

Abstract
Neurologic conditions including stroke, Alzheimer’s disease, Parkinson’s disease and Huntington’s disease are leading causes of death and long-term disability in the United States, and efforts to develop novel therapeutics for these conditions have historically had poor success in translating from bench to bedside. Hypoxia Inducible Factor-1alpha (HIF-1α) mediates a broad, evolutionarily conserved, endogenous adaptive program to hypoxia, and manipulation of components of the HIF pathway are neuroprotective in a number of human neurological diseases and experimental models. In this review, we discuss molecular components of one aspect of hypoxic adpatation in detail, and provide perspective on which targets within this pathway appear to be ripest for preventing and repairing neurodegeneration. Further, we highlight the role of HIF prolyl hydroxylases as emerging targets for the salutary effects of metal chelators on ferroptosis in vitro as well in animal models of neurological diseases.
Keywords: Metal chelators, neurodegeneration, hypoxia inducible factors, transcription, prolyl hydroxylases
Adaptive mechanisms to stress: combating neurodegeneration
Over the past twenty years, our laboratory has used two primary approaches to understand therapeutic strategies for preventing oxidative stress-mediated neurodegeneration. First, we have studied the endogenous mechanisms that lead to or prevent cell death induced by depletion of the antioxidant glutathione in primary neurons in vitro. Detailed biochemical analysis of this form of death, recently termed ferroptosis [1], has revealed novel redox regulated transcriptional pathways that can compensate for persistent glutathione depletion and oxidative stress in order to foster cell survival [2, 3]. In parallel, investigations into the mechanism of classical “antioxidants” in our in vitro model has surprisingly revealed remarkable and recurrent bias toward transcriptional adaptive mechanisms often involving more than 100 genes [4–7]. In this review, we will discuss evidence from these distinct investigations, and how that evidence has led us to focus our attention on an evolutionarily conserved adaptive response and its role in neuroprotection.
Oxygen-dependent metabolism
It is thought that self-replicating RNA became enclosed in a phospholipid membrane about 3.8 billion years ago, forming the basic unit of life called the cell (reviewed by [8]. These cells were able to obtain the energy required to replicate themselves and perform other necessary tasks from the sea of organic molecules from which they originated. However, such a self-limiting situation drove cells to evolve mechanisms through which they could autonomously generate, store, and use energy.
The principal pathways of energy metabolism are remarkably similar in all present-day cells, indicating that they emerged early in the process of evolution and have been conserved. For instance, all cells use ATP as the currency of energy to drive the synthesis of cell constituents and to carry out other energy-requiring activities. The mechanisms by which cells generate ATP are thought to have evolved sequentially: first glycolysis, then photosynthesis, and finally oxidative metabolism. In glycolysis, glucose is broken down to produce 2 ATP molecules. In photosynthesis, energy from the sun is harvested to produce glucose, which creates molecular oxygen (O2) as a byproduct and causes that oxygen to become abundant in Earth’s atmosphere. In oxidative metabolism, oxygen is used to break down glucose much more efficiently than in glycolysis, yielding 36 ATP molecules instead of 2.
Because of this efficiency, almost all present-day cells—including archaea, bacteria, and eukaryotic cells in humans—use oxidative reactions as their principal source of energy. In order for cells to rely on oxygen for metabolism, they need to be able to sense a decrease in oxygen availability—a phenomenon called hypoxia—and to then trigger a response program that helps them to cope with that decrease. One simple way to do so is to have sensor enzymes that use oxygen to inhibit the response program; when oxygen supply drops, the sensors are inhibited and the response program is then rapidly activated.
Cells as simple as bacteria express prolyl 4-hydroxylase domain-containing enzymes (PHDs) that use oxygen to add a hydroxyl group to proline residues on specific substrate proteins. This hydroxylation is a highly evolutionarily conserved sensory mechanism for oxygen; in fact, prolyl 4-hydroxylation is the single most prevalent posttranslational modification in humans [9]. In all animal cells, even those of the simplex animal, Trichoplax adhaerens, one of the proteins subject to prolyl 4-hydroxylation is called Hypoxia Inducible Factor (HIF) [10].
Hypoxia Inducible Factor
HIF was first identified as a transcriptional activator that binds to the hypoxia response element (HRE) in the promoter region of erythropoietin, and has since been shown to coordinate many evolutionarily conserved adaptive responses to hyoxia [11]. HIF target genes include VEGF, EPO, GLUT1, PFK1, BNIP3, Neuroglobin and dozens of other genes that work in concert at cellular, local tissue, and systemic levels, to restore oxygen delivery, to enhance glucose uptake and glycolysis, to reduce mitochondrial content and the rate of oxidative phosphorylation, and to regulate cell survival either by promoting adaptation or by engaging programmed cell death under prolonged or severe stress conditions. HIF-1 regulates gene expression not only by direct binding to target gene promoters at the consensus sequence (5′-RCGTG-3′), but also by counting among its targets numerous transcription factors, histone demethylases, and microRNAs that can in turn affect gene expression [12].
HIF is a heterodimer consisting of a constitutively present β subunit and a short-lived, oxygen-regulated α subunit. HIF family members are diagrammed in Figure 1. Three α isoforms have been identified in humans, and HIF-1α is highly evolutionarily conserved, sharing approximately 90% homology with mouse and rat. All α and β subunits contain an N-terminus basic Helix-Loop-Helix (bHLH) domain for DNA binding and a PAS domain for dimerization. HIF-1α and -2α subunits contain two oxygen dependent degradation domain, and a C-terminal transactivation domain (C-TAD) that binds the co-activators CBP/p300; HIF-3α is truncated to lack the C-TAD, and thus is speculated to act as a competitive inhibitor of HIF-1α and HIF-2α.
Figure 1. Schematic of oxygen-dependent HIF-1α regulation.
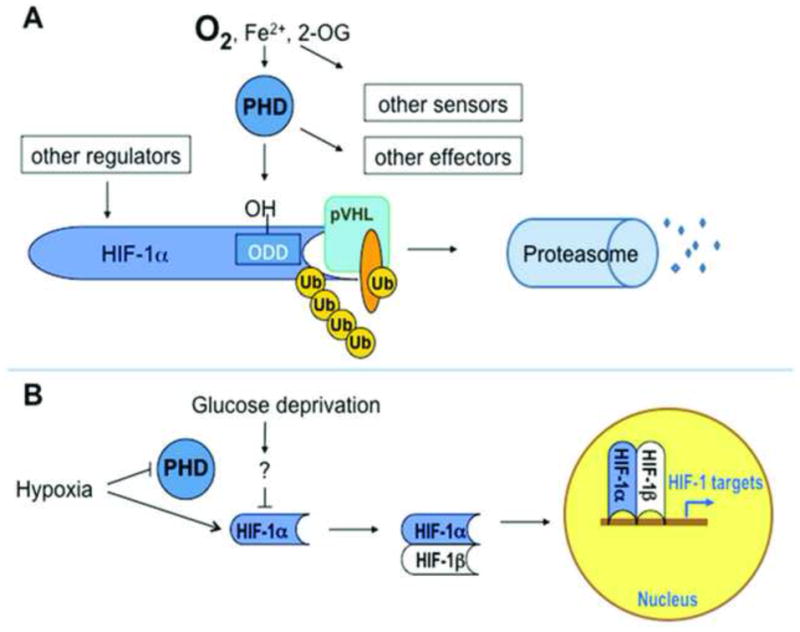
A. Under normoxic conditions, PHDs hydroxylate P564 on HIF-1α, allowing it to be recognized by the E3 ubiquitin ligase von Hippel-Lindau protein (VHL), ubiquitinated, and targeted for proteasomal degradation. As members of a large family of iron- and 2-oxoglutarate dependent dioxygenases, PHDs integrate multiple signals of metabolic homeostasis, and are one of many such sensors; further, PHDs have HIF-independent substrates, and HIF protein levels and transcriptional activity are regulated in many PHD-independent ways. B. Under hypoxia, PHDs are inhibited, allowing HIF-1α to elude degradation, dimerize with its β partner in the nucleus, bind transcriptional coactivators and hypoxia response elements in promoter regions of target genes, and enhance transcription rates. Glucose deprivation has been reported to decrease hypoxic stabilization of HIF-1α; the mechanisms by which this occurs are unclear.
HIF subunits do not only activate transcription of HRE-regulated genes, though. HIF-2α was recently shown to bind the RNA-binding protein RBM4 and the cap-binding eIF4E2 in order to initiate translation of select mRNAs; this process occurs under conditions of hypoxia in which the normal cap-dependent translation machinery is inhibited [13]. HIF-1α binds to the transcription factor Sp1 to block activation of the gene encoding the mismatch DNA repair protein MSH2, and also binds to the Notch intracellular domain (NICD) in order to potentiate transcriptional activation of Notch target genes [12]. HIF-β subunits, first identified as the aryl hydrocarbon receptor nuclear translocator (ARNT), are necessary for xenobiotic response mediated by the aryl hydrocarbon receptor (AhR).
HIF-1α is the most widely studied of the α isoforms, and is expressed in all human cell types, while HIF-2α shows more restricted expression. Both HIF-1α and HIF-2α show some selectivity in their cassettes of target genes. For example, HIF-1α preferentially induces glycolytic enzyme genes whereas HIF-2α induces several genes involved in neurological disorders and cancer invasion [14]. HIF-1α appears to mediate acute responses to hypoxia, while HIF-2α is activated primarily during chronic hypoxia.
Constitutive deletion of the Hif-1a gene in mice causes lethality by day eleven of embryonic development (E11), resulting from cardiovascular malformation and defective cephalic vascularization, indicating that HIF-1α is essential for embryonic vascularization. Neural cell-specific HIF-1α-deficient mice exhibit hydrocephalus accompanied by a reduction in neural cells and an impairment of spatial memory, indicating that expression of HIF-1α in neural cells is essential for normal development of the brain.
Oxygen-Dependent Degradation of HIF-1α
HIF-α protein half-life is regulated by oxygen-dependent degradation (Figure 1.A). Under normoxic conditions, prolyl hydroxylase domain enzymes (PHDs) hydroxylate P564 on HIF-1α, allowing the domain to be recognized by Von Hippel-Lindau (VHL) protein, an E3 ubiquitin ligase, and thereby targeted for degradation by the 26s proteasome [15–17]. Under hypoxia, a decrease in PHD activity leads to HIF-1α accumulation, heterodimerization with β-subunits, recruitment of the histone acetyltransferases p300 and CBP, and transactivation of target gene expression [18].
A number of other proteins contribute to the canonical PHD-VHL-Proteasome degradative pathway. ARD1 acetylates lysine532 of HIF-1α, located in the ODD domain. This modification appears to enhance recruitment of VHL, but is not required for HIF-1α degradation under normal conditions [19]. VHL binds hydroxylated PHD in complex with Elongin B, Elongin C, Cul2, and Rbx1, all of which are required for ubiquitination [20].
HIF Prolyl Hydroxylases (PHDs)
Prolyl Hydroxylase Domain (PHD) enzymes are highly conserved iron-dependent, 2-oxoglutarate-dependent dioxygenases. PHDs are the primary oxygen sensors that keep HIF-1α protein levels low during normoxia and allow the protein to be rapidly stabilized upon hypoxia [16, 17]. Pharmacological and molecular studies have demonstrated that PHD inhibition is broadly neuroprotective and mediates the salutary effects of iron chelating drugs. FDA-approved drugs that inhibit PHDs have been identified and are poised for clinical trials [21–23].
PHDs, also known as egl nine homologs (EGLNs), exist in three isoforms named PHD1 (EGLN2), PHD2 (EGLN1) and PHD3 (EGLN3). They are part of a superfamily of iron-dependent, 2-oxoglutarate-dependent dioxygenases; other members of this family include the collagen prolyl hydroxylases, which regulate the extracellular matrix, and the jumonji-domain containing histone demethylases, which regulate gene expression through chromatin structure modifications. PHDs hydroxylate both P564 and P402 on HIF-1α, but under conditions of normoxia, P564 is hydroxylated prior to P402 and primarily regulates oxygen-dependent degradation [24]. PHD2 is the most abundant PHD and the most important in setting steady-state levels of HIF-α subunits [25]. PHDs, especially PHD2 and PHD3, are transcriptionally upregulated by HIF-1, and are therefore important not only for basal regulation of HIF but for feedback-inhibition during prolonged hypoxia or rapid degradation upon reoxygenation [26]. Although all three PHD isoforms recognize the LXXLAP motif, they show some isoform-selective preferences for flanking regions on substrates, which provides some direction for attempts to develop isoform-specific inhibitors [27].
Several isoform-specific, HIF-independent functions of the PHDs have been identified. Some alternative substrates have been identified that show selectivity among the PHD isoforms. The β-(2)adrenergic receptor, a G-protein coupled receptor important for cardiac function, is hydroxylated specifically by PHD3, ubiquitinated by VHL, and proteasomally degraded under hypoxia [28]. Rbp1, the large and enzymatically active subunit of RNA polymerase II, is recruited to DNA under oxidative stress conditions in a manner requiring its hydroxylation at Pro1465 and subsequent non-degradative ubiquitination by VHL; PHD1 is necessary for Pro1465 hydroxylation while PHD2 inhibits this hydroxylation [29]. PHD1 also specifically regulates Cyclin D1 in a hydroxylase-dependent, transcription-dependent, HIF-independent manner: PHD1 inactivation decreases Cyclin D1 levels and suppresses mammary gland cell proliferation and tumor formation in vivo [30].
Indeed, PHDs are not only gatekeepers for the oxygen-dependent degradation of HIF-1α, but are integrated sensors of cellular metabolism [31]. Proline hydroxylation is enzymatically coupled to the decarboxylation of 2-oxoglutarate (2-OG), a process which yields succinate and CO2. PHD activity therefore requires 2-OG and is inhibited in the presence of high concentrations of tricarboxylic acid cycle intermediates such as pyruvate, isocitrate, oxaloacetate, succinate, or fumarate. Since PHD enzyme activity also requires iron, PHDs serve moreover as sensors of iron homeostasis [32].
Although it is well-established that PHDs, particularly PHD2, are critical for normoxic degradation of HIF-1α, it remains debated exactly how hypoxia inhibits PHD activity. At least three distinct models have been proposed to explain this phenomenon. The first model proposes that PHD enzymatic activity could be reduced as substrate levels drop; this is a simple model in which PHDs are the primary oxygen sensors regulating HIF, and is consistent with Km for PHD observed in vitro [33]. The second model argues that O2 consumption by an intact electron transport chain redistributes O2 intracellularly into mitochondria [34, 35]. Models one and two are not mutually exclusive, however, since subcellular O2 gradients could shift the apparent Km of PHDs for O2 in vivo. The third model purports that mitochondrial peroxide production increases under hypoxia, and that H2O2 dismutated from O2− produced at Reiske proteins is a specific signaling molecule released from mitochondria in order to inhibit PHDs [36]. Work by several groups suggests that the stabilization of HIF-1α by hypoxia requires signaling through mitochondria and intracellular kinases, in contrast to HIF-1α stabilization by anoxia or by PHD inhibiting-iron chelators [37, 38]. Models two and three invoke conflicting data on whether mitochondrial ROS production is necessary for or irrelevant to hypoxic HIF stabilization.
Von Hippel-Lindau Protein
VHL was first identified as a tumor-suppressor gene that is mutated in Von Hippel-Lindau syndrome, a dominantly inherited familial cancer syndrome that precipitates a variety of malignant and benign tumors. The most common of these tumors are central nervous system hemangioblastomas (benign, highly-vascularized tumors), pheochromocytomas (tumors originating from the chromaffin cells of the adrenal glands), and clear-cell renal-cell carcinomas (ccRCCs). Somatic mutations affecting VHL are also seen in the majority of cases of sporadic ccRCC.
VHL was shown to mediate HIF-1α degradation, which explains the angiogenic phenotype of many VHL-null tumors [39]. VHL was subsequently shown to have a number of substrates other than HIF-α, including fibronectin and collagen IV α2 chain in the extracellular matrix, microtubules, the RNA Polymerase II subunits Rbp1 and Rbp7, atypical protein kinase Cλ, VHL-interacting deubiquitinating enzymes VDU-1 and VDU-2, p27, p53, and Jade-1 [40]. VHL-mediated ubiquitination does not always lead to substrate degradation, though; VHL stabilizes p53 and Jade-1, for example [41].
The Ubiquitin-Proteasome Degradation Pathway
An ATP-dependent proteolytic system independent of lysosomes was first described in reticulocytes [42], and the Nobel Prize in Chemistry 2004 was awarded jointly to Aaron Ciechanover, Avram Hershko and Irwin Rose for the discovery of ubiquitin-mediated protein degradation.
Proteins are targeted for proteasomal degradation by the covalent addition of at least four ubiquitin molecules to a lysine residue on the client protein [43]. The addition of ubiquitin requires three enzymatic steps. First, a ubiquitin-activating enzyme (E1) couples the hydrolysis of ATP with adenylylation of a ubiquitin molecule [44]. This adenylylated ubiquitin is then transferred to a cysteine of a second enzyme, ubiquitin-conjugating enzyme (E2). Finally, a ubiquitin ligase (E3) recognizes the specific protein to be ubiquitinated and transfers the ubiquitin from E2 to the target protein. Addition of further ubiquitin groups can be mediated by an E3 or an E4 polyubiquitin chain conjugation factor. This conjugation is significant, since the way in which ubiquitin chains are constructed can determine the fate of ubiquitinated proteins [45]. Polyubiquitinated proteins are recognized by the 19S regulatory cap, then deubiquitinated and unfolded, so that they can be translocated through a narrow gate into the central channel of the 20S catalytic core of the proteasome. The beta-subunits of the 20S core act as proteases to degrade client proteins, releasing short peptides typically 7–9 amino acids in length.
ATP is required at several steps of the ubiquitin-proteasome degradative pathway (reviewed by [46]. First, ATP hydrolysis is required for activation of ubiquitin by E1, but not for later steps of ubiquitin conjugation. ATP binding, but not hydrolysis, is required for the assembly of the 26S proteasome from the 19S and 20S particles, as well as for target protein binding by the 19S subunit, gate opening, translocation, and proteolysis. ATP hydrolysis is required for the unfolding of substrate proteins and, in some cases but not all, for proteolysis and deubiquitination [47, 48].
ATP is required for proteasomal degradation and can also be depleted under conditions of ischemia (when blood supply drops, leading to a scarcity of oxygen, glucose, and other energy substrates). Nevertheless, it is worth noting that proteasomal degradation continues during brief ischemia, and can even mediate rapid preconditioning tolerance to later harmful ischemia [49]. In contrast, prolonged ischemia can inactivate proteasomal degradation, leading to the accumulation of ubiquitinated proteins. Such accumulation may contribute to cell stress, or form aggregates that may in turn be degraded by the autophagasome/lysosome pathway [49]. A major regulator of ATP homeostasis, AMPK is negatively regulated by the proteasome such that inhibition of proteasomal degradation can stimulate the energy conservation program mediated by AMPK [50].
Regulation of HIF-1α by ODD-independent mechanisms
HIF-1α synthesis, stability, and ability to activate target gene transcription can be regulated through many known ODD-independent mechanisms under different physiological and pathological conditions.
Regulation of HIF-1α activity
Once HIF-1α is stabilized, its ability to bind p300/CBP is negatively regulated by an O2− and Fe2+-dependent asparaginyl hydroxylase, a Factor Inhibiting HIF (FIH), which targets Asn803 within the C-TAD [51]. In addition to CPB and p300, other histone acetyltransferases (HATs) that coactivate transcription have been shown to interact with and potentiate HIF, including SRC1 and TIF2. On the other hand, the HAT CITED2 competes with HIF-1 for binding to p300/CBP and is transcriptionally upregulated in hypoxia by HIF-1, and thus may provide a negative feedback on HIF1 activity during extended hypoxia [52]. CITED2 is also a negative regulator of NF-κB mediated gene transcription, and thus may serve a general role in feedback regulation for homeostatic programs.
HIF-1α phosphorylation by casein kinase 1delta at Ser247 within the PAS domain inhibits dimerization with HIF-α and subsequent transcriptional activity, without affecting HIF-1α stability or nuclear accumulation [53]. HIF-1α phosphorylation by p42 / p44 mitogen-activated protein kinases (MAPK1/3) at Ser641/643, and this phosphorylation enhances HIF-1-dependent nuclear localization and gene transcription [54, 55]. The regulatory associated protein of mTOR (Raptor) interacts with HIF-1α at an mTOR signaling (TOS) motif located in the N terminus of HIF-1α. Raptor facilitates binding of HIF-1α to the co-activator CBP/p300 thus enhancing HIF-1α-mediated transcription [56]. S-nitrosylation of Cys800 and the redox responsive protein Ref-1/APE-1 also promote HIF-1 transcriptional activity in a manner dependent on p300 [57–59].
ODD-independent Regulation of HIF-1α protein stability
The half-life of HIF-1α is regulated in an O2-independent manner by the competitive binding of either the heat shock protein 90 (HSP90) or the receptor of activated protein kinase C (RACK1) to the PAS domain of HIF-1α. RACK1 is an E3 ubiquitin ligase that interacts with Elongin C via a binding site that shows significant sequence similarity to VHL, thereby promoting HIF-1α ubiquitination and degradation in a manner that is independent of PHD2 and VHL [60]. RACK1-mediated HIF-1α degradation can be blocked not only by HSP90, but by Calcineurin, a calcium-activated phosphatase that dephosphorylates RACK1 and thus blocking its dimerization and binding to Elongin C, suggesting that HIF-1α can be stabilized by increases in intracellular calcium [61].
GSK3 can directly phosphorylate Ser551, Thr555, and Ser589 within the ODD domain, leading to recognition by a ubiquitin ligase called F-box and WD protein (Fbw7) and targeting HIF-1α for proteasomal degradation independent of prolyl hydroxylation and VHL binding [62, 63]. HIF Associated Factor (HAF), an E3 ligase for HIF-1α (but not HIF-2α), plays an important role in oxygen-independent HIF-1α regulation in cancer by degrading HIF-1α in response to growth factor signaling, and perhaps other stimuli even in the presence of hypoxia [64]. The small ubiquitin-related modifier (SUMO) can be conjugated to HIF-1α at Lys391 and Lys477 and enhance HIF-1α stability [65], but the mechanism by which this stabilization occurs and the physiological or pathological context in which SUMOylation regulates HIF-1 has not been determined. In response to oxidative stress, a small ubiquitin-like protein called neural precursor cell expressed developmentally down- regulated 8 (NEDD8) becomes conjugated to the PAS domain of HIF-1α and stabilizes HIF-1α [66]. The role of ROS in hypoxic HIF-1α stabilization is controversial.
Regulation of HIF-1α transcription and translation
HIF-1α is regulated by positive and negative feedback loops; in addition to hypoxic induction of PHD3 described above, Hif1a itself is among the target genes of HIF-1α. Hypoxic induction of HIF-1α mRNA requires the activity of the PI3K/Akt pathway but not ERK1/2 pathway [67].
NF-κB, a major mediator of immune responses, binds to the promoter of HIF-1α and increases HIF-1α mRNA levels under hypoxia [67]. Crosstalk between NF-κB and HIF-1α is complex; for example, NF-κB has long been known to be induced by hypoxia [68], and more recently it was shown that NF-κB protein levels are increased under hypoxia downstream of HIF PHD1 hydroxylation of IKKβ [69].
The NO donors NOC18 or S-nitrosoglutathione induce HIF-1α expression and transcriptional activity without inhibiting HIF-1α hydroxylation, ubiquitination, and degradation, indicating an effect on HIF-1α protein synthesis that was confirmed by pulse labeling studies and shown to require PI3K and MAPK signaling through the translational regulatory proteins 4E-BP1, p70 S6 kinase, and eIF-4E [70]. The tumor suppressor p53 binds HIF-1α and promotes its degradation in hypoxia via Mdm-2-mediated ubiquitination and proteasomal degradation [71]. This can be overcome by Jun activation domain-binding protein-1 (Jab-1), which was previously known as a coactivator of the AP-1 transcription factor before it was shown to activate HIF-1 transcriptional activity by interfering with p53 binding to HIF-1 [72].
Growth factor signaling boosts the rate of translation of HIF-1α, and a select group of other mRNAs (reviewed by [73]). Receptor tyrosine kinases in the plasma membrane are activated by extracellular growth factor ligand binding, which initiates two parallel cascades of phosphorylation activation: PI3K → Akt/PKB → mTOR and RAS→MEK→ERK. mTOR and ERK converge on two mechanisms of promoting translation: activation of S6K and subsequent activation of the ribosomal protein S6, as well as inhibition of eukaryotic translation initiation factor 4E binding protein (4E-BP1), to disinhibit cap-dependent translation mediated by eIF-4E. Additionally, ERK activates MNK, which directly phosphorylates eIF-4E to increase its activity. It is not known precisely how mTOR signaling promotes the translation of HIF-1α, but HIF-1α is likely to be particularly sensitive to fluctuations in the rate of protein synthesis due to its short half-life (approximately 5 minutes) under normoxic conditions. The many levels of regulation on HIF-1α, and its involvement in cellular responses to stimuli other than hypoxia such as inflammation and growth factor signaling, all support the notion that HIF-1α is a critical central regulator of adaptive transcriptional responses.
HIF PHD inhibition as a neuroprotective strategy
Because HIF-1α is primarily degraded under normoxia in a manner requiring prolyl hydroxylase enzymes (PHDs), HIF can be pharmacologically induced by structurally diverse small molecule PHD inhibitors. HIF PHD inhibition consistently produces neuroprotection in diverse neurological disease models, including stroke [5, 74], Alzheimer’s Disease [75]; [76]), allergic encephalomyelitis [77], Parkinson’s Disease [78], oxidative stress [79], excitotoxicity [80], trophic factor deprivation [81] and mitochondrial dysfunction [82] (Table 1). PHDs have been identified as the critical targets of iron chelators such as desferoxamine that are clinically beneficial in many neurological disorders and that recapitulate the neuroprotective effects of hypoxic preconditioning on later ischemic challenge [5]. Novel PHD inhibitors poised for clinical trials have been identified through high-throughput screening of FDA-approved drug libraries [83]. PHD inhibition produces neuroprotection via both HIF-dependent and HIF-independent mechanisms [84]. Thus, PHD inhibition is a promising therapeutic approach that engages multiple downstream effector pathways, of which HIF is one.
Table 1. Small molecules with HIF PHD inhibitory activity are neuroprotective in a host of models of neurological conditions in vitro and in vivo.
Desferoxamine inhibits HIF PHD activity by depleting iron, a critical cofactor for these and other iron, 2-oxoglutarate, and oxygen dependent dioxygenases. Dihyroxybenzoic acid and dimethyloxaloglycine are competitive inhibitors of 2-oxoglutarate, a co-substrate in the HIF PHD catalytic cycle.
| Neurological diseases |
HIF PHD inhibitors |
In vitro model | In vivo model | References |
|---|---|---|---|---|
| Stroke | Deferoxamine (DFO) | Oxygen glucose deprivation (OGD) | [124];[125]; [126] | |
| Glutathione depletion model | [79]; [5]; [89]; [127] | |||
| NMDA toxicity | [128]; [129] | |||
| Nerve growth factor (NGF) deprivation | [130] | |||
| Hypoxia-ischemic injury | [131] | |||
| Middle cerebral artery occlusion (MCAO) | [124]; [5]; [132]; [133]; [103]; [134]; [135] | |||
| Ethyl 3, 4 dihydroxy benzoate (DHB) | Glutathione depletion model | [5]; [89]; [127] | ||
| H2O2-induced oxidative injury | [136] | |||
| Nerve growth factor (NGF) deprivation | [81] | |||
| Middle cerebral artery occlusion (MCAO) | [5]; [103] | |||
| Dimethyl-oxalyl glycine (DMOG) | Oxygen glucose deprivation(OGD) | [137] | ||
| Glutathione depletion model | [127] | |||
| Nerve growth factor (NGF) deprivation | [81] | |||
| Middle cerebral artery occlusion (MCAO) | [74]; [137] | |||
| Parkinson’s disease | Deferoxamine (DFO) | 6-hydroxydopamine (6OHDA) | [138] | |
| 1-methyl-4-phenylpyridinium (MPP+) | [104] | |||
| Rotenone | [104] | |||
| 6-hydroxydopamine (6OHDA) | [139] | |||
| Ethyl 3, 4 dihydroxy benzoate (DHB) | 1-methyl-4-phenylpyridinium (MPP+) | [84] | ||
| 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) | [84];[140] | |||
| Dimethyl-oxalyl glycine (DMOG) | 1-methyl-4-phenylpyridinium (MPP+) | [84] | ||
| Alzheimer’s disease | Deferoxamine (DFO) | SHSY-5Y cells overexpressing the human APP Swedish mutation | [141] | |
| APP/PS1 mice | [141] | |||
| P301L mice | [142] | |||
| Huntington’s disease | Deferoxamine (DFO) | 3-Nitropropionic acid (3-NP) | [143];[82] | |
| Dimethyl-oxalyl glycine (DMOG) | [82] | |||
| Friedreich’s ataxia | Deferoxamine (DFO) | H2O2-induced oxidative injury in FRDA fibroblasts | [144] | |
| frataxin expression in FRDA lymphoblasts | [145] |
Prolyl hydroxylation is substantially more sensitive than asparaginyl hydroxylation to inhibition by iron chelators [85]. Therefore, FDA-approved iron chelators or more specific PHD inhibitors could therapeutically activate the many downstream effectors of PHD signaling. There is also potential for off-target effects upon pathophysiological or pharmacological HIF manipulation. For these reasons, probing the mechanisms by which HIF is regulated by ischemia and by candidate drugs is as important for safety and efficacy as measuring HIF activity itself. Identifying the physiological, pathological and pharmacological conditions permissive to HIF stabilization and their mechanism of action is an important first step in understanding the therapeutic benefits or potential side effects of manipulating HIF signaling.
HIF PHD inhibitors abrogate ferroptosis, a novel form of non-apoptotic death
Recently a novel form of iron-dependent, non-apoptotic death was defined by Brent Stockwell and coworkers [1]. Using Ras mutant cancer cells, the group elegantly demonstrated that cancer cell death induced by the chemotherapeutic agent, erastin had unique morphological, biochemical and genetic features when compared to cell death induced by the non-specific tyrosine kinase inhibitor, staurosporine, the ROS hydrogen peroxide, or the autophagy inducer rapamycin. Specifically, ferroptosis could be blocked by iron chelators (DFO), Erk signaling pathway inhibitors (e.g. U0126), and cycloheximide, a protein synthesis inhibitor. Moreover, an unbiased short interfering RNA screen involving 1,087 genes identified 6 genes that are required for errastin induced ferroptosis: ribosomal protein L8 (RPL8), iron response element binding protein 2 (IREB2), ATP synthase Fo complex subunit C3 (ATP5G3), citrate synthase (CS), tetratricopeptide repeat doman 35 (TTCC35), and acetyl-CoA synthetase family member (ACSF2) (Table 2). In this model, cell death occurs downstream of inhibition of the plasma membrane transporter for cystine, Xc−. Inhibiton of cystine transport leads to depletion of antioxidant levels via starvation of cysteine, the rate limiting amino acid precursor in glutathione synthesis. Erastin thus induces oxidative stress which ultimately leads to death. The findings are intriguing because it is well established that iron chelators [79], Erk signaling pathway inhbitors [86, 87] and cycloheximide [88] can abrogate oxidative glutamate toxicity in neurons. In the neuronal paradigm, glutamate, like erastin, inhibits cystine uptake leading to an iron dependent, protein synthesis dependent form of cell death. And while it as yet unclear whether the genes necessary for erastin induced ferroptosis also are required for oxidative glutamate toxicity in neurons we have shown that an on target effect of iron chelators in preventing oxidative glutamate toxicity (likely a neuronal form of ferroptosis) are the HIF Prolyl hydroxylases [5], specifically HIF Proly Hydroxylase 1 [89]. Thus we would argue that the salutary effects of iron chelators in preventing ferroptosis can be assigned to inhibition of a specific family of iron, 2-oxoglutarate and oxygen dependent dioxygenases, the HIF prolyl hydroxylases, and not to direct inhibition of Fenton Chemistry or reactive oxygen species formation.
Table 2. Features of Ferroptosis (from [1]).
Ferroptosis appears to be a biochemically, morphologically and genetically distinct form of cell death that is sensitive to iron chelation in cancer cells. While ferroptosis has not been characterized fully in neurons, our studies suggest that several agents known to block ferroptosis in cancer cells, also block ferroptosis in primary neurons (e.g. Erk inhibitors, cycloheximide, and iron chelators). Our studies suggest that iron chelators target the HIF prolyl hydroxylases to inhibit ferroptosis [89]; [123].
| Ferroptosis | |
|---|---|
| Characteristic features | Occurrence |
| Morphology | Smaller mitochondria with increased membrane density |
| Cell death mechanism | Iron dependent ROS accumulation |
| Precursor requirement | Requires initial lipid precursor |
| Possible cell death rescue mechanisms | Iron chelation and genetic inhibition of cellular iron uptake |
| Regulatory genes | RPL8, IREB2, ATP5G3, CS, TTC35, ACSF2, |
| Reported inducers | RSLs such as erastin and RSL-3 |
| Characterized inhibitor | Ferrostatin-1 |
| Other inhibitory compounds | Iron chelator DFO, antioxidant trolox, MEK inhibitor U0126, and protein synthesis inhibitor cycloheximide |
| Reported incidences of ferroptosis | Glutamate induced neurotoxicity and cell death of certain cancer cells |
| Bioenergetics failure (A necrotic feature) | No |
| Cytoplasmic and organelle swelling (A necrotic feature) | No |
| Plasma membrane rupture (A necrotic feature) | No |
| Apoptotic bodies (An apoptotic feature) | No |
| Chromatin condensation (An apoptotic feature) | No |
| Formation of double membrane vesicle (An autophagic cell death feature) | No |
Cerebral Ischemia
Epidemiology and Pathology of Cerebral Ischemia
Stroke is a leading cause of death and long-term disability in the US [90]. Every year, about 795,000 people in the United States have a stroke, and about 23% of people who survive a stroke eventually have another [90]. In 2010, the estimated cost of stroke in the United States was $53.9 billion, including the cost of health care services, medications, and lost productivity [90]. Cerebral ischemia is the most common form of stroke, accounting for approximately 87% of all strokes. Cerebral ischemia is defined as insufficient blood flow to the brain to supply an adequate amount of oxygen and nutrients. Cerebral ischemia may result from occlusion or constriction of blood vessels. At present, there is only one FDA-approved drug treatment for cerebral ischemia: the thrombolytic agent tissue plasminogen activator (tPA), which is recommended for IV administration only within the first 4.5 hours following onset of stroke symptoms.
Unlike hypoxia, in which cells can maintain ATP levels by shifting metabolism away from mitochondrial respiration and toward glycolysis, ischemia involves a reduction in glucose supply. Glucose is generally understood to be the obligatory energy substrate for the brain; in an intact brain, astrocytes take up glucose and utilize it for glycolysis, then shuttle pyruvate and lactate into neurons, which then use these substrates for oxidative phosphorylation [91]. More recently it has been shown that ketone bodies can also be used by the brain as energy substrates, and indeed are taken up by the brain at an increased rate during cerebral ischemia [92, 93]. Nonetheless, in the ischemic core where oxygen and glucose supply are lowest, ATP may be severely depleted within minutes; in a rat model of forebrain ischemia, within 10 minutes glucose concentration dropped from 3.64 to 0.21 μM/g and ATP concentration dropped from 2.64 to 0.18 μM/g, with corresponding increases in lactate and AMP [94].
ATP depletion in the ischemic core causes rapid necrotic cell death. Although many ATP-requiring processes such as gene transcription, DNA repair, protein synthesis and trafficking, and proteasomal degradation can be shut down briefly without causing lasting harm, neurons need a constant supply of ATP in order to maintain ionic gradients that support membrane polarization. Loss of membrane potential leads to excessive glutamate release, activation of extrasynaptic NMDA receptors, aberrant calcium influx, mitochondrial damage, and downstream intracellular signaling events culminating in excitotoxic cell death (reviewed by [95]). In addition to glutamate-mediated mechanisms, a number of other ion channels can contribute to ion dyshomeostasis and activate cell death pathways, including sodium-calcium exchangers, hemichannels, volume-regulated anion channels, acid-sensing channels, transient receptor potential channels, and nonselective cation channels [96]. Aberrant calcium influx can cause not only to excitotoxic release of neurotransmitters but also activation of calcium-dependent intracellular death pathways (e.g. through calpain), and mitochondrial permeability transition leading to caspase-dependent apoptotic cell death.
In the tissue surrounding a focal ischemic core, termed the penumbra, collateral blood flow produces gradients of oxygen and glucose that allow cells to avoid rapid energetic crisis and necrotic cell death, but many of these cells die in the 24–72 hours following the ischemic event. This death is largely apoptotic rather than necrotic and follows a complex cascade of excitotoxicity, inflammation and oxidative stress [97]. Current strategies for developing novel therapeutics to reduce the loss of brain tissue and motor/cognitive function are focused on restoring homeostasis and promoting neuronal survival within the penumbral regions. In experimental models of cerebral ischemia, hypoxic preconditioning has been shown to be extremely effective in reducing infarct volume and behavioral deficits ([98]; reviewed by [99]). Elucidating the mechanisms by which hypoxic preconditioning affords protection has been a major focus of ischemia research. Endogenous sensors of hypoxia and their downstream effectors of broad, evolutionarily conserved adaptive responses to hypoxia are prime targets for therapeutic manipulation [100]. Key among these targets is the transcriptional activator Hypoxia-Inducible Factor (HIF).
HIF-1α in cerebral ischemia
HIF-1α protein levels are increased in mouse and primate brains after experimental cerebral ischemia [101, 102], but it is unclear whether HIF-1α limits or contributes to ischemic pathology.
In vivo models of cerebral ischemia show that HIF-1α can prevent injury [103] and improve functional recovery [104], or increase infarct and edema volume [105, 106]. In a rat model of focal ischemia, a biphasic induction of HIF-1α was observed both in vivo and in vitro at 1–12 hours after stroke and then again after 48 hours, and selective inhibition of HIF-1α at the early but not late timepoint was neuroprotective [107]. In vitro models of cerebral ischemia also show HIF-1α either protects or exacerbates cell death via expression of target genes that encode both pro-survival and pro-apoptotic proteins [108, 109], and demonstrate divergent roles of HIF-1α in distinct CNS cell types [110].
Hypoxia and ischemia activate other stress response pathways that engage in cross-talk with the HIF pathway, including ER stress-induced activation of the unfolded protein response (UPR) and suppression of protein synthesis [111]. Accumulation of misfolded proteins in the ER lumen leads to PERK-mediated phosphorylation of eIF2α, inhibiting protein synthesis to prevent further accumulation of unfolded proteins in the ER. Therefore the rates of translation of HIF-1α, its target genes, and its interactors are subject to additional layers of regulation under hypoxic or ischemic conditions. In vitro studies using oxygen and glucose deprivation (OGD) have indicated that glucose is required for hypoxic HIF-1α stabilization in human mesangial and hepatoma (Hep3B) cells [112] and in human renal carcinoma (UOK262) cells [113]. In considering how glucose deprivation could curtail hypoxic HIF-1α stabilization, these researchers considered that glucose not only supports ATP production through glycolysis and mitochondrial oxidative phosphorylation, but also supplies the pentose phosphate pathway (also known as the hexose monophosphate shunt) which produces NADPH. They proposed a model by which glucose fuels NADPH production through the pentose phosphate pathway/hexose monophosphate shunt to provide a substrate for NADPH-dependent oxidases, which in turn produce reactive oxygen species that inhibit prolyl hydroxylases. According to this model, glucose deprivation enhances HIF-1α degradation by relieving a ROS-mediated inhibition on PHD enzyme activity. However, it remains unclear 1) whether PHDs in fact mediate the effects of glucose and 2) whether this model is applicable to neuronal cells. More recent work demonstrated that NEDD8 conjugation to the PAS domain mediates ROS-induced HIF stability, calling into question whether proline hydroxylation is truly the target through which glucose deprivation affects HIF-1α levels [114].
Quantitative mechanistic reporters for HIF-1α as a surrogate for dynamically monitoring PHD activity in cells
Although the potential therapeutic value of HIF PHD inhibition and HIF-1α in cerebral ischemia has been widely appreciated for over a decade, prior studies of HIF-1α regulation in neurons have been limited by the difficulty of performing quantitative analysis of immunoblotting employing commercially available HIF-1α antibodies. This is particularly true in neuronal cells, where HIF-1α levels are very low, and complicates the study of post-transcriptional modifications of HIF-1α that are key for understanding the mechanisms by which it is regulated. Further, given the neuroprotective properties of prolyl hydroxylase inhibition, a practical, quantitative reporter for PHD enzyme activity in vivo would be extremely valuable. To address this gap, we have developed a sensitive, specific reporter to quantitatively examine distinct parts of the canonical HIF degradation pathway in human neuronal cells, representing a substantial departure from the status quo, namely the approach of looking only at HIF-1α levels and HIF-1 mediated gene transcription [115].
Hypoxia Response Element (HRE)-promoter-driven luciferase reporters have been widely used to measure HIF transactivation of target gene expression. For example, HRE-luciferase expressed in an immortalized mouse striatal neuron cell line, HT22, was used for high-throughput screening of drugs that activate HIF-mediated transcription [116]. These constructs are valuable tools for assaying an endpoint of HIF activation, but do not provide mechanistic insight regarding HIF regulation. For measuring HIF-1α protein accumulation, several reporters containing luciferase and GFP have been developed [117–119]. The main advantage of using luciferase rather than GFP is the substantial increase in sensitivity with an enzymatic assay; luciferase is three orders of magnitude more sensitive and has a correspondingly much wider dynamic range of linearity than fluorescent proteins. Quantitative assays of luciferase activity can be confirmed with immunoblotting with a highly specific and sensitive commercially available monoclonal antibody against firefly luciferase that are much more quantitative than currently available antibodies against HIF-1α.
A full-length HIF-1α reporter such as that used by Moroz et al. which includes the transactivation domain introduces the confound of exaggerating HIF-mediated gene transcription, thus engaging feedback loops that may alter HIF-1α regulation. In contrast, a reporter containing only the ODD lacks the dimerization and DNA-binding domains, allowing it to report on the oxygen-dependent regulation of HIF-1α protein levels without interfering with endogenous HIF signaling. Some studies have inferred PHD activity from HIF-1α levels without directly assaying PHD enzyme activity [112] [113]. This is problematic given that HIF-1α protein stability is regulated in PHD-independent ways. PHD enzymatic assays employing GST-fusion constructs have been performed in cell lysates from HEK293 cells [120], but not in neuronal cells. In vitro assays of PHD enzyme activity have been developed, but require large amounts of recombinant enzyme and expensive reagents (reviewed by [121]). Therefore it would be highly valuable to be able to biochemically detect not only stabilization but prolyl-hydroxylation and ubiquitination of a HIF-1α reporter in order to identify mechanisms by which HIF-1α is stabilized under given experimental conditions.
A fusion protein containing the oxygen dependent degradation domain of HIF-1α (a.a. 530–652) fused to firefly luciferase under control of the cytomegalovirus (CMV) promoter has been previously used as a reporter for HIF-1α stabilization in high-throughput screening for novel activators of the hypoxic response [83], and for in vivo imaging [122] [117]. These studies employing the ODD-luc reporter have looked only at accumulation of the reporter as measured by luciferase activity assay, without investigating the mechanism by which it accumulates or is degraded. We have used the ODD-luc reporter as a quantitative reporter for the oxygen-dependent degradation of HIF-1α in SH-SY5Y human neuroblastoma cells, taking advantage of luciferase activity assays and the wider dynamic range of luciferase antibodies relative to antibodies for endogenous HIF-1α. We have confirmed that ODD-luc is regulated by PHD, VHL, and the proteasome in a manner similar to endogenous HIF, and found that ODD-luc is a practical, specific, and quantitative reporter for the oxygen-dependent degradation of HIF in human neuronal cells [115]. Further, immunoblotting for P564 hydroxylation on ODD-luc provides a sensitive and quantitative direct assay of PHD enzyme activity in human neuronal cells. We expect this reporter to provide important information regarding PHD activity in vitro and in vivo following ischemia (and in other neurological disorders) and we expect that it will facilitate strategies to stabilize HIF in ischemia. These efforts will provide additional, needed clarification regarding the salutary and deleterious roles of HIF in ischemia, and how HIF PHD inhibition can be used to bias outcomes favorably.
Concluding remarks
In this review, we have tried to highlight the important evolutionary role that hypoxic adaptation has played in metazoans. We have reviewed the molecular mechanisms subserving post transcriptional and transcriptional adaptation to hypoxia, focusing primarily on the nervous system. Finally, we highlight the fact that HIF activation and HIF prolyl hydroxylase inhibition are not interchangeable, and the articulate potential roles for inhibitors of the HIF prolyl hydroxylases in a range of neurological conditions, particularly stroke.
Highlights.
Homeostatic responses to hypoxia have evolved over billions of years.
Central sensors in adaptive responses to hypoxia are the prolyl-4 hydroyxlases- oxygen, iron, and 2-oxoglutarate dependent dioxygenases
Molecular and pharmacological inhibition of the HIF prolyl hydroxylases in normoxia by “antioxidant” iron chelators not only stabilizes HIF-1, but also protects against oxidative stress induced ferroptosis in vitro, but also against cerebral ischemia in vivo.
HIF prolyl hydroxylase inhibition protects via HIF dependent and independent pathways.
Acknowledgments
Grant funding supporting this work comprises NYS DOH C019772 (R.R.R.), the Thomas Hartman Foundation (R.R.R), the Miriam and Sheldon G. Adelson Medical Research Foundation (R.R.R), and the Burke Foundation.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryu H, Lee J, Zaman K, Kubilis J, Ferrante RJ, Ross BD, Neve R, Ratan RR. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 2003;23:3597–3606. doi: 10.1523/JNEUROSCI.23-09-03597.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryu H, Lee J, Olofsson BA, Mwidau A, Dedeoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:4281–4286. doi: 10.1073/pnas.0737363100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, Bush AI, Langsetmo I, Seeley T, Gunzler V, Ratan RR. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, Mahishi L, Cooper AJ, Antonyak MA, Cerione RA, Li B, Starkov A, Chaturvedi RK, Beal MF, Coppola G, Geschwind DH, Ryu H, Xia L, Iismaa SE, Pallos J, Pasternack R, Hils M, Fan J, Raymond LA, Marsh JL, Thompson LM, Ratan RR. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2010;2:349–370. doi: 10.1002/emmm.201000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sleiman SF, Langley BC, Basso M, Berlin J, Xia L, Payappilly JB, Kharel MK, Guo H, Marsh JL, Thompson LM, Mahishi L, Ahuja P, MacLellan WR, Geschwind DH, Coppola G, Rohr J, Ratan RR. Mithramycin is a gene-selective Sp1 inhibitor that identifies a biological intersection between cancer and neurodegeneration. J Neurosci. 2011;31:6858–6870. doi: 10.1523/JNEUROSCI.0710-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper GM. The Cell: A Molecular Approach. Sunderland (MA): Sinauer Associates; 2000. [Google Scholar]
- 9.Gorres KL, Raines RT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. 2010;45:106–124. doi: 10.3109/10409231003627991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loenarz C, Coleman ML, Boleininger A, Schierwater B, Holland PW, Ratcliffe PJ, Schofield CJ. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12:63–70. doi: 10.1038/embor.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 12.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 13.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, Payette J, Holcik M, Pause A, Lee S. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486:126–129. doi: 10.1038/nature11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65:3299–3306. doi: 10.1158/0008-5472.CAN-04-4130. [DOI] [PubMed] [Google Scholar]
- 15.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 16.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 17.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 18.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 19.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 20.Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, Harper JW, Conaway JW. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 21.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- 22.Ratan RR, Siddiq A, Aminova L, Langley B, McConoughey S, Karpisheva K, Lee HH, Carmichael T, Kornblum H, Coppola G, Geschwind DH, Hoke A, Smirnova N, Rink C, Roy S, Sen C, Beattie MS, Hart RP, Grumet M, Sun D, Freeman RS, Semenza GL, Gazaryan I. Small molecule activation of adaptive gene expression: tilorone or its analogs are novel potent activators of hypoxia inducible factor-1 that provide prophylaxis against stroke and spinal cord injury. Ann N Y Acad Sci. 2008;1147:383–394. doi: 10.1196/annals.1427.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smirnova NA, Rakhman I, Moroz N, Basso M, Payappilly J, Kazakov S, Hernandez-Guzman F, Gaisina IN, Kozikowski AP, Ratan RR, Gazaryan IG. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 2010;17:380–391. doi: 10.1016/j.chembiol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 26.Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, Jelkmann W, Jaakkola P, Metzen E. Hypoxia-inducible factor-1 (HIF-1) promotes its degradation by induction of HIF-alpha-prolyl-4-hydroxylases. Biochem J. 2004;381:761–767. doi: 10.1042/BJ20040620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Landazuri MO, Vara-Vega A, Viton M, Cuevas Y, del Peso L. Analysis of HIF-prolyl hydroxylases binding to substrates. Biochem Biophys Res Commun. 2006;351:313–320. doi: 10.1016/j.bbrc.2006.09.170. [DOI] [PubMed] [Google Scholar]
- 28.Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2:ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikhaylova O, Ignacak ML, Barankiewicz TJ, Harbaugh SV, Yi Y, Maxwell PH, Schneider M, Van Geyte K, Carmeliet P, Revelo MP, Wyder M, Greis KD, Meller J, Czyzyk-Krzeska MF. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 2008;28:2701–2717. doi: 10.1128/MCB.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, Polyak K, Brown M, Richardson AL, Hahn WC, Kaelin WG, Jr, Bommi-Reddy A. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell. 2009;16:413–424. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boulahbel H, Duran RV, Gottlieb E. Prolyl hydroxylases as regulators of cell metabolism. Biochem Soc Trans. 2009;37:291–294. doi: 10.1042/BST0370291. [DOI] [PubMed] [Google Scholar]
- 32.Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9:11–22. doi: 10.1016/j.cmet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 34.Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, Hagen T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–31284. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 36.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 37.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandau KB, Zhou J, Kietzmann T, Brune B. Regulation of the hypoxia-inducible factor 1alpha by the inflammatory mediators nitric oxide and tumor necrosis factor-alpha in contrast to desferroxamine and phenylarsine oxide. J Biol Chem. 2001;276:39805–39811. doi: 10.1074/jbc.M107689200. [DOI] [PubMed] [Google Scholar]
- 39.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 40.Nyhan MJ, O’Sullivan GC, McKenna SL. Role of the VHL (von Hippel-Lindau) gene in renal cancer: a multifunctional tumour suppressor. Biochem Soc Trans. 2008;36:472–478. doi: 10.1042/BST0360472. [DOI] [PubMed] [Google Scholar]
- 41.Zhou MI, Wang H, Ross JJ, Kuzmin I, Xu C, Cohen HT. The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J Biol Chem. 2002;277:39887–39898. doi: 10.1074/jbc.M205040200. [DOI] [PubMed] [Google Scholar]
- 42.Etlinger JD, Goldberg AL. A soluble ATP-dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. Proc Natl Acad Sci U S A. 1977;74:54–58. doi: 10.1073/pnas.74.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme. Mechanism and role in protein-ubiquitin conjugation. J Biol Chem. 1982;257:2543–2548. [PubMed] [Google Scholar]
- 45.Kuhlbrodt K, Mouysset J, Hoppe T. Orchestra for assembly and fate of polyubiquitin chains. Essays Biochem. 2005;41:1–14. doi: 10.1042/EB0410001. [DOI] [PubMed] [Google Scholar]
- 46.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 47.Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell. 2005;20:687–698. doi: 10.1016/j.molcel.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 48.Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN. ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell. 2006;24:39–50. doi: 10.1016/j.molcel.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meller R. The role of the ubiquitin proteasome system in ischemia and ischemic tolerance. Neuroscientist. 2009;15:243–260. doi: 10.1177/1073858408327809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zungu M, Schisler JC, Essop MF, McCudden C, Patterson C, Willis MS. Regulation of AMPK by the ubiquitin proteasome system. Am J Pathol. 2011;178:4–11. doi: 10.1016/j.ajpath.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 52.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalousi A, Mylonis I, Politou AS, Chachami G, Paraskeva E, Simos G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J Cell Sci. 2010;123:2976–2986. doi: 10.1242/jcs.068122. [DOI] [PubMed] [Google Scholar]
- 54.Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. doi: 10.1074/jbc.274.46.32631. [DOI] [PubMed] [Google Scholar]
- 55.Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem. 2006;281:33095–33106. doi: 10.1074/jbc.M605058200. [DOI] [PubMed] [Google Scholar]
- 56.Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282:20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 57.Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105–109. doi: 10.1016/s0014-5793(03)00807-x. [DOI] [PubMed] [Google Scholar]
- 58.Carrero P, Okamoto K, Coumailleau P, O’Brien S, Tanaka H, Poellinger L. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–415. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lando D, Pongratz I, Poellinger L, Whitelaw ML. A redox mechanism controls differential DNA binding activities of hypoxia-inducible factor (HIF) 1alpha and the HIF-like factor. J Biol Chem. 2000;275:4618–4627. doi: 10.1074/jbc.275.7.4618. [DOI] [PubMed] [Google Scholar]
- 60.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO, Semenza GL. Calcineurin promotes hypoxia-inducible factor 1alpha expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem. 2007;282:37064–37073. doi: 10.1074/jbc.M705015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flugel D, Gorlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol. 2007;27:3253–3265. doi: 10.1128/MCB.00015-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flugel D, Gorlach A, Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119:1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bae SH, Jeong JW, Park JA, Kim SH, Bae MK, Choi SJ, Kim KW. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324:394–400. doi: 10.1016/j.bbrc.2004.09.068. [DOI] [PubMed] [Google Scholar]
- 66.Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem. 2011;286:6963–6970. doi: 10.1074/jbc.M110.188706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell. 2007;18:4691–4697. doi: 10.1091/mbc.E07-04-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res. 1994;54:1425–1430. [PubMed] [Google Scholar]
- 69.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004;279:2550–2558. doi: 10.1074/jbc.M308197200. [DOI] [PubMed] [Google Scholar]
- 71.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 72.Bae MK, Ahn MY, Jeong JW, Bae MH, Lee YM, Bae SK, Park JW, Kim KR, Kim KW. Jab1 interacts directly with HIF-1alpha and regulates its stability. J Biol Chem. 2002;277:9–12. doi: 10.1074/jbc.C100442200. [DOI] [PubMed] [Google Scholar]
- 73.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 74.Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. doi: 10.1038/jcbfm.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 76.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 77.Bowern N, Ramshaw IA, Clark IA, Doherty PC. Inhibition of autoimmune neuropathological process by treatment with an iron-chelating agent. J Exp Med. 1984;160:1532–1543. doi: 10.1084/jem.160.5.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaur D, Andersen J. Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res Rev. 2004;3:327–343. doi: 10.1016/j.arr.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 79.Zaman K, Ryu H, Hall D, O’Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li D, Bai T, Brorson JR. Adaptation to moderate hypoxia protects cortical neurons against ischemia-reperfusion injury and excitotoxicity independently of HIF-1alpha. Exp Neurol. 2011;230:302–310. doi: 10.1016/j.expneurol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lomb DJ, Straub JA, Freeman RS. Prolyl hydroxylase inhibitors delay neuronal cell death caused by trophic factor deprivation. J Neurochem. 2007;103:1897–1906. doi: 10.1111/j.1471-4159.2007.04873.x. [DOI] [PubMed] [Google Scholar]
- 82.Niatsetskaya Z, Basso M, Speer RE, McConoughey SJ, Coppola G, Ma TC, Ratan RR. HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington’s disease and Alzheimer’s disease. Antioxid Redox Signal. 2010;12:435–443. doi: 10.1089/ars.2009.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smirnova NA, Rakhman I, Moroz N, Basso M, Payappilly J, Kazakov S, Hernandez-Guzman F, Gaisina IN, Kozikowski AP, Ratan RR, Gazaryan IG. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 2010;17:380–391. doi: 10.1016/j.chembiol.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee DW, Rajagopalan S, Siddiq A, Gwiazda R, Yang L, Beal MF, Ratan RR, Andersen JK. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. J Biol Chem. 2009;284:29065–29076. doi: 10.1074/jbc.M109.000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tian YM, Yeoh KK, Lee MK, Eriksson T, Kessler BM, Kramer HB, Edelmann MJ, Willam C, Pugh CW, Schofield CJ, Ratcliffe PJ. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286:13041–13051. doi: 10.1074/jbc.M110.211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 87.Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, Kim E, Antonyak MA, Cerione RA, Iismaa SE, Willis D, Cho S, Ratan RR. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 2012;32:6561–6569. doi: 10.1523/JNEUROSCI.3353-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de Simone G, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 92.Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008;28:1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Faria MH, Muniz LR, Vasconcelos PR. Ketone bodies metabolism during ischemic and reperfusion brain injuries following bilateral occlusion of common carotid arteries in rats. Acta Cir Bras. 2007;22:125–129. doi: 10.1590/s0102-86502007000200009. [DOI] [PubMed] [Google Scholar]
- 94.Hofer RE, Wagner SRt, Pasternak JJ, Albrecht RF, 2nd, Gallagher WJ, Lanier WL. Fructose-1,6-bisphosphate and fructose-2,6-bisphosphate do not influence brain carbohydrate or high-energy phosphate metabolism in a rat model of forebrain ischemia. J Neurosurg Anesthesiol. 2009;21:31–39. doi: 10.1097/ANA.0b013e31818acfa4. [DOI] [PubMed] [Google Scholar]
- 95.Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–1373. doi: 10.1038/nn.2951. [DOI] [PubMed] [Google Scholar]
- 96.Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 97.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bruer U, Weih MK, Isaev NK, Meisel A, Ruscher K, Bergk A, Trendelenburg G, Wiegand F, Victorov IV, Dirnagl U. Induction of tolerance in rat cortical neurons: hypoxic preconditioning. FEBS Lett. 1997;414:117–121. doi: 10.1016/s0014-5793(97)00954-x. [DOI] [PubMed] [Google Scholar]
- 99.Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1: 26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ratan RR, Siddiq A, Aminova L, Lange PS, Langley B, Ayoub I, Gensert J, Chavez J. Translation of ischemic preconditioning to the patient: prolyl hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke therapy. Stroke. 2004;35:2687–2689. doi: 10.1161/01.STR.0000143216.85349.9e. [DOI] [PubMed] [Google Scholar]
- 101.Stowe AM, Plautz EJ, Nguyen P, Frost SB, Eisner-Janowicz I, Barbay S, Dancause N, Sensarma A, Taylor MD, Zoubina EV, Nudo RJ. Neuronal HIF-1 alpha protein and VEGFR-2 immunoreactivity in functionally related motor areas following a focal M1 infarct. J Cereb Blood Flow Metab. 2008;28:612–620. doi: 10.1038/sj.jcbfm.9600560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chavez JC, LaManna JC. Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J Neurosci. 2002;22:8922–8931. doi: 10.1523/JNEUROSCI.22-20-08922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu Y, Li X, Xie W, Jankovic J, Le W, Pan T. Neuroprotection of deferoxamine on rotenone-induced injury via accumulation of HIF-1 alpha and induction of autophagy in SH-SY5Y cells. Neurochem Int. 2010;57:198–205. doi: 10.1016/j.neuint.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 105.Chen W, Jadhav V, Tang J, Zhang JH. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008;31:433–441. doi: 10.1016/j.nbd.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Higashida T, Peng C, Li J, Dornbos D, 3rd, Teng K, Li X, Kinni H, Guthikonda M, Ding Y. Hypoxia-inducible factor-1alpha contributes to brain edema after stroke by regulating aquaporins and glycerol distribution in brain. Curr Neurovasc Res. 2011;8:44–51. doi: 10.2174/156720211794520251. [DOI] [PubMed] [Google Scholar]
- 107.Yeh SH, Ou LC, Gean PW, Hung JJ, Chang WC. Selective inhibition of early--but not late--expressed HIF-1alpha is neuroprotective in rats after focal ischemic brain damage. Brain Pathol. 2011;21:249–262. doi: 10.1111/j.1750-3639.2010.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aminova LR, Chavez JC, Lee J, Ryu H, Kung A, Lamanna JC, Ratan RR. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280:3996–4003. doi: 10.1074/jbc.M409223200. [DOI] [PubMed] [Google Scholar]
- 109.Guo S, Miyake M, Liu KJ, Shi H. Specific inhibition of hypoxia inducible factor 1 exaggerates cell injury induced by in vitro ischemia through deteriorating cellular redox environment. J Neurochem. 2009;108:1309–1321. doi: 10.1111/j.1471-4159.2009.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vangeison G, Carr D, Federoff HJ, Rempe DA. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci. 2008;28:1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 112.Osada-Oka M, Hashiba Y, Akiba S, Imaoka S, Sato T. Glucose is necessary for stabilization of hypoxia-inducible factor-1alpha under hypoxia: contribution of the pentose phosphate pathway to this stabilization. FEBS Lett. 2010;584:3073–3079. doi: 10.1016/j.febslet.2010.05.046. [DOI] [PubMed] [Google Scholar]
- 113.Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, Linehan WM, Neckers L. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ryu JH, Li SH, Park HS, Park JW, Lee B, Chun YS. Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. J Biol Chem. 2011;286:6963–6970. doi: 10.1074/jbc.M110.188706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karuppagouder SS, Basso M, Sleiman SF, Ma TC, Speer RE, Smirnova NA, Gazaryan IG, Ratan RR. In vitro Ischemia Suppresses Hypoxic Induction of Hypoxia Inducible Factor-1α by Inhibition of Synthesis and Not Enhanced Degradation. Journal of Neuroscience Research. 2013 doi: 10.1002/jnr.23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ratan RR, Siddiq A, Aminova L, Langley B, McConoughey S, Karpisheva K, Lee HH, Carmichael T, Kornblum H, Coppola G, Geschwind DH, Hoke A, Smirnova N, Rink C, Roy S, Sen C, Beattie MS, Hart RP, Grumet M, Sun D, Freeman RS, Semenza GL, Gazaryan I. Small molecule activation of adaptive gene expression: tilorone or its analogs are novel potent activators of hypoxia inducible factor-1 that provide prophylaxis against stroke and spinal cord injury. Ann N Y Acad Sci. 2008;1147:383–394. doi: 10.1196/annals.1427.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Safran M, Kim WY, O’Connell F, Flippin L, Gunzler V, Horner JW, Depinho RA, Kaelin WG., Jr Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc Natl Acad Sci U S A. 2006;103:105–110. doi: 10.1073/pnas.0509459103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moroz E, Carlin S, Dyomina K, Burke S, Thaler HT, Blasberg R, Serganova I. Real-time imaging of HIF-1alpha stabilization and degradation. PLoS One. 2009;4:e5077. doi: 10.1371/journal.pone.0005077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Arquier N, Vigne P, Duplan E, Hsu T, Therond PP, Frelin C, D’Angelo G. Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem J. 2006;393:471–480. doi: 10.1042/BJ20050675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, Hagen T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–31284. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smirnova NA, Hushpulian DM, Speer RE, Gaisina IN, Ratan RR, Gazaryan IG. Catalytic mechanism and substrate specificity of HIF prolyl hydroxylases. Biochemistry (Mosc) 2012;77:1108–1119. doi: 10.1134/S0006297912100033. [DOI] [PubMed] [Google Scholar]
- 122.Goldman SJ, Chen E, Taylor R, Zhang S, Petrosky W, Reiss M, Jin S. Use of the ODD-luciferase transgene for the non-invasive imaging of spontaneous tumors in mice. PLoS One. 2011;6:e18269. doi: 10.1371/journal.pone.0018269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32:1347–1361. doi: 10.1038/jcbfm.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J, Scharff A, Dirnagl U, Meisel A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J Cereb Blood Flow Metab. 2002;22:520–525. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Hamrick SE, McQuillen PS, Jiang X, Mu D, Madan A, Ferriero DM. A role for hypoxia-inducible factor-1alpha in desferoxamine neuroprotection. Neurosci Lett. 2005;379:96–100. doi: 10.1016/j.neulet.2004.12.080. [DOI] [PubMed] [Google Scholar]
- 126.Li YX, Ding SJ, Xiao L, Guo W, Zhan Q. Desferoxamine preconditioning protects against cerebral ischemia in rats by inducing expressions of hypoxia inducible factor 1 alpha and erythropoietin. Neurosci Bull. 2008;24:89–95. doi: 10.1007/s12264-008-0089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ma TC, Langley B, Ko B, Wei N, Gazaryan IG, Zareen N, Yamashiro DJ, Willis DE, Ratan RR. A screen for inducers of p21(waf1/cip1) identifies HIF prolyl hydroxylase inhibitors as neuroprotective agents with antitumor properties. Neurobiology of disease. 2012;49C:13–21. doi: 10.1016/j.nbd.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Regan RF, Panter SS. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma. 1996;13:223–231. doi: 10.1089/neu.1996.13.223. [DOI] [PubMed] [Google Scholar]
- 129.Almli LM, Hamrick SE, Koshy AA, Tauber MG, Ferriero DM. Multiple pathways of neuroprotection against oxidative stress and excitotoxic injury in immature primary hippocampal neurons. Brain Res Dev Brain Res. 2001;132:121–129. doi: 10.1016/s0165-3806(01)00302-9. [DOI] [PubMed] [Google Scholar]
- 130.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol. 2000;48:285–296. [PubMed] [Google Scholar]
- 132.Mu D, Chang YS, Vexler ZS, Ferriero DM. Hypoxia-inducible factor 1alpha and erythropoietin upregulation with deferoxamine salvage after neonatal stroke. Exp Neurol. 2005;195:407–415. doi: 10.1016/j.expneurol.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 133.Freret T, Valable S, Chazalviel L, Saulnier R, Mackenzie ET, Petit E, Bernaudin M, Boulouard M, Schumann-Bard P. Delayed administration of deferoxamine reduces brain damage and promotes functional recovery after transient focal cerebral ischemia in the rat. Eur J Neurosci. 2006;23:1757–1765. doi: 10.1111/j.1460-9568.2006.04699.x. [DOI] [PubMed] [Google Scholar]
- 134.Hanson LR, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ, Marti DL, Hoekman JD, Matthews RB, Frey WH, 2nd, Panter SS. Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J Pharmacol Exp Ther. 2009;330:679–686. doi: 10.1124/jpet.108.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao Y, Rempe DA. Prophylactic neuroprotection against stroke: low-dose, prolonged treatment with deferoxamine or deferasirox establishes prolonged neuroprotection independent of HIF-1 function. J Cereb Blood Flow Metab. 2010;31:1412–1423. doi: 10.1038/jcbfm.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chu PW, Beart PM, Jones NM. Preconditioning protects against oxidative injury involving hypoxia-inducible factor-1 and vascular endothelial growth factor in cultured astrocytes. Eur J Pharmacol. 2010;633:24–32. doi: 10.1016/j.ejphar.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 137.Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1alpha. Neurobiology of disease. 2012;45:733–742. doi: 10.1016/j.nbd.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zheng H, Blat D, Fridkin M. Novel neuroprotective neurotrophic NAP analogs targeting metal toxicity and oxidative stress: potential candidates for the control of neurodegenerative diseases. J Neural Transm Suppl. 2006:163–172. doi: 10.1007/978-3-211-33328-0_18. [DOI] [PubMed] [Google Scholar]
- 139.Youdim MB, Stephenson G, Ben Shachar D. Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Ann N Y Acad Sci. 2004;1012:306–325. doi: 10.1196/annals.1306.025. [DOI] [PubMed] [Google Scholar]
- 140.Chinta SJ, Rajagopalan S, Ganesan A, Andersen JK. A possible novel anti-inflammatory mechanism for the pharmacological prolyl hydroxylase inhibitor 3,4-dihydroxybenzoate: implications for use as a therapeutic for Parkinson’s disease. Parkinsons Dis. 2012;2012:364684. doi: 10.1155/2012/364684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guo C, Wang T, Zheng W, Shan ZY, Teng WP, Wang ZY. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:562–575. doi: 10.1016/j.neurobiolaging.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 142.Fine JM, Baillargeon AM, Renner DB, Hoerster NS, Tokarev J, Colton S, Pelleg A, Andrews A, Sparley KA, Krogh KM, Frey WH, Hanson LR. Intranasal deferoxamine improves performance in radial arm water maze, stabilizes HIF-1alpha, and phosphorylates GSK3beta in P301L tau transgenic mice. Exp Brain Res. 2012;219:381–390. doi: 10.1007/s00221-012-3101-0. [DOI] [PubMed] [Google Scholar]
- 143.Yang YT, Ju TC, Yang DI. Induction of hypoxia inducible factor-1 attenuates metabolic insults induced by 3-nitropropionic acid in rat C6 glioma cells. Journal of neurochemistry. 2005;93:513–525. doi: 10.1111/j.1471-4159.2005.03032.x. [DOI] [PubMed] [Google Scholar]
- 144.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, Cortopassi G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8:425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 145.Li K, Besse EK, Ha D, Kovtunovych G, Rouault TA. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008;17:2265–2273. doi: 10.1093/hmg/ddn127. [DOI] [PMC free article] [PubMed] [Google Scholar]