

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of Kaposi's sarcoma. K-Rta and K-bZIP are two major viral transcription factors that control reactivation of this virus. Here we report a genome-wide analysis of transcriptional capacity by evaluation of a comprehensive library of 83 putative KSHV promoters. In reporter assays, 34 viral promoters were activated by K-Rta, whereas K-bZIP activated 21 promoters. When K-Rta and K-bZIP were combined, 3 K-Rta responsive promoters were repressed by K-bZIP. The occupancy of K-Rta and K-bZIP across KSHV promoters was analyzed by chromatin immunoprecipitation with a viral promoter-chip in BCBL-1 cells. In addition, acetylation of local histones was examined to determine accessibility of promoters during latency and reactivation. Finally, 10 promoters were selected to study the dynamics of transcription factor recruitment. This study provides a comprehensive overview of the responsiveness of KSHV promoters to K-Rta and K-bZIP, and describes key chromatin changes during viral reactivation.
Keywords: Kaposi's sarcoma, KSHV, HHV-8, Transcription, Chromatin immunoprecipitation, Gene expression, K-Rta/ORF50, K-bZIP/K8, Microarray, Chromatin
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV) is the most recently discovered member of the human gamma-herpesviruses (Ganem, 2006). Like other members of this viral subfamily, KSHV establishes latency in B cells with a capacity to reactivate in response to certain stimuli, including phorbol esters such as 12-O-tetradecanoyl-phorbol-13-acetate (TPA). This reactivation of lytic replication leads to an ordered sequence of gene expression and viral DNA replication, enabling the assembly and release of infectious virus particles (Sun et al.,1999). One KSHV gene in particular, a strong transactivator called K-Rta/ORF50, has the capacity to initiate the entire reactivation cascade when expressed in cells harboring latent virus (Gradoville et al., 2000; Lukac et al., 1998; Sun et al., 1998). This critical role for K-Rta in the life cycle of the virus has produced a large body of work aimed at elucidating its effects on various viral promoters, cellular binding partners, regulation of transcription, and putative DNA binding motifs (Carroll et al., 2007; Chang et al., 2005; Chen et al., 2000; Song et al., 2003; Wang et al., 2001). To date, much of this work has depended on reporter assays and DNA binding experiments, in which the proteins and selected promoter targets are examined in transient expression assays using cell lines such as 293, BJAB, SLK, and others. Although, the results have been highly informative regarding the molecular mechanisms of K-Rta action, these studies were often carried out in the absence of other viral proteins which could modify the action of K-Rta. K-bZIP, a positional homologue of BZLF1 in Epstein–Barr virus (Gruffat et al., 1999; Lin et al., 1999), has been implicated in a broad array of viral processes, including regulation of transcription, cell cycle, host antiviral response, and DNA replication (AuCoin et al., 2004; Izumiya et al., 2003a, 2003b, 2005b, 2007; Rossetto et al., 2007; Wang et al., 2004; Wu et al., 2001). K-bZIP serves as a coregulator of K-Rta (Izumiya et al., 2003a). However, the extent of co-recruitment and the potential regulatory effects of K-bZIP and K-Rta have yet to be investigated in the context of KSHV reactivation from latency.
The stable association of latent viral episomes with host cell nuclei is critical for viral persistence, and this association is presumed to involve epigenetic control of chromatin. One of the levels of such regulation involves covalent modification of the amino-termini of the histone proteins or the “histone code” (Strahl and Allis, 2000). Among these modifications, acetylation of histone 3 on lysine 9 (H3acK9), and the methylation at lysine 4 (H3meK4), are strongly associated with “open” chromatin, undergoing active transcription (Berger, 2007). H3acK9 modification-specific antibodies are particularly valuable in the study of the active chromatin state. One of the well-established chemical triggers of gamma-herpesvirus reactivation is sodium butyrate treatment, which is believed to act through an inhibition of the histone deacetylase complex (HDAC). Different doses of sodium butyrate potentiate varying kinetic patterns of viral gene expression in BCBL-1 cells (Yu et al., 1999). In addition, sodium butyrate treatment synergizes with exogenous expression of K-Rta to drive reactivation in a cell line model based on recombinant KSHV (rKSHV.219), (Vieira and O'Hearn, 2004). Early studies on the latent origins of DNA replication of both EBV and KSHV have also shown local patterns of increased histone 3 acetylation (Day et al., 2007; Stedman et al., 2004), as well as demethylation of the K-Rta promoter, a step linked to histone acetylation, associated with an increase in spontaneous reactivation (Chen et al., 2001). Additionally, the transient expression of CBP (CREB-binding protein), a histone acetyl-transferase (HAT), leads to K-Rta transactivation in a HAT-dependent manner (Lu et al., 2003).
In this work, we performed a screen of all the putative promoters of the KSHV genome, with the intent to define the respective roles of K-Rta and K-bZIP during the reactivation cascade. For promoters strongly activated by K-Rta, the effects of cotransfection with K-bZIP were also tested. To study recruitment of these two viral proteins, the same promoter regions were subsequently printed on glass slides to serve as a substrate for hybridization analysis by ChIP-on-chip assay (Ren et al., 2000). This latter assay served as a tool to examine recruitment of both factors across all the putative promoters of the KSHV genome in primary effusion lymphoma (PEL) cells undergoing reactivation from latency. In addition, the association of viral promoters with acetylated histone 3 at lysine 9 (H3acK9), as a marker for open chromatin, was examined. Finally, a set of ten viral promoters were selected for an analysis of temporal regulation of recruitment and chromatin remodeling of the KSHV episome during reactivation.
Results
K-Rta responsive promoters
K-Rta is a master switch protein of KSHV reactivation, and has been shown to activate many KSHV promoters such as PAN, K9, K12, K14, ORF57, K-bZIP, vIL-6, vMIP-1α, and ORF8 (Chang et al., 2002; Chen et al., 2000; Deng et al., 2002; Duan et al., 2001; Liao et al., 2003; Zhang et al., 1998; Ziegelbauer et al., 2006). The potency of K-Rta transactivation varies widely across different promoters and experimental conditions, from five-fold to over a thousandfold. Accordingly, we developed an approach to simultaneously evaluate all promoters in the same setting, to reveal which viral promoters represent the most potent potential targets of K-Rta. The strategy was based on the understanding that the open reading frames of herpesvirus genes contain little intervening non-coding DNA, such that promoter regions are generally found immediately upstream of each ORF. Although the promoter segments in our library would miss any transcriptional control elements located within the coding region itself, this approach is an effective way to define the potential for transactivation by individual viral genes and provides the basal information to investigate the more complex control of transcription in the physiologically relevant context. Open reading frames that were known to have alternative or distant transcriptional start sites at the time of the library design were included, such as the LTc and LTi promoters of LANA (Matsumura et al., 2005; Staudt and Dittmer, 2006). The predominant pattern of nonstandard transcription for KSHV appears to be common polyadenylation site usage among neighboring genes, in which the functional mRNA for each open reading frame still initiates from a directly upstream promoter (Zheng, 2003). Thus, this library of putative promoters represents a powerful tool to reveal basal regulatory interactions for the majority of the genes of KSHV. These experiments were conducted in 293 cells because of their consistently high transfection efficiency. This line is also permissive for KSHV infection and reactivation, thus presenting a valid cellular context for the study of viral replication.
To survey K-Rta responsive promoters, the reporter constructs were each cotransfected with a K-Rta expression plasmid. Protein expression was confirmed by immunoblotting (Fig. 1, right upper panel). A 10-fold threshold was chosen for significance to eliminate experimental background. By this criterion, K-Rta activated 34 of 83 promoters tested (Fig. 1). Under a more stringent 30-fold cutoff, 19 of the 83 promoters showed strong activation by K-Rta. These genes mainly encode transactivator/regulators (K-bZIP and ORF57), immune-evasion/cellular homologs (ORF4, K2, ORF70/K3, K6, K9, K12, K14, K15 and ORF75), DNA replication-related proteins (ORF6, ORF9, K-bZIP, ORF59, and Ori-RNA), proteins related to nucleotide synthesis/repair (ORF21 and ORF61), and several structural proteins (ORF8, ORF22, ORF19, ORF43, ORF47, ORF64 and ORF67). Among the most highly activated promoters, the well established targets K-bZIP, ORF57, ORF59, and PAN all displayed greater than 200-fold activation. Additionally, twelve putative late promoters, whose gene expression was previously shown to be significantly inhibited by Cidofovir (Lu et al., 2004), were also responsive to K-Rta. These late genes are ORF19, ORF21, ORF22, ORF23, ORF28, ORF42, K10, ORF61, ORF64, ORF67, K12, and ORF75. The majority of these predicted late gene targets were activated less than 30-fold (7 of 12, 58%), whereas three (ORF21, ORF22, ORF28) were activated between 30 and 40-fold. By contrast, the ORF61 and K12 promoters were activated 76 and 99-fold, respectively. This screen served to identify a number of novel potential targets for K-Rta activation, especially among late genes, and agrees well with previous work on early gene targets.
Fig. 1.
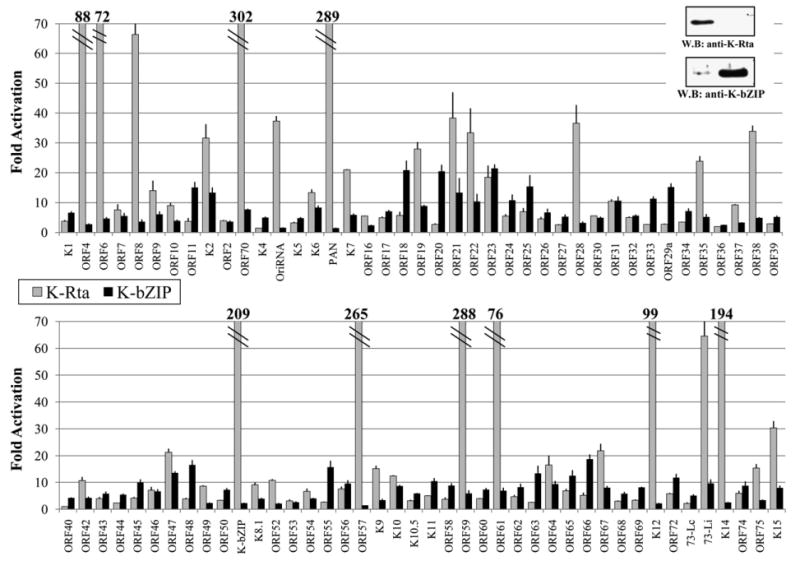
Transactivation by K-Rta (light), or K-bZIP (dark). 293 cells were cotransfected with KSHV promoter reporter constructs along with K-Rta, K-bZIP, or a vector control. Fold activation over vector transfected lysates are shown for each promoter. Upper right panel shows confirmation of expression by immunoblotting.
K-bZIP responsive promoters
Using transient expression assays, we examined responsiveness of all KSHV putative promoters to K-bZIP in 293 cells, and found that 21 of the 83 KSHV promoters displayed at least some activation by K-bZIP, using a 10-fold cutoff (Fig. 1). The highest activation was observed for the ORF23 promoter at 21-fold over vector control. K-bZIP alone did not activate the promoters of any of the eight established DNA replication related genes (ORF6, ORF9, ORF40-41, ORF44, K-Rta, K-bZIP, ORF56, and ORF59) nor immediate-early genes (K-bZIP and ORF57). Interestingly, K-bZIP appeared to transactivate promoters of viral structural genes such as tegument proteins (ORF19 and ORF63), glycoproteins gH and gL (ORF22 and ORF47) and capsid proteins (ORF11, ORF25, and ORF65). These results suggest a model in which K-bZIP has transcriptional activation functions in later time points of gene expression. Indeed, 10 of the promoters activated by K-bZIP were among those determined to be Cidofovir-sensitive (Lu et al., 2004). The potential of K-bZIP to act as a modest transactivator across a broad range of KSHV promoters is a novel finding, which may be important in considering its larger role in viral reactivation.
Combinatorial effects of K-bZIP and K-Rta on major targets of K-Rta transactivation
Building on these results, transcriptional effects of K-Rta and K-bZIP in combination were examined in cotransfection assays. Experiments were performed for all 34 of the K-Rta target promoters showing 10-fold or greater levels of response; each promoter reporter plasmid was cotransfected with K-Rta alone, or both K-Rta and K-bZIP. Protein expression was confirmed by immunoblotting using specific antibodies (Fig. 2A). Consistent with previous work, early lytic promoter activation mediated by K-Rta (ORF57 and K-bZIP promoters) was significantly reduced by K-bZIP co-expression (Izumiya et al., 2003a; Liao et al., 2003), as was the activation of the K15 promoter (Fig. 2B). Surprisingly, the majority of the other K-Rta responsive promoters (23 of 31, 74%) were instead enhanced by K-bZIP, displaying two or greater-fold increase in reporter activity over K-Rta alone. The greatest extent of enhancement was observed for the K9 and ORF67 promoters, at 6.9 and 9.1-fold over K-Rta alone, respectively. A strong majority of the Cidofovir-sensitive late targets of K-Rta (10 of 12, 83%) were among those displaying significant transcriptional cooperativity with K-bZIP, and even the ORF42 and K12 promoters showed a potentially significant increase, although it did not pass our 2-fold criterion for defining synergy. Interestingly, the capacity of K-bZIP for enhancement of transcriptional activation of K-Rta is not well predicted by its own direct action on promoters; less than a quarter (5 of 23, 22%) of the synergistically activated promoters made the 10-fold threshold for K-bZIP activation in the single-factor screening. The only other promoter which could be significantly activated individually by both K-bZIP and K-Rta, ORF47, still showed positive transcriptional cooperativity, but fell short of the 2-fold mark. The broad diversity of K-Rta target promoters enhanced by K-bZIP raises the possibility that the mode of action may be indirect, rather than through individual sequence-specific protein–DNA interactions between K-bZIP and each of the synergized promoters. Reciprocally, the promoters responsive to K-bZIP alone, but not to K-Rta, do not show significant synergistic effect by K-Rta (data not shown); this finding suggests that the two factors do not act on the same motifs and that their direct protein–protein interactions alone are insufficient to direct transcriptional synergy.
Fig. 2.
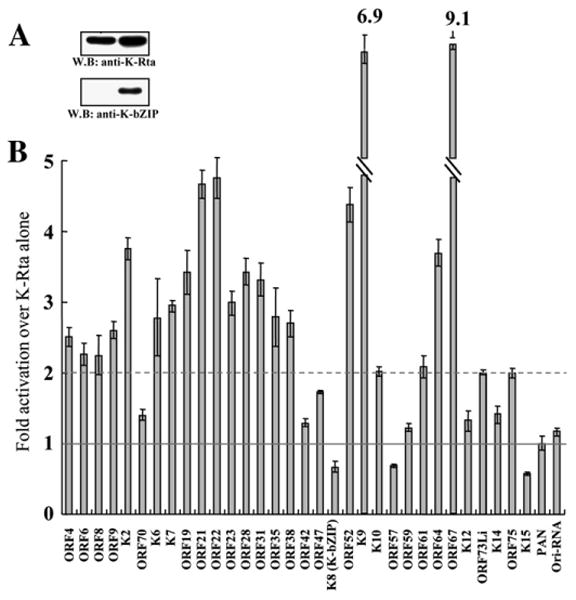
Functional interaction between K-Rta and K-bZIP on K-Rta responsive promoters. (A) Confirmation of expression levels for both factors. (B) Effects of K-bZIP expression on K-Rta transactivation. Luciferase value of K-Rta alone was normalized as 1 (solid line), the broken line at 2-fold represents a doubling of K-Rta activity attributable to K-bZIP cotransfection. Values below 1-fold represent K-bZIP repression of K-Rta transactivation.
Genome-wide promoter array analysis of K-Rta recruitment by chromatin immunoprecipitation
As demonstrated by the reporter assay screen, K-Rta has many transactivation targets in the viral genome. In order to examine which of these potential targets of K-Rta are actually occupied during reactivation, we designed a custom viral promoter array chip based on the same putative promoter fragments examined by reporter assay. Although not a comprehensive map of the entire viral genome, this array enabled analysis of transcription factor recruitment to the putative and established promoters for every known KSHV gene simultaneously. In brief, fixed chromatin complexes from latent or reactivating KSHV-positive primary effusion lymphoma cells were immunoprecipitated with a purified polyclonal antibody to K-Rta or IgG as a specificity control. We employed the well-characterized K-Rta-inducible TREx BCBL1-Rta cells (Nakamura et al., 2003) for these experiments in order to ensure a robust and specific reactivation from latency. The initial focus was on a delayed-early time point of 12 h after induction. In order to minimize overexpression of K-Rta, the cell culture medium was changed after 4 h to remove residual doxycycline. This pulse induction was confirmed to be sufficient to produce strong expression of the late-class glycoprotein, K8.1 by 24 h post induction (Fig. 4). Enriched IP-DNA was subjected to linear amplification, labeled, and hybridized to the custom viral promoter chip against labeled input DNA. The Cy3–Cy5 ratios for each spot on the viral promoter array served as an indication of the relative enrichment of that promoter fragment by the antibody, and therefore as a measure of target protein occupancy on the promoter at the time of fixation (Fig. 3B).
Fig. 4.
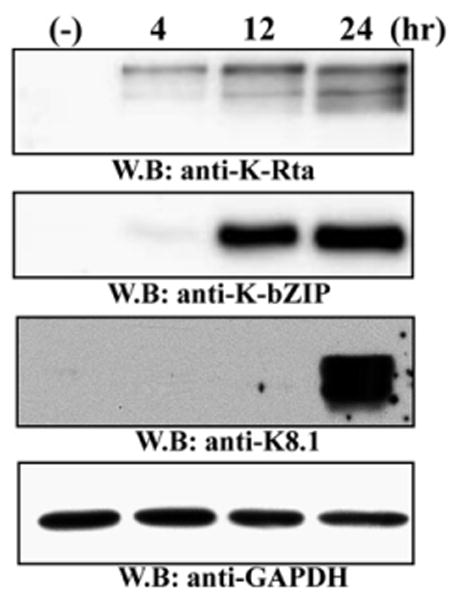
Immunoblotting for K-Rta and K-bZIP expression during reactivation of TREx BCBL1-Rta cells following dox treatment. Expression levels of K-Rta, K-bZIP, and K8.1 are shown for all time points considered in this study. K-Rta expression is detectable by 4 h after treatment, while both K-Rta and K-bZIP are expressed strongly by 12 h. GAPdH served as a loading control.
Fig. 3.
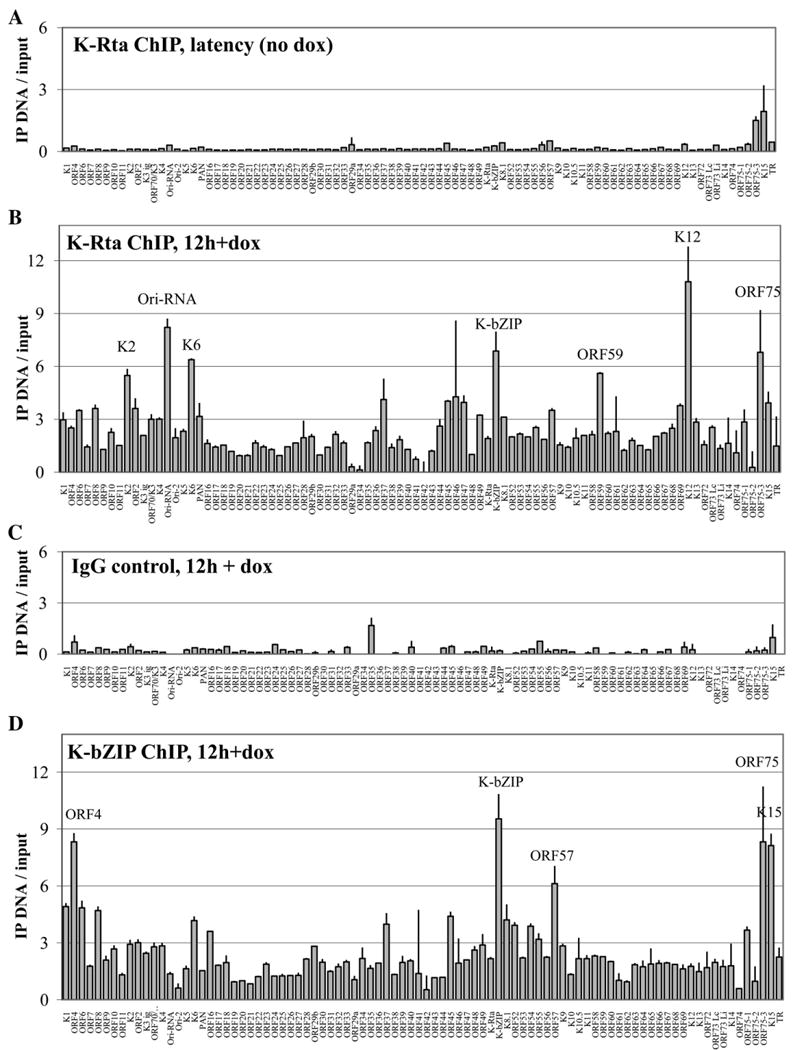
Genomewide promoter association profiles for K-Rta and K-bZIP in TREx BCBL1-Rta cells. Normalized background-subtracted Cy3–Cy5 ratios of linear-amplified IP-DNA over input DNA are shown with standard deviations determined by paired replicate spots. (A) K-Rta promoter occupancy in latent cells with no doxycycline addition. (B) K-Rta distribution at 12 h after induction with dox. (C) Control IgG ChIP-on-chip for 12-hour induced cells. (D) K-bZIP association at 12 h after induction with dox. Promoters are ordered by genomic position; all fragments which were enriched greater than 5-fold are labeled for clarity.
Of the 22 promoters that were more than 3-fold enriched in the K-Rta immunoprecipitate for the 12 h time-point, most (14 of 22, 64%) were also significant potential targets for K-Rta transcriptional activation in 293 cells by the reporter assay screen in Fig.1. However, of the 7 most highly enriched promoters (>5-fold enrichment) for K-Rta association; vIL-6 (K2), Ori-RNA, vMIP-1a (K6), K-bZIP, Kaposin (K12), ORF59, and ORF75, only two (K-bZIP and ORF59) were among the 6 major transactivation targets of K-Rta (>100-fold in 293 cells), despite the fact that we expect all of these major targets to be transcribed at this time point in this system (Nakamura et al., 2003). The relative scarcity of K-Rta on promoters with known binding motifs despite ample protein expression (Fig. 4) suggests that another level of regulation is involved in the context of reactivating latent genomes, that may limit the accessibility of some potential targets of K-Rta binding and transactivation. In contrast, no viral promoter was more than 2-fold enriched by the K-Rta ChIP assay without reactivation (Fig. 3A). Similarly, an IgG control ChIP did not show any enrichment in the same 12-hour induced PEL cells (2-fold cutoff, Fig. 3C).
In order to address specific disagreements between transactivation potential of K-Rta in 293 cells and recruitment of K-Rta in BCBL-1, several additional reporter assays were performed in the same K-Rta inducible BCBL-1 cell line used for the ChIP experiments. Five promoters that displayed K-Rta recruitment in infected cells (>3.5-fold), yet failed to respond to K-Rta in 293 cells, were examined in this context alongside seven known K-Rta transcription targets as positive controls. Reporter plasmids were transfected into TREx-K-Rta BCBL-1. At 36 h post-transfection, K-Rta expression was induced with doxycycline, and luciferase activity was measured 12 h after K-Rta induction. This time-point was chosen to correlate directly with the K-Rta recruitment experiment (Fig. 3A). The majority of the promoters of interest (4 of 5) displayed enhanced reporter activity in BCBL-1 as compared to 293 cells, with 3 promoters (ORF37p, ORF45p, ORF46p) achieving the 10-fold cutoff in these cells (Fig. 5A). This finding suggests that either B-cell specific transcriptional factor(s) or viral factor(s) are required for effective transactivation of these promoters. The expression level of K-Rta under these conditions is shown in Fig. 5B. In particular, the ORF37 promoter was activated over 50-fold, for which we detected less than 10-fold activation by K-Rta in 293 cells. Similarly, the promoters of K6, K12, and the Ori-RNA were also notably enhanced in BCBL-1. The only putative promoter which appeared to be an occupancy site of K-Rta during reactivation, without appreciable K-Rta transactivation by reporter assay, was ORF2, which displayed no significant reporter activity in either context. Interestingly, all seven examined promoters that were activated more than 10-fold in 293 cells were again activated in BCBL-1 at least 10-fold; this finding supports our initial approach.
Fig. 5.
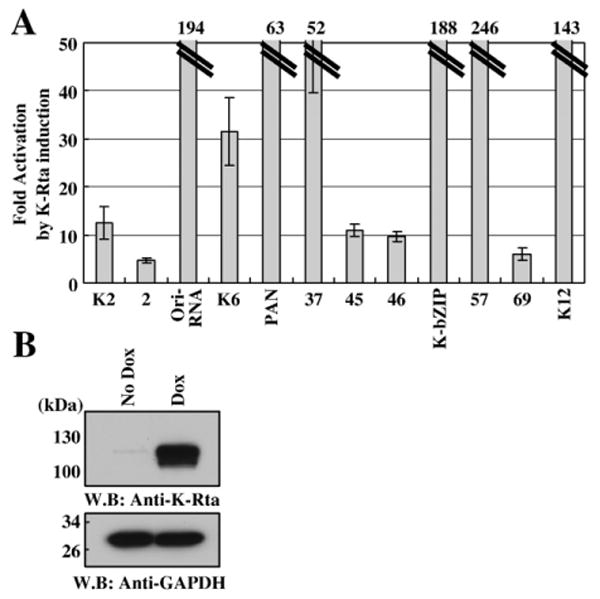
Transactivation of selected promoters in TREx BCBL1-Rta cells. Promoter reporter constructs were transfected into TRExBCBL1-Rta cells which were subsequently induced to express K-Rta and initiate reactivation by dox treatment for 12 h. (A) Fold induction of relative luciferase units between untreated (no K-Rta) and dox-treated cells. (B) Expression level of K-Rta by western blot in uninduced or induced TREx BCBL1-Rta cells at 12 h. GAPdH is included as a loading control.
Genome-wide promoter array analysis of K-bZIP recruitment by chromatin immunoprecipitation
When the ChIP-on-chip assay was performed on induced TREx BCBL1-Rta cells using the purified antibody raised against K-bZIP, the binding profile was more restricted than the one observed for K-Rta. Strikingly, all three repression target promoters (K15, ORF57, and K-bZIP) were more than 6-fold enriched for K-bZIP occupancy (Fig. 3D). The K-bZIP promoter was the most strongly enriched target, at more than 9.5-fold over input hybridization levels. Additionally, we also measured greater than 6-fold enrichment of the ORF4 and ORF75 promoters, although neither of these showed significant (2 or greater fold) potential for transcriptional synergy or repression between K-bZIP and K-Rta in the reporter assay screen. Using a less stringent (4-fold) cutoff, K-bZIP was found to be associated with six additional promoters at 12 h post reactivation: K1, ORF6, ORF8, K6, ORF45, and K8.1. Interestingly, none of the eleven K-bZIP ChIP association targets were significant (greater than 10-fold) direct transactivation targets of K-bZIP by the reporter assay screen. Indeed, among this set, the highest transactivation attributable to K-bZIP alone was the ORF45 promoter, at 10-fold over vector alone. This finding raises the hypothesis that the coactivation potential of K-bZIP is not mediated through direct binding.
KSHV promoter association with acetylated histones during latency and reactivation
To further investigate the state of the viral chromatin in PEL cells during reactivation, a ChIP assay was performed using an antibody against acetylated lysine at position 9 of histone 3, which is a well-established marker of “open” or active promoters. Acetylated histone association was profiled for 12-hour doxycycline treated cells (Fig. 6B), as well as for latent cells not treated with doxycycline (Fig. 6A). In reactivation, the most highly enriched promoters for H3acK9 (>12-fold) at the 12-hour time point were somewhat unexpected: K2 (vIL-6), ORF2 (DHFR), MIR2 (K5), ORF16 (vBCL-2), ORF47 (gL), and K-bZIP. Of these six most highly associated promoters, only K2 and K-bZIP promoters were associated with (>5-fold) K-Rta occupancy at this time point. Examination of the H3acK9-associated promoters at a less stringent 5-fold cutoff revealed a total of 48 enriched promoters, more than half of the entire complement of KSHV. These included the great majority of the K-Rta-associated promoters (18 of 21, 86%, >3-fold), as well as all the promoters on which K-bZIP was detected (11 of 11, >4-fold). Interestingly, this analysis also identified two clusters of late gene promoters that were depleted for the acetylated histone mark; the region from ORF19–ORF33, as well as ORF61–ORF65. These areas agree well with regions of Cidofovir-sensitive late genes, and as such may represent regions of the genome tightly regulated by chromatin structure during reactivation.
Fig. 6.
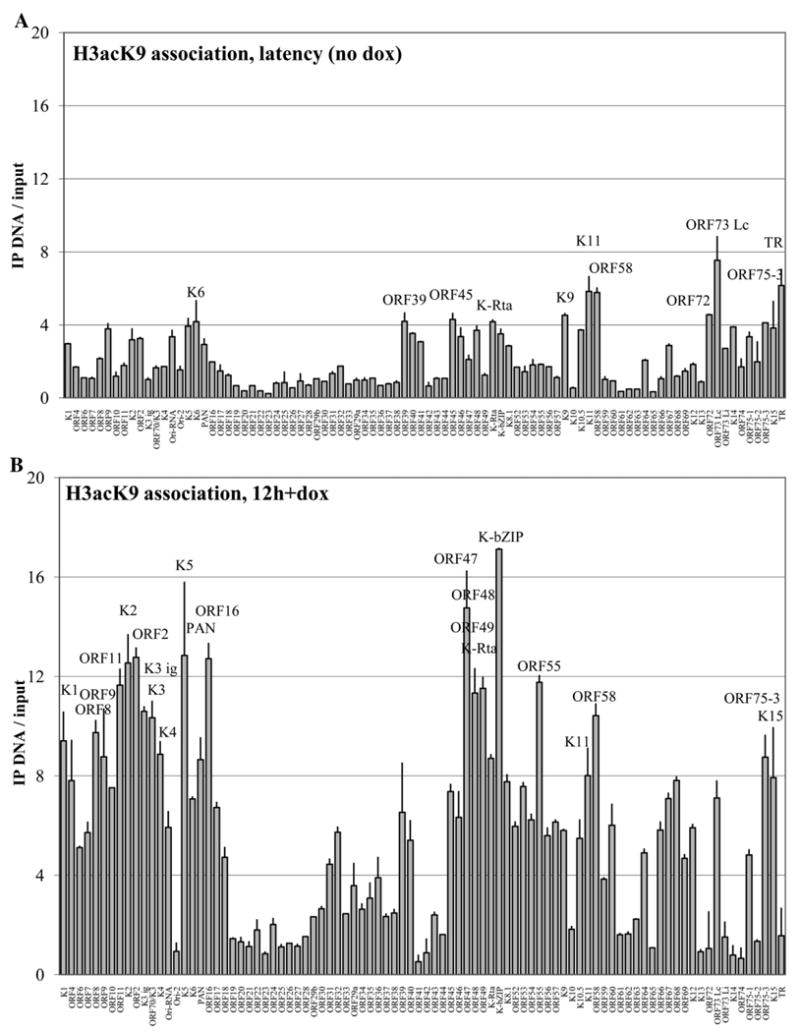
Genomewide KSHV promoter association profiles with acetylated histone 3 at lysine 9 in TREx BCBL1-Rta cells. (A) Latent genome promoter association with acetylated histone 3, no dox treatment; fragments over 4-fold enriched are labeled. (B) Reactivation profile, 12 h after doxycycline treatment; promoter fragments above 8-fold are labeled. Normalized Cy3–Cy5 ratios of background-subtracted IP-DNA over input DNA are shown with standard deviations determined by paired replicate spots.
Strikingly, when the state of viral chromatin was examined in latently infected cells, only two array promoter fragments were enriched for H3acK9 association more than six-fold over input DNA (Fig. 6A). The strongest enrichment (7.5-fold) was on the ORF73-Lc (LANA) constitutive latent promoter, followed by the fragment corresponding to the KSHV terminal repeat sequence (6.2-fold), which serves as the latent origin of replication. Of the 11 array spots that displayed a greater than 4-fold increase, nine (82%) were promoters enriched more than 5-fold in the H3acK9 ChIP assay after doxycycline induction. This finding is suggestive of a low but detectable background level of spontaneous reactivation in the TREx BCBL1-Rta cells under these conditions.
Studies of dynamics of K-Rta and K-bZIP recruitment
To verify the observations made with the ChIP-on-chip method, 10 promoters were selected for individual analysis, using real-time quantitative PCR. This set focused on the previously characterized targets of K-Rta binding (K2, K6, PAN, K-bZIP, ORF57, K12). For those promoters that have published K-Rta response elements (RRE), real-time PCR primer pairs were designed to directly target those regions (Table 2). The greater sensitivity of the PCR-based method also enabled analysis of a dilution of the immunoprecipitated DNA directly, without additional PCR-based linear amplification. TREx BCBL1-Rta cells were fixed at 4, 12, and 24 h after doxycycline treatment, and ChIP was performed with specific antibodies to study the recruitment of both viral transcription factors, as well as to examine association with acetylated H3K9. Comparison of the qPCR samples harvested at 12 h with those analyzed by ChIP-on-chip revealed agreement between these two methods for all 10 promoters assayed (Fig. 7). The vIL6 (K2), vMIP-1a (K6), PAN, and Kaposin (K12) promoters were the major sites of K-Rta occupancy at 12 h by both assays, although the fold enrichment was much greater in the qPCR assay (25–120-fold) as compared to the ChIP-on-chip assay (3–12-fold, Fig. 3B). The difference may be due to the sensitivity of the detection methodologies, or the comparative enrichment over input DNA (ChIP-on-chip) versus the comparison of fold enrichment over an IgG ChIP control (qPCR method). Likewise, K-bZIP binding across the validation set agreed well, with ORF57 and K-bZIP promoters displaying the highest levels of recruitment, and negligible occupancy on promoters absent from the ChIP-on-chip hybridization (PAN, K12) (Figs. 7B and C).
Fig. 7.
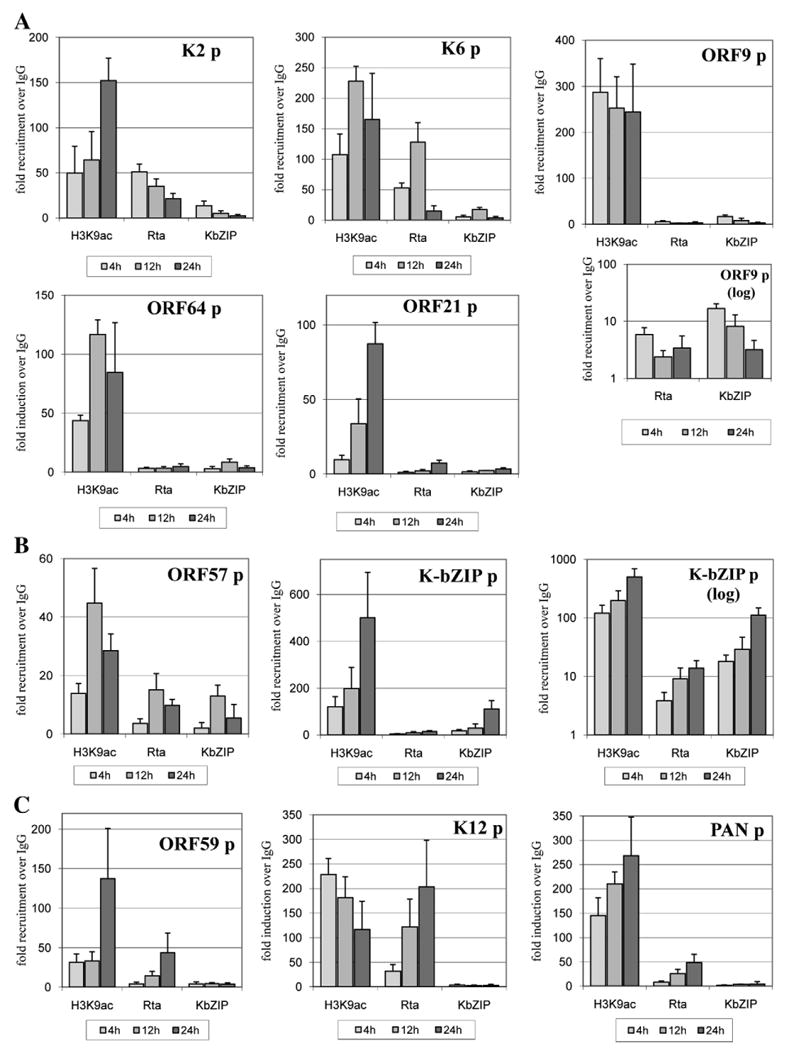
Time-course analysis of factor recruitment to selected KSHV promoters during reactivation. All values are expressed as fold increase over IgG control pull-down, assayed by real-time PCR at 4, 12, and 24 h after doxycycline treatment. ChIP was performed for IgG, K-Rta, K-bZIP, and H3acK9. (A) Five KSHV promoters were examined on which K-bZIP and K-Rta synergize. (B) Profiles for two K-Rta target promoters repressed by K-bZIP expression. (C) Recruitment profiles for three additional K-Rta target promoters on which K-bZIP had a little to no transcriptional effect. The ChIP elutions were assayed directly by real-time PCR after a 1:50 dilution. ORF9 and K-bZIP promoters are displayed in both linear and log scale due to the strength of the H3acK9 association with these fragments.
The flexibility of the real-time qPCR approach enabled further investigation of the temporal regulation of K-bZIP and K-Rta recruitment. The primer set included 5 promoters on which K-bZIP showed greater than 2-fold transcriptional synergy of K-Rta activity (ORF9, K2, K6, ORF21, ORF64), as well as two K-bZIP-repressed promoters (K-bZIP, ORF57), and 3 promoters on which K-bZIP did not affect K-Rta activation (PAN, ORF59, K12). All 10 promoters selected for more detailed analysis displayed potential for activation by K-Rta greater than 10-fold (Fig. 1), and 5 of them were among the most potent K-Rta transactivation targets by reporter assay, at greater than 90-fold activation in 293 cells. These time-course analyses in the PEL cells revealed that K-Rta was recruited to an increasingly larger set of its potential transactivation targets as the reactivation progressed over time. At 4 h after induction, only 5 of the examined K-Rta target promoters displayed recruitment of greater than 5-fold over the IgG control. These included strong early promoter targets PAN and K12, as well as less potent transactivation targets ORF9, K2, and K6. By 12 h, 7 of 10 were observed at this stringency, whereas 9 of the 10 showed K-Rta recruitment by 24 h (Fig. 7). The additional recruitment at later time points included promoters of early and delayed-early genes such as ORF59 and K-bZIP, and at 24 h, the late gene promoter of ORF21. Another late gene promoter, ORF64, did not exceed 4.5-fold K-Rta enrichment by 24 h. Altogether, these results demonstrate that K-Rta distribution across the selected set of target promoters is regulated temporally during the progression of KSHV reactivation from latency.
The genome-wide ChIP-on-chip results for H3acK9 demonstrated a correlation between the increase in promoters associated with open chromatin and the progression of reactivation. Delayed and late gene promoters such as ORF59, ORF64 and ORF21 showed comparatively less enrichment from an H3acK9 pull-down (less than 50-fold), especially at 4 and 12 h time points, although all three increased appreciably by 24 h (Figs. 7A, C). In contrast, very early target promoters PAN, K-bZIP, and K6 exhibited greater than 100-fold enrichment for H3acK9 as early as 4 h, and increased thereafter to 200 or greater fold by 12 h. For all ten promoters examined, significant levels of H3acK9 association were observed before or concurrent with K-Rta or K-bZIP binding. The ORF9 promoter, which could be activated 14-fold by K-Rta in reporter assays, was strongly and consistently associated with H3acK9 (>200-fold), but did not exceed a 6-fold enrichment for K-Rta recruitment during the 24-hour time course analyzed. In contrast, the potent K-Rta target promoter, ORF57, was only 5 to 15-fold enriched for K-bZIP and K-Rta binding while in the context of comparatively low (14–45-fold over IgG) association with H3acK9 (Fig. 7B). This result indicates that viral chromatin structure may determine the accessibility of potential transactivation targets over time.
As a next step, a potential correlation between mode of activation and recruitment of K-bZIP was examined. The ten promoters selected for more detailed analysis included five which appeared to be synergistically activated by K-bZIP when tested in combination with K-Rta. Of those, four (K2, K6, ORF9, and ORF64, 80%), displayed detectable K-bZIP occupancy (8–17-fold) during the 4 and/or 12 h time points, with a loss of association by 24 h post reactivation (Fig. 7A). Additionally, each of these promoters displayed a stable or increasing association with acetylated histone 3 at lysine 9, across the 12 and 24 h time points. The additional late gene synergy target, the ORF21 promoter, only displayed K-Rta occupancy at 24 h, indicating that synergistic activation observed in reporter assays was likely due to indirect effects. K-Rta was most heavily recruited to the K2 and K6 early synergy promoters at 4 and 12 h (35–128-fold over IgG), tapering off by 24 h. Among three promoters on which K-bZIP had no effect (positive or negative) on K-Rta transactivation in 293 cells, none appeared to be significantly bound by K-bZIP in the reactivating PEL cells (all less than 5-fold over IgG; PAN, ORF59, K12), showing specificity of this experiment (Fig. 7C). On the other hand, two viral promoters on which K-bZIP produces a repressive effect on K-Rta transactivation, ORF57 and K-bZIP, were the most heavily enriched sites for K-bZIP occupancy within the set of 10 promoters assayed by real-time qPCR-ChIP (Fig. 7B). The peak recruitment of K-bZIP to its own promoter was at 24 h (111-fold over IgG), whereas the ORF57 promoter had a peak at 12 h after induction. Interestingly, the comparative levels of K-bZIP occupancy at these two promoters across the 4, 12, and 24 h time points paralleled the recruitment of K-Rta in a manner suggestive of co-recruitment in a regulatory complex.
Discussion
This work establishes an overview of K-bZIP and K-Rta interactions with the entire complement of putative promoters contained within the KSHV genome. Although some capacity for K-bZIP-mediated transactivation has been previously shown with GAL4 fusions of N-terminal deletions of K-bZIP via Ini1/hSNF5 (Hwang et al., 2003), as well as a potential for recruitment of CREB-binding protein (Hwang et al., 2001), this is the first time that full-length K-bZIP has been demonstrated to activate viral promoters. The experimental investigation of the combined effects of K-bZIP and K-Rta together revealed an unexpected and significant capacity for transcriptional cooperativity between these two viral gene products. This finding was in marked contrast to our earlier findings, based on a detailed study of the K-bZIP, ORF57, and PAN promoters, in which the former two were repressed by K-bZIP expression, and the latter unaffected (Izumiya et al., 2003a). Synergy between ZEBRA and Rta of EBV in reactivation from latency is well established (Carey et al.,1992). Additionally, in our analyses K15 was identified as an additional viral promoter target for K-bZIP-mediated repression. K15 is a complex spliced transmembrane protein, which may play an important role in modulation of host cell gene expression during reactivation (Brinkmann et al., 2007; Choi et al., 2000). Although the specific role of K15 in reactivation has not yet been established, microarray analysis of the TREx BCBL1-Rta cells by others places this gene in the same expression class as K-bZIP and ORF57, on the basis of the timing by which the transcript level initially doubles after reactivation (Nakamura et al., 2003). All three of these viral proteins represent regulatory factors with potential to alter the balance of viral and cellular factors in the critical early stages of reactivation. Intriguingly, when K-bZIP was deleted in the context of a recombinant BACmid clone, the expression of several viral transcripts was enhanced during reactivation, although reactivation overall was deficient unless K-Rta was strongly over-expressed (Kato-Noah et al., 2007). This finding supports a key role for K-bZIP in modulating gene expression mediated by K-Rta, a function which is critically important to reactivation under normal conditions of infection.
K-bZIP was recruited preferentially to viral promoters on which it has a repressive potential during reactivation of KSHV in naturally infected PEL cells. The two modes of K-bZIP interaction with K-Rta appear to be distinct in terms of their recruitment to individual promoters over time. In the case of the repressive complex for the ORF57 and K-bZIP promoters, the association is strong across the time-course examined. Similarly, the K15 promoter shows significant occupancy by both K-Rta and K-bZIP as demonstrated in the by ChIP-on-chip assay. In fact, all 3 repression targets of K-bZIP are among the 5 predominant sites of K-bZIP occupancy in TREx BCBL1-Rta cells during reactivation. Because K-bZIP was previously demonstrated to directly bind the PRDIII-I region of the IFN-beta promoter (Lefort et al., 2007), we examined the ORF57, K15, and K-bZIP promoters for these sites. Although limited sequence similarities exist in all three promoters, closer matches were also found in non-associated promoters such as K12, indicating that that IRF-3 response elements cannot fully explain the binding specificity of K-bZIP in the context of the viral genome. In contrast to the highly enriched repression sites, the viral promoters with demonstrated synergistic potential involve a lower level transient recruitment of K-bZIP in the context of reactivation in PEL cells. One explanation for this relationship may be through recruitment by K-bZIP of chromatin remodeling systems such as SWI/SNF through hSNF5 interaction, or histone acetylase activity via CBP; either of these mechanisms could lead to increased accessibility of those promoters at later time points by a “hit-and-run” mode of regulation. Alternatively, K-bZIP could mediate coactivation of viral promoters indirectly by sequestering limiting factors important for normal host gene transcription, freeing more core transcription resources for direction by other specific viral transcription factors such as K-Rta. Indeed, we see a broad repression of host gene transcription in cells which overexpress K-bZIP (unpublished data), and others have reported specific examples of this for CBP-mediated transcription (Hwang et al., 2001). The argument for indirect synergy is also intriguing when examining the cooperative reporter assay results for the PAN and K7 promoters, which are overlapping and collinear. Despite being entirely contained within the larger PAN promoter fragment and sharing the same K-Rta response element, the K7 promoter construct was synergistically activated by K-bZIP, whereas the PAN promoter was not. If binding to a specific DNA motif was the sole determinant of K-bZIP coactivation, we would expect the same effect from K-bZIP cotransfection on both promoter constructs. Interestingly, K-bZIP occupancy remains low or even decreases on some promoters (K2, ORF9), despite a dramatic increase in overall K-bZIP expression levels from 4 to 12 h. This suggests that the distribution of K-bZIP across the viral genome is not static, but rather governed by other cofactors or local conditions which shift over the course of reactivation. It is conceivable that other early or delayed-early genes may act on K-bZIP directly or through competition for partners or sites by 12 h. Furthermore, by 24 h, the onset of late gene expression and viral DNA replication may dramatically change the environment in which viral genomes and K-bZIP interact. The limited time course examined in this study was insufficient to address these questions, however the finding serves to illustrate the dynamic nature of factor recruitment in a physiological context.
Examination of K-Rta recruitment across the entire complement of viral promoters during reactivation also offers novel insight into functions of this viral protein and its distribution across KSHV promoters during reactivation. Although we observed significant occupancy at well-established target promoters (K-bZIP, PAN, ORF59, etc), the most highly enriched promoters for K-Rta binding include several on which it has only a moderate transactivation potential, such as K2, K6, ORF37 (SOX), ORF75, K15, and the lytic origin region (OriRNA). Because K-Rta has been demonstrated to act both through direct DNA binding, as well as through host transcription factors such as Oct-1, RBP-JK and K-RBP (Carroll et al., 2007; Chen et al., 2001; Wang et al., 2001; West and Wood, 2003), this finding may reflect differences in the mode of recruitment. ORF57 and K6 promoters share a significantly conserved motif for indirect K-Rta binding, which includes overlapping RBP-JK and SP1 sites. The PAN and K12 promoters share another motif that lacks RBP-JK consensus and are directly bound by purified K-Rta in vitro (Chang, Shedd, and Miller, 2005). Comparing K12 and PAN promoters, we found a much higher occupancy of K-Rta on the K12 promoter across the same time points, despite comparable levels of H3acK9 association, which would otherwise suggest comparable promoter accessibility. This result is in contrast to in vitro gel-shift studies, in which K-Rta was demonstrated to have a higher affinity for the PAN Rta-response element than the K12 one, and further, that K-Rta binding to the K12 RRE was enhanced by a 2-base mutation to more closely resemble the PAN promoter (Song, Deng, and Sun, 2003). Our finding may therefore reflect augmentation of K-Rta binding by host factors on the K12 promoter that do not similarly influence the PAN promoter. When this manuscript was under review, another study examined the initial transactivation targets of K-Rta using a fusion of the estrogen receptor with ORF50 in combination with an inhibitor of translation (Bu et al., 2008). Of the eight transcripts proposed as direct targets of K-Rta in that work, seven showed appreciable promoter response to K-Rta in our reporter assays (K2, K9, PAN, ORF37, ORF52, ORF57, and K14); only ORF56 putative promoter failed to do so. The lack of luciferase activity for the putative promoters of ORF56 may be explained by upstream transcriptional initiation. Our ORF56 promoter fragment may be too short to respond well to K-Rta, as it only includes 81 bp of sequence upstream of the two reported transcriptional start sites mapped in JSC-1 cells, due to initiation more than 350 bp upstream of the open reading frame (Majerciak et al., 2006). As for ORF37, it has recently been shown that this gene is transcribed from a site, 5′ of ORF35 (Haque et al., 2006), corresponding to our ORF35 putative promoter which responded well to K-Rta activation in our reporter assay. Intriguingly, our proximal ORF37 promoter fragment was strongly activated by K-Rta in BCBL-1 reporter assays, but not in 293 cells, raising the possibility that the transcriptional initiation of this region may differ significantly by cell line.
Investigations of KSHV chromatin associations are not without precedent. However, this work represents the first time such an analysis has been performed across all putative promoter regions of the viral genome. The genomic profile obtained in latent PEL cells for acetylated histone 3 at lysine 9 association agrees with previous viral ChIP experiments using a limited set of primers to represent key areas of the genome (Stedman et al., 2004). Both studies demonstrated enrichment of the ORF73 promoter and terminal repeat regions by H3acK9 immunoprecipitation. Our focus, however, was on early and delayed-early regulation. During reactivation, we observed broad and increasing H3acK9 association with most of the viral promoters examined. One notable exception to this pattern was the ORF57 promoter, which peaked at 12 h after induction and subsequently decreased. Intriguingly, this decrease in H3acK9 association corresponded to a decrease in K-bZIP and K-Rta occupancy despite ample expression of the factors at that time point as detected by immunoblotting. Given the known potential of K-bZIP to repress K-Rta transactivation of the ORF57 promoter, this behavior is suggestive of a role for chromatin remodeling in the mechanism of repression, leading to a restriction of access to the promoter. In contrast, ORF59, a delayed-early gene, was not substantially increased in promoter association with H3acK9 until 24 h post induction, at which point it coincided with strongly increased K-Rta occupancy. This observation is also supportive of histone acetylation as a potential temporal regulatory mechanism for the kinetics of K-Rta transcriptional targets, although different approaches may be necessary to determine if modification of viral chromatin actually controls the distribution of K-Rta, or if changes in viral chromatin structure are dictated by K-Rta recruitment. Similarly, the KSHV promoter profile for H3acK9 association during reactivation also has implications for regulation of late gene expression. Our studies revealed two islands of putative late genes (ORF19–28, and ORF61–65), which remained relatively depleted for acetylated histone H3 during reactivation, in sharp contrast to the significant H3acK9 enrichment over much of the rest of the viral genome. Given the potential of K-bZIP and K-Rta to activate many of these same late gene promoters, the lack of occupancy for both factors during early and delayed early time points on these late promoter islands further supports the notion that these regions are regulated by a “closed” chromatin structure. This also raises the possibility of a role for cellular factors such as CTCF in maintaining boundaries between the early and late regions of the genome, as reported for Epstein–Barr virus (Chau et al., 2006).
In summary, our findings advance the understanding of the roles of K-bZIP and K-Rta in KSHV reactivation by revealing a novel capacity for transcriptional synergy across a wide range of late gene promoters. Additionally, this comprehensive analysis of KSHV transcription demonstrates the specificity of the repressive role of K-bZIP on the ORF57 and K-bZIP promoters. The identification of K15 as an additional potential target of this activity is also a novel finding, which better informs our understanding of repression mediated by K-bZIP, and may additionally suggest a role for K15 in the early lytic reactivation cascade. The observation that K-bZIP is found most abundantly on viral promoters that it represses underscores its evolving role as a modulator of K-Rta activity, essential in orchestrating the complex sequence of events after the lytic cycle is initiated. In addition, the K-Rta chromatin and transactivation profiles represent the first methodologically unbiased comparison of K-Rta action and association across the entire complement of putative KSHV promoters. Although the examined set of upstream regions cannot represent all possible regulatory regions of the viral genome, our study represents an important initial survey of the chromatin state and occupancy sites of these two critical KSHV factors during lytic reactivation. The examination of changes in viral factor and acetylated histone recruitment across the full set of promoters in induced PEL cells sheds new light on the regulation of reactivation, and offers significant insight into the role of chromatin structure in the control of KSHV replication.
Materials and methods
Cell culture
Human embryonic kidney epithelial 293 cells were grown in monolayer cultures in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) or serum free medium (Pro293a-CDM: Cambrex Bio Science, Walkersville, MD) in the presence of 5% CO2. TREx BCBL1-Rta cells (Nakamura et al., 2003) were maintained in RPMI 1640 supplemented with 15% fetal bovine serum, 2 mM L-glutamine, and antibiotics, and induced for K-Rta expression and subsequent KSHV reactivation by treatment with 100 ng/mL doxycycline for 4 h.
KSHV promoter library and hybridization array
The KSHV promoter library was created based on published work where available, otherwise putative KSHV promoter regions of approximately 500 bp upstream of AUG of respective open reading frame were amplified by PCR using Pfu-turbo (Stratagene) with corresponding primers. Amplified fragments were digested using restriction enzyme sites (Fermentas), introduced via primer sequences and inserted upstream of the firefly luciferase coding region (Luc) of pGL3 basic vector (Promega). Either purified BCBL-1 genomic DNA or a KSHV cosmid library (Izumiya et al., 2005) were used as the PCR template. Cloned promoter sequences were confirmed by sequencing, and differences identified between cloned promoter sequence and published KSHV genomic DNA sequence (Russo et al., 1996) were summarized as a footnote in Table 1. These plasmids were then sent for commercial amplification and spotting onto glass slides (KamTek Inc., Gaithersburg, MD). The 83 individual viral promoter fragments were spotted in duplicate to each of the array chips, along with 17 non-viral control fragments. Hybridization specificity was confirmed using known KSHV promoters as probes.
Table 1.
Location of cloned KSHV promoters
| Promoters | Location | Promoters | Location | Promoters | Location | Promoters | Location |
|---|---|---|---|---|---|---|---|
| K1 | 1–100 | ORF22 | 36423–37111 | ORF45 | 68577–69223 | ORF64 | 103388–103995 |
| ORF4 | 642–1141 | ORF23 | 40519–41105 | ORF46 | 69405–69892 | ORF65 | 112444–112906 |
| ORF6 | 2686–3209 | ORF24 | 42791–43478 | ORF47 | 69916–70409 | ORF66 | 113765–114279 |
| ORF7 | 6146–6627 | ORF25 | 42080–42776 | ORF48 | 71382–71870 | ORF67 | 114509–115052 |
| ORF8 | 8192–8698 | ORF26 | 46401–46930 | ORF49 | 72539–73166 | ORF68 | 114230–114767 |
| ORF9 | 10777–11362 | ORF27 | 47211–47865 | ORF50 | 70899–71595 | ORF69 | 115979–116668 |
| ORF10 | 13921–14520 | ORF28 | 48318–48990 | K–bZIP | 74691–74849 | K12 | 118717–119351 |
| ORF11 | 15171–15784 | ORF29b | 49855–50379 | K8.1 | 75409–75914 | ORF72 | 123567–124218 |
| K2 | 18554–19054 | ORF30 | 49977–50622 | ORF52 | 77198–77647 | ORF73 Lc | 127773–128309 |
| ORF02 | 19706–20203 | ORF31 | 50258–50762 | ORF53 | 77666–78153 | ORF73 Li | 127496–127812 |
| K3 | 21835–22291 | ORF32 | 50890–51402 | ORF54 | 77106–77666 | K14 | 127238–127873 |
| ORF70 | 1–100 | ORF33 | 52228–52760 | ORF55 | 79449–80058 | ORF74 | 128817–129371 |
| K4 | 642–1141 | ORF29a | 54678–55320 | ORF56 | 78792–79435 | ORF75 | 134442–135061 |
| Ori-RNA | 23613–24288 | ORF34 | 54112–54673 | ORF57 | 81556–82089 | K15 | 136281–137039 |
| K5 | 26485–27134 | ORF35 | 55144–55638 | K9 | 85225–85875 | ||
| K6 | 27425–28127 | ORF36 | 55645–55975 | K10.5 | 91397–91904 | Controls and others | |
| PAN | 27426–28680 | ORF37 | 56744–57272 | K10 | 88202–88879 | Ori region | 25062–25705 |
| K7 | 27949–28620 | ORF38 | 58145–58687 | K11 | 93369–94017 | Terminal repeat | 801 bp NotI fragment |
| ORF16 | 29461–30142 | ORF39 | 60234–60766 | ORF58 | 95549–96029 | ORF75–2 | 134890–135676 |
| ORF17 | 32482–33105 | ORF40 | 59800–60307 | ORF59 | 96742–97248 | ORF75–3 | 135643–136218 |
| ORF18 | 31806–32423 | ORF41 | 61285–61826 | ORF60 | 97789–98304 | Human Ch.19 | 162122–162594 |
| ORF19 | 34844–35482 | ORF42 | 63308–63754 | ORF61 | 100195–100733 | Sumo–1 coding sequence Ubc9 coding sequence |
|
| ORF20 | 35573–36226 | ORF43 | 64954–65527 | ORF62 | 101195–101757 | Yeast genomic DNA | 4950–5393 (Acc#U18916) |
| ORF21 | 34707–35379 | ORF44 | 64307–64891 | ORF63 | 100525–101207 | Yeast genomic DNA E. coli genomic DNA |
13798–14065
(Acc#U11581) 3473366–3473971c (Acc#U00096) |
Cloning of putative KSHV promoters. Putative KSHV promoters were cloned in pGL3 vector. Location of cloned promoters were listed, and number is based on published sequence (Acc#U75698). Sequences were verified by sequencing analysis. Point mutations and deletion of direct repeat were observed following promoters, ORF16 promoter (deletion of 29787–29929), ORF19p (nt 35221 G to T), ORF20p (nt 35598 T to C), ORF23p (nt 40548 T to C), ORF24p (nt 43296 T to C), ORF30p (nt 50157 G to A and 50394 A to C), ORF31p (nt 50394 A to C), ORF32p (nt 51276 A to C), ORF47p (nt 70182 C to T), ORF60p (nt 98125 G to A), ORF61p (nt 100324 G to A and 100241 C to T) and ORF69p (nt 116039 A to C).
Reporter assays
293 cells were seeded in 12-well plates at 1×105/well in 1.0 mL of DMEM supplemented with 10% FBS and incubated at 37 °C with 5% CO2. An equal amount of plasmid DNA, including the reporter and control/expression plasmid, were transfected using the Fugene6 reagent by following the manufacturer's protocol (Roche). Cell lysates were prepared with 1× Passive Lysis Buffer (Promega), 48 h after transfection.
TREx BCBL1-Rta cells were seeded in 12-well plates at 5×106/well in 1.0 mL of medium. Reporter plasmid (0.5 μg/well) was transfected with Fugene6 HD by following the manufacturer's protocol (Roche). Thirty-six hours after transfection, K-Rta expression was induced with doxycycline, and cell lysates were prepared with 1× Passive Lysis Buffer after 12 h induction of K-Rta. Luciferase assay was performed according to the manufacturer's protocol using a Lumat LB 9501 Luminometer (Wallac Inc., CA). At least two independent experiments were performed in the same setting.
Western blotting
293 or TREx BCBL1-Rta cells were lysed in 1× RIPA buffer (10 mM Tris–HCl [pH 7.5],1.0% NP-40, 0.1% sodium deoxycholate,150 mM NaCl, 1 mM EDTA) supplemented with Complete™ protease inhibitor cocktail (Roche) and frozen at −80 °C. Samples of these cell lysates were balanced for total protein level using the Bio-Rad Protein Assay reagent in comparison with a standard curve was generated using bovine serum albumin (BSA, New England Biolabs), and 50 μg of total cell lysates were subjected to a SDS-polyacrylamide gel electrophoresis (PAGE), and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore) using a semidry transfer apparatus (BioRad). Following transfer, membranes were incubated in 4% blocking solution (1× TBST plus 4% dry milk power) for 30 min at room temperature. Antibodies specific to K-Rta, K-bZIP, or GAPdH (Chemicon) were applied to the membranes at 1:2000 dilution and incubated at room temperature for 1 h with gentle rocking. Unbound antibody was removed by 3 sequential washes with 1× TBST for 5 min each, after which an HRP-conjugated goat anti-rabbit secondary antibody (Amersham) was applied at 1:4000 dilution for 1 h at room temperature with gentle rocking. After three additional washes with 1× TBST, the membrane was visualized using ECL Plus reagent (GE Healthcare) and a digital image was captured using a FluorChem biological imaging cabinet (Alpha Innotech).
Chromatin immunoprecipitation
TREx BCBL1-Rta cells (∼3×107/time point) were seeded in RPMI plus 15% serum, at a density of 5×105/mL, allowed to recover for ∼15 h, then pulsed with 100 ng/mL doxycycline for 4 h, after which time the media was changed to prevent overexpression of K-Rta. At various time points following the treatment, the cultures were crosslinked and fixed by addition of 1:10 volume formaldehyde solution (0.1 M NaCl, 1 mM EDTA, 50 mM HEPES, pH 7.5, 11% HCHO with CH3OH) for 10 min at room temperature. Fixed cells were washed once in cold PBS, then in Buffer I (0.25% Triton X100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5), Buffer II (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5), then pellets were frozen at −80 °C until all samples were collected. The thawed pellets were resuspended in with 0.8 mL ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–Cl, pH 8.1, plus Complete™ protease inhibitor cocktail (Roche)), then disrupted with two treatments on a Diagenode Biorupter sonicator (15 min, high intensity, 10 second on/off interval). Sonicate was diluted 5× in dilution buffer plus inhibitors (1% Triton X100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–Cl, pH 8.1), then split into tubes for immunoprecipitation. Chromatin complexes were incubated with primary antibodies to K-Rta, K-bZIP (both purified rabbit polyclonal), pre-immune rabbit serum, or an acetyl-specific monoclonal antibody to histone 3, lysine 9 (Abcam) overnight at 4 °C, then recovered with a 1:1 mix of TE-equilibrated protein A/protein G-agarose beads, after addition of 5 μg sonicated salmon sperm DNA. After 2 h at 4 °C, the beads were recovered and washed sequentially in TSE buffer (0.1% SDS, 1% Triton X100, 2 mM EDTA, 20 mM Tris, pH 8.1) plus 150 mM NaCl, TSE buffer plus 500 mM NaCl, Buffer III (0.25 M LiCl, 1% NP-40, 1% Deoxycholate, 1 mM EDTA, 10 mM Tris, pH 8.1), and twice with cold TE buffer. Each wash included 5 min rocking at 4 °C. The bound DNA was then eluted in 10× bead volume (1% SDS, 0.1 M NaHCO3) at room temperature for 10 min with agitation. Elutions were incubated overnight at 65 °C to reverse crosslinks, diluted 3-fold with 0.2 M NaCl, then DNA was precipitated with 2.5 volumes of ethanol, washed with 70% ethanol, then purified on a Qiaquick PCR cleanup column as per manufacturer's recommendations (Qiagen).
Viral promoter ChIP-on-chip
DNA enriched for association with K-Rta, K-bZIP, pre-immune serum, or acetylated Histone 3, was concentrated in a Speedvac (Savant) and subjected to one or two rounds of linear amplification as necessary using the Whole Genome Amplification kit (Sigma) as per manufacturer's recommendations, omitting the fragmentation step (as the enriched DNA has already been fragmented by sonication). These DNA fragment populations were labeled with Cy3 or Cy5 using a 3DNA Array 900DNA Kit (Genisphere) as per manufacturer's protocol, with omission of the digestion step, and a 16 h initial hybridization in formamide-containing buffer at 52 °C, followed by a 4 h hybridization with the 3DNA Capture Reagent at 50 °C in an ozone-free tent. The slides were then scanned with an Agilent DNA Microarray Scanner B at a 10 μm resolution. These images were spotted and quantified using the Scanalyze software (EisenSoftware), and normalized (0.5) calibrated ratios were averaged to determine fold enrichment over the input DNA sample for each pair of replicate spots.
Real time qPCR for ChIP analysis
Eluted DNA from the ChIP assays were diluted 1:50 in ddH20 and used as a template for qPCR using the primer sets listed in Table 2. 5 μL of template were used per 25 μL reaction, all samples were analyzed in triplicate using SYBR-green 2× master mix (Bio-Rad) on an iCycler iQ thermal cycler (Bio-Rad). After an initial denaturation and activation incubation of 3 min at 95 °C, 45 cycles of 3-step cycling were performed with an annealing temperature of 60 °C. Melt curve analysis was then performed to verify product specificity. Relative fold induction over IgG for each immunoprecipitate and time-point were determined by examining the change in threshold cycle number (delta Ct), with standard deviation calibrated to account for variance in both IgG and IP-DNA signal, then propagated logarithmically. All primer sets were assayed for all time points of at least 2 independent experimental repeats.
Table 2.
Primer sets for PCR amplification for detection of KSHV promoters in ChIP assays
| Forward primer | Reverse primer | Genomic pos. | Size | K-bZIP | Class | |
|---|---|---|---|---|---|---|
| ORF9p | 5′-GAAACGGGGGAGTGACAGT-3′ | 5 ′-GCGGACGGTAATTTGTGTCT-3′ | 11222–11357 | 136 | Synergy | Early |
| K2pa | 5′-CCCAAACCTTGTCAATTTGC-3′ | 5 ′-GAACTCGCCAAAAAGTGAGC-3′ | 18279–18425 | 147 | Synergy | Early |
| K6pa | 5′-TTAAGCCAGGTTTCCATTGC-3′ | 5 ′-TACAGAGACACGCCTCAAGC-3′ | 27764–27886 | 123 | Synergy | Early |
| PANpa | 5′-AAGGGTCAGCTTGAAGGATG-3′ | 5 ′-GGCAGTCCCAGTGCTAAACT-3′ | 28544–28678 | 135 | No effect | Early |
| ORF21p | 5′-AATTACGCAGTCGGCAATTC-3′ | 5 ′-CCGATCTATGGCGGTTTCTA-3′ | 35154–35260 | 107 | Synergy | Late |
| KbZIPpa | 5′-CAGTTTGGTGCAAAGTGGAG-3′ | 5 ′-TCGACAACGGAGGAAATACC-3′ | 74685–74814 | 130 | Repression | Early |
| ORF57pa | 5′-TTCCATTAGGGTGAGCGAAG-3′ | 5′-CCACTGGTACCACAAACGAA-3′ | 81847–81953 | 107 | Repression | Early |
| ORF59p | 5′-CACACTTCCACCTCCCCTAA-3′ | 5 ′-CGCACAGAGAAATCACAGGA-3′ | 96755–96849 | 95 | No effect | Del. early |
| ORF64p | 5′-CGCTCAGGCTAACATTCCTC-3′ | 5 ′-CAGTGGGACAAAACGGAGTT-3′ | 103820–103958 | 139 | Synergy | Late |
| K12pa | 5′-GTCGGTCTCCCCTCTTCTTT-3′ | 5′-CTAGGTCCACGCTCACCTCT-3′ | 118770–118891 | 122 | No effect | Late |
Also listed are amplicon size (in bp), expression class (early, delayed early, late), and observed mode of K-bZIP/K-Rta interaction by reporter assay.
Designates promoter fragment containing a published K-Rta response element.
Acknowledgments
We are grateful to Clifford G. Tepper and Jeffrey P. Gregg (UC Davis School of Medicine; Dept. of Biochemistry and Molecular Medicine, and Dept. of Pathology and Laboratory Medicine, respectively), and the UC Davis Cancer Center Gene Expression Resource, where the microarray work was performed. We also thank Stephenie Y. Liu and Ryan R. Davis for their expert technical assistance in performing the labeling, hybridization, and array scanning; and Latricia Fitzgerald for support and input on the chromatin immunoprecipitation assays.
This work was supported by grants from the California HIV/AIDS Research Program (ID07-D-165 to Y.I.), and the National Institutes of Health (NIH) (CA111185 to H.J.K.). Additional funding was provided by the Cancer Center Support Grant (P30 CA93373-04 to Y.I.). The UC Davis Cancer Center Gene Expression Resource is supported by Cancer Center Support Grant 2 P30 CA93373 from the National Cancer Institute. This publication was made possible by Grant Number UL RR024146 from the National Center for Research Resources, a component of the NIH, and NIH Roadmap for Medical Research.
References
- AuCoin DP, Colletti KS, Cei SA, Papouskova I, Tarrant M, Pari GS. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP) Virology. 2004;318(2):542–555. doi: 10.1016/j.virol.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Brinkmann MM, Pietrek M, Dittrich-Breiholz O, Kracht M, Schulz TF. Modulation of host gene expression by the K15 protein of Kaposi's sarcoma-associated herpesvirus. J Virol. 2007;81(1):42–58. doi: 10.1128/JVI.00648-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu W, Palmeri D, Krishnan R, Marin R, Aris VM, Soteropoulos P, Lukac DM. Identification of direct transcriptional targets of the Kaposi's sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J Virol. 2008;82(21):10709–10723. doi: 10.1128/JVI.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey M, Kolman J, Katz DA, Gradoville L, Barberis L, Miller G. Transcriptional synergy by the Epstein–Barr virus transactivator ZEBRA. J Virol. 1992;66(8):4803–4813. doi: 10.1128/jvi.66.8.4803-4813.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM. Direct interactions of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J Virol. 2007;81(16):8451–8467. doi: 10.1128/JVI.00265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PJ, Shedd D, Gradoville L, Cho MS, Chen LW, Chang J, Miller G. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J Virol. 2002;76(7):3168–3178. doi: 10.1128/JVI.76.7.3168-3178.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PJ, Shedd D, Miller G. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J Virol. 2005;79(14):8750–8763. doi: 10.1128/JVI.79.14.8750-8763.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau CM, Zhang XY, McMahon SB, Lieberman PM. Regulation of Epstein–Barr virus latency type by the chromatin boundary factor CTCF. J Virol. 2006;80(12):5723–5732. doi: 10.1128/JVI.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ueda K, Sakakibara S, Okuno T, Yamanishi K. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J Virol. 2000;74(18):8623–8634. doi: 10.1128/jvi.74.18.8623-8634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci U S A. 2001;98(7):4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Lee BS, Shim SN, Li M, Jung JU. Identification of the novel K15 gene at the rightmost end of the Kaposi's sarcoma-associated herpesvirus genome. J Virol. 2000;74(1):436–446. [PMC free article] [PubMed] [Google Scholar]
- Day L, Chau CM, Nebozhyn M, Rennekamp AJ, Showe M, Lieberman PM. Chromatin profiling of Epstein–Barr virus latency control region. J Virol. 2007;81(12):6389–6401. doi: 10.1128/JVI.02172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Song MJ, Chu JT, Sun R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) J Virol. 2002;76(16):8252–8264. doi: 10.1128/JVI.76.16.8252-8264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Wang S, Liu S, Wood C. Characterization of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch Virol. 2001;146(2):403–413. doi: 10.1007/s007050170185. [DOI] [PubMed] [Google Scholar]
- Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu Rev Pathol. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol. 2000;74(13):6207–6212. doi: 10.1128/jvi.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruffat H, Portes-Sentis S, Sergeant A, Manet E. Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) encodes a homologue of the Epstein–Barr virus bZip protein EB1. J Gen Virol. 1999;80(Pt 3):557–561. doi: 10.1099/0022-1317-80-3-557. [DOI] [PubMed] [Google Scholar]
- Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. Genetic organization and hypoxic activation of the Kaposi's sarcoma-associated herpesvirus ORF34-37 gene cluster. J Virol. 2006;80(14):7037–7051. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Gwack Y, Byun H, Lim C, Choe J. The Kaposi's sarcoma-associated herpesvirus K8 protein interacts with CREB-binding protein (CBP) and represses CBP-mediated transcription. J Virol. 2001;75(19):9509–9516. doi: 10.1128/JVI.75.19.9509-9516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Lee D, Gwack Y, Min H, Choe J. Kaposi's sarcoma-associated herpesvirus K8 protein interacts with hSNF5. J Gen Virol. 2003;84(Pt 3):665–676. doi: 10.1099/vir.0.18699-0. [DOI] [PubMed] [Google Scholar]
- Izumiya Y, Lin SF, Ellison T, Chen LY, Izumiya C, Luciw P, Kung HJ. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J Virol. 2003a;77(2):1441–1451. doi: 10.1128/JVI.77.2.1441-1451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Lin SF, Ellison TJ, Levy AM, Mayeur GL, Izumiya C, Kung HJ. Cell cycle regulation by Kaposi's sarcoma-associated herpesvirus K-bZIP: direct interaction with cyclin-CDK2 and induction of G1 growth arrest. J Virol. 2003b;77(17):9652–9661. doi: 10.1128/JVI.77.17.9652-9661.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Ellison TJ, Yeh ET, Jung JU, Luciw PA, Kung HJ. Kaposi's sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J Virol. 2005;79(15):9912–9925. doi: 10.1128/JVI.79.15.9912-9925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Izumiya C, Van Geelen A, Wang DH, Lam KS, Luciw PA, Kung HJ. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J Virol. 2007;81(3):1072–1082. doi: 10.1128/JVI.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato-Noah T, Xu Y, Rossetto CC, Colletti K, Papouskova I, Pari GS. Overexpression of the Kaposi's sarcoma-associated herpesvirus transactivator K-Rta can complement a K-bZIP deletion BACmid and yields an enhanced growth phenotype. J Virol. 2007;81(24):13519–13532. doi: 10.1128/JVI.00832-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J Virol. 2007;81(20):10950–10960. doi: 10.1128/JVI.00183-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Tang Y, Lin SF, Kung HJ, Giam CZ. K-bZIP of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J Virol. 2003;77(6):3809–3815. doi: 10.1128/JVI.77.6.3809-3815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SF, Robinson DR, Miller G, Kung HJ. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein–Barr virus. J Virol. 1999;73(3):1909–1917. doi: 10.1128/jvi.73.3.1909-1917.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol. 2003;77(21):11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Suen J, Frias C, Pfeiffer R, Tsai MH, Chuang E, Zeichner SL. Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J Virol. 2004;78(24):13637–13652. doi: 10.1128/JVI.78.24.13637-13652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252(2):304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- Majerciak V, Yamanegi K, Zheng ZM. Gene structure and expression of Kaposi's sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J Virol. 2006;80(24):11968–11981. doi: 10.1128/JVI.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Fujita Y, Gomez E, Tanese N, Wilson AC. Activation of the Kaposi's sarcoma-associated herpesvirus major latency locus by the lytic switch protein RTA (ORF50) J Virol. 2005;79(13):8493–8505. doi: 10.1128/JVI.79.13.8493-8505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol. 2003;77(7):4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290(5500):2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Rossetto C, Gao Y, Yamboliev I, Papouskova I, Pari G. Transcriptional repression of K-Rta by Kaposi's sarcoma-associated herpesvirus K-bZIP is not required for oriLyt-dependent DNA replication. Virology. 2007;369(2):340–350. doi: 10.1016/j.virol.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc Natl Acad Sci U S A. 1996;93(25):14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Deng H, Sun R. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol. 2003;77(17):9451–9462. doi: 10.1128/JVI.77.17.9451-9462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt MR, Dittmer DP. Promoter switching allows simultaneous transcription of LANA and K14/vGPCR of Kaposi's sarcoma-associated herpesvirus. Virology. 2006;350(1):192–205. doi: 10.1016/j.virol.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Stedman W, Deng Z, Lu F, Lieberman PM. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J Virol. 2004;78(22):12566–12575. doi: 10.1128/JVI.78.22.12566-12575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95(18):10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol. 1999;73(3):2232–2242. doi: 10.1128/jvi.73.3.2232-2242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325(2):225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Wu MH, Geng Y, Wood C. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J Virol. 2001;75(24):11961–11973. doi: 10.1128/JVI.75.24.11961-11973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J Virol. 2004;78(16):8615–8629. doi: 10.1128/JVI.78.16.8615-8629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JT, Wood C. The role of Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene. 2003;22(33):5150–5163. doi: 10.1038/sj.onc.1206555. [DOI] [PubMed] [Google Scholar]
- Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol. 2001;75(3):1487–1506. doi: 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol. 1999;80(Pt 1):83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chiu J, Lin JC. Activation of human herpesvirus 8 (HHV-8) thymidine kinase (TK) TATAA-less promoter by HHV-8 ORF50 gene product is SP1 dependent. DNA Cell Biol. 1998;17(9):735–742. doi: 10.1089/dna.1998.17.735. [DOI] [PubMed] [Google Scholar]
- Zheng ZM. Split genes and their expression in Kaposi's sarcoma-associated herpesvirus. Rev Med Virol. 2003;13(3):173–184. doi: 10.1002/rmv.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelbauer J, Grundhoff A, Ganem D. Exploring the DNA binding interactions of the Kaposi's sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J Virol. 2006;80(6):2958–2967. doi: 10.1128/JVI.80.6.2958-2967.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]