

Abstract
Circular permutation of the NADPH-dependent oxidoreductase Old Yellow Enzyme from Saccharomyces pastorianus (OYE1) can significantly enhance the enzyme’s catalytic performance. Termini relocation into four regions of the protein (sectors I–IV) near the active site has proven effective in altering enzyme function. To better understand the structural consequences and rationalize the observed functional gains in these OYE1 variants, we selected representatives from sectors I–III for further characterization by biophysical methods and X-ray crystallography. These investigations not only show trends in enzyme stability and quaternary structure as a function of termini location but also provide a possible explanation for the catalytic gains in our top-performing OYE variant (new N-terminus at residue 303; sector III). Crystallographic analysis indicates that termini relocation into sector III affects the loop β6 region (amino acid positions: 290–310) of OYE1, which forms a lid over the active site. Peptide backbone cleavage greatly enhances local flexibility, effectively converting the loop into a tether and consequently increasing the environmental exposure of the active site. Interestingly, such an active site remodeling does not negatively impact the enzyme’s activity and stereoselectivity; neither does it perturb the conformation of other key active site residues with the exception of Y375. These observations were confirmed in truncation experiments, deleting all residues of the loop β6 region in our OYE variant. Intrigued by the finding that circular permutation leaves most of the key catalytic residues unchanged, we also tested OYE permutants for possible additive or synergistic effects of amino acid substitutions. Distinct functional changes in these OYE variants were detected upon mutations at W116, known in native OYE1 to cause inversion of diastereoselectivity for (S)-carvone reduction. Our findings demonstrate the contribution of loop β6 toward determining the stereoselectivity of OYE1, an important insight for future OYE engineering efforts.
Keywords: circular permutation, Old Yellow Enzyme, X-ray crystallography, biocatalysis, protein engineering, oxidoreductases
Introduction
Members of the Old Yellow Enzyme (OYE) family [EC 1.3.1.31] are popular biocatalysts for the asymmetric reduction of alkenes. Operating at ambient temperatures and under benign environmental conditions, these flavin oxidoreductases give direct access to chiral synthons with up to two stereogenic centers by catalyzing a highly stereoselective trans-hydrogenation. In addition, their inherent substrate promiscuity enables conversion of a wide range of substrates including α,β-unsaturated ketones, aldehydes and carboxylic acids, nitriles, and nitroalkenes, as well as nitrate esters, nitroaromatics, and triazines.1−10 The redox reaction proceeds via a Michael-type hydride transfer step from reduced flavin mononucleotide (FMN) to the substrate’s β-carbon with concomitant protonation at its α-position. After product release, the oxidized flavin cofactor is regenerated via reduction by NADPH (Scheme 1).
Scheme 1. Flavin-Dependent ene Reduction.

Driven by the need for novel and effective biocatalysts for applications in biotechnology and the pharmaceutical industry, an ever-growing number of putative OYEs from different organisms and environments have been reported.4,7,11−14 At the same time, protein engineering by mutagenic methods and directed evolution has been employed to tailor these enzymes.15−17 Complementing these protein engineering efforts, we have been exploring the impact of circular permutation (CP) on the catalytic performance of enzymes. In contrast to the traditional protein engineering approaches that modify enzyme function through substitution of amino acids, CP keeps the amino acid composition unchanged but reorganizes the protein’s primary sequence. During the process of CP, the native N- and C-termini of a protein are covalently linked with a short peptide, and new termini are introduced through backbone cleavage elsewhere in the polypeptide sequence.18 Examples of such termini relocation are found in nature.19−21 In addition, the method has frequently been used in the laboratory, initially in protein folding experiments22−26 and more recently for protein engineering.27−32 An analysis of the effects of CP on natural and laboratory-based evolution of proteins has revealed potential functional benefits through changes in local protein conformation and backbone flexibility near the newly created termini. These changes to protein dynamics may reshape access tunnels and active site binding pockets as well as accelerate or even eliminate rate-limiting conformational changes in the catalytic reaction cycle.
To explore and possibly exploit some of these changes to protein structure and function in flavin oxidoreductases for creating novel biocatalysts, we recently applied the CP approach to OYE1 from Saccharomyces pastorianus.32 Earlier structural studies of OYE1 had identified several loop regions and helix α1 (Figure 1) that partially cover the bound FMN and shape the active site located in the central part of the (βα)8-barrel protein.33−35 On the basis of these structural features, we hypothesized that CP could have a significant functional impact on the enzyme. The idea was tested by preparing and functionally characterizing a synthetic gene library of 228 circular permutants of OYE1.32 More importantly, we identified 70 circular permutated OYE (cpOYE) variants with activity equal to or better than wild type for reduction of ketoisophorone 1 to (R)-levodione 2 (Scheme 1). Some of these variants showed rate enhancements of over an order of magnitude, and none of the tested cpOYEs compromising the parental enzyme’s high enantioselectivity.
Figure 1.

Structure overlay of OYE1 (white; PDB: 1OYA), cpOYE154 (gray; PDB: 4RNX), and cpOYE303 (blue; PDB: 4RNU). Super positioning of the three structures was based on the FMN cofactor, shown in sticks. The three helices at the dimer interface (α4, α5, and α6) and the native OYE1 termini are labeled. The two inserts focus on the new termini regions in cpOYE154 (right) and cpOYE303 (left). Red arrows mark the locations of backbone cleavage (shown on OYE1) upon circular permutation, illustrating the portions of protein sequence invisible in the CP variants due to a lack of electron density.
Similar functional gains were found with other substrates, including stereoselective conversion of (S)-carvone 3 to the (1R/4S)-diastereomer of dihydrocarvone 4. Interestingly, the new termini locations in these 70 cpOYE clustered in four specific sectors of the protein sequence: sector I stretches over most of the exterior helical region α1 (amino acid residues 125–160; numbering based on OYE1), sector II includes loop β5 and parts of subsequent helix α5 (residues 250–265), sector III covers loop β6 (residues 290–310), and sector IV colocates with a short loop near the native C-terminus (residues 375–380) (Figure 1). All four sectors cover loop regions and secondary structure elements near the active site, and as anticipated, three of them (sectors I–III) have been identified in biochemical and structural studies as possible candidates undergoing substantial conformational changes during the catalytic cycle.33−40 Because several cpOYE variants with new termini in these three sectors show significant functional improvements, we investigated the impact of CP on the ternary and quaternary structures as well as the stability of these biocatalysts. Variants from sector IV were not included in these studies because the observed functional gains were modest at best.
To better understand the structural and functional consequences of termini relocation in cpOYE variants, we selected representatives from each sector and examined them in a series of biophysical studies. Separately, four sets of X-ray crystallographic data were collected for representatives from sectors I and III. The latter revealed only minimal structural perturbation of amino acid conformations in positions other than residues immediately flanking the new N- and C-termini. The mostly conserved active site environment helped to rationalize the preservation of parental stereoselectivity in cpOYE variants. The similar orientation of key catalytic residues further raised questions about possible additive or synergistic effects of amino acid substitutions and CP on the function of these biocatalysts. Our initial exploration of such effects focused on substitutions to the tryptophan at position 116 (W116) in OYE1. W116 is known to affect the diastereoselectivity of (S)-carvone reduction in OYE1.14,41,42 Although the tested cpOYE variants did not show the predicted change in stereoselectivity, our findings provide new insight regarding the contribution of loop regions on OYE1 function.
Results and Discussion
Primary Characterization of cpOYE Variants
We initially studied structural changes of selected cpOYE variants by circular dichroism (CD) spectroscopy and size-exclusion chromatography. Nine candidates were chosen on the basis of either the location of their new termini or enhancements in catalytic performance using the results from our previous study of cpOYE1 variants.32 More specifically, representatives from each of the three sectors (sector I, residues 125–160; sector II, residues 250–265; sector III, 290–310) that yielded catalytically active permutants were investigated. Probing for differences in secondary structure, the far-UV CD spectra of these enzyme variants showed no significant change (Supporting Information (SI) Figure S1). The overall mixed β/α signature found in OYE1 was conserved in all cpOYE candidates.
The same experimental setup was used to assess the thermostability of enzyme variants by measuring spectral change at 222 nm as a function of temperature (Table 1 and SI Figure S2). Although the temperature of unfolding (TM) for native OYE1 was measured at 49 °C, the thermal robustness of CP variants varied quite significantly. The three candidates with new termini in sector I (cpOYE146, -154, and -160) exhibited significantly higher TM values, raising the half point of unfolding by 15 to 23 °C. The temperature range for the folded-to-unfolded transition of these variants is only slightly broader than OYE1, suggesting that the termini relocation does enhance the structural integrity of sector I variants (SI Figure S2). The underlying reason for the increased stability is not clear. Although size exclusion experiments (see below) indicate some protein with quaternary conformation greater than dimer, the small fraction (<10% of total purified protein) is insufficient to account for the observed TM gains. The sole representative from sector II, cpOYE260, closely matched the stability of wild type enzyme.
Table 1. Stability and Quaternary Structure of OYE1 and Selected cpOYE Variants.
| OYE variant | TM (°C) (±1 °C) | oligomeric state(s) |
|---|---|---|
| OYE1 | 49 | dimer |
| cpOYE146 | 64 | monomer/dimer/oligomer |
| cpOYE154 | 72 | monomer/dimer/oligomer |
| cpOYE160 | 66 | monomer |
| cpOYE260 | 47 | monomer |
| cpOYE291 | 43 | dimer |
| cpOYE292 | 47 | dimer |
| cpOYE303 | 57 | dimer |
| cpOYE305 | 57 | dimer |
| cpOYE307 | 55 | dimer |
The five remaining candidates (cpOYE291, -292, -303, -305, and -307) were selected from sector III, the region with variants that showed the most substantive catalytic improvements.32 Sector III is located entirely in loop β6, an extended loop region that flanks the FMN binding site and whose tip (N294 to L297) forms part of the active site binding pocket. Interestingly, CP variants with their new termini in the N-terminal stem of the loop exhibit TM values slightly lower than wild type enzyme (cpOYE291 and -292). In contrast, variants with new termini in the C-terminal portion of the loop trend in the opposite direction, yielding enzymes with elevated temperatures of unfolding by 6–8 °C. These overall changes are small but show correlation with the termini location. Although residues flanking positions 291 and 292 are facing the protein’s interior environment, amino acid residues 303–307 are surface-exposed. The latter, although less disruptive to the protein’s tertiary structure than the former, would be expected to result in great local flexibility, a prediction that is consistent with the notably broader temperature transition for protein unfolding compared to cpOYE291 and -292. In summary, our thermodenaturation data demonstrate that CP does not automatically translate into destabilization of a protein. To the contrary, results for selected OYE1 variants indicate that termini relocation can have a substantial stabilizing effect.
Finally, we used size-exclusion chromatography to examine potential quaternary structure changes upon sequence reorganization by CP. Such changes can impact protein stability as observed in dimeric variants of the normally monomeric lipase B from Candida antarctica (CALB).43 Separately, enzyme activity can be affected by the quaternary structure change, as seen for the OYE1 homologue 12-oxophytodienoate reductase 3 (OPR3).44 Our analysis confirmed the dimeric solution state for wild type OYE1, consistent with previously reports for the native enzyme (Table 1 and SI Figure S3).33 For representatives of sector I, quaternary structure varied from mostly monomeric (cpOYE160) to mono/dimeric mixtures (cpOYE146 and -154), reflected in broad signal peaks. In addition, chromatograms for these three variants also show higher-molecular-weight species, hinting the presence of small amounts of trimeric or tetrameric assemblies. In contrast, the elution profile for cpOYE260 (sector II) suggests almost exclusively monomeric enzyme, whereas the SEC data for cpOYE variants with termini in sector III show native-like dimeric quaternary structure. These latter findings can be rationalized on the basis of the OYE1 dimer interface involving helices α4, α5, and α6 (Figure 1). In cpOYE260, the new termini are located at the very beginning of helix 5, which is likely to affect the local structure integrity and, consequently, disrupt the dimer interface. Interestingly, the same reorganization of the protein sequence in the adjacent loop β6−α6 region (sector III) leaves the oligomeric state of the enzyme unchanged. The less disruptive effects of new termini in sector III is explained by the 3–10-residue spacing between α6 and the backbone cleavage sites.
Structure Analysis by X-ray Crystallography
To further explore the observed differences in catalytic performance and biophysical properties in our cpOYE collection, we expanded our structural study to analyze three representatives by X-ray crystallography. One candidate from each sector (I, cpOYE154; II, cpOY260; III, cpOYE303) was chosen for crystallization. Following overexpression and purification of the individual proteins, crystal growth conditions were screened for each candidate. We obtained diffraction quality crystals for cpOYE154 and cpOYE303. Both variants contained a bound FMN and diffracted X-ray to 1.25 and 2.69 Å resolution, respectively (Table 2, PDB ID: 4RNX and 4RNU). Separately, cpOYE303 crystals were soaked in a solution of p-hydroxybenzaldehyde (HBA), a tight-binding competitive inhibitor of OYE1, yielding structural information on the enzyme·FMN·inhibitor complex at 2.47 Å resolution (PDB ID: 4RNV). The crystallographic asymmetric unit contains four complexes. Interestingly, we found that HBA could be fitted best into the electron density in two orientations, depending on the molecule: a first orientation with its aldehyde moiety in hydrogen bonding distance to amino acids H191/N194 (Figure 2A) and a second, flipped orientation similar to that seen for wild type OYE1 (Figure 2B).33 For our third cpOYE candidate, all attempts failed to find conditions for producing suitable crystals of cpOYE260. The inability to grow crystals of this variant might be linked to its observed quaternary structure changes and lower temperature of unfolding (Table 1). The resulting disruption of critical interprotein contacts and elevated structural heterogeneity could interfere with crystal growth.
Table 2. Summary of X-ray Data Collection and Refinement Statistics.
| data collection | cpOYE303 (4RNU) | cpOYE154 (4RNX) | cpOYE303T (4RNW) | cpOYE303inh (4RNV) |
|---|---|---|---|---|
| space group | P1 | P1 | P21 | P1 |
| cell dimensions | a = 45.67 Å, b = 87.69, c = 113.71 | a = 44.09 Å, b = 57.98 Å, c = 85.90 Å | a = 64.25 Å, b = 73.00 Å, c = 84.74 Å | a = 45.6 Å, b = 88.1 Å, c = 105.6 Å |
| α = 69.34°, β = 82.60°, γ = 90.06° | α = 102.46°, β = 98.36°, γ = 111.19° | α = 90°, β = 101.27°, γ = 90° | α = 89.45°, β = 81.43°, γ = 89.88° | |
| beamline (SERCAT) | APS 22-BM | APS 22-BM | APS 22-ID | APS 22-BM |
| wavelength (Å) | 1 | 1 | 1 | 1 |
| resolution (Å)a | 38.78–2.69 (2.79–2.69) | 27.11–1.25 (1.29–1.25) | 28.98–1.55 (1.61–1.55) | 34.8–2.47 (2.58–2.47) |
| Rmergeb | 0.107(0.829) | 0.075(0.681) | 0.120(0.799) | 0.098 (0.607) |
| ⟨I/σI⟩c | 13.8(1.9) | 17.0(1.9) | 13.7(1.9) | 10.8 (1.8) |
| completeness (%)a | 92.0(92.9) | 95.1(92.0) | 98.0(96.6) | 98.7 (97.9) |
| redundancya | 6.0(5.4) | 4.2(4.0) | 5.8(5.4) | 3.2 (3.1) |
| obs reflections | 253 086 | 831 001 | 629 067 | 182 865 |
| unique reflectionsa | 42 123 (4241) | 196 866 (19 029) | 108 578 (10 639) | 56 814 (5641) |
| Refinement | ||||
| resolution (Å) | 2.69 | 1.25 | 1.55 | 2.47 |
| no. reflections | 41 949 | 196 804 | 108 290 | 56 763 |
| Rwork/Rfreed,e | 0.241/0.279 | 0.134/0.162 | 0.190/0.216 | 0.219/0.251 |
| twin law/fraction | –h, k, k – l/0.11 | |||
| no. atoms | ||||
| protein | 11 891 | 6257 | 6038 | 12 144 |
| solvent | 112 | 1145 | 1007 | 267 |
| other heteroatoms | 144 | 62 | 92 | 160 |
| B factors (Å2) | ||||
| protein | 81.5 | 12.1 | 18 | 52.9 |
| solvent | 44.5 | 28.4 | 31.4 | 35.8 |
| other heteroatoms | 81.9 | 7.4 | 13.7 | 45.5 |
| rms Deviations | ||||
| bond lengths (Å) | 0.002 | 0.003 | 0.005 | 0.002 |
| bond angles (deg) | 0.5 | 0.9 | 1.0 | 0.6 |
Values in parentheses correspond to highest resolution shell.
Rmerge = Σ|I – ⟨I⟩|/Σ I, where I is the observed intensity and ⟨I⟩ is the averaged intensity from multiple observations.
⟨I/σI⟩ = averaged ratio of the intensity (I) to the error of the intensity (σI).
Rwork = Σ|Fobs – Fcal|/Σ|Fobs|, where Fobs and Fcal are the observed and calculated structure factors, respectively.
Rfree was calculated using a randomly chosen subset (5%) of the reflections not used in refinement.
Figure 2.
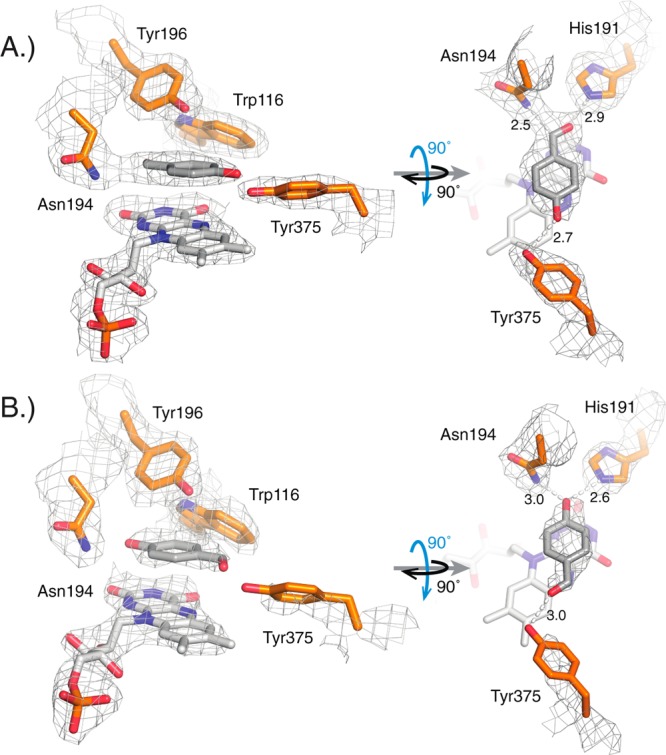
Structural study of the active site of cpOYE303 (PBD: 4RNV) with key amino acid side chains (orange), FMN (white), and substrate analog p-hydroxybenzaldehyde (HBA; gray). (A) In two of the four protein complexes per asymmetric unit, the best fit to the observed electron density orients the aldehyde group of HBA in hydrogen-bonding distance to H191 and N194, whereas in the other two complexes (B), the ligand is flipped with its hydroxyl moiety pointing toward H191 and N194, as seen for OYE1.33 The gray mesh represents the 2mFo-DFc map contoured at the 1.0 σ level for key protein side chains, FMN and HBA. Hydrogen bonding distances (in angstroms) are indicated.
The three crystallized cpOYE variants show high overall structural similarities with the parental OYE1, yet exhibit significant conformational perturbation near the old and new termini regions. Superposition of cpOYE154 and cpOYE303 with wild type enzyme (PDB: 1OYA)33 resulted in root-mean-square deviation (RMSDs) of 0.38 and 0.44 Å (over the entire protein length), respectively (Figure 1). For cpOYE154, the protein sequence could be modeled continuously from residue 154 to 144 (all residue numbering based on OYE1) visualizing the entire new N-terminal region while missing only nine amino acids at the C-terminus (amino acid positions 145–153). In the case of cpOYE303, the electron density map matched residues 306–291, with the exception of two amino acid residues in the linker region connecting the native termini. The notably higher crystallographic thermal B-factor in the linker region of cpOYE154 (SI Figure S4) and the gap in electron density for cpOYE303 suggests that future redesigns might be possible with shorter or no linker at all. The protein backbone seems well adapted to accommodate the additional steric strain introduced upon ligation of the native termini. For cpOYE303, there is also a lack of electron density in the new termini regions, leaving out three N-terminal residues (positions 303–305) and 11 C-terminal amino acids (positions 292–302). Given the region’s proximity to the enzyme active site, we speculated that the binding of the competitive inhibitor HBA to cpOYE303 could help with orienting additional residues. The presence of HBA did slightly improve the overall structure resolution but did not affect the N- and C-terminal regions. In fact, the indistinguishable structures of cpOYE303 with and without HBA in the active site led us to base all further discussion of full-length cpOYE303 in this manuscript on the coordinates for the HBA-bound enzyme.
Focusing on the active site, CP did not perturb the position of key catalytic residues in either OYE variant but affected the solvent accessibility of the catalytic center (Figure 3). Supported by clearly defined electron density for FMN, the orientation of the bound cofactor is identical in the two variants compared with OYE1. Furthermore, we analyzed both variants for changes to the positions and side chain conformations of amino acid residues within 12 Å of the FMN N5 atom. In addition to the obvious absence of the highly flexible residues near the new termini in CP variants, the comparison with native OYE1 (based on 1OYA33) revealed only small changes in the side chain orientation of Y375. For cpOYE154, the phenol moiety of Y375 is rotated by −20° around χ2, whereas in cpOYE303, the same residue swings in the opposite direction by 20° and 40° around χ1 and χ2, respectively. Interestingly, the orientation of Y375 in cpOYE303 closely resembles the side chain position in structures with bound substrate (analogs) (Figure 4A/B). Furthermore, β-factor analysis indicated no significant changes in backbone flexibility near the active sites (SI Figure S4).
Figure 3.
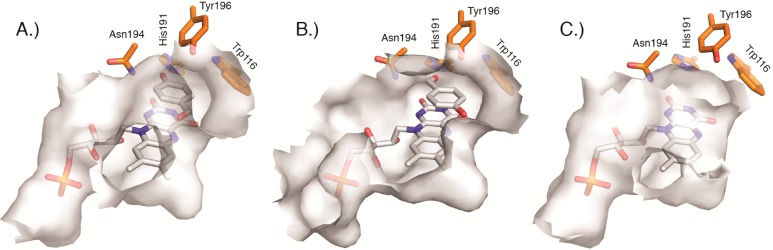
Comparison of active site binding pockets for (A) OYE1 (PDB: 1OYA), (B) cpOYE303 (PDB: 4RNU), and (C) cpOYE154 (PDB: 4RNX). Gray-shaded surfaces mark the interior protein surface. Key amino acids in the active sites (W116, H191, N194, and Y196) are orange. The bound FMN cofactor and substrate analog HBA (present in A and B) are shown as white sticks.
Figure 4.
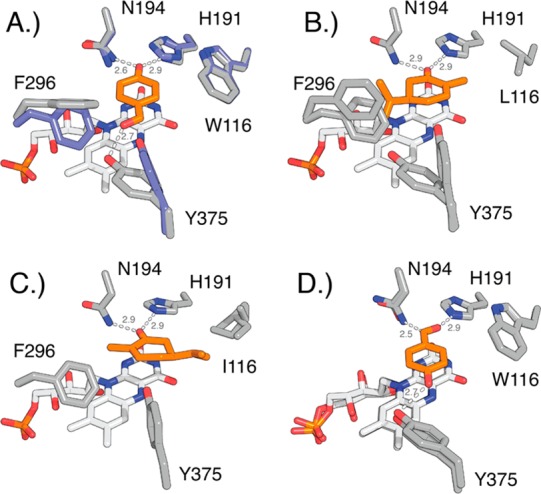
Conformational changes at positions 116, 296, and 375 upon substrate binding in native OYE1 and OYE variants. Ligands are highlighted in orange. (A) Overlay of OYE1 (blue, PDB: 1OYA) and OYE1 with bound HBA (gray, PDB: 1K03) shows the reorientation of F296 (90° rotation of phenyl ring) and Y375 (side chain rotation). (B) OYE1(W116L) with bound (R)-carvone (PDB: 4GWE) as model for carvone binding in normal orientation. (C) OYE1(W116I) with bound (S)-carvone (PDB: 4GE8) shows substrate in flipped orientation. The repositioning of the isopropenyl group near I116 eliminates the need for conformational changes of F296 and Y375. (D) Overlay of cpOYE303 holoenzyme (PDB: 4RNU) and cpOYE303 with bound HBA (PDB: 4RNV). Hydrogen bonding interactions and distances (in angstroms) are indicated.
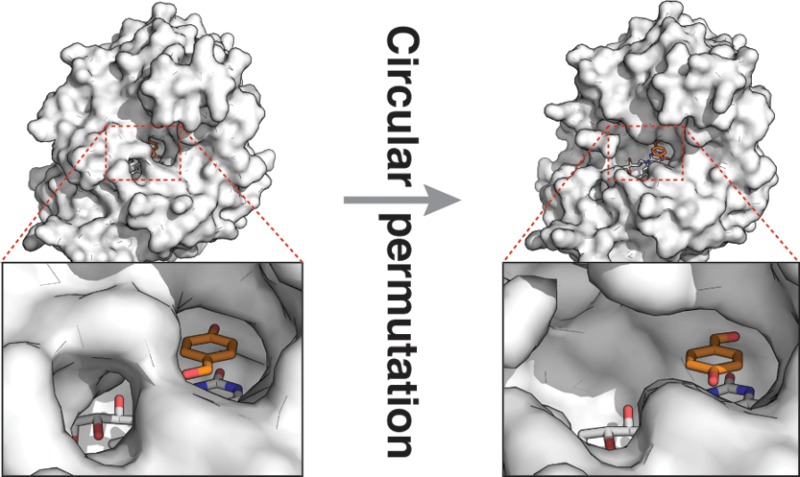
In contrast, the mapping of the enzymes’ interior surface, including the substrate and FMN binding pocket, revealed quite significant structural changes. The substrate-bound structure for OYE1 shows only a narrow access channel to the protein surface near Y196 (Figure 3A). The same channel is slightly enlarged in cpOYE154 (Figure 3C). Although direct comparison of the two structures is complicated by the absence of substrate (analog) in the latter, the increased active site accessibility in the CP variant could account for observed moderate gains in catalytic activity.32 In cpOYE303, the termini relocation into loop β6 has a much more dramatic impact on the shape and solvent accessibility of the catalytic center (Figure 3B). In wild type enzyme, this loop forms a lid that sequesters bound FMN and substrate from the environment. Moreover, conformational changes in the loop region are thought to be part of the catalytic cycle. Upon CP, peptide bond cleavage at position 302/303 disrupts the orderly arrangement of the loop and significantly widens the access tunnel to the active site without interfering with residues in the substrate binding pocket or the flavin cofactor. Such changes enable faster exchange of substrate and product but do not perturb catalytic action.
Termini Truncation in cpOYE303
The invisible residues at the newly created amino and carboxy termini of cpOYE303 were the focus of further biochemical and biophysical investigations. The absence of electron density is typically associated with increased protein backbone flexibility, a common phenomenon in termini regions. The same effect was observed in previous CP experiments with CALB and is at least in part responsible for the functional improvements of the biocatalyst.27,43,45 Nevertheless, subsequent studies with CALB variants have shown that the contributions of these invisible regions can be more complex and critical to function. We therefore decided to evaluate the functional role of the two terminal regions in cpOYE303 by creating a truncated variant (cpOYE303T). Nucleotides encoding for the three invisible residues at the N-terminus and the 11 amino acids at the C-terminus of the gene sequence in cpOYE303 were deleted, followed by heterologous expression of cpOYE303T in Escherichia coli.
Protein preparation as well as basic biochemical and biophysical characterization did not reveal any significant difference in stability or catalytic performance of cpOYE303T compared with its parent cpOYE303. Expression levels were robust and indistinguishable from cpOYE303. Furthermore, stability as measured by thermal unfolding in the CD spectrophotometer seems unaffected by the deletion of terminal residues. The catalytic performance of the truncated variant was tested on the reduction of ketoisophorone 1 to (R)-levodione 2. No difference was detectable with respect to catalytic activity or enantioselectivity of cpOYE303T over cpOYE303. In short, our experiments did not find evidence for any functional changes upon truncation of the invisible termini, suggesting no essential role for residues in this region. Consistent with the high degree of functional similarity between full-length and truncated cpOYE303, the crystal structures of the two enzymes are perfectly superimposable. At 1.55 Å resolution, the structure of cpOYE303T (PDB ID: 4RNW) provides a more refined view of the engineered biocatalyst, confirming the absence of change to the positioning of active site residues, including FMN seen in cpOYE154 and cpOYE303. Overall, the findings demonstrate the remarkable robustness of OYE1. The enzyme’s (β/α)8 barrel fold not only accommodates backbone cleavage near the active site but also can tolerate complete removal of the loop β6 region without detectable detrimental effects on function or stability. In contrast, the cleavage or elimination of the loop region in cpOYE303 and cpOYE303T, respectively, enhances the overall catalytic performance with several test substrates. As mentioned above, we rationalize these functional gains by its involvement in a rate-limiting conformational change as part of the catalytic cycle. Detailed steady-state experiments and rapid enzyme kinetics studies to investigate the role of loop β6 on catalysis in native OYE1 and cpOYE303 variants are ongoing. Consistent with previously reported data, these studies point to rate enhancements in the enzyme’s oxidative half reaction.32
Secondary Engineering of CP Variants
Beyond CP, we were interested in exploring the impact of amino acid substitutions on the catalytic performance of cpOYE303. The combination of CP with traditional site-directed and random mutagenesis could give rise to novel OYE variants exhibiting potential functional benefits from additive or even synergistic effects. To test the idea, we decided to focus on W116, probably one of more extensively studied amino acid position in OYE1.14,41,42 The indol moiety of W116 lines the active site, and amino acid changes in this position were shown to affect the diastereoselectivity of (S)-carvone 3 reduction in OYE1. Stewart and co-workers identified substitutions of Ala, Val, or Ile for W116 as the most effective switches of the enzyme’s stereoselectivity. Thus, we prepared the W116I variants of cpOYE154 and cpOYE303 and measured their catalytic performance with (S)-carvone 3 (Figure 5). Our positive control OYE1(W116I) showed the expected reversal in diastereoselectivity from the normal R/S product 4 to its S/S diastereomer 5. Similarly, the W116I substitution in cpOYE154 switched the stereoselectivity, although at only 67% de. We suspect that the moderate diastereoselectivity for 5 is linked to the low levels of activity of cpOYE154(W116I). Low signal-to-noise in the GC analysis complicates the integration of product peaks and, consequently, the determination of a precise de value. The cause for the dramatic decline in activity upon introducing W116I in this variant is unclear because the parental cpOYE154 exhibits excellent stability, and substitutions in position 116 are generally well tolerated in OYE1 and other variants. Overall, the results for cpOYE154 supports the idea that amino acid changes affecting stereoselectivity can be transferred into the CP variant, although in a nonadditive fashion.
Figure 5.
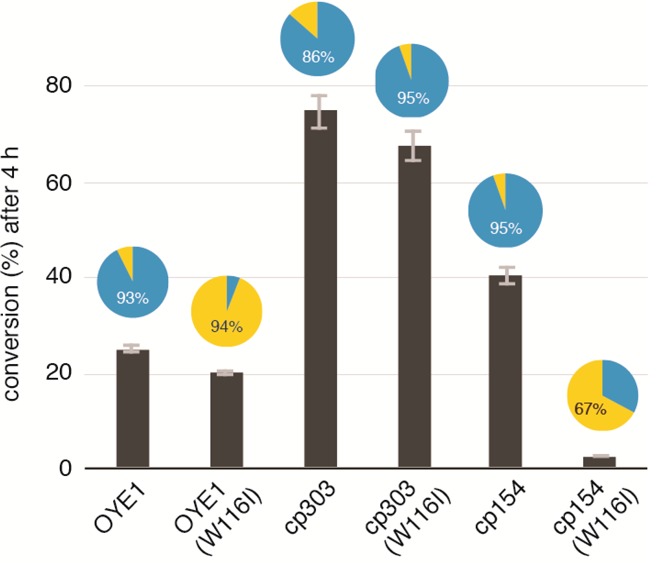
Summary of catalytic activity and diastereoselectivity of OYE1, cpOYE154, and cpOYE303 as well as their respective W116I variants. The percent conversion of (S)-carvone (3) is indicated by column height, and diastereoselectivity based on formation of (R/S)-4 (blue) versus (S/S)-5 (yellow) is shown in the pie diagrams.
For cpOYE303, the consequences of W116I are in sharp contrast to the above results. The amino acid substitution does not significantly alter the rate of conversion and, at ∼95% de for (R/S)-isomer 4, even slightly improves the parental enzyme’s diastereoselectivity (∼86%). Given the unexpected findings for cpOYE303(W116I), we examined our results in the context of the current understanding of diastereoselectivity-determining factors in the OYE1-catalyzed reduction of (S)-carvone. The present model is based on a combination of crystallographic and experimental data and points toward three key amino acid positions: W116, F296, and Y375 (Figure 4).14,41
In OYE1, binding of carvone in a catalytically productive orientation (Cβ near N5)46 is facilitated by hydrogen bonding of its carbonyl group with H191 and N194. These interactions allow for two distinct modes of substrate binding. In the first mode, the isopropenyl side chain of carvone points toward F296 and Y375 (Figure 4B), which Stewart refers to as the “normal” mode because it produces the natural (R/S)-diastereomer 4. The second mode has the carvone in a “flipped” orientation, with its side chain directed toward position 116 (Figure 3C). The latter conformation is usually blocked by W116, but substitutions with Ala, Val, or Ile create a hydrophobic pocket that can accommodate the isopropenyl moiety, leading to formation of (S/S)-diastereomer 5. Although OYE1(W116A/V/I) can accommodate substrate in both binding modes, experiments found that (S)-carvone preferentially binds in the “flipped” orientation. The observation was rationalized by the extra entropic cost associated with reorientation of the F296 and Y375 side chains upon “normal” substrate binding (Figure 4A/B), which is not necessary in the “flipped” mode (Figure 4C).
In cpOYE303 (Figure 4D), F296 has effectively been removed because of an increase in the flexibility of the nearby protein termini, which enables the Y375 side chain to assume a preferred orientation that closely resembles the conformation in the “normal” binding mode, even in the absence of substrate. The well-defined electron density map for the Y375 side chain supports this conclusion (Figure 2).47−49 cpOYE303(W116I) therefore represents an OYE variant without restraints arising from steric repulsion (W116) and unfavorable energetics of side chain reorganization (F296/Y375). As a consequence, one might expect a decline in diastereoselectivity because the “normal” and “flipped” binding modes are equally accessible; however, our experiments show almost exclusively formation of the (R/S)-product 4, suggesting a clear preference for the “normal” enzyme–substrate interactions.
We conclude that the switch from 94% de for (S/S)-product 5 in OYE1(W116I) to 95% de (R/S)-product 4 in cpOYE303(W116I) is to a significant part controlled by F296. Located at the tip of loop β6, the residue’s phenyl side chain seems to restrict rotational movement of neighboring Y375 whose reorientation is necessary for substrate binding in native OYE1. The deletion of the entire loop upon CP eliminates these constraints and allows for preorganization of Y375. Such an arrangement may also lead to tighter substrate binding, which could contribute to the overall enhancements in catalytic rates of the oxidative half reaction observed in stopped-flow experiments. On the basis of our current findings, a more detailed investigation of the functional role of loop β6 by site-directed mutagenesis of F296 and flanking residues in the loop region could prove insightful. Local amino acid changes may also allow for a fine-tuning of OYE1 diastereoselectivity for carvone and other chiral synthons.
Acknowledgments
This work was funded in part by the U.S. National Science Foundation (CBET-1159434). J.R.H. and X.C. were supported by NIH GM049245-21. The Department of Biochemistry at the Emory University School of Medicine supported the use of the Southeast Regional Collaborative Access Team (SER-CAT) synchrotron beamlines (22-ID and 22-BM) at the Advanced Photon Source of Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U.S. Department of Energy under Contract W-31-109-Eng-38. We thank the members of the Lutz lab for their critical and helpful comments and suggestions on the manuscript and would like to acknowledge the assistance of Leann Quertinmont, Samantha Iamurri. and Dr. Roberto Orru for independently verifying biophysical and catalytic data reported in this paper.
Supporting Information Available
The following file is available free of charge on the ACS Publications website at DOI: 10.1021/cs501702k.
Materials and methods and additional figures (PDF)
Author Present Address
§ Nephron Pharmaceuticals Corp., 4500 12th Street, West Columbia, South Carolina 29172, United States
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Kawai Y.; Inaba Y.; Tokitoh N. Tetrahedron-Asymmetry 2001, 12, 309–318 10.1016/S0957-4166(01)00029-5. [DOI] [Google Scholar]
- Swiderska M. A.; Stewart J. D. J. Mol. Catal., B: Enzymol. 2006, 42, 52–54 10.1016/j.molcatb.2006.06.023. [DOI] [Google Scholar]
- Stuermer R.; Hauer B.; Hall M.; Faber K. Curr. Opin Chem. Biol. 2007, 11, 203–213 10.1016/j.cbpa.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Hall M.; Stueckler C.; Hauer B.; Stuermer R.; Friedrich T.; Breuer M.; Kroutil W.; Faber K. Eur. J. Org. Chem. 2008, 1511–1516 10.1002/ejoc.200701208. [DOI] [Google Scholar]
- Kosjek B.; Fleitz F. J.; Dormer P. G.; Kuethe J. T.; Devine P. N. Tetrahedron-Asymmetry 2008, 19, 1403–1406 10.1016/j.tetasy.2008.05.023. [DOI] [Google Scholar]
- Toogood H. S.; Fryszkowska A.; Hare V.; Fisher K.; Roujeinikova A.; Leys D.; Gardiner J. M.; Stephens G. M.; Scrutton N. S. Adv. Synth. Catal. 2008, 350, 2789–2803 10.1002/adsc.200800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adalbjornsson B. V.; Toogood H. S.; Fryszkowska A.; Pudney C. R.; Jowitt T. A.; Leys D.; Scrutton N. S. ChemBioChem 2010, 11, 197–207 10.1002/cbic.200900570. [DOI] [PubMed] [Google Scholar]
- Toogood H. S.; Gardiner J. M.; Scrutton N. S. ChemCatChem 2010, 2, 892–914 10.1002/cctc.201000094. [DOI] [Google Scholar]
- Durchschein K.; Hall M.; Faber K. Green Chem. 2013, 15, 1764–1772 10.1039/c3gc40588e. [DOI] [Google Scholar]
- Toogood H. S.; Scrutton N. S. Curr. Opin. Chem. Biol. 2014, 19, 107–115 10.1016/j.cbpa.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Kitzing K.; Fitzpatrick T. B.; Wilken C.; Sawa J.; Bourenkov G. P.; Macheroux P.; Clausen T. J. Biol. Chem. 2005, 280, 27904–27913 10.1074/jbc.M502587200. [DOI] [PubMed] [Google Scholar]
- Hall M.; Stueckler C.; Ehammer H.; Pointner E.; Oberdorfer G.; Gruber K.; Hauer B.; Stuermer R.; Kroutil W.; Macheroux P.; Faber K. Adv. Synth. Catal. 2008, 350, 411–418 10.1002/adsc.200700458. [DOI] [Google Scholar]
- Fryszkowska A.; Toogood H.; Sakuma M.; Gardiner J. M.; Stephens G. M.; Scrutton N. S. Adv. Synth. Catal. 2009, 351, 2976–2990 10.1002/adsc.200900574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeu Y. A.; Sullivan B.; Walton A. Z.; Stewart J. D. Adv. Synth. Catal. 2012, 354, 1949–1960 10.1002/adsc.201200213. [DOI] [Google Scholar]
- Hulley M. E.; Toogood H. S.; Fryszkowska A.; Mansell D.; Stephens G. M.; Gardiner J. M.; Scrutton N. S. ChemBioChem 2010, 11, 2433–2447 10.1002/cbic.201000527. [DOI] [PubMed] [Google Scholar]
- Bougioukou D. J.; Kille S.; Taglieber A.; Reetz M. T. Adv. Synth. Catal. 2009, 351, 3287–3305 10.1002/adsc.200900644. [DOI] [Google Scholar]
- van den Heuvel R. H.; van den Berg W. A.; Rovida S.; van Berkel W. J. J. Biol. Chem. 2004, 279, 33492–33500 10.1074/jbc.M312968200. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Lutz S. Trends Biotechnol. 2011, 29, 18–25 10.1016/j.tibtech.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. J. Mol. Evol. 1999, 49, 161–164 10.1007/PL00006529. [DOI] [PubMed] [Google Scholar]
- Hisano T.; Kasuya K. I.; Tezuka Y.; Ishii N.; Kobayashi T.; Shiraki M.; Oroudjev E.; Hansma H.; Iwata T.; Doi Y.; Saito T.; Miki K. J. Mol. Biol. 2006, 356, 993–1004 10.1016/j.jmb.2005.12.028. [DOI] [PubMed] [Google Scholar]
- Weiner J. 3rd; Bornberg-Bauer E. Mol. Biol. Evol. 2006, 23, 734–743 10.1093/molbev/msj091. [DOI] [PubMed] [Google Scholar]
- Luger K.; Hommel U.; Herold M.; Hofsteenge J.; Kirschner K. Science 1989, 243, 206–210 10.1126/science.2643160. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Bertelsen E.; Benvegnu D.; Alber T. Biochemistry 1993, 32, 12311–12318 10.1021/bi00097a006. [DOI] [PubMed] [Google Scholar]
- Hennecke J.; Sebbel P.; Glockshuber R. J. Mol. Biol. 1999, 286, 1197–1215 10.1006/jmbi.1998.2531. [DOI] [PubMed] [Google Scholar]
- Iwakura M.; Nakamura T.; Yamane C.; Maki K. Nat. Struct. Biol. 2000, 7, 580–585 10.1038/76811. [DOI] [PubMed] [Google Scholar]
- Haglund E.; Lindberg M. O.; Oliveberg M. J. Biol. Chem. 2008, 283, 27904–27915 10.1074/jbc.M801776200. [DOI] [PubMed] [Google Scholar]
- Qian Z.; Lutz S. J. Am. Chem. Soc. 2005, 127, 13466–13467 10.1021/ja053932h. [DOI] [PubMed] [Google Scholar]
- Maatta J. A. E.; Airenne T. T.; Nordlund H. R.; Janis J.; Paldanius T. A.; Vainiotalo P.; Johnson M. S.; Kulomaa M. S.; Hytonen V. R. ChemBioChem 2008, 9, 1124–1135 10.1002/cbic.200700671. [DOI] [PubMed] [Google Scholar]
- Whitehead T. A.; Bergeron L. M.; Clark D. S. Protein Eng. Des. Sel. 2009, 22, 607–613 10.1093/protein/gzp034. [DOI] [PubMed] [Google Scholar]
- Reitinger S.; Yu Y.; Wicki J.; Ludwiczek M.; D’Angelo I.; Baturin S.; Okon M.; Strynadka N. C.; Lutz S.; Withers S. G.; McIntosh L. P. Biochemistry 2010, 49, 2464–2474 10.1021/bi100036f. [DOI] [PubMed] [Google Scholar]
- Guntas G.; Kanwar M.; Ostermeier M. PLoS One 2012, 7, e35998. 10.1371/journal.pone.0035998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A. B.; Govindarajan S.; Lutz S. J. Am. Chem. Soc. 2013, 135, 14425–14432 10.1021/ja4074886. [DOI] [PubMed] [Google Scholar]
- Fox K. M.; Karplus P. A. Structure 1994, 2, 1089–1105 10.1016/S0969-2126(94)00111-1. [DOI] [PubMed] [Google Scholar]
- Karplus P. A.; Fox K. M.; Massey V. FASEB J. 1995, 9, 1518–1526. [DOI] [PubMed] [Google Scholar]
- Brown B. J.; Hyun J. W.; Duvvuri S.; Karplus P. A.; Massey V. J. Biol. Chem. 2002, 277, 2138–2145 10.1074/jbc.M108453200. [DOI] [PubMed] [Google Scholar]
- Massey V.; Schopfer L. M. J. Biol. Chem. 1986, 261, 1215–1222. [PubMed] [Google Scholar]
- Niino Y. S.; Chakraborty S.; Brown B. J.; Massey V. J. Biol. Chem. 1995, 270, 1983–1991 10.1074/jbc.270.5.1983. [DOI] [PubMed] [Google Scholar]
- Vaz A. D. N.; Chakraborty S.; Massey V. Biochemistry 1995, 34, 4246–4256 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- Reich S.; Hoeffken H. W.; Rosche B.; Nestl B. M.; Hauer B. ChemBioChem 2012, 13, 2400–2407 10.1002/cbic.201200404. [DOI] [PubMed] [Google Scholar]
- Reich S.; Kress N.; Nestl B. M.; Hauer B. J. Struct. Biol. 2014, 185, 228–233 10.1016/j.jsb.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Padhi S. K.; Bougioukou D. J.; Stewart J. D. J. Am. Chem. Soc. 2009, 131, 3271–3280 10.1021/ja8081389. [DOI] [PubMed] [Google Scholar]
- Pompeu Y. A.; Sullivan B.; Stewart J. D. ACS Catal. 2013, 3, 2376–2390 10.1021/cs400622e. [DOI] [Google Scholar]
- Qian Z.; Horton J. R.; Cheng X.; Lutz S. J. Mol. Biol. 2009, 393, 191–201 10.1016/j.jmb.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breithaupt C.; Kurzbauer R.; Lilie H.; Schaller A.; Strassner J.; Huber R.; Macheroux P.; Clausen T. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 14337–14342 10.1073/pnas.0606603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z.; Fields C. J.; Lutz S. ChemBioChem 2007, 8, 1989–1996 10.1002/cbic.200700373. [DOI] [PubMed] [Google Scholar]
- Fraaije M. W.; Mattevi A. Trends Biochem. Sci. 2000, 25, 126–132 10.1016/S0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Borek D.; Majewski W.; Minor W. Acta Crystallogr. 2003, 59, 228–234 10.1107/S0108767303005488. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. Acta Crystallogr., Part D: Biol. Crystallogr. 2010, 66, 213–221 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Part D: Biol. Crystallogr. 2004, 60, 2126–2132 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.