

Abstract
Smoking is a major risk factor for diabetes and cardiovascular disease and may contribute to non-alcoholic fatty liver disease (NAFLD). The health risk associated with smoking is exaggerated by obesity and is the leading causes of morbidity and mortality worldwide. We recently demonstrated that combined treatment with nicotine and a high-fat diet (HFD) triggers greater oxidative stress, activates hepatocellular apoptosis, and exacerbates HFD-induced hepatic steatosis. Given that hepatocellular apoptosis plays a pivotal role in the pathogenesis of NAFLD, using this model of exacerbated hepatic steatosis, we elucidated the signal transduction pathways involved in HFD plus nicotine-induced liver cell death. Adult C57BL6 male mice were fed a normal chow diet or HFD with 60% of calories derived from fat and received twice daily IP injections of 0.75 mg/kg BW of nicotine or saline for 10 weeks. High resolution light microscopy revealed markedly higher lipid accumulation in hepatocytes from mice received HFD plus nicotine, compared to mice on HFD alone. Addition of nicotine to HFD further resulted in an increase in the incidence of hepatocellular apoptosis and was associated with activation of caspase 2, induction of inducible nitric oxide synthase (iNOS), and perturbation of the BAX/BCL-2 ratio. Together, our data indicate the involvement of caspase 2 and iNOS –mediated apoptotic signaling in nicotine plus HFD-induced hepatocellular apoptosis. Targeting the caspase 2-mediated death pathway may have a protective role in development and progression of NAFLD.
Keywords: Nicotine, high-fat diet, oxidative stress, caspase 2, iNOS, apoptosis, hepatic steatosis
Introduction
Cigarette smoking is the leading preventable cause of death and disability from various diseases worldwide [1]. Smoking is a major risk factor for cardiovascular disease, chronic obstructive pulmonary disease and lung cancer [2–4]. There is increasing evidence that smoking also contributes to nonalcoholic fatty liver disease (NAFLD) [5–8]. Importantly, the health risk associated with smoking is exaggerated by obesity, and smoking and obesity are the leading causes of morbidity and mortality worldwide [9, 10].
NAFLD is the most common form of liver pathologies and includes the whole spectrum of fatty liver, ranging from simple steatosis to nonalcoholic steatohepatitis (NASH), which can progress to liver cirrhosis and hepatocellular carcinoma [11, 12]. Oxidative stress coupled with hepatocellular apoptosis is believed to play a pivotal role in pathogenesis of NAFLD [12–14]. In fact, emerging data now show that hepatocellular apoptosis is a prominent feature in patients with NAFLD and NASH and correlates with disease severity [15, 16]. In a recent study, we have shown that nicotine in combination with high-fat diet (HFD) triggers greater oxidative stress, activates hepatocellular apoptosis, and amplifies HFD-induced hepatic steatosis [17]. The additive effects of nicotine on hepatocellular apoptosis was further associated with activation of c-Jun-NH2-terminal kinase (JNK) and downstream caspases 9 and 3 [17]. However, we do not know what triggers JNK activation or the signaling events that link JNK activation to downstream caspase activation leading to hepatocellular apoptosis.
Caspase 2 plays an important role in inducing apoptosis in various cell systems working upstream of JNK activation and mitochondria-dependent apoptotic pathway, characterized by perturbation of BAX/BCL2 ratio and activation of the initiator caspase 9 [18–21]. Of importance, oxidative stress can trigger caspase 2 activation [18, 20, 21]. Nitric oxide (NO) production through up-regulation of inducible nitric oxide synthase (iNOS) can also be a potential target of JNK signaling [22, 23]. Increased nitric oxide (NO) production, through upregulation of iNOS, has also been implicated in cellular injury and apoptosis in a variety of cell systems, including hepatocytes [22, 23, 24]. One possible mechanism by which NO can induce apoptosis is through perturbation of the BAX/BCL2 rheostat and the subsequent activation of the mitochondria-dependent death pathway [22, 24]. We, therefore, hypothesize that nicotine in combination with a HFD induces hepatocellular apoptosis through activation of caspase 2 and iNOS mediated apoptotic signaling. To this end, we fed C57BL/6J mice a HFD deriving 60% of calories from fat in the presence of nicotine for 10 weeks to induce hepatocellular apoptosis and hepatic steatosis [17].
Materials and methods
Animals and experimental protocol
Male 10-week old C57BL/6 mice weighing 22–24 g obtained from Taconic Farms (Germantown, NY, USA) were used for all experiments. Mice were housed (2–4 per cage) under controlled temperature (22°C) and photoperiod (12-h light and 12-h dark cycle) with free access to water and food. Groups of five mice were fed either a normal chow diet (NCD) with 5% fat (2.03 kcal/gm; laboratory rodent diet no. 5001; Lab diet, Richmond, IN) or HFD with 60% of calories derived from fat consisting of 26.2% protein, 26.3% carbohydrate, and 34.9% fat, mostly lard (5.24 kcal/g; D12492; Research Diets, New Brunswick, NJ) for 10 weeks as described previously [17]. Mice on either diet (n=5) received twice daily IP injections of nicotine (0.75 mg/kg BW) or saline for 10 weeks. Mice were fasted overnight before euthanasia with a lethal injection of sodium pentobarbital (200 mg/kg BW). Livers were removed and were either fixed in 2.5% glutaraldehyde for high-resolution light microscopy or 4% formalin for routine histological and immunohistochemical or immunofluorescence studies. Animal handling and experimentation were in accordance with the recommendation of the American Veterinary Medical Association and were approved by the Charles R. Drew University School of Medicine and Science Institutional Animal Care and Use Committee (IACUC).
Liver pathology
Liver pathology was evaluated by high-resolution light microscopy using glutaraldehyde fixed, osmium tetroxide post-fixed, epoxy embedded, and toluidine blue stained sections as described before [17, 25].
Assessment of apoptosis
In situ detection of cells with DNA strand breaks was performed in formalin-fixed, paraffin-embedded liver sections by the terminal deoxynucleotidyl transferase (TdT)-mediated deoxy-UTP nick end labeling (TUNEL) technique [17, 25] using an ApopTag-peroxidase kit (Chemicon International, Inc., San Francisco, CA, USA). The rate of hepatocellular apoptosis was expressed as the percentage of the TUNEL-positive apoptotic nuclei per total (apoptotic plus non-apoptotic) nuclei present within a reference area of 62,500 μm2 [17, 25].
Immunohistochemical and immunofluorescence analyses
Formalin fixed, paraffin-embedded liver sections were immunostained as described previously [17, 25]. Given that nicotine treatment neither affect liver morphology nor alter the incidence of hepatocellular apoptosis in mice feed with NCD [17], we only used liver sections from NCD-fed mice without nicotine as controls. Primary antibodies included rabbit polyclonal iNOS (1:100) from BD Transduction Laboratories, San Diego, CA, active caspase 2 (1:50), BAX (1:200), and BCL-2 (1:200) from Santa Cruz Biotechnology, Santa Cruz, CA. Immunoreactivity was detected using biotinylated anti-rabbit IgG secondary antibody followed by avidin-biotinylated horseradish peroxidase complex, and visualized with diaminobenzidine tetrahydrochloride (DAB) as per the manufacturer’s instructions (VECTASTAIN Elite ABC Rabbit IgG kit, Burlingame, CA). Slides were counterstained with hematoxylin. Negative controls were run for every assay and processed in an identical manner, except the primary antibody was substituted by the rabbit IgG. Enumeration of active caspase 2-postive hepatic cells was carried out in liver sections using an American Optical Microscope with an X40 objective and a pair of X10 eyepieces. A square grid fitted within one eyepiece provided a reference of 62,500 μm2. In certain cases, immunoreactivity was quantified by computerized densitometry using the ImagePro Plus, version 5.1 software (Media Cybernetics, Silver Spring, MD) coupled to an Olympus BHS microscope equipped with a VCC video camera as described previously [25].
Induction of iNOS in hepatic cells undergoing apoptosis was detected by fluorescence microscopy using double immunostaining as previously described [17, 25]. In brief, after fluorescein staining for TUNEL, slides were incubated in a humidified chamber for overnight at 4°C with a rabbit polyclonal iNOS (1:100) antibody followed by goat-anti-rabbit Texas Red-labeled secondary antibody for 45 min at room temperature, washed and mounted in ProLong Antifade (Molecular Probes). For controls, sections were treated only with secondary antibody, and no signals were detected. Sections were viewed with a Zeiss-Axioscop fluorescence microscope.
Western blotting
Western blotting was performed using liver lysates as described previously [17, 25, 26]. In brief, proteins (50–80 μg) were separated on a 4–12% SDS-polyacrylamide gel with 2-(N-morpholine) ethane sulfonic acid or 3 (N-morpholino) propane sulfonic acid buffer purchased from Invitrogen (Carlsbad, CA, USA) at 200V. Gel was transferred on an Immuno-blot PVDF Membrane (Bio-Rad, Hercules, CA, USA) overnight at 4°C. Membranes were blocked in blocking solution (0.3% Tween 20 in Tris-buffered saline and 10% nonfat dry milk) for 1 h at room temperature then probed using rabbit polyclonal active caspase 2 (1:200), BAX (1:200), and BCL-2 (1:200) antibodies from Santa Cruz Biotechnology for 1 h at room temperature or overnight at 4°C with constant shaking. Following 3 X 10-min washes in TBS-T buffer, membranes were then incubated in anti-rabbit IgG secondary antibody (Amersham Biosciences, Piscataway, NJ, USA) at a 1:2000 dilution. For immunodetection, membranes were washed three times in TBS-T wash buffer, incubated with ECL solutions per manufacturer’s specifications (Amersham Biosciences), and exposed to Hyper film ECL. The membranes were stripped and re-probed with a rabbit polyclonal GAPDH (1:2000) from Millipore, Billerica, MA, for normalization of the loading. Band intensities were determined using Quantity One software from Bio-Rad.
Statistical analysis
Statistical analyses were performed using the SigmaStat 2.0 Program (Jandel Corporation, San Rafael, CA, USA). Data were presented as mean ± SEM. We used one-way Analysis of Variance (ANOVA) to assess the statistically significant difference among various treatment groups. If overall ANOVA revealed significant differences, post-hoc (pairwise) comparisons were performed using Tukey’s tests. Differences were considered significant if p< 0.05.
Results
Nicotine exacerbates HFD-induced hepatic steatosis and induces hepatocellular apoptosis in obese mice
We recently reported on the effects of nicotine plus HFD on body weight and food intake [26]. There was no difference in body weight in mice on a NCD with or without nicotine. Mice fed with HFD exhibited a progressive increase in body weight relative to mice fed with NCD in the absence or presence of nicotine. By 10 weeks of combined treatment with nicotine and HFD, mean body weight was reduced by 17.6% relative to mice fed with HFD alone. This reduction in body in the combined treatment group was also associated with reduced food intake. In the current study, we examined the effect on nicotine plus HFD on hepatic steatosis and hepatocellular apoptosis. High-resolution light microscopy using glutaraldehyde fixed, osmium tetroxide post-fixed, epoxy embedded, and toluidine blue stained liver sections from mice fed NCD in the absence (Fig. 1a) or presence (Fig. 1b) of nicotine exhibited normal morphology. Mice fed a HFD showed accumulation of many smaller lipid droplets (Fig. 1c). Notably, addition of nicotine to HFD caused a striking increase in larger lipid droplets (Fig. 1d) compared to those from mice fed HFD and saline (Fig. 1c). We next examined the additive effects of nicotine and HFD on liver cell apoptosis. Compared with mice fed NCD with or without nicotine (Fig. 1e, panel I), or HFD (Fig. 1e, panel II) where little or no apoptosis is detected, combined treatment with nicotine and HFD resulted in a marked increase in the incidence apoptosis (Fig. 1e, panel III). As shown in Fig. 1f, a very low incidence, expressed as the percentage of TUNEL-positive nuclei per total (apoptotic plus non-apoptotic) nuclei, of apoptosis was noted in mice on a NCD in the presence or absence of nicotine. While HFD alone resulted a modest but significant (p<0.05) increase in the incidence of hepatocellular apoptosis, HFD plus nicotine induced a further significant (p<0.05) increase in the incidence of hepatocellular apoptosis.
Fig. 1.
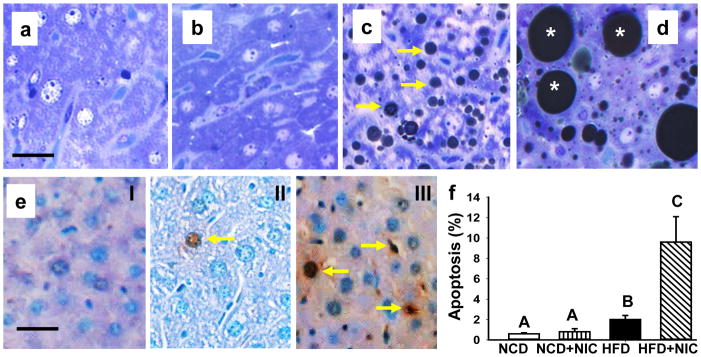
Representative light microscopic images of glutaraldehyde-fixed, osmium tetroxide post-fixed, epoxy-embedded, and toluidine-blue stained liver sections from mice fed with NCD in the absence (a) or presence (b) of nicotine or HFD without (c) or with (d) nicotine. Note a marked increase in lipid accumulation of varying sizes in the liver after combined treatments with nicotine and HFD (d). e. In situ detection of hepatocyte apoptosis detected by TUNEL in control mice fed NCD (panel I), HFD in the absence (panel II) or presence (arrow; panel III) of nicotine. Scale bar = 20 μm. f. Quantitation of hepatocellular apoptosis. Apoptotic rate was expressed as the percentage of TUNEL-positive nuclei per total nuclei (apoptotic plus non-apoptotic nuclei) counted in a unit reference area. Values are means ± SEM (n=5). Means with unlike superscripts are significantly (p<0.05) different.
Involvement of caspase 2 and iNOS in nicotine plus HFD-induced hepatocellular apoptosis
Because caspase 2 works upstream of JNK activation and mitochondria-dependent apoptotic pathway [18, 19], we carried out Western blot analysis of active caspase 2 (Fig. 2a). Densitometric analysis revealed a significant increase (p <0.05) in active caspase 2 levels in the combined treatment group as compared to either treatment alone (Fig. 2b). We also visualized caspase 2 activation by immunohistochemistry. Compared with mice on a NCD (Fig. 2c. panel I), where no active caspase 2-positive cells were noted, occasional active caspase-2-positive cells could be seen in the liver of mice fed HFD (Fig. 2c. panel II). When combined, nicotine plus HFD resulted in a marked increase in the number of active caspase 2-positive liver cells (Fig. 2c. panel III). Morphometric analysis further confirmed the histological findings and revealed a significant increase (p<0.05) in the number of active caspase 2-positive liver cells (12.6 ± 2.5) after combined treatment with nicotine and HFD compared with mice on a HFD alone (2.8 ± 0.4). The number of caspase 2-positive liver cells was very low (0.9 ± 0.4) in NCD-fed mice.
Fig. 2.

a. Western blot analysis shows increased expression of hepatic active caspase 2 in the combined treatment group as compared to mice fed NCD or HFD alone. GAPDH in the immunoblot is shown as a loading control. b. Quantitation of band intensities. Data for active caspase 2 were normalized to GAPDH. Values are means ± SEM (n=5). Means with unlike superscripts are significantly (p<0.05) different. c. Visualization of caspase 2 activation in hepatocytes by immunohistochemistry in mice fed NCD (panel I) or HFD in the absence (panel II) or presence (panel III) of nicotine. d. iNOS induction in the hepatocytes from mice fed NCD (panel I) or HFD in the absence (panel II) or presence (panel III) of nicotine. f. Double immunofluorescence staining shows TUNEL (green), iNOS (red), and colocalization of TUNEL and iNOS (yellow) in apoptotic hepatocytes. Scale bar=20 μm.
We next examined the role of iNOS in nicotine plus HFD-induced hepatocellular apoptosis. Compared with mice on NCD, where no iNOS immunoreactivity is detected (Fig. 2d. panel I), we found a marked increase in the expression of iNOS in selective hepatic cells in mice fed with HFD (Fig. 2d. panel II). A further increase in the number of iNOS-positive hepatocytes was noted in the combined treatment group (Fig. 2d. panel III). Image analysis further showed that HFD alone led to a significant (p<0.01) increase in hepatocyte iNOS staining intensity (by 21.2-fold) compared with mice on a NCD. Combined with nicotine, HFD caused an additional (1.4-fold) increase in iNOS staining intensity. Double immunofluorescence staining of iNOS and TUNEL further confirmed induction of iNOS only in those hepatic cells undergoing apoptosis (Fig. 2e. panels I–III).
We next examined the expression profiles of BAX and BCL-2 via immunoblotting and immunohistochemical techniques. Western blot analysis revealed a significant (p<0.05) increase in BAX and a decrease in BCL-2 expression in the combined treatment group compared with those from mice fed NCD or HFD alone (Supplementary (S) Fig. 1a–c). We also found a marked increase in BAX immunoreactivity in hepatocytes after combined treatments with nicotine and HFD (S1d. panel III) compared with those from mice fed NCD (S1d. panel I) or HFD alone (S1d. panel II). Consistent with its anti-apoptotic role, we found a marked a decrease in the BCL-2 expression after combined treatments with nicotine and HFD (S1d. panel VI) compared with those from mice fed NCD (S1d. panel IV) or HFD alone (S1d. panel V).
Discussion
In a recent study, using the model of diet-induced obesity in C57BL6J mice, we demonstrated that nicotine in combination with a HFD triggered greater oxidative stress, activated hepatocellular apoptosis, and amplified HFD-induced hepatic steatosis [17]. In the current study, using the same model system, we show that nicotine plus HFD triggers hepatocellular apoptosis through activation of caspase 2, induction of iNOS, and perturbation of the BAX/BCL-2 ratio.
Consistent with a role of caspase 2 in apoptotic signaling in hepatic cells of patients with different stages of NAFLD ranging from simple steatosis to NASH [16], in the present study, we show activation of caspase 2 as evidenced by immunoblotting and immunohistochemical staining of liver sections for active caspase 2 in the combined treatment group compared with that of mice on a HFD alone. Emerging evidence now suggests that oxidative stress can trigger activation of caspase 2 in various cell systems [18–21]. Our group has shown that indeed nicotine when combined with a HFD triggers a higher degree of hepatic oxidative stress compared to either insult alone [17]. Thus, the signal for activation of caspase 2 most likely emanates from the generation of severe oxidative stress in the combined treatment group.
The downstream signaling pathways that link caspase 2 activation and hepatocellular apoptosis have not been previously identified. One possibility is that increased oxidative stress can induce hepatocellular apoptosis in response to HFD plus nicotine is through activation of JNK signaling [17, 22, 25, 27], resulting in the activation of the mitochondria-dependent intrinsic pathway signaling [17, 22, 25, 27]. Consistent with this possibility, we recently demonstrated activation of JNK and caspases 9 (the key initiator caspase in the intrinsic pathway signaling) and 3 in nicotine plus HFD-induced hepatocellular apoptosis [17].
The downstream signaling events that couple JNK activation with hepatocellular apoptosis have not been well-identified. One possible mechanism by which JNK could induce apoptosis is through induction of iNOS, resulting in NO production and subsequent activation of mitochondria-dependent death pathway. There are previous studies demonstrating a role for JNK in iNOS induction [22–24]. It is worth noting that iNOS plays a crucial role in the development and progression of hepatic steatosis in diet-induced obese mice [24, 28, 29]. Increased NO production, through up-regulation of iNOS, has also been implicated in cellular injury and apoptosis in various cell systems, including hepatic cells [22, 24]. Consistent with a role of iNOS in hepatocyte apoptosis, in the present study, we found additive effects of HFD and nicotine on iNOS induction in selective hepatic cells. Co-staining of TUNEL and iNOS further confirmed induction of iNOS only in those hepatic cells undergoing apoptosis. iNOS induction is further associated with stimulation of the mitochondria-dependent apoptotic pathway, characterized by the perturbation of the BAX/BCL-2 ratio. Together, these data indicate that caspase 2 and NO-mediated mitochondria dependent pathway plays an important role in the regulation of nicotine plus HFD-induced hepatocellular apoptosis (Fig. 3).
Fig. 3.

Key signaling pathways involved in nicotine plus HFD-induced hepatocellular apoptosis. Nicotine when combined with a HFD induces oxidative stress and results in activation of caspase 2 and JNK and induction of iNOS. It is likely that induction of iNOS, through increased nitric oxide production, perturbs BAX/BCL-2 ratio and triggers cytochrome c release from mitochondria, resulting in activation of caspases 9 and 3 and hepatocellular apoptosis [17, 22].
In summary, we have provided insights into the molecular mechanisms by which nicotine exacerbates HFD-induced hepatocellular apoptosis. This study further emphasizes the suitability of this model to study NAFLD, with a goal of developing better therapeutic remedies against fatty liver. Targeting the caspase 2 activation and interrupting NO production may have a protective role in development and progression of hepatic steatosis.
Supplementary Material
References
- 1.He J, Gu D, Wu X, Reynolds K, Duan X, Yao C, Wang J, Chen CS, Chen J, Wildman RP, Klag MJ, Whelton PK. Major causes of death among men and women in China. N Engl J Med. 2005;353:1124–1134. doi: 10.1056/NEJMsa050467. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ. New concepts in chronic obstructive pulmonary disease. Ann Rev Med. 2003;54:113–129. doi: 10.1146/annurev.med.54.101601.152209. [DOI] [PubMed] [Google Scholar]
- 3.Zaher C, Halbert R, Dubios R, George D, Nonikov D. Smoking-related diseases: the importance of COPD. Int J Tuberc Lung. 2004;8:1423–1428. [PubMed] [Google Scholar]
- 4.Hudson NL, Mannino DM. Tobacco use: a chronic illness? J Community Health. 2010;35:549–553. doi: 10.1007/s10900-010-9241-x. [DOI] [PubMed] [Google Scholar]
- 5.Hamabe A, Uto H, Imamura Y, Kusano K, Mawatari S, Kumagai K, Kure T, Tamai T, Moriuchi A, Sakiyama T, Oketani M, Ido A, Tsubouchi H. Impact of cigarette smoking on onset of nonalcoholic fatty liver disease over a 10-year period. J Gastroenterol. 2011;46:769–778. doi: 10.1007/s00535-011-0376-z. [DOI] [PubMed] [Google Scholar]
- 6.Zein CO, Unalp A, Colvin R, Liu Y-C, McCullough AJ. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J Hepatol. 2011;54:753–759. doi: 10.1016/j.jhep.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azzalini L, Ferrer E, Ramalho LN, Moreno M, Dominguez M, Colmenero J, Peinado VI, Barbera JA, Arroyo V, Gines P, Caballeria J, Bataller R. Cigarette smoking exacerbates nonalcoholic fatty liver disease in obese rats. Hepatology. 2010;5:1567–1576. doi: 10.1002/hep.23516. [DOI] [PubMed] [Google Scholar]
- 8.Yuan H, Shyy JY, Martins-Green M. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J Hepatol. 2009;51:535–547. doi: 10.1016/j.jhep.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 10.Chiolero A, Faeh D, Paccaud F, Cornuz J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. 2008;87:801–809. doi: 10.1093/ajcn/87.4.801. [DOI] [PubMed] [Google Scholar]
- 11.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–838. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trauner M, Arrease M, Wangner M. Fatty liver and lipotoxicity. Biochem Biophys Acta. 2010;1801:299–310. doi: 10.1016/j.bbalip.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Kojima H, Sakurai S, Uemura M, Fukui H, Morimoto H, Tamagawa Y. Mitochondrial abnormality and oxidative stress in nonalcoholic steatohepatitis. Alcohol Clin Exp Res. 2007;31(Suppl 1):S61–S66. doi: 10.1111/j.1530-0277.2006.00288.x. [DOI] [PubMed] [Google Scholar]
- 14.Mantena S, King A, Andringa K, Eccleston H, Bailey S. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol. 2008;44:1259–1272. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puri P, Mirshahl F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 16.Ferreira DM, Castro RE, Machado MV, Evangelista T, Silvestre A, Costa A, Coutinho J, Carepa F, Cortez-Pinto H, Rodrigues CM. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia. 2011;54:1788–1798. doi: 10.1007/s00125-011-2130-8. [DOI] [PubMed] [Google Scholar]
- 17.Friedman TC, Sinha-Hikim I, Parveen M, Najjar SM, Liu Y, Mangubat M, Shin C-S, Lyzlov A, Ivey R, Shaheen M, French SW, Sinha-Hikim AP. Additive effects of nicotine and high-fat diet on hepatic steatosis in male mice. Endocrinology. 2012;153:5809–5820. doi: 10.1210/en.2012-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 19.Braga M, Sinha Hikim AP, Datta S, Ferrini M, Brown D, Kovacheva EL, Gonzalez-Cadavid NF, Sinha-Hikim I. Involvement of oxidative stress and caspase 2-mediated intrinsic pathway signaling in age-related increase in muscle cell apoptosis in mice. Apoptosis. 2008;13:822–832. doi: 10.1007/s10495-008-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad V, Chandele A, Jagtap JC, Kumar S, Shastry P. ROS-triggered caspase 2 activation and feedback amplification loop in b-carotene-induced apoptosis. Free Radic Biol Med. 2006;41:431–442. doi: 10.1016/j.freeradbiomed.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Tamm C, Zhivotovsky B, Ceccatelli S. Caspase-2 activation in neural stem cells undergoing stress-induced apoptosis. Apoptosis. 2008;13:354–363. doi: 10.1007/s10495-007-0172-7. [DOI] [PubMed] [Google Scholar]
- 22.Sinha-Hikim I, Braga M, Shen R, Sinha Hikim AP. Involvement of c-Jun NH2-terminal kinase and nitric oxide-mediated mitochondria-dependent intrinsic pathway signaling in cardiotoxin-induced muscle cell death: role of testosterone. Apoptosis. 2007;12:1965–1978. doi: 10.1007/s10495-007-0120-6. [DOI] [PubMed] [Google Scholar]
- 23.Biswas N, Mahato SK, Chowdhury AA, Manna A, Vinayagam J, Chatterjee S, Jaisankar P, Chaudhuri U, Bandyopadhyay S. CB3E induces iNOS expression by ROS-dependent JNK and ERK activation for apoptosis of leukemia cells. Apoptosis. 2012;17:612–626. doi: 10.1007/s10495-011-0695-9. [DOI] [PubMed] [Google Scholar]
- 24.Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, Inamori M, Saito S, Iwasaki T, Terauchi Y, Maeyama S, Nakajima A. Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcohol Clin Expt Res. 2010;34 (Suppl 1):S18–24. doi: 10.1111/j.1530-0277.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 25.Sinha-Hikim I, Sinha-Hikim AP, Shen R, Kim H, French SW, Vaziri ND, Crum A, Rajavashisth TB, Norris KC. A novel cystine based antioxidant attenuates oxidative stress and hepatic steatosis in diet-induced obese mice. Exp Mol Pathol. 2011;91:419–428. doi: 10.1016/j.yexmp.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha-Hikim I, Friedman TC, Shin C-S, Lee D, Ivey R, Sinha-Hikim AP. Nicotine in combination with a high-fat diet causes intramyocellular mitochondrial abnormalities in male mice. Endocrinology. 2014;155:865–872. doi: 10.1210/en.2013-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh R, Wang Y, Schattenberg JM, Xian Y, Czaja MJ. Chronic oxidative stress sensitizes hepatocytes to death from 4-hydroxynonenal by JNK/c-Jun overactivation. Am J Physiol Gastrointest Liver Physiol. 2009;297:G907–917. doi: 10.1152/ajpgi.00151.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kathirvel E, Morgan K, Nandgiri G, Sandoval BC, Caudill MA, Bottiglieri T, French SW, Morgan TR. Betaine improves nonalcoholic fatty liver and associated hepatic insulin resistance: a potential mechanism for hepatoprotection by betaine. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1068–1077. doi: 10.1152/ajpgi.00249.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He S, Rehman H, Wright GL, Zhong Z. Inhibition of inducible nitric oxide synthase prevents mitochondrial damage and improves survival of steatotic liver grafts. Transplantation. 2010;89:291–298. doi: 10.1097/TP.0b013e3181c99185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.