electrophoresis/ MALDI-TOF/TOF and data validation by routine clinical assays. Electrophoresis. 2006;27:1641–1650. doi: 10.1002/elps.200500857. [DOI] [PubMed] [Google Scholar]
- 16.Toillon RA, Lagadec C, Page A, Chopin V, et al. Proteomics demonstration that normal breast epithelial cells can induce apoptosis of breast cancer cells through insulin-like growth factor-binding protein-3 and maspin. Mol Cell Proteomics. 2007;6:1239–1247. doi: 10.1074/mcp.M600477-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Whiteaker JR, Zhang H, Zhao L, Wang P, et al. Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. Journal of proteome research. 2007;6:3962–3975. doi: 10.1021/pr070202v. [DOI] [PubMed] [Google Scholar]
- 18.Menon R, Omenn GS. Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer research. 70:3440–3449. doi: 10.1158/0008-5472.CAN-09-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz DM, Bollner C, Thomas G, Atkinson M, et al. Identification of differentially expressed proteins in triple-negative breast carcinomas using DIGE and mass spectrometry. Journal of proteome research. 2009;8:3430–3438. doi: 10.1021/pr900071h. [DOI] [PubMed] [Google Scholar]
- 20.Adam PJ, Boyd R, Tyson KL, Fletcher GC, et al. Comprehensive proteomic analysis of breast cancer cell membranes reveals unique proteins with potential roles in clinical cancer. J Biol Chem. 2003;278:6482–6489. doi: 10.1074/jbc.M210184200. [DOI] [PubMed] [Google Scholar]
- 21.Chan HL, Gharbi S, Gaffney PR, Cramer R, et al. Proteomic analysis of redox- and ErbB2-dependent changes in mammary luminal epithelial cells using cysteine- and lysine-labelling two-dimensional difference gel electrophoresis. Proteomics. 2005;5:2908–2926. doi: 10.1002/pmic.200401300. [DOI] [PubMed] [Google Scholar]
- 22.Li DQ, Wang L, Fei F, Hou YF, et al. Identification of breast cancer metastasis-associated proteins in an isogenic tumor metastasis model using two-dimensional gel electrophoresis and liquid chromatography-ion trap-mass spectrometry. Proteomics. 2006;6:3352–3368. doi: 10.1002/pmic.200500617. [DOI] [PubMed] [Google Scholar]
- 23.Malorni L, Cacace G, Cuccurullo M, Pocsfalvi G, et al. Proteomic analysis of MCF-7 breast cancer cell line exposed to mitogenic concentration of 17beta-estradiol. Proteomics. 2006;6:5973–5982. doi: 10.1002/pmic.200600333. [DOI] [PubMed] [Google Scholar]
- 24.Ou K, Yu K, Kesuma D, Hooi M, et al. Novel breast cancer biomarkers identified by integrative proteomic and gene expression mapping. Journal of proteome research. 2008;7:1518–1528. doi: 10.1021/pr700820g. [DOI] [PubMed] [Google Scholar]
- 25.Sarvaiya HA, Yoon JH, Lazar IM. Proteome profile of the MCF7 cancer cell line: a mass spectrometric evaluation. Rapid Commun Mass Spectrom. 2006;20:3039–3055. doi: 10.1002/rcm.2677. [DOI] [PubMed] [Google Scholar]
- 26.Zhang D, Tai LK, Wong LL, Chiu LL, et al. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER- 2/neu-positive breast cancer. Mol Cell Proteomics. 2005;4:1686–1696. doi: 10.1074/mcp.M400221-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Chen H, Lee JS, Liang X, Zhang H, et al. Hoxb7 inhibits transgenic HER-2/neu-induced mouse mammary tumor onset but promotes progression and lung metastasis. Cancer research. 2008;68:3637–3644. doi: 10.1158/0008-5472.CAN-07-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harsha HC, Molina H, Pandey A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat Protoc. 2008;3:505–516. doi: 10.1038/nprot.2008.2. [DOI] [PubMed] [Google Scholar]
- 29.Chaerkady R, Harsha HC, Nalli A, Gucek M, et al. A quantitative proteomic approach for identification of potential biomarkers in hepatocellular carcinoma. Journal of proteome research. 2008;7:4289–4298. doi: 10.1021/pr800197z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 31.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, et al. A gene-expression signature as a predictor of survival in breast cancer. The New England journal of medicine. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 32.Bair E, Hastie T, Paul T, Tibshirani R. Prediction by Supervised Principal Components. J Am Stat Assoc. 2006;101:119–137. [Google Scholar]
- 33.Guy CT, Webster MA, Schaller M, Parsons TJ, et al. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huff T, Rosorius O, Otto AM, Muller CS, et al. Nuclear localisation of the G-actin sequestering peptide thymosin beta4. J Cell Sci. 2004;117:5333–5341. doi: 10.1242/jcs.01404. [DOI] [PubMed] [Google Scholar]
- 35.Philp D, Huff T, Gho YS, Hannappel E, Kleinman HK. The actin binding site on thymosin beta4 promotes angiogenesis. Faseb J. 2003;17:2103–2105. doi: 10.1096/fj.03-0121fje. [DOI] [PubMed] [Google Scholar]
- 36.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 37.Wen XF, Yang G, Mao W, Thornton A, et al. HER2 signaling modulates the equilibrium between pro- and antiangiogenic factors via distinct pathways: implications for HER2-targeted antibody therapy. Oncogene. 2006;25:6986–6996. doi: 10.1038/sj.onc.1209685. [DOI] [PubMed] [Google Scholar]
- 38.Walsh N, Dowling P, O’Donovan N, Henry M, et al. Aldehyde dehydrogenase 1A1 and gelsolin identified as novel invasion-modulating factors in conditioned medium of pancreatic cancer cells. J Proteomics. 2008;71:561–571. doi: 10.1016/j.jprot.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Thor AD, Edgerton SM, Liu S, Moore DH, 2nd, Kwiatkowski DJ. Gelsolin as a negative prognostic factor and effector of motility in erbB-2-positive epidermal growth factor receptor-positive breast cancers. Clin Cancer Res. 2001;7:2415–2424. [PubMed] [Google Scholar]
- 40.Orlandi A, Ferlosio A, Ciucci A, Francesconi A, et al. Cellular retinol binding protein-1 expression in endometrial hyperplasia and carcinoma: diagnostic and possible therapeutic implications. Mod Pathol. 2006;19:797–803. doi: 10.1038/modpathol.3800586. [DOI] [PubMed] [Google Scholar]
- 41.Schmitt-Graeff A, Koeninger A, Olschewski M, Haxelmans S, et al. The Ki67+ proliferation index correlates with increased cellular retinol-binding protein-1 and the coordinated loss of plakophilin-1 and desmoplakin during progression of cervical squamous lesions. Histopathology. 2007;51:87–97. doi: 10.1111/j.1365-2559.2007.02724.x. [DOI] [PubMed] [Google Scholar]
- 42.Tagliabue E, Balsari A, Campiglio M, Pupa SM. HER2 as a target for breast cancer therapy. Expert opinion on biological therapy. 10:711–724. doi: 10.1517/14712591003689972. [DOI] [PubMed] [Google Scholar]
- 43.Landis MD, Seachrist DD, Montanez-Wiscovich ME, Danielpour D, Keri RA. Gene expression profiling of cancer progression reveals intrinsic regulation of transforming growth factor-beta signaling in ErbB2/Neu-induced tumors from transgenic mice. Oncogene. 2005;24:5173–5190. doi: 10.1038/sj.onc.1208712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Astolfi A, Landuzzi L, Nicoletti G, De Giovanni C, et al. Gene expression analysis of immune-mediated arrest of tumorigenesis in a transgenic mouse model of HER-2/neu-positive basal-like mammary carcinoma. The American journal of pathology. 2005;166:1205–1216. doi: 10.1016/S0002-9440(10)62339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solassol J, Jacot W, Lhermitte L, Boulle N, et al. Clinical proteomics and mass spectrometry profiling for cancer detection. Expert Rev Proteomics. 2006;3:311–320. doi: 10.1586/14789450.3.3.311. [DOI] [PubMed] [Google Scholar]
- 46.Sigdel TK, Sarwal MM. The proteogenomic path towards biomarker discovery. Pediatric transplantation. 2008;12:737–747. doi: 10.1111/j.1399-3046.2008.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.