

Abstract
Two new steroids, 3α,17α,19,20-tetrahydroxy-4α-methylpregn-8-ene (1) and 3α,12α,17α,20-tetrahydroxy-4α-methylpregn-8-ene (2) and three new sesquiterpenoids, 12-hydroxy-α-cadinol (3), 3α,12-dihydroxy-δ-cadinol (4), and 3α,6α-dihydroxyspiroax-4-ene (5), have been isolated from cultures of the fungus Phellinus igniarius. Their structures were characterized based on extensive spectroscopic data. In preliminary in vitro assays, compounds 3 and 4 exhibited the vascular-activities against phenylephrine-induced vasoconstriction with the relaxing rates of 11.0 % and 7.0 % at 3 × 10−4 M, respectively.
Electronic supplementary material
The online version of this article (doi:10.1007/s13659-014-0045-z) contains supplementary material, which is available to authorized users.
Keywords: Pregnene steroids, Sesquiterpenes, Phellinus igniarius, Cytotoxicity, Vascular-activities
Introduction
Fungi are biosynthetically talented organisms capable of producing a wide range of chemically diverse and biologically intriguing small molecules. Phellinus igniarius, belonging to Polyporaceae family, is widely distributed in Yunnan and Sichuan Provinces of China [1]. It preferably grows on stems of aspen, robur, and birch. Its fruiting body was used to treat fester, abdominalgia, bloody gonorrhea and antidiarrheal in traditional Chinese medicine [2]. Previous chemical investigations on both fruiting bodies and cultures of this fungus reported various secondary metabolites with interesting structures and significant bioactivities [3–7]. Phelligridins D and E showed selective cytotoxicity against a human lung cancer cell line (A 549) and a liver cancer cell line (Bel 7402) [3], while phelligridins H–J, being pyrano[4,3-c] [2] benzopyran-1,6-dione and furo[3,2-c]pyran-4-one derivatives, showed cytotoxic activity against human cancer cell lines and protein tyrosine phosphatase 1B inhibition [4]. A pyrano[4,3-c] [2] benzopyran-1,6-dione derivative and a novel 26-membered macrocyclic metabolite phelligridimer A with antioxidant activities were also isolated from the fruiting bodies [5, 6]. Moreover, several tremulane sesquiterpenes were obtained from the cultures of this fungus, some of which showed significant vascular-relaxing activities against phennylephrine-induced vasoconstriction [7]. To seek for more active molecules, further investigation of this fungus has resulted in the isolation of two pregnene steroids (1 and 2) and three sesquiterpenes (3–5) (Fig. 1). Compounds 1 and 2 are unusual 4-methyl homopregnane derivatives [8], and compound 5 is a rare spiroaxane sesquiterpene which was firstly isolated from the marine sponge Axinella cannabina in 1973 [9]. Based on the results of previous biological assays [10, 11], compounds 1, 2, and 5 were tested for their cytotoxicity in vitro against five human tumor cell lines, while compounds 3 and 4 were tested for their vascular-activities against phenylephrine-induced vasoconstriction. This paper describes the isolation, structure elucidation and results of biological activities.
Fig. 1.
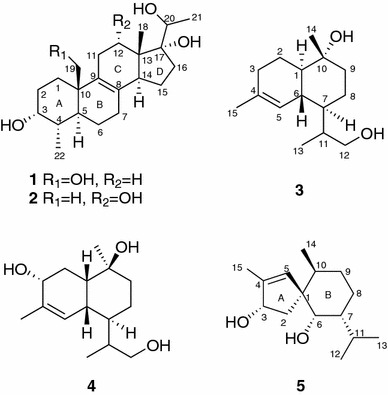
Structures of compounds 1–5
Results and Discussion
Compound 1 was isolated as a white amorphous powder. The HREIMS data (m/z 364.2611 [M]+) indicated the molecular formula C22H36O4, requiring five degrees of unsaturation. The IR absorption bands at 3441 and 1631 cm−1 suggested the presence of hydroxy and double bond groups, respectively. The 1H NMR spectrum of 1 (Table 1) displayed signals of three methyls (δH 0.74, 0.94, and 1.18). The 13C NMR (Table 1) and DEPT spectra of 1 indicated 22 carbon resonances, including an oxygenated methylene carbon (δC 66.3), two oxygenated methines (δC 72.3, 73.1), one oxygenated quaternary carbon (δC 86.4), and a tetrasubstituted double bond (δC 130.9 and 135.1). Apart from one double bond, the remaining four degrees of unsaturation indicated that 1 possessed a four-ring system. Inspection of 1H-1H COSY correlations resulted in the fragments as shown (Fig. 2). In the HMBC spectrum (Fig. 2), correlations from δH 3.73 (br s, H-3) to δC 26.7 (t, C-1), 30.5 (t, C-2) and 41.8 (d, C-5), δH 0.94 (3H, d, J = 6.0 Hz, Me-22) to δC 72.3 (d, C-3), 37.3 (d, C-4) and C-5, δH 3.86 (H, d, J = 10.7 Hz, H-19a) and 3.63 (H, d, J = 10.7 Hz, H-19) to C-1 and C-5 gave an evidence for a six-member ring A. Besides, correlations of δH 1.69 (H, overlap, H-6a) and δH 2.10 (H, m, H-7a) with δC 130.9 (s, C-8), H-19 with δC 135.1 (s, C-9) and δC 41.7 (s, C-10) indicated the fragments of C-6-C-7-C-8, C-9-C10. Moreover, C-8 and C-9 were connected by the double bond. Hence, another six-member ring B was established. Likewise, in the HMBC spectrum, the correlations from δH 0.74 (3H, s, Me-18) to δC 30.4 (t, C-12) and 48.7 (d, C-14), from H-7 to C-14 and from δH 2.28 (2H, m, H-11) to C-9 indicated the presence of ring C. The last ring D was clearly established by HMBC correlations of Me-18 and δH 1.75 (H, overlap, H-16b) with δC 86.4 (s, C-17). Finally, the backbone of a 6/6/6/5 ring system related to that of 3,17,20-trihydroxy-4-methylpregn-8-en-7-one [7] was deduced. In the ROESY spectrum (Fig. 2), correlations between H-22/H-5 and H-3/H-4 were observed, suggesting that H-3 and H-4 were both β oriented. Thus, the structure of compound 1 was elucidated to be 3α,17α,19,20-tetrahydroxy-4α-methylpregn-8-ene, as shown.
Table 1.
1H and 13C NMR data for 1 and 2 (δ in ppm, J in Hz)
| No. | 1 | 2 | ||
|---|---|---|---|---|
| 1H | 13C | 1H | 13C | |
| 1 | 1.92, m | 26.7, CH2 | 1.61, overlap | 32.1, CH2 |
| 1.50, m | 1.48, m | |||
| 2 | 1.52, m | 30.5, CH2 | 1.76, m | 30.5, CH2 |
| 1.80, m | 1.71, m | |||
| 3 | 3.73, br s | 72.3, CH | 3.69, brd (2.5) | 72.4, CH |
| 4 | 1.60, overlap | 37.3, CH | 1.54, m | 36.8, CH |
| 5 | 1.59, overlap | 41.8, CH | 1.59, overlap | 41.7, CH |
| 6 | 1.69, overlap | 21.7, CH2 | 1.73, m | 21.9, CH2 |
| 1.35, m | 1.30, m | |||
| 7 | 2.10, m | 28.1, CH2 | 2.11, overlap | 28.4, CH2 |
| 2.04, m | 2.04, m | |||
| 8 | 130.9, C | 129.0, C | ||
| 9 | 135.1, C | 133.9, C | ||
| 10 | 41.7, C | 37.5, C | ||
| 11 | 2.23, m | 25.7, CH2 | 2.33, dt (19.0,4.0) | 33.4, CH2 |
| 2.09, overlap | ||||
| 12 | 1.80, m | 30.4, CH2 | 4.17, d (4.0) | 73.4, CH |
| 1.70, overlap | ||||
| 13 | 46.7, C | 49.9, C | ||
| 14 | 2.80,m | 48.7, CH | 3.23,m | 41.6, CH |
| 15 | 1.74, overlap | 24.0, CH2 | 1.83, m | 22.9, CH2 |
| 1.40, m | 1.46, m | |||
| 16 | 2.13, m | 38.9, CH2 | 2.07, overlap | 38.5, CH2 |
| 1.75, overlap | 1.79, m | |||
| 17 | 86.4, C | 88.6, C | ||
| 18 | 0.74, s | 14.6, CH3 | 0.63, s | 14.4, CH3 |
| 19 | 3.86, d (10.7) | 66.3, CH2 | 0.96, s | 18.5, CH3 |
| 3.63, d (10.7) | ||||
| 20 | 3.78, q (6.4) | 73.1, CH | 3.76, q (6.4) | 72.6, CH |
| 21 | 1.18, d (6.4) | 18.8, CH3 | 1.29, d (6.4) | 18.4, CH3 |
| 22 | 0.94, d (6.0) | 17.1, CH3 | 0.95, d (6.5) | 16.7, CH3 |
Data (δ) were measured in methanol-d4. The assignments were based on DEPT, 1H-1H COSY, HSQC, and HMBC experiments
Fig. 2.
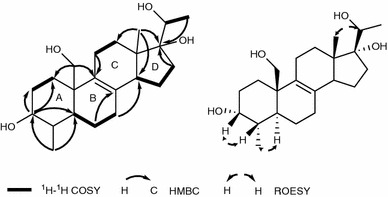
Key1H-1H COSY, HMBC and ROESY correlations of compound 1
Compound 2, also purified as a white amorphous powder, had the same molecular formula of C22H36O4 as that of compound 1, according to its HREIMS at m/z 364.2605 ([M]+). The 13C NMR (Table 1) spectroscopic data were similar to those of compound 1. The main differences were the missing of an oxygenated methylene and the presence of an oxygenated methine in 2, which revealed the change of substitution of a hydroxy group. It was further confirmed by the HMBC correlations of δH 0.63 (3H, s, Me-18) with the oxygenated methine carbon. The mentioned information suggested that the hydroxy group was located at C-12. On the basis of the ROESY experiment, H-12 was elucidated to be β oriented by correlations of H-12 with H-18. Therefore, compound 2 was established to be 3α,12α,17α,20-tetrahydroxy-4α-methylpregn-8-ene, as shown.
Compound 3, a colorless oil, had a molecular formula of C15H26O2 on the basis of HREIMS at m/z 238.1931 ([M]+). The 13C NMR (Table 2) and DEPT spectra of 3 indicated 15 carbon resonances, including three methyls, five methylenes (one oxygenated at δC 67.1), five methines (one sp2 carbon at δC 123.3), and two quaternary carbon (one oxygenated at δC 72.9 and one sp2 carbon at δC 136.0). Detailed analysis of NMR data suggested that compound 3 should be a cadinane-type sesquiterpene with a similar structure to that of 15-hydroxy-α-cadinol [12]. The main difference between the two compounds was that the hydroxy should be placed at C-12 in 3 rather than at C-15 in 15-hydroxy-α-cadinol, which was confirmed by the correlations of δH 2.15 (H, m, H-11) and 0.78 (3H, d, J = 7.0 Hz, Me-13) with δC 67.1 (t, C-12) in the HMBC spectrum. Further 2D NMR data supported that the other parts of the structure of 3 were the same as those of 15-hydroxy-α-cadinol [12]. Therefore, compound 3 was deduced to be 12-hydroxy-α-cadinol.
Table 2.
1H NMR and 13C NMR data for compounds 3–5 (δ in ppm, J in Hz)
| No. | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| 1 | 1.23, overlap | 51.0, CH | 1.67, dt (14.0, 2.0) | 46.3, CH | 57.8, C | |
| 2 | 2.06, m | 23.9, CH2 | 1.89, dd (11.5, 6.8) | 31.8, CH2 | 2.45, dd (13.9, 7.8) | 42.8, CH2 |
| 1.22, overlap | 1.56, overlap | 1.56, overlap | ||||
| 3 | 1,94 ~ 2.01, m | 32.0, CH2 | 4.04, t (8.2) | 72.1, CH | 4.58, t (6.7) | 78.2, CH |
| 4 | 136.0, C | 138.0, C | 144.9, C | |||
| 5 | 5.50, s | 123.3 CH | 5.76, d (5.8) | 129.6, CH | 5.48, s | 130.7, CH |
| 6 | 1.79, m | 40.5, C | 2.44, m | 35.8, CH | 3.33, overlap | 76.3, CH |
| 7 | 1.33, m | 43.0, CH | 1.50, m | 43.9, CH | 1.03, m | 46.9, CH |
| 8 | 1.49, overlap | 23.2, CH2 | 1.43, overlap | 23.0, CH2 | 1.65, overlap | 25.2, CH2 |
| 1.21, overlap | 1.43, overlap | 1.36, m | ||||
| 9 | 1.74, dt (13.0, 3.8) | 42.7, CH2 | 1.54, overlap | 35.7, CH2 | 1.56, overlap | 33.1, CH2 |
| 1.47, overlap | 1.44, overlap | 1.23, m | ||||
| 10 | 72.9, C | 72.5, C | 1.66, overlap | 32.6, CH | ||
| 11 | 2.15, m | 35.4, CH | 1.94, m | 37.3, CH | 1.55, overlap | 30.2, CH |
| 12 | 3,39 ~ 3.46, m | 67.1, CH2 | 3.74, dd (10.7, 4.7) | 65.2, CH2 | 0.89, d (6.7) | 21.5, CH3 |
| 3.31, overlap | ||||||
| 13 | 0.78, d (7.0) | 10.6, CH3 | 0.98, d (6.9) | 16.3, CH3 | 0.87, d (6.7) | 21.1, CH3 |
| 14 | 1.07, s | 20.5, CH3 | 1.18, s | 29.3, CH3 | 0.80, d (6.8) | 17.8, CH3 |
| 15 | 1.66, s | 24.0, CH3 | 1.74, s | 19.8, CH3 | 1.75, s | 14.0, CH3 |
Data (δ) were measured in methanol-d4. The assignments were based on DEPT, 1H-1H COSY, HSQC, and HMBC experiments
Compound 4, a white amorphous powder, possessed the molecular formula C15H26O3, on the basis of its HREIMS at m/z 254.1904 ([M]+), 16 mass units higher than that of 3. The 1D NMR spectroscopic data (Table 2) were quite similar to those of 3, with the main difference being an oxygenated methane replacing methylene signal confirmed by the HMBC correlations of δH 4.04 (H, t, J = 8.2 Hz, H-3) with δC 138.0 (s, C-4) and 129.6 (d, C-5). In the ROESY spectrum, the presence of correlations of H-1/H-3, H-1/H-6 indicated that H-1, H-3 and H-6 were at the same face assigned as β orientation. The correlations of H-6 with H-12 suggested that H-7 was at α orientation. In addition, the correlations of H-1 with H-9b, Me-14 with H-9a revealed that Me-14 was at α orientation. Hence, compound 4 was identified as 3α,12-dihydroxy-δ-cadinol, as shown.
Compound 5, a colorless oil, had the molecular formula of C15H26O2 based on its HRESIMS at m/z 268.1814 ([M + Na]+), which implied the presence of three degrees of unsaturation. The IR spectrum showed absorption bands at 3440 and 1632 cm−1, indicating the presence of hydroxy and double bond groups, respectively. The 13C NMR (Table 2) and DEPT spectra indicated 15 carbon resonances, classified as four methyls, three methlyenes, six methines (two oxygenated at δC 76.3 and 78.2; one sp2 carbon at δC 130.7), and two quaternary carbons (one sp2 carbon at δC 144.9; one sp3 quaternary carbon at δC 57.8). In consideration of one degree of unsaturation occupied by one double bond, compound 5 was revealed to possess a two-ring system. Analysis of the 1H-1H COSY spectrum resulted in the deduction of fragments of C-2-C-3, C-7-C-11 and C-6-C-7-C-8-C-9 as shown (Fig. 3). In the HMBC spectrum (Fig. 3), correlations from δH 0.87 (3H, d, J = 6.7 Hz, Me-13) to δC 78.2 (d, C-3), δH 2.45 (H, dd, J = 13.9, 7.8 Hz, H-2a) and 5.48 (H, s, H-5) to δC 57.8 (s, C-1) supported the foundation of a five-member ring A. Similarly, the other ring B was clearly shown by HMBC correlations of δH 3.33 (H, overlap, H-6), 1.56 (H, overlap, H-9a) and 0.89 (3H, d, J = 6.7 Hz, Me-12) with C-1, Me-12 with δC 33.1 (t, C-9). Hence, the two-ring system connected by the spirocarbon C-1 was deduced, which possessed the same skeleton as that of 15-hydroxy-6α,12-epoxy-7β,10αH,11βH-spiroax-4-ene [13]. In the ROESY spectrum (Fig. 3), correlations of H-5/Hax-7 and Hax-7/Heq-6 revealed that H-6, H-7 were β oriented, while the correlations of Heq-6/H-2β and H-2β/H-3 suggested H-3 was also β oriented. However, the presence of correlation of Hax-10/H ax-8α indicated H-10 was α oriented. Finally, compound 5 was established as 3α,6α-dihydroxy-spiroax-4-ene.
Fig. 3.
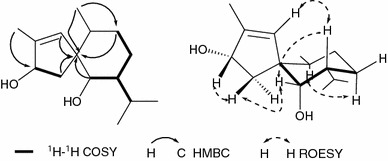
Key1H-1H COSY, HMBC and ROESY correlations of compound 5
Compounds 1, 2, and 5 were evaluated for their cytotoxicity against five human cancer cell lines. None was found to possess significant activity with IC50 values less than 40 μM. Compounds 3 and 4 were tested for the vascular-activities against phenylephrine-induced vasoconstriction. They exhibited the vascular-activities with the relaxing rates of 11.0 % and 7.0 % at 3 × 10−4 M, respectively. It’s worth mentioning that this is the first time to report the vascular-activity of cadinane-type sesquiterpenes.
Experimental
General Experimental Procedures
Optical rotations were measured on a Jasco-P-1020 polarimeter. IR spectra were obtained using a Bruker Tensor 27 FT-IR spectrometer with KBr pellets. NMR spectra were acquired with instrument of a Bruker Avance Ш 600 with deuterated solvent signals as internal standards. HREIMS were measured on a Waters Autospec Premier P776 spectrometer. HRESIMS were recorded on an API QSTAR Pulsar spectrometer. Silica gel (200–300 mesh and 80–100 mesh, Qingdao Marine Chemical Inc., China), Sephadex LH-20 and RP-18 gel (20–45 µM, Fuji Silysia Chemical Ltd., Japan) were used for column chromatography (CC). Preparative HPLC was performed on an Agilent 1100 series with a Zorbax SB-C18 (5 μM, 9.4 × 150 mm) column. Fractions were monitored by thin layer chromatography (TLC) (Qingdao Marine Chemical Inc., China) and spots were visualized by heating silica gel plates immersed in H2SO4 in EtOH, in combination with the Agilent 1200 series HPLC system (Eclipse XDB-C18 column, 5 μM, 4.6 × 150 mm).
Fungal Material and Cultivation Conditions
Fruiting bodies of Phellinus igniarius were collected at Changbai Mountain National Nature Reserve, Antu, Jilin Province, China in 2008 and identified by Prof. Yu-Cheng Dai (Beijing Forestry University). A specimen (No. KIB20081017) was deposited at Kunming Institute of Botany, Chinese Academy of Sciences. The culture medium was composed of glucose (5 %), pork peptone (0.15 %), yeast powder (0.5 %), KH2PO4 (0.05 %) and MgSO4 (0.05 %). The initial PH was adjusted to 6.0 and the fermentation was carried out on a shaker at 150 rpm for 25 days.
Extraction and Isolation
The cultures (20 L) were filtered through cheesecloth to separate broth and mycelium. The broth was extracted four times with ethyl acetate, while the mycelium was extracted three times with CHCl3-MeOH (1:1). The organic layer of both parts were evaporated together to yield a crude extract (9 g). Then this residue was subjected on reverse-phased C18 column eluted with gradient mixture of MeOH and H2O (30:70–100:0, v/v). Fractions were collected and monitored by TLC. Similar fractions were pooled to give twelve sub-fractions (A–L). Sub-fraction L was isolated by reverse-phased C18 column eluted with mixture of MeOH and H2O (45:55, v/v), then purified by Sephadex LH-20 CC (Me2CO) to yield compound 1 (1.4 mg). Sub-fraction G was subjected to Sephadex LH-20 CC (MeOH) and silica gel CC eluted with a petroleum ether-acetone gradient system (6:1, v/v) to give compound 2 (0.8 mg). Sub-fraction I was separated by repeated CC on silica gel and purified by preparative HPLC (MeCN/H2O, from 0:100 to 40:60, 10 mL/min, 40 min) to obtain compound 3 (1.8 mg). Sub-fraction A, isolated by Sephadex LH-20 CC (MeOH) and reverse-phased C18 column eluted with mixture of MeOH and H2O (30:70, v/v) to yield compound 4 (0.7 mg). Sub-fraction J was subjected to silica gel CC eluted with a petroleum ether-acetone gradient system (4:1, v/v) and Sephadex LH-20 CC (Me2CO) to yield compound 5 (1.4 mg).
3α,17α,19,20-Tetrahydroxy-4α-methylpregn-8-ene (1): amorphous powder, −7.2 (c 0.13 MeOH); IR (KBr) νmax 3441, 2922, 2852, 1631, 1465, 1384, 1105, 1036 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HREIMS: m/z 364.2611 (calcd for C22H36O4, [M]+, 364.2614).
3α,12α,17α,20-Tetrahydroxy-4α-methylpregn-8-ene (2): amorphous powder, −9.7 (c 0.07 MeOH); IR (KBr) νmax 3443, 2927, 1634, 1457, 1381, 1065 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 1; HREIMS: m/z 364.2605 (calcd for C22H36O4, [M]+, 364.2614).
12-Hydroxy-α-cadinol (3): colorless oil, +18.7 (c 0.18 MeOH); IR (KBr) νmax 3441, 2926, 1631, 1452, 1382, 1120, 1035 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 2; HREIMS: m/z 238.1931 (calcd for C15H26O2, [M]+, 238.1933).
3α,12-Dihydroxy-δ-cadinol (4): amorphous powder, +9.6 (c 0.06 MeOH); IR (KBr) νmax 3424, 2929, 1631, 1436, 1384, 1030 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 2; HREIMS: m/z 254.1904 (calcd for C15H26O3, [M]+, 254.1882).
3α,6α-Dihydroxy-spiroax-4-ene (5): colorless oil, −4.2 (c 0.14 MeOH); IR (KBr) νmax 3440, 2926, 2870, 1632, 1459, 1384, 1060 cm−1; for 1H (600 MHz) and 13C NMR (150 MHz) data (methanol-d4), see Table 2; HRESIMS: m/z 268.1814 (calcd for C15H26O2Na, [M + Na]+, 268.1831).
Cytotoxicity Assay
Human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, lung cancer A-549 cells, breast cancer MCF-7 and colon cancer SW480 cell lines were used in the cytoxic assay. All cell lines were cultured in RPMI-1640 or DMEM medium (Hyclone, USA), supplemented with 10 % fetal bovine serum (Hyclone, USA) in 5 % CO2 at 37 °C. The cytotoxicity assay was performed according to the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method in 96-well microplates [14].
Vasodilating Activity Assays
Sprague–Dawley rats, weighing 250–350 g, were anaesthetized with pentobarbital sodium (40 mg/kg, i.p.), and the thoracic aorta was removed and placed in Krebs–Henseleit solution (KHS). An aortic ring of about 2–3 mm in length was suspended between two stainless steel hooks in a 5 mL water-jacketed bath containing KHS of the following composition (in mmol/L): NaCl, 120; KCl, 4.7; MgSO4·7H2O, 1.2; KH2PO4, 1.2; CaCl2·2H2O, 2.5; NaHCO3, 25; and glucose, 10. The bathing solution was maintained at 37 ± 0.5 °C and was bubbled with 95 % O2 and 5 % CO2 (pH 7.4) throughout the experiments. One of stainless steel hooks was then connected to a force–displacement transducer (Chengdu instrument factory, Sichuan, China). The initial tension was adjusted to 1.5 g and an equilibration period of 90 min was allowed before commencing the experiments. The resting tension acting in the artery was readjusted periodically until stabilization was achieved. After equilibration, the reactivity of the thoracic aorta was ensured by KCl (60 mmol/L)-induced contraction. When a steady contraction was reached, 10−5 mol/L Acetylcholine (ACh) was added to induce endothelium-dependent relaxation. This step was necessary to verify the integrity of the endothelium.
In order to investigate the effects of various agents on phenylephrine hydrochloride (PE)-induced contraction, when a steady contraction induced by PE (10−6 mol/L) was reached, various agents (3 × 10−4 mol/L) was added to the organ bath. The resulting relaxation was expressed as a percentage (%) of the PE-induced steady contraction in the absence of treatment with various agents.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This project was supported by the National Natural Sciences Foundation of China (U1132607, 81102346) and Natural Sciences Foundation of Yunnan Province (2011FB099).
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.X.L. Mao. The Macrofungi in China. Henan Science and Technology Publishing House: Zhengzhou 477 (2000)
- 2.Mo SY, Yang YC, Shi JG. China J. Chin. Mater. Med. 2003;28:339–341. [PubMed] [Google Scholar]
- 3.Mo SY, Wang S, Zhou G, Yang Y, Li Y, Chen X, Shi JG. J. Nat. Prod. 2004;67:823–828. doi: 10.1021/np030505d. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Shang XY, Wang SJ, Mo SY, Li S, Shi JG, He L. J. Nat. Prod. 2007;70:296–299. doi: 10.1021/np060476h. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Mo SY, Wang SJ, Li S, Yang YC, Shi JG. Org. Lett. 2005;7:1675–1678. doi: 10.1021/ol0475764. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Wang SJ, Mo SY, Li S, Yang YC, Shi JG. Org. Lett. 2005;7:4733–4736. doi: 10.1021/ol0520875. [DOI] [PubMed] [Google Scholar]
- 7.Wu XL, Lin S, Zhu CG, Yue ZG, Yu Y, Dai JG, Shi JG. J. Nat. Prod. 2010;73:1294–1300. doi: 10.1021/np100216k. [DOI] [PubMed] [Google Scholar]
- 8.Habermehl G, Hundrieser HJ. Naturwissenschaften. 1983;70:566–568. doi: 10.1007/BF00376676. [DOI] [Google Scholar]
- 9.Cafieri F, Fattorusso E, Magno S, Santacroce C, Sica D. Tetrahedron. 1973;29:4259. doi: 10.1016/0040-4020(73)80267-4. [DOI] [Google Scholar]
- 10.Ding JH, Feng T, Li ZH, Li L, Liu JK. Nat. Prod. Bioprospect. 2012;2:200–205. doi: 10.1007/s13659-012-0060-x. [DOI] [Google Scholar]
- 11.Yang XY, Feng T, Wang GQ, Ding JH, Li ZH, Li Y, He SH, Liu JK. Phytochemistry. 2014;104:89–94. doi: 10.1016/j.phytochem.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Kuo YH, Chen CH, Chien SC, Lin YL. J. Nat. Prod. 2002;65:25–28. doi: 10.1021/np0101402. [DOI] [PubMed] [Google Scholar]
- 13.Liu DZ, Jia RR, Wang F, Liu JK, Naturforsch Z. B. 2008;63:111–113. [Google Scholar]
- 14.Mosmann TJ. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.