

Abstract
Background
The combination of imaging technologies and luciferase-based bioluminescent bacterial reporter strains provide a sensitive and simple non-invasive detection method (photonic bioimaging) for the study of diverse biological processes, as well as efficacy of therapeutic interventions, in live animal models of disease. The engineering of bioluminescent bacteria required for photonic bioimaging is frequently hampered by lack of promoters suitable for strong, yet stable luciferase gene expression.
Results
We devised a novel method for identification of constitutive native promoters in Gram-negative bacteria. The method is based on a Tn5/7 transposon that exploits the unique features of Tn5 (random transposition) and Tn7 (site-specific transposition). The transposons are designed such that Tn5 transposition will allow insertion of a promoter-less bacterial luxCDABE operon downstream of a bacterial gene promoter. Cloning of DNA fragments from luminescent isolates results in a plasmid that replicates in pir+ hosts. Sequencing of the lux-chromosomal DNA junctions on the plasmid reveals transposon insertion sites within genes or operons. The plasmid is also a mini-Tn7-lux delivery vector that can be used to introduce the promoter-lux operon fusion into other derivatives of the bacterium of interest in an isogenic fashion. Alternatively, promoter-containing sequences can be PCR-amplified from plasmid or chromosomal DNA and cloned into a series of accompanying mini-Tn7-lux vectors. The mini-Tn5/7-lux and mini-Tn7-lux vectors are equipped with diverse selection markers and thus applicable in numerous Gram-negative bacteria. Various mini-Tn5/7-lux vectors were successfully tested for transposition and promoter identification by imaging in Acinetobacter baumannii, Escherichia coli, and Burkholderia pseudomallei. Strong promoters were captured for lux expression in E. coli and A. baumannii. Some mini-Tn7-lux vectors are also equipped with attB sites for swapping of the lux operon with other reporter genes using Gateway technology.
Conclusions
Although mini-Tn5-lux and mini-Tn7-lux elements have previously been developed and used for bacterial promoter identification and chromosomal insertion of promoter-lux gene fusions, respectively, the newly developed mini-Tn5/7-lux and accompanying accessory plasmids streamline and accelerate the promoter discovery and bioluminescent strain engineering processes. Availability of vectors with diverse selection markers greatly extend the host-range of promoter probe and lux gene fusion vectors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0354-3) contains supplementary material, which is available to authorized users.
Keywords: Imaging, Luciferase, Bioluminescent bacteria, Host range, Mini-Tn5/7-Lux vectors, Lux fusion vectors, Gram-negative bacteria
Background
The combination of recent advances in imaging technologies and development of luciferase-based bioluminescent reporter strains provide a sensitive and simple non-invasive detection method (biophotonic imaging) for the study of diverse biological processes, as well as efficacy of therapeutic interventions, in live animal models of human and animal disease [1-6]. In vivo bioluminescence can be employed to determine initial locations of infections and spatial migration of bioluminescently labeled pathogens over a period of several days to weeks. This technology has been applied to study chronic soft-tissue Pseudomonas aeruginosa and Staphylococcus aureus biofilm infections [7-10], P. aeruginosa and Proteus mirabilis urinary tract infections [11], as well as catheter-associated endovascular infections [12], and others [13-16]. Biophotonic imaging also allows assessments of the in vivo efficacy of antibiotic therapy in real time in living animals [9,11,13,17-20]. Some caveats of biophotonic imaging are: 1) luciferase-catalyzed reactions require energy (in the form of ATP and FMNH2), oxygen and a specific fatty acid substrate [21] and therefore allow the detection of only live, metabolically active cells. Because of the oxygen requirement of luciferases, bacterial cells expressing luciferase in strictly anaerobic environments such as the gut were in some instances found to be non-luminescent [1,2,16]. However, such instances are rare and bioluminescence can be detected in harvested organs exposed to oxygen [16]. Furthermore, other authors reported luciferase expression in anaerobic bacteria, e.g. Bifidobacterium breve grown in vitro and in vivo [22], and luciferase-tagged bacteria in anaerobic environments such as tumors [23]; 2) to ensure stable maintenance during the course of infections in animals, the bioluminescent reporter must be integrated into the chromosome of the respective bacteria. Replicating plasmids carrying the lux operon have been evaluated for bioimaging studies, but their use is limited because they only allow short-term (<48 h) infections to be accurately monitored in vivo in animals due to plasmid loss or dilution in the absence of antibiotic selection [24]. Chromosomal integration of plasmids via homologous recombination has been employed for construction of bioluminescent strains but the resulting strains are potentially unstable in the absence of antibiotic selection [25]. Initially, stable chromosomal integration was achieved by random transposition of a mini-Tn5-luxCDABE element [7,11] or another suitable transposon carrying the lux operon [13], followed by antibiotic resistance selection and screening for cells exhibiting strong expression of luciferase activity from a chromosomal promoter. Consequences of employing random transposition are: 1) need for investment of considerable efforts to determine transposon insertion sites and fitness of the mutant bacteria; 2) integrated transposons cannot easily be recovered or transferred between different mutant backgrounds for meaningful comparative analyses because most bacteria lack efficient chromosomal gene transfer procedures, except for those for which transducing phages are available or that are naturally transformable [13]; and 3) lack of a universal promoter for lux gene expression across either Gram-negative or Gram-positive bacteria necessitates development of new bioluminescent strains for each bacterial species to be studied with this technology.
In some bacteria the first two issues have been largely addressed and can be circumvented by use of site-specific insertion elements [25-30]. However, construction of bioluminescent reporter strains is still one of the limiting factors of biophotonic imaging. The major unmet need is lack of suitable promoters for luciferase expression in different bacteria. In Gram-positive bacteria development of synthetic promoters for luciferase gene expression have been successful in some cases [22,27]. However, previous attempts by our laboratory to engineer synthetic promoters based on, for example, the Escherichia coli lac operon-trp operon hybrid promoter Ptac [31] for use in non-enteric bacteria were largely unsuccessful mostly because of the instability of many of the synthetic promoters. We have successfully used the P1 integron promoter [32,33] for driving luciferase gene expression in Burkholderia species [29] indicating that this promoter may be useful for high-level constitutive gene expression in other non-enteric bacteria.
The purpose of this study was to create a simple to use, yet highly versatile series of plasmids for use in Gram-negative bacteria that facilitate promoter discovery and capture, as well as the creation of stable, bioluminescent strains of bacteria. To do this, we combined several features of transposons Tn5 [34] and Tn7 [35].
Tn5 transposes randomly in bacteria. Minimal requirements for transposition are a transposase that can be provided in trans, mosaic ends (MEs) and an antibiotic resistance selection marker [36]. A mini-Tn5 transposon contains the 19 bp MEs flanking the selection marker and is located on a delivery plasmid that contains the transposase gene tnpA outside of the mini-Tn5 element [36]. Cargo cloned on the mini-Tn5 can be randomly transposed into bacterial chromosomes. In contrast to Tn5, Tn7 transposes site-specifically in Gram-negative bacteria, notably to chromosomal attTn7 sites in the presence of the site-specific transposition pathway composed of TnsABCD [35]. Most Gram-negative bacteria contain only a single attTn7 site associated with the essential glmS gene (encoding glucosamine-6-phosphate synthase) [37-41]. However, some contain multiple glmS genes and thus multiple attTn7 sites [37,42,43]. In one instance, Proteus mirabilis, one glmS- and one non-glmS-associated attTn7 site was documented [44]. Minimal requirements for Tn7 transposition are a transposase that can be provided in trans, Tn7 left and right ends (Tn7L and Tn7R) and an antibiotic resistance selection marker [35,37,45]. Cargo cloned on the mini-Tn7 element can be site- and orientation-specifically transposed into bacterial chromosomes in the presence of a plasmid that transiently expresses the Tn7 transposase subunits TnsABCD [37].
In this study, we constructed and tested mini-Tn5/7-lux elements with diverse selection markers that allow promoter identification by random Tn5-mediated transposition into the chromosomes of diverse target bacteria and screening for cells exhibiting strong expression of luciferase activity from a chromosomal promoter. The promoters can then be captured by self-ligation of chromosomal DNA fragments which creates a plasmid carrying a mini-Tn7 element that serves as a template for promoter identification by DNA sequencing, or by PCR amplification of promoter-containing fragments. In some instances, e.g. in the presence of short chromosomal DNA inserts or when recombination-deficient recipient strains are available, the mini-Tn7-lux elements can be transposed into other bacteria without further modification. Alternatively, promoter-containing DNA fragments can be subcloned into a series of accompanying mini-Tn7-lux delivery vectors with diverse selection markers.
Results
Overview of the mini-Tn5/7-lux promoter identification, capture, and mini-Tn7-lux tagging procedures
The overall procedure involves promoter identification and capture (steps 1–3) (Figure 1) and, a unique aspect of the new procedure, methods for bacterial lux tagging by site-specific chromosomal insertion of promoter-lux fusions using mini-Tn7-lux elements (Figure 2). Promoter identification and capture comprises three steps. Step 1 involves Tn5 transposition into the chromosome of the bacterial host of interest. This is achieved by conjugal transfer of the mini-Tn5/7-lux delivery plasmid, followed by selection of Kmr or other antibiotic resistance markers and screening for light-emitting exconjugants. Because the mini-Tn5/7-lux delivery plasmid is non-replicative in the host and Tn5 transposase is only transiently transcribed from the plasmid backbone, the resulting bioluminescent bacteria have the mini-Tn5/7-lux element stably integrated such that the lux operon is transcribed from the promoter(s) of the target gene containing the mini-Tn5/7-lux insertion. This gene can either be a single transcriptional unit or part of an operon. To identify the mini-Tn5/7-lux insertion site, genomic DNA is digested with an enzyme that does not cleave within the transposed element (step 2). Since mini-Tn5/7-lux contains an oriR6K and an antibiotic resistance selection marker, religation of the DNA fragment containing the transposon results in a plasmid which can be recovered by transformation of a pir+E. coli host and selecting antibiotic resistant transformants (step 3). Sequencing of plasmid DNA with a luxC-specific primer (P2385) will reveal the transposon insertion site. Tn5 transposition is not affected by the target bacterium’s recombination status because the mini-Tn5/7-lux plasmid does not carry any chromosomal DNA.
Figure 1.
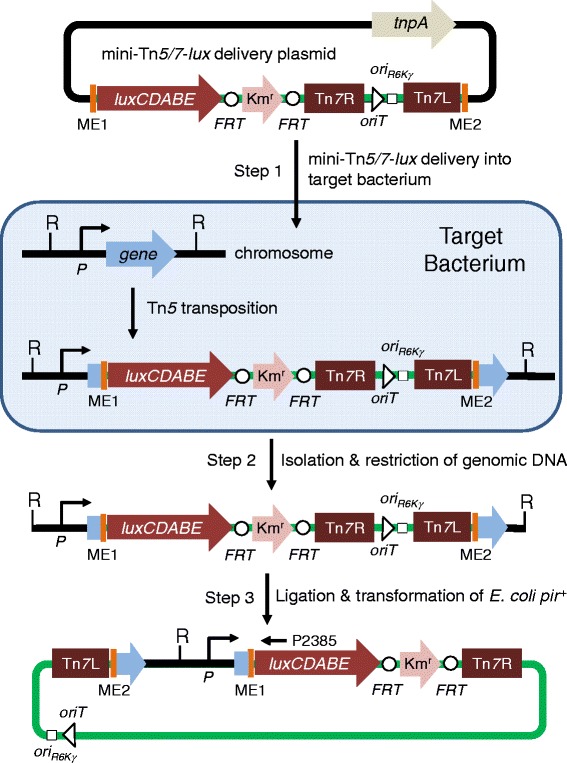
Promoter identification and capture using mini-Tn 5/7-lux elements. Step 1 involves random Tn5 transposition into the chromosome of the bacterial host of interest (target bacterium) after conjugal transfer of the mini-Tn5/7-lux delivery plasmid. This plasmid also contains the Tn5 transposase encoding tnpA gene. TnpA acts on the Tn5 mosaic ends (ME), which results in random mini-Tn5/7-lux transposition into the bacterial chromosome, including insertion in a gene where the Photorhabdus luminescens luxCDABE operon is transcribed from the gene’s promoter (P). The mini-Tn7 element, i.e. sequences flanked by the Tn7 left (Tn7L) and right (Tn7R) ends, located on the mini-Tn5/7-lux delivery plasmid does not transpose in this step because the delivery plasmid does not encode Tn7 transposase. Mini-Tn5/7-lux chromosomal insertion is stable because the chromosomally-integrated elements do neither encode Tn5 nor Tn7 transposase. In step 2, chromosomal DNA of in this example kanamycin resistance (Kmr) and light-emitting transformants is isolated and digested with a restriction enzyme (R) that does not cleave within the transposed element. In step 3, chromosomal DNA fragments are religated and a plasmid containing the R6K origin of replication (ori R6K) and origin of conjugal transfer (oriT) is recovered by transformation of a pir + E. coli host and selecting Kmr transformants. Sequencing of plasmid DNA with a luxC-specific primer (P2385) will reveal the transposon insertion site and putative promoter sequences. The Kmr selection marker contained on the chromosomally integrated mini-Tn7-lux elements is flanked by Flp recombinase target (FRT) sites for optional marker excision with Saccharomyces cerevisiae Flp recombinase.
Figure 2.
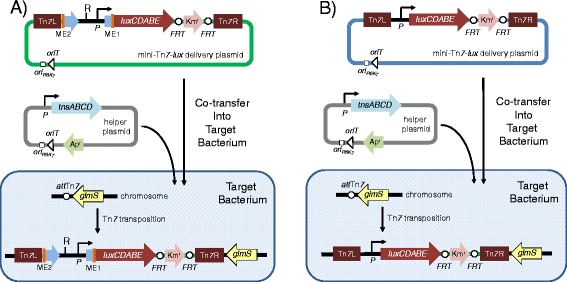
Methods for site-specific insertion of promoter- lux fusions using mini-Tn 7-lux elements. Chromosomal integration of promoter-lux fusions can be achieved in two ways. (A ) The plasmid recovered in step 3 of the promoter identification and capture procedure illustrated in Figure 1 is a functional mini-Tn7 delivery plasmid which in some instances (e.g. when the plasmid contains short regions of promoter-containing chromosomal DNA or when RecA-deficient target strains are available) may be used to directly transpose site-specifically into the glmS gene-associated Tn7 attachment site (attTn7) in the chromosome of the bacterium under study to obtain luminescent derivatives. Site-specific mini-Tn7-lux insertion is achieved by co-transfer of the mini-Tn7-lux delivery plasmid and a helper plasmid that encodes the site-specific TnsABCD transposition pathway, which acts on the Tn7 left (Tn7L) and right (Tn7R) ends. Both plasmids contain the origin of transfer (oriT) for conjugal transfer into the target bacterium and the conditional R6K origin of replication (ori R6K), which limits their replication to E. coli hosts expressing the plasmid R6K π protein. The mini-Tn5 element (sequences flanked my the mosaic ends, ME) contained on the mini-Tn7-lux delivery plasmid does not transpose because the delivery plasmid does not encode Tn5 transposase. (B ) Alternatively, the promoter identified by sequencing the mini-Tn5/7-lux-chromosomal junction sequences located on the plasmid rescued in step 3 of the procedure illustrated in Figure 1 can be cloned into other mini-Tn7-lux elements. These are then transposed into the target bacterium for obtaining bioluminescent bacteria by site-specific mini-Tn7-lux transposition as described above. In both scenarios, A and B, the Kmr selection marker contained on the chromosomally integrated mini-Tn7-lux elements is flanked by Flp recombinase target (FRT) sites for optional marker excision with Saccharomyces cerevisiae Flp recombinase. The ampicillin resistance (Apr) marker is used for selection and maintenance of the helper plasmid in E. coli.
The captured promoter transcribing the lux gene operon can be used to derive bioluminescent bacteria by tagging with mini-Tn7-lux elements in two ways. The choice of method is in part affected by the target bacterium’s recombination status and the size of promoter-containing DNA fragment. First, the mini-Tn5/7-lux delivery plasmid used for promoter identification and capture is designed such that the plasmid recovered in step 3 of the promoter identification and capture procedure illustrated in Figure 1 is a functional mini-Tn7 delivery plasmid which in some instances (e.g. when the plasmid contains short regions of promoter-containing chromosomal DNA or when RecA-deficient target strains are available) may be used to directly transpose site-specifically into the glmS gene-associated Tn7 attachment site (attTn7) in the chromosome of the bacterium under study to obtain luminescent derivatives. For site-specific transposition, mini-Tn7 elements require the Tn7 transposase complex, which is encoded by a helper plasmid containing the tnsABCD genes specifying the site-specific Tn7 transposition pathway. Second, the plasmid recovered in step 3 of the promoter identification and capture procedure illustrated in Figure 1 may be used as a source for promoter-containing DNA fragments that can be PCR amplified, cloned into other mini-Tn7-lux elements, and be employed for obtaining bioluminescent bacteria after site-specific mini-Tn7-lux transposition as described above. This procedure is advised when the plasmid obtained in the promoter recovery step contains larger (several kb) regions of promoter-containing chromosomal DNA or when RecA-deficient target strains are not available. In these instances, chromosomal integration via homologous recombination is favored over site-specific mini-Tn7 integration. Examples for both mini-Tn7-lux tagging scenarios are presented below. Direct tagging with a mini-Tn7-lux element containing the captured promoter transcribing the lux operon is illustrated in a recA E. coli strain. A wild-type Acinetobacter baumannii strain is presented as an example for a bacterium tagged with a mini-Tn7-lux element where lux operon transcription is driven by a promoter which was identified during using mini-Tn5/7-lux mediated identification and capture, and then subcloned in a mini-Tn7 element harboring a promoter-less lux operon.
It should be reiterated at this point that, as noted above, most Gram-negative bacteria contain only one chromosomal glmS-associated attTn7 site [37-41] with the exception of Proteus mirabilis [44]. In contrast, the majority of Burkholderia species examined to date contain multiple glmS genes and thus multiple attTn7 sites, ranging from two sites in B. thailandensis [37] and B. mallei [42] to three sites in B. pseudomallei [43]. Although insertions in these bacteria can occur at all sites, most insertions are usually at one, preferred attTn7 site. In B. mallei, analysis of 24 randomly selected insertions showed that 96% of the insertions were at the glmS1-associated attTn7 site. By contrast, only 8% of the insertions were at the glmS2-associated attTn7 site. Only 4% of the transformants had insertions at both glmS1 and glmS2 [42]. In B. pseudomallei, >65% of observed insertions occur at the glmS2-associated attTn7 site, but there is no obvious preference for either the glmS1- or glmS3-associated attTn7 sites. While double insertions in two separate attTn7 sites are fairly common (10 to 20% with some strains), triple insertions are rarely observed [43]. Presence of multiple attTn7 sites is not an impediment because sites of insertions can be readily differentiated by multiplex PCR. An example for insertion site analysis in B. pseudomallei is illustrated in Additional file 1: Figure S3.
Tn5/7-lux-based promoter capture in E. coli DH5α
To assess the feasibility of the mini-Tn5/7-lux procedure, we constructed the first member of the mini-Tn5/7-lux family, pTn5/7-LuxK3 (Figure 3). Transposition of pTn5/7-LuxK3 into DH5α after conjugal transfer and selection of Kmr exconjugants produced approximately 1% brightly luminescent clones amongst the colonies that were examined for luminescence. A bright isolate (KVT9) was chosen for further study. Genomic DNA was isolated, digested with EcoRI and self-ligated fragments were used to transform E. coli pir+ strain CC118(λpir+) [46]. Kmr transformants were screened for light emission and a plasmid containing the captured promoter region, pTn7DH51LuxK3, was isolated. The mini-Tn5/7-lux insertion site was determined to be located in the rbsC gene, the third gene of the rbsDACBK operon required for the transport and initial steps of ribose metabolism [47] (Figure 4A). The mini-Tn7-Prbs-lux element contained on pTn7DH51LuxK3 was transposed into the DH5α chromosome and mini-Tn7 transposition was confirmed by PCR. Since DH5α is a recA mutant strain all exconjugants examined contained the mini-Tn7-Prbs-lux element inserted at the attTn7 site instead of recombination-mediated insertion at the rbsDACBK locus. Imaging showed that the resulting strain (KVT11) emitted similar levels of light when compared to the original mini-Tn5/7-lux transposition clone KVT9 (Figure 4B). To ascertain that the rbsDACBK operon promoter (Prbs) located in the 167 bp kup-rbsD intergenic region was responsible for lux operon transcription in strain KVT9 and KVT11, the predicted rbs promoter region was PCR-amplified on a 182-bp fragment and directionally cloned upstream of the lux operon contained on pTn5/7LuxK4 resulting in pTn7PrbsLuxK4. Transposition of the resulting mini-Tn7-Prbs-lux element into DH5α resulted in strain KVT12 which was more bioluminescent than KVT9 and KVT11 presumably because the rbs promoter was placed closer to the lux operon on the mini-Tn7-Prbs-lux element inserted in KVT12 (Figure 4B).
Figure 3.
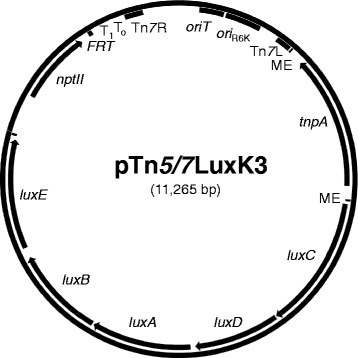
Map of mini-Tn 5/7 - lux delivery plasmid pTn 5/7 LuxK3. The mini-Tn5/7-lux element flanked by the Tn5 mosaic ends (ME) carries a promoter-less P. luminescens luxCDABE operon, the kanamycin resistance encoding nptII gene, the Tn7 left (Tn7L) and right (Tn7R) ends, the R6K origin of replication (ori R6K), and an origin of conjugative transfer (oriT). The Tn5 transposase-encoding tnpA gene is located outside of the transposable element. Other abbreviations: FRT, single Flp recombinase target site; T0T1, transcriptional terminators T0 and T1 from bacteriophage λ and E. coli rrnB operon, respectively.
Figure 4.
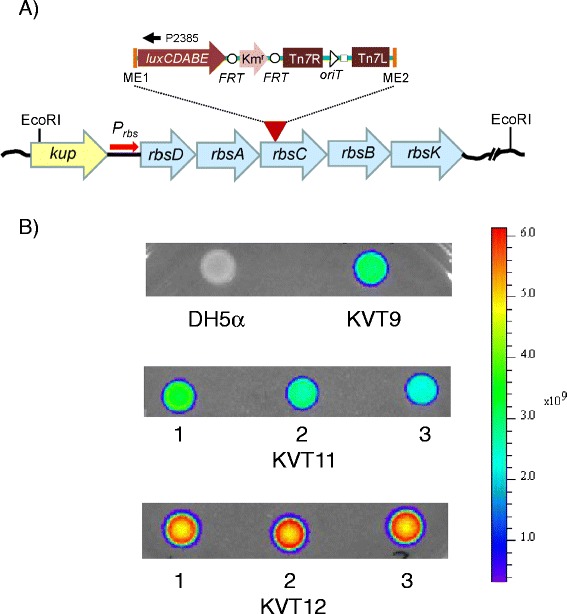
Mini-Tn 5/7 - lux aided promoter identification and capture in E. coli . A) The promoter-less mini-Tn5/7-lux element (indicated by the red arrowhead and expanded inset above it) from pTn5/7LuxK3 was transposed into the DH5α chromosome. Chromosomal DNA from a Kmr and luminescent exconjugant (KVT9; panel B) was isolated, digested with EcoRI, religated and transformed into CC118(λpir +). The lux operon-chromosomal DNA junction on the plasmid was sequenced with the luxC-specific primer P2385. The mini-Tn5/7-lux transposon was inserted in rbsC, the third gene of the rbsDACBK operon required for the transport and initial metabolic steps of ribose. The kup gene located upstream of the rbsDACBK operon encodes a potassium transporter. B) The recovered plasmid was used to transpose the mini-Tn7-P rbs-lux element residing on it to the attTn7 site on the DH5α chromosome resulting in strain KVT11. In a parallel effort, The rbs operon promoter (P rbs) located in the 167 bp kup-rbsD intergenic region was PCR-amplified and cloned on a 173 bp StuI-DraIII fragment into pTn5/7LuxK4 where it replaced the tnpA gene and flanking MEs to drive transcription of the lux operon. The resulting mini-Tn7-P rbs-lux element was transposed into the attTn7 site on the DH5α chromosome resulting in strain KVT12. Ten μl samples of an overnight culture of the indicated strains were spotted on an LB plate, grown overnight at 37°C and light emission was measured using a Xenogen IVIS imager.
Construction of next generation lux vectors
After successful testing in E. coli, we sought to expand the versatility of the mini-Tn5/7-lux system for use in a broad-range of bacteria and by inclusion of other desirable properties such as incorporation of attB1 and attB2 sites flanking the lux operon to facilitate its exchange for other reporter genes such as gfp via Gateway technology [48]. This resulted in a family of versatile plasmids with diverse selection markers (gentamicin, kanamycin, tetracycline and trimethoprim) most of which can be excised in vivo using Flp recombinase, a lux operon with or without exchangeable promoters and the Tn5 transposase tnpA gene with flanking MEs either transcribed from its own promoter or the constitutive S12 gene promoter from B. thailandensis (Table 1). A graphical representation of the genealogy of the various plasmids is presented in Additional file 1: Figure S1 and detailed maps of two representative plasmids, pTn7oLuxK4 and pTn5/7LuxK6, are shown in Figure 5.
Table 1.
Plasmids
| Plasmid | GenBank accession no. | Pertinent features a,b | Source |
|---|---|---|---|
| pTn7xLuxG0 | KF532964 | Gmr; luxCDEBA operon transcribed from P PA4974 contained on DraIII fragment | This Study |
| pTn7oLuxG0 | KF532961 | Gmr; DraIII fragment containing P PA4974 replaced with DraIII fragment containing P ompA c | This Study |
| pTn7xLuxG3 | KF532965 | Kmr; pTn7xLuxG0 with BamHI and PstI sites deleted by partial digestion | This Study |
| pTn7xLuxK3d | KC332283 | Kmr; pTn7xLuxG3 with Gmr encoding aacC1 gene replaced with Kmr encoding nptII gene from pFKM4 | This Study |
| pTn7oLuxK3d | KC332281 | Kmr; pTn7xLuxK3 with DraIII fragment containing P PA4974 replaced with DraIII fragment containing B. pseudomallei P ompA | This Study |
| pTn7tLuxK3d | KC332282 | Kmr; pTn7xLuxK3 with DraIII fragment containing P PA4974 replaced with DraIII fragment containing B. pseudomallei P tolC | This Study |
| pTn5/7LuxK3d | KC332286 | Kmr; pTn7xLuxK3 with StuI-DraIII fragment containing P PA4974 replaced with StuI-DraIII fragment containing Tn5 transposase gene tnpA and flanking mosaic ends | This Study |
| pTn7oLuxG4 | KF532962 | Gmr; pTn7oLuxG0 with BamHI multiple cloning site fragment deleted | This Study |
| pTn7oLuxK4 | KC332284 | Kmr; pTn7oLuxG4 with Gmr encoding aacC1 gene replaced with Kmr encoding nptII gene from pFKM4 | This Study |
| pTn7oLuxT4 | KF532963 | Tpr; pTn7oLuxG4 with Gmr encoding aacC1 gene replaced with Tpr encoding dhfRII gene from pFTP2 | This Study |
| pTn5/7LuxG4 | KF813061 | Gmr; pTn7oLuxG4 with StuI-DraIII fragment containing P ompA replaced with StuI-DraIII fragment containing Tn5 transposase gene tnpA and flanking mosaic ends | This Study |
| pTn5/7LuxK4 | KF532957 | Kmr; pTn7oLuxK4 with StuI-DraIII fragment containing P ompA replaced with StuI-DraIII fragment containing Tn5 transposase gene tnpA and flanking mosaic ends | This Study |
| pTn5/7LuxT4 | KF813062 | Tpr; pTn7oLuxT4 with with StuI-DraIII fragment containing P ompA replaced with StuI-DraIII fragment containing Tn5 transposase gene tnpA and flanking mosaic ends | This Study |
| pTn5/7LuxK5 | KF532958 | Kmr; pTn5/7LuxK4 with tnpA gene transcribed from P S12 | This Study |
| pTn5/7LuxT5 | KF532959 | Tpr; pTn5/7LuxT4 with tnpA gene transcribed from P S12 | This Study |
| pTn5/7LuxT6 | KC332285 | Tpr; pTn5/7LuxT5 with attB1 inserted at DraIII site | This Study |
| pTn5/7LuxG6 | KC332289 | Gmr; pTn5/7LuxT6 with Tpr encoding dhfRII gene replaced with Gmr encoding aacC1 gene from pFGM1 | This Study |
| pTn5/7LuxK6 | KC332287 | Kmr; pTn5/7LuxT6 with Tpr encoding dhfRII gene replaced with Kmr encoding nptII gene from pFKM4 | This Study |
| pTn5/7LuxTc6 | KC332288 | Tcr; pTn5/7LuxT6 with Tpr encoding dhfRII gene replaced with Tcr encoding tetA gene from pFTC2 | This Study |
| pTn7P DH51-lux | N/A | Kmr; captured DH5α rbsC::Tn5/7LuxK3 insertion on mini-Tn7 delivery plasmid obtained by religation of chromosomal Acc65I fragment | This Study |
| pTn7P rbsLuxK4 | N/A | Kmr; E. coli rbs promoter (P rbs) cloned into pTn7LuxK4 | This Study |
| pTn7P H1ALuxK5 | N/A | Kmr; A. baumannii A1S_0945 gene promoter (P H1A) cloned into pTn7LuxK5 | This Study |
aAbbreviations: Ap, ampicillin; FRT, Flp recombinase target; Gm, gentamicin; Km, kanamycin; r, resistance/resistant; Tc, tetracycline; Tp, trimethoprim.
bSee supplemental methods for details of plasmid constructions.
cPromoters used are: P PA4974, P. aeruginosa gene PA4974 promoter; P ompA, B. pseudomallei ompA promoter; P tolC, B. pseudomallei tolC promoter; P S12, B. thailandensis ribosomal S12 gene promoter.
dPlasmids are missing a FRT site flanking the nptII gene.
Figure 5.
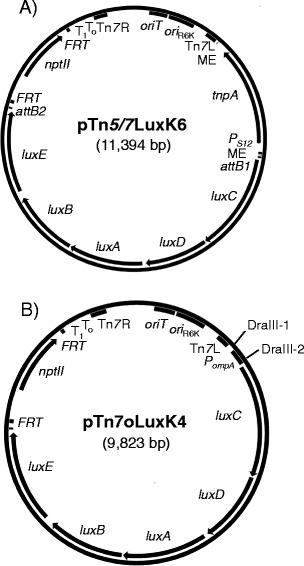
Maps of next generation mini-Tn 5/7 - lux and mini-Tn 7 - lux delivery vectors. A) pTn5/7LuxK6. As in pTn5/7LuxK3 (Figure 2) the mini-Tn5/7-lux element is flanked by the Tn5 mosaic ends (ME) and carries a promoter-less P. luminescens luxCDABE operon, in this example the kanamycin resistance encoding nptII gene, the Tn7 left (Tn7L) and right (Tn7R) ends, the R6K origin of replication (ori R6K), and an origin of conjugative transfer (oriT). The new generation of vectors is further functionalized by 1) transcription of the Tn5 transposase-encoding tnpA gene located outside of the transposable element by the constitutive S12 gene promoter (P S12) from Burkholderia thailandensis; 2) flanking of the antibiotic resistance marker by functional Flp recombinase target (FRT) sites; and 3) flanking of the lux operon by attB sites for Gateway recombineering. The pTn5/7LuxG6, pTn5/7LuxT6 and pTn5/7LuxTc6 contain gentamicin, trimethoprim and tetracycline resistance markers, respectively. Other abbreviations are defined in the Figure 2 legend. B) pTn7oLuxK4. This vector contains many features of the pTn5/7Lux series but does not contain the Tn5 transposase gene or mosaic ends. Its unique features include two DraIII sites that because of the 3-nucleotide ambiguity (indicated by bold letters) in the DraIII recognition sites (DraIII-1 5′-CACTATGTG and DraIII-2 5′- CACCGCGTG) can be used for directional cloning of promoter-containing DNA fragments for transcription of the lux operon genes. In the illustrated example the lux operon is transcribed from the B. pseudomallei ompA promoter (P ompA). This is also indicated by the “o” in the plasmid name. The pTn7oLuxG4 and pTn7oLuxT4 contain P ompA and the gentamicin (aacC1) and trimethoprim (dhfRII) resistance markers, respectively. Other abbreviations are as in A).
Tn5/7-lux based promoter capture in A. baumannii
To demonstrate promoter identification and recovery in a bacterium other than E. coli, pTn5/7LuxK5 was conjugated into A. baumannii strain ATCC19606. From the pool of recovered Kmr exconjugants, about 1% exhibited strong light emission. Four luminescent strains (IFD1-4) were retained for further studies. To identify mini-Tn5/7-lux insertion sites in these strains, chromosomal DNA was isolated and digested with Acc65I, EcoRI and NotI. These enzymes were empirically chosen because they cleave chromosomal DNA with various frequencies in different bacteria based on G + C content and their capacity for heat inactivation. For instance, in A. baumannii the chromosomal DNA cleavage frequency decreased from Acc65I to EcoRI to NotI. Plasmids harboring the mini-Tn5/7-lux elements were obtained after self-ligation and transformation of E. coli pir-116+ strain MaH1 (Table 2). The transposon-chromosomal junction sequences were determined by sequencing and aligning the sequences thus obtained to the chromosomal sequence (EMBL accession no. CP000521) of A. baumannii strain ATCC17978. These analyses revealed single mini-Tn5/7-lux insertions in isolate IFD1 (gene A1S_2680), IFD2 (gene A1S_2773) and IFD4 (gene A1S_2621) and two insertions in strain IFD3 (genes A1S_0947 and A1S_2736). In an attempt to insert the respective mini-Tn7-lux elements located on the respective delivery plasmids into the A. baumannii chromosomal Tn7 attachment site they were conjugally transferred from E. coli RHO3 into strain ATCC19606 together with the helper plasmid pTNS3 [43]. Although luminescent exconjugants were observed, PCR screening of 80 colonies revealed no mini-Tn7-lux insertion at the attTn7 site but rather only strains in which the mini-Tn7-lux elements had integrated at the respective chromosomal loci via RecA-mediated homologous recombination. To minimize homologous recombination events, we identified potential promoter regions upstream of the previously identified mini-Tn5/7-lux insertion sites. These analyses revealed the presence of multiple potential promoters in the A1S_0944–A1S_0945 intergenic region that is located upstream of the mini-Tn5/7-lux insertion site in A1S_047 encoding a putative vanillate O-demethylase oxygenase subunit. The region containing the putative gene 0945 promoter region (PH1A) was PCR-amplified and directionally cloned into pTn5/7LuxK5 to create pTn7PH1ALuxK5. KmrE. coli MaH1 transformants containing the desired promoter insertion were identified by screening for increased luminescence as the pTn5/7LuxK5 vector with a promoter-less lux operon confers negligible luminescence. The mini-Tn7-PH1A-lux element was then transposed into the ATCC19606 chromosome. The resulting strain IFD5 containing the mini-Tn7-PH1A-lux transposon integrated at the chromosomal attTn7 site emitted slightly less light than the original strain IFD3, which contained two separate mini-Tn5/7-lux insertions (Figure 6).
Table 2.
Bacterial strains
| Strain | Genotype or relevant features | Source |
|---|---|---|
| E. coli | ||
| DH5α | F−φ80 lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK −mK +) phoA glnV44 | [49] |
| MaH1 | F−φ80 lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK −mK +) phoA glnV44 attTn7::pir116+ | [50] |
| RHO3 | thi-1 thr-1 leuB26 tonA21 lacY1 supE44 recA integrated RP4-2 Tcr::Mu (λpir +)Δasd::FRT ΔaphA::FRT | [51] |
| RHO5 | attTn7::pir116 + thi-1 thr-1 leuB26 tonA21 lacY1 supE44 recA integrated RP4-2 Tcr::Mu (λpir +)Δasd::FRT ΔaphA::FRT | [50] |
| KVT9 | DH5α rbsC::mini-Tn5/7LuxK3 | This Study |
| KVT11 | DH5α attTn7::mini-Tn7-P DH51-lux | This Study |
| KVT12 | DH5α attTn7::mini-Tn7-Prbs-lux | This Study |
| B. pseudomallei | ||
| Bp82 | 1026b ΔpurM | [52] |
| Bp82.27 | Bp82 Δ(amrRAB-oprA) | Laboratory collection |
| Bp82.68 | Bp82.27 attTn7::mini-Tn7-P ompA -lux (glmS1) | This Study |
| Bp82.69 | Bp82.27 attTn7::mini-Tn7-P tolC -lux (glmS1) | This Study |
| Bp82.70 | Bp82.27attTn7::mini-Tn7-P PA4974-lux (glmS2 + glmS3) | This Study |
| Bp82.77 | Bp82.27attTn7::mini-Tn7-P ompA -lux (glmS1 + glmS2) | This Study |
| Bp82.78 | Bp82.27 attTn7::mini-Tn7-P ompA -lux (glmS1) | This Study |
| Bp82.79 | Bp82.27 attTn7::mini-Tn7-P ompA -lux (glmS2 + glmS3) | This Study |
| Bp82.80 | Bp82.27 attTn7::mini-Tn7-P ompA -lux (glmS2) | This Study |
| Bp82.81 | Bp82.27 attTn7::mini-Tn7-P ompA -lux (glmS3) | This Study |
| A. baumannii | ||
| ATCC19606 | Prototroph | American Type Culture Collection |
| IFD1 | ATCC19606 A1S_2680::mini-Tn5/7LuxK5 | This Study |
| IFD2 | ATCC19606 A1S_2773::mini-Tn5/7LuxK5 | This Study |
| IFD3 | ATCC19606 A1S_0947::mini-Tn5/7LuxK5 and A1S_2736::Tn5/7LuxK5 | This Study |
| IFD4 | ATCC19606 A1S_2621::mini-Tn5/7LuxK5 | This Study |
| IFD5 | ATCC19606 attTn7::mini-Tn7-P H1A-LuxK5 | This Study |
Figure 6.

Mini-Tn 5/7 - lux aided promoter identification and capture in A. baumannii . The promoter-less mini-Tn5/7-lux element from pTn5/7LuxK5 was transposed into the A. baumannii strain ATCC19606 chromosome. Chromosomal DNA from a Kmr and luminescent exconjugant (IFD3) was isolated, digested with Acc65I, religated and transformed into E. coli pir-116 + strain MaH1. DNA from a Kmr transformants was sequenced with the luxC-specific primer P2385. The mini-Tn5/7-lux transposon was inserted in a gene annotated as A1S_0947 in strain ATCC17978 and the upstream A1S_0944-A1S_0945 intergenic region containing several possible promoters was PCR-amplified and cloned on a 329-bp StuI-DraIII fragment into pTn5/7LuxK5. The mini-Tn7-P H1A-lux element was transposed into the attTn7 site on the ATCC19606 chromosome resulting in strain IFD5. Ten μl samples of an overnight culture of the indicated strains were spotted on an LB plate, grown overnight at 37°C and light emission was measured using a Xenogen IVIS imager.
Luminescence from mini-Tn7-lux elements in bacteria with multiple attTn7 sites is insertion site dependent
Noting that bacteria which contain multiple mini-Tn7-lux insertions due to the presence of multiple Tn7 insertion (attTn7) sites exhibit differential luminescence we decided to examine light emission from bacteria which naturally contain more than one mini-Tn7 insertion site and in which luminescence is thus either insertion site-dependent or due to multiple insertions.
During isolation of B. pseudomallei strains containing mini-Tn7-lux elements we noticed that the resulting strains emitted various amount of light and this seemed to be promoter and insertion site dependent. In this study we therefore compared various promoters, including P. aeruginosa PPA4974 [7], B. pseudomallei PompA [28] and PtolC [25] cloned upstream of the lux operon residing on pTn7oLuxK4 and inserted at diverse attTn7 sites in the genome of B. pseudomallei strain Bp82.27, an aminoglycoside susceptible Δ(amrAB-oprA) derivative of the select agent excluded strain Bp82 [52]. Kanamycin resistant transformants were patched onto Km-containing LB plates and light production was compared. In this system, light production from a lux operon transcribed by PompA was consistently strongest followed by PPA4974 and PtolC (Figure 7A and B). Mini-Tn7-lux insertion into the glmS1-associated attTn7 site consistently conferred the greatest luminescence while strains carrying single mini-Tn7-PompA-lux insertions into the glmS2-associated or glmS3-associated attTn7 sites produced consistently less luminescence (Figure 7C). A strain carrying simultaneous insertions of mini-Tn7-PompA-lux in all three B. pseudomallei attTn7 sites was not observed.
Figure 7.

Promoter strength and insertion site location determine bioluminescence signal strength in a bacterium with multiple Tn 7 insertion sites. Mini-Tn7 elements in which lux operon expression is directed from the indicated promoters were transposed into the genome of B. pseudomallei strain Bp82.27 and insertion at either glmS1-, glmS2- or glmS3-associated attTn7 sites was determined by PCR. Colonies were patched on LB agar plates with 35 μg/ml kanamycin and incubated at 37°C. Patches were imaged using a Bio-Rad Universal Hood II ChemiDocXRS using high sensitivity chemiluminescence settings. A) and B) Bp82.27 with mini-Tn7-lux insertions derived from transposition from pTn7oLuxK3, pTn7tLuxK3 and pTn7xLuxK3. The promoters directing lux operon expression in these constructs are B. pseudomallei P ompA, B. pseudomallei P tolC and P. aeruginosa P PA4974, respectively. Patches were grown overnight at 37°C and either imaged using white light and a 0.007 s exposure time (A) or imaged for luminescence using a 30 s exposure time (B). C) Insertion site dependence of bioluminescence intensity. The mini-Tn7-P ompA -lux element from pTn7oLuxK4 was transposed into the Bp82.27 genome. Insertion sites were determined by PCR. Patches were grown overnight at 37°C and imaged for luminescence using a 30 s exposure time.
Discussion
The mini-Tn5/7-lux vectors were employed to successfully identify, capture and clone promoters capable of producing significant amounts of light in E. coli and A. baumannii under laboratory conditions. While in this study efforts were focused on vector construction and evaluation in vitro, future efforts must include studies aimed at promoter activity evaluation in suitable in vivo model systems, especially with pathogens such as A. baumannii and others. In E. coli, the ribose operon promoter was the strongest promoter we identified in this study. This was somewhat surprising because expression from this promoter is normally repressed by the ribose operon repressor RbsR and induced in the presence of the inducer D-ribose [47]. In this promoter-capture proof-of-concept study, we only examined Prbs-lux gene expression in LB-grown cells which must represent at least partially inducing conditions but the utility of this promoter for in vivo imaging of E. coli infections remains uncertain absent of expression studies in bacteria grown in vivo, e.g. animal infection or cell culture experiments, or in vitro studies employing various conditions encountered by bacteria during infections (e.g. cell-density, defined nutrient sources, etc.). The same is true for the A. baumannii PH1A promoter identified and characterized using in vitro laboratory conditions, i.e. LB-grown bacteria. In addition to E. coli and A. baumannii, mini-Tn5/7-lux vectors were also used to identify strong promoters capable of driving lux operon expression in LB-grown cells of B. pseudomallei. Luminescent isolates could be readily identified suggesting that the system will be useful for promoter identification in diverse bacteria. Two promoters that were identified in B. pseudomallei were the put (proline utilization) and paa (phenylacetic acid degradation) operon promoters but since promoters for construction of bioluminescent B. pseudomallei suitable for in vitro and in vivo bioimaging studies, e.g. PompA [28] and PtolC [25], are already available, the put and paa operon promoters were not further pursued.
Promoter identification in E. coli, A. baumannii and B. pseudomallei using the mini-Tn5/7-lux system identified a frequent scenario encountered with bacteria which is insertion in promoter-distal genes in operons. In practice, this makes direct use of mini-Tn7-lux elements with captured promoter regions for isolation of bioluminescent bacteria problematic in recA+ strain backgrounds as the sometimes large regions of homology carried by the transposable element promote recombination into the chromosome instead of site-specific integration via Tn7-transposition. While this undoubtedly diminishes the novelty of the mini-Tn5/7-lux system, i.e. the combination of the mini-Tn5-lux and mini-Tn7-lux systems which existed separately before, the newly developed method has several advantages over the separate systems: 1) the newly constructed pTn7Lux vectors exhibit expanded repertoire and utility with respect to cloning of promoter-containing DNA fragments when compared to previously constructed mini-Tn7-lux vectors; and 2) in some instances, e.g. where short promoter-containing chromosomal DNA regions are present or recombination-deficient strains are either available or can be readily constructed, the combination of the Tn5 and Tn7 transposon allows quick isolation and site-specific insertion of the promoter-lux fusion constructs in naturally occurring Tn7 attachment site(s) in strains transiently expressing the Tn7 TnsABCD transposase complex.
In the course of the present study we also noted that in the few instances where bacteria contain more than one chromosomal attTn7 site one must be aware of copy number and position effects on reporter gene expression. For instance, incorporation of the same mini-Tn7-PompA-lux reporter into one or more of the three attTn7 sites in the B. pseudomallei genome resulted in differential levels of light emission. In general, insertions into the glmS1-associated attTn7 site emitted more light than mini-Tn7 insertions in either of the other two attTn7 sites. Although we have no experimental evidence that would explain these observations, insertion site effects may at least be partially responsible for differential lux transcription from constructs integrated at different Tn7 integration sites. The three attTn7 sites found in B. pseudomallei are located in the intergenic regions of glmS1, glmS2 and glmS3 and the respective downstream genes which in all cases are divergently transcribed from glmS (Additional file 1: Figure S3) [43]. The mini-Tn7-lux elements insert at these sites such that the lux gene is in the same orientation as these downstream genes which may lead to partial read-through lux transcription from the downstream gene promoters. This may be exacerbated by the fact that insertions at glmS1 occur in the predicted transcriptional terminator that seems to be shared by glmS1 and the divergently transcribed downstream gene. In contrast, insertions at the glmS2- and glmS3-associated attTn7 sites do not disrupt the transcriptional terminators of the respective divergently transcribed genes. To minimize transcriptional read-through effects from promoters of adjacent genes, transcriptional terminators could be included inside the Tn7 left and right ends, but this was not pursued in the present studies. As expected, isolates with double insertions produced more light than those with single insertions and levels were comparable with isolates that contained single insertions at the glmS1-associated attTn7 site. Isolates with mini-Tn7 insertions at all three attTn7 sites resulting from a single transposition experiment are generally rare and were not observed in this study.
Conclusions
We created a suite of vectors that comprise a versatile system for promoter identification, capturing, cloning and construction of bioluminescent Gram-negative bacterial strains that contain the reporter genes stably integrated in the bacterial chromosome. The mini-Tn5/7-lux vectors incorporate the random transposition property of Tn5 catalyzed by transient expression of Tn5 transposase TnpA in a wide range of bacteria and combines it with the site-specific transposition property of Tn7 catalyzed by transient expression of the Tn7 TnsABCD transposase complex in Gram-negative bacteria. The system was created with versatility and customization in mind. For example, the vectors are equipped with diverse selection markers to expand their host range to bacteria, which may exhibit intrinsic resistance to some antibiotics commonly used for selection of recombinants. Antibiotic resistance markers are flanked by 48-bp FRT sites which allow exchange of the resident antibiotic marker with other FRT cassettes using unique XbaI restriction sites in each FRT. All vectors possess unique StuI and DraIII restriction sites that allow for the deletion of tnpA and its flanking mosaic ends for orientation-controlled insertion of promoter sequences for lux operon transcription. In this study we exclusively tested Tn5/7Lux and Tn7Lux vectors for purposes of promoter identification, capturing and cloning for construction of bioluminescent clones. However, their uses extend well beyond these applications. For instance, attB1 and attB2 sites bordering luxCDABE facilitate exchange of the resident lux operon for other reporter genes such as gfp via Gateway BP clonase recombination. Vectors on which gfp-transcription is driven from the same promoter(s) identified and used for lux gene expression can then be employed for construction of fluorescent instead of luminescent strains. Availability of isogenic bioluminescent and fluorescent strains of the same species has several applications. For instance, they can be employed in bioluminescence, fluorescence, and optical density based real-time assays can to determine the bacteriostatic or bacteriocidal effects of antibiotics [53]. Furthermore, such strains can be used to differentiate effects of antimicrobials on metabolism. Luciferase activity is dependent on availability of metabolites such as ATP, FMNH2 and a specific fatty acid substrate [21] and its activity thus adversely affected by inhibitors of metabolism whereas GFP activity is not prone to such inhibition. Lastly, strain labeling with luciferase or GFP reporters – or dual labeling with both – broadens the repertoire for imaging of various biological processes [5,54].
These capabilities allow for tailoring the plasmids to investigators’ needs. The tools developed in this study should prove to be useful as their customizability allows for an extremely wide array of uses in diverse Gram-negative bacteria.
Methods
Bacterial strains, media and growth conditions
Table 2 lists the bacterial strains used in this study. Bacteria were routinely grown in liquid or agar solidified Lennox Luria Bertani (LB) (MO BIO Laboratories, Carlsbad, CA). E. coli conjugation strains RHO3 and RHO5 were grown in LB medium supplemented with diaminopimelic acid (DAP; LL-, DD-, and meso-isomers) which was used at 400 μg/ml for agar plates and 200 μg/ml for liquid cultures. Lennox (5 g/L NaCl) LB cultures of B. pseudomallei Bp82 were supplemented with 80 μg/ml adenine. Media were supplemented with antibiotics at the following final concentrations. For E. coli, gentamicin (Gm), 10 μg/mL and 15 μg/ml for broth cultures and agar plates, respectively; kanamycin (Km), 35 μg/mL; tetracycline (Tc), 10 μg/ml; trimethoprim (Tp), 100 μg/mL. For B. pseudomallei, Km, 35 μg/ml for Bp82.27 and 500 μg/ml for Bp82. For A. baumannii, Km was used at a concentration of 35 μg/ml.
DNA manipulation
Chromosomal DNA was isolated using the Puregene Core Kit A (Gentra Systems, Qiagen, Valencia, CA) and plasmid DNA was purified from bacterial cultures using the GeneJET Plasmid MiniPrep Kit (Fermentas, Glen Burnie, MD). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA) and used according to the manufacturer’s recommendations. Ligation reactions were conducted using T4 DNA ligase from Invitrogen (Life Technologies, Carlsbad, CA) and the supplied T4 DNA ligase buffer. DNA sequencing was conducted using an ABI 3130xL Genetic Analyzer (Applied Biosystems, Carlsbad, CA) at the Colorado State University Proteomics and Metabolomics Facility.
Plasmid construction
Plasmid construction details are provided in Additional file 1: Methods and Table S1. The main plasmids constructed in this study are listed in Table 1.
Transformation and conjugation procedures
Plasmid transformation of E. coli was done either by using standard electroporation or chemical transformation procedures [55]. Bacterial conjugations were conducted as bi-parental matings with E. coli mobilizer strains RHO3 or RHO5 using previously described methods [51,56]. A modified mating procedure was used for conjugations with A. baumannii. Cultures of donor and recipient were grown overnight. Thirty μl of the donor culture was sub-cultured into 3 ml of LB broth and the culture was grown at 37°C with shaking to an OD600 of 0.6-0.7. Meanwhile, 3 ml of pre-warmed 20 mM NaNO3 was added to the overnight recipient culture which was then incubated at 42°C without shaking for at least three hours. Donor and recipient cultures were then harvested by centrifugation, washed twice with fresh LB, concentrated 5-fold, and 60 μl of donor and 10 μl of recipient culture were combined on a filter disk. The remainder of the procedure follows previously described protocols.
Construction and identification of mini-Tn7-lux containing B. pseudomallei strains
The mini-Tn7-lux delivery vectors pTn7oLuxK3, pTn7tLuxK3, pTn7xLuxK3 containing a luxCDABE operon transcribed from PompA, PtolC and PPA4974, respectively, and a Kmr selection marker were transformed into the E. coli mobilizer strain RHO3. The mini-Tn7-lux elements were delivered into the Bp82.27 recipient strain using multi-parental conjugation with RHO3 containing the Tn7 transposase helper plasmid pTNS3 [43]. Kmr transformants were selected and mini-Tn7-lux insertion sites determined by PCR using primer sets P479 & P1509, P479 & P1510, and P479 & P1511 to check for insertion at glmS1, glmS2, or glmS3, respectively [43]. Table 3 lists oligonucleotides used in this study. Alternatively, a newly developed multiplex PCR was used to detect mini-Tn7 inserts in B. pseudomallei and using the primer set P479 & 1510 & 2595 & 2596 (see Additional file 1: Methods for details and Additional file 1: Figure S3 for a representative example of multiplex PCR results).
Table 3.
Oligonucleotides
| Oligo name | Description | Sequence (5′ → 3′) a |
|---|---|---|
| 478 | Tn7R | CACAGCATAACTGGACTGATTTC |
| 479 | Tn7L | ATTAGCTTACGACGCTACACCC |
| 536 | oriT-UP | TCCGCTGCATAACCCTGCTTC |
| 537 | oriT-DN | CAGCCTCGCAGAGCAGGATTC |
| 572 | PstSUp2 | GCTATACGTGTTTGCTGATCAAGATGC |
| 1354 | AB_glmSF | GGCGGTCAGTTGTATGTCTT |
| 1509 | BPGLMS1 | GAGGAGTGGGCGTCGATCAAC |
| 1510 | BPGLMS2 | ACACGACGCAAGAGCGGAATC |
| 1511 | BPGLMS3 | CGGACAGGTTCGCGCCATGC |
| 2372 | attTn7-1 | GATGCTGGTGGCGAAGCTGTC |
| 2373 | attTn7-2 | GATGACGGTTTGTCACATGGAG |
| 2385 | LUXpro-UP | ATTGCACTAAATCATCACTTTC |
| 2550 | Stu-PrbsEco-for | TAAAGGCCTGCCAGACGCCTCCTTTCT |
| 2551 | Dra-PrbsEco-rev | TAACACCGCGTGTTCTCCATCAGCGAAACGT |
| 2584 | H1-A StuI F | ATTAGGCCTTCTTGCGAATCTTCTTCAATCTC |
| 2586 | H1A DraIII R3 | ATTCACCGCCTGACAAACTGAGATCCAACTCATACCT |
| 2595 | BPGLMS1-New4 | ACCTGATTGCGTTCGTCGTCC |
| 2596 | BPGLMS3-New | ATCACGCTGCTTTGGCTGG |
aItalicized letters indicate restriction sites: DraIII (2551 and 2586) and StuI (2550 and 2584).
Luminescence imaging
Relative luminescence was imaged using a Bio-Rad Universal Hood II ChemiDocXRS using high sensitivity chemiluminescence settings and a 10–30 s exposure time. Quantification of light production was performed using an IVIS Spectrum (Xenogen, Alameda, CA). An open emission filter with no excitation was utilized to measure the signal.
Tn5/7-lux promoter capture procedure
To recover random mini-Tn5/7-lux chromosomal insertions, the Tn5/7-lux vector containing the appropriate antibiotic resistance marker was first transformed into the E. coli donor strain RHO3. A bi-parental mating was then performed with the donor and desired recipient strain and antibiotic resistant exconjugants were selected. Exconjugants were either patched onto an LB agar plate with the respective antibiotic used for selection of exconjugants or inoculated into 96-well microtiter plates containing LB medium with 10% glycerol and antibiotic supplement. After overnight growth at 37°C, patches or wells were observed over a period of four days to identify bright and stable luminescent clones. Chromosomal DNA was isolated from selected mini-Tn5/7-lux containing colonies and 1 μg digested in separate reactions using restriction enzymes Acc65I, EcoRI or NotI. (Note: these enzymes were empirically chosen to work with most bacteria we study but others can be used as well.) The digestions were terminated by heat-inactivation at 68°C and cleaned either by using the Gen Elute Gel Extraction Kit (Sigma-Aldrich) or by heat-inactivating the restriction enzyme and drop dialyzing the DNA on a Millipore“V” series (VMWP) filter with a 0.05 μm pore size over distilled and deionized water for 20 min. Digested DNA (1 μg) was ligated overnight using T4 DNA Ligase and ligations were drop dialyzed for 20–25 min. The entire sample was removed from the filter and immediately used for transformation of the pir-116+E. coli strain MaH1. (Note: other pir+ strains are equally suited for transformation. We routinely employ this strain because it increases the copy number of plasmid with the R6K origin of replication and thus yields more plasmid DNA [53].) Transformants were grown overnight and plasmid DNA was isolated. The mini-Tn5/7-lux insertion site was then determined by sequencing using primer P2385.
The resulting plasmid now constitutes a mini-Tn7 delivery plasmid (Figure 1) which was used for two purposes.
First, the plasmid was sometimes used to isolate chromosomal mini-Tn7-lux insertions in the host of interest. This was achieved by electroporation into the pir-116+E. coli mobilizer strain RHO5 (electroporation was chosen over transformation due to the unknown, but presumably quite large, size of the recovered mini-Tn5/7-lux containing plasmid). The mini-Tn7-lux plasmid was then introduced into the target bacterium chromosome by co-conjugation with the Tn7 site-specific transposition pathway expressing pTNS3. Exconjugants were selected on LB plates with appropriate antibiotics and screened for light production. The presence of mini-Tn7-lux insertions was verified by PCR employing species specific primer pairs, e.g. P2372 & P2373 for E. coli, P478 & P1354 for A. baumannii, and P479 & P1509, P479 & P1510, and P479 & P1511 for B. pseudomallei. To distinguish Tn7 insertions at attTn7 sites from homologous recombination events a PCR using primer pairs P536 & P537 was also performed to confirm the absence of the plasmid-borne oriT in the recipient chromosome. All confirmatory PCRs were done using DNA templates obtained via from boiling preparations. Briefly, individual colonies were transferred to 30 μl of sterile distilled and deionized water and the cell suspension was boiled for 10 min. The resulting lysates were then centrifuged for 30 s at 12,000 × g at room temperature in a microcentrifuge and the supernatants transferred to a clean microcentrifuge tube. Six μl of supernatant were used a template in 50 μl PCR mixes containing the respective primers and Taq DNA polymerase (New England Biolabs).
Second, the mini-Tn7-lux plasmid was used as source for promoter-containing DNA fragments. Putative promoter regions were first predicted based on the genomic context of the insertion and using the Berkeley Drosophila Genome Project Neural Network Promoter Prediction and prokaryotic settings (http://www.fruitfly.org/seq_tools/promoter.html). Putative promoters were mapped onto the genome and the most likely promoter region was chosen based on number of possible promoters in the area and how close they were to the Tn5 insertion. In general, oligonucleotides were designed to PCR amplify the promoter region and add DraIII and StuI restriction sites to either end to control the direction of the promoter upon cloning into the desired Tn5/7-lux vector (in each case the cloned promoter region replaced the Tn5 transposase gene tnpA and flanking MEs). Transformants were chosen based on degree of luminescence and the presence of the correct plasmids was confirmed by a DraIII + StuI restriction digest and/or PCR amplification of the promoter region from the plasmid, followed by DNA sequencing. Mini-Tn7-lux insertions were then obtained and confirmed as described above.
Acknowledgments
Funding was provided by indirect cost recovery returns made possible by several extramural National Institutes of Health, National Institute of Allergy and Infectious Diseases research grants and the Ruth L. Kirschstein National Research Service Award F32 AI088884 to Brian Kvitko. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to thank Carolina Lopez for constructing pPS2305 and pFTC2, and Katie Quinn for constructing pFTP2. We also thank Colin Manoil, University of Washington, for providing pLG107 and Brad Borlee, Colorado State University, for the modified conjugation protocol.
Additional file
Additional methods; Table S1 Auxiliary plasmids used for vector construction; Figure S1 Genealogy of mini-Tn 7- lux and mini-Tn 5/7- lux vector creation from pTn 7 xLuxG0; Figure S2 Maps of mini-Tn 7 delivery vectors; Figure S3 Tn 7 insertion at multiple sites in B. pseudomallei and detection by multiplex PCR.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
STB, BHK, RKS and HPS conceived and designed the experiments. RKS was STB’s undergraduate advisor. STB and BHK conducted experiments. BHK and HPS wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Steven T Bruckbauer, Email: steven.bruckbauer@gmail.com.
Brian H Kvitko, Email: bkvitko@gmail.com.
RoxAnn R Karkhoff-Schweizer, Email: Roxann.Karkhoff-Schweizer@ColoState.EDU.
Herbert P Schweizer, Email: hschweizer@ufl.edu.
References
- 1.Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA. Photonic detection of bacterial pathogens in living hosts. Mol Microbiol. 1995;18:593–603. doi: 10.1111/j.1365-2958.1995.mmi_18040593.x. [DOI] [PubMed] [Google Scholar]
- 2.Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002;4:235–60. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 3.Piwnica-Worms D, Schuster DP, Garbow JR. Molecular imaging of host-pathogen interactions in intact small animals. Cell Microbiol. 2004;6:319–31. doi: 10.1111/j.1462-5822.2004.00379.x. [DOI] [PubMed] [Google Scholar]
- 4.Kadurugamuwa JL, Francis KP. Bioluminescent imaging of bacterial biofilm infections in vivo. Methods Mol Biol. 2008;431:225–39. doi: 10.1007/978-1-60327-032-8_18. [DOI] [PubMed] [Google Scholar]
- 5.Kong Y, Shi Y, Chang M, Akin AR, Francis KP, Zhang N, et al. Whole-body imaging of infection using bioluminescence. Curr Prot Microbiol. 2011;Chapter 2:Unit 2C 4. doi: 10.1002/9780471729259.mc02c04s21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baban CK, Cronin M, Akin AR, O’Brien A, Gao X, Tabirca S, et al. Bioluminescent bacterial imaging in vivo. J Vis Exp. 2012;69:e4318. doi: 10.3791/4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kadurugamuwa JL, Sin L, Albert E, Yu J, Francis K, DeBoer M, et al. Direct continuous method for monitoring biofilm infection in a mouse model. Infect Immun. 2003;71:882–90. doi: 10.1128/IAI.71.2.882-890.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadurugamuwa JL, Sin LV, Yu J, Francis KP, Kimura R, Purchio T, et al. Rapid direct method for monitoring antibiotics in a mouse model of bacterial biofilm infection. Antimicrob Agents Chemother. 2003;47:3130–7. doi: 10.1128/AAC.47.10.3130-3137.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadurugamuwa JL, Sin LV, Yu J, Francis KP, Purchio TF, Contag PR. Noninvasive optical imaging method to evaluate postantibiotic effects on biofilm infection in vivo. Antimicrob Agents Chemother. 2004;48:2283–7. doi: 10.1128/AAC.48.6.2283-2287.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelsman AF, van der Mei HC, Francis KP, Busscher HJ, Ploeg RJ, van Dam GM. Real time noninvasive monitoring of contaminating bacteria in a soft tissue implant infection model. J Biomed MaterRes B Appl Biomater. 2009;88(1):123–9. doi: 10.1002/jbm.b.31158. [DOI] [PubMed] [Google Scholar]
- 11.Kadurugamuwa JL, Modi K, Yu J, Francis KP, Purchio T, Contag PR. Noninvasive biophotonic imaging for monitoring of catheter-associated urinary tract infections and therapy in mice. Infect Immun. 2005;73:3878–87. doi: 10.1128/IAI.73.7.3878-3887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong YQ, Willard J, Kadurugamuwa JL, Yu J, Francis KP, Bayer AS. Real-time in vivo bioluminescent imaging for evaluating the efficacy of antibiotics in a rat Staphylococcus aureus endocarditis model. Antimicrob Agents Chemother. 2005;49:380–7. doi: 10.1128/AAC.49.1.380-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Francis KP, Yu J, Bellinger-Kawahara C, Joh D, Hawkinson MJ, Xiao G, et al. Visualizing pneumococcal infections in the lungs of live mice using bioluminescent Streptococcus pneumoniae transformed with a novel gram-positive lux transposon. Infect Immun. 2001;69:3350–8. doi: 10.1128/IAI.69.5.3350-3358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy J, Francis KP, DeBoer M, Chu P, Gibbs K, Contag CH. Extracellular replication of Listeria monocytogenes in the murine gall bladder. Science. 2004;303:851–3. doi: 10.1126/science.1092712. [DOI] [PubMed] [Google Scholar]
- 15.Kuklin NA, Pancari GD, Tobery TW, Cope L, Jackson J, Gill C, et al. Real-time monitoring of bacterial infection in vivo: development of bioluminescent staphylococcal foreign-body and deep-thigh-wound mouse infection models. Antimicrob Agents Chemother. 2003;47:2740–8. doi: 10.1128/AAC.47.9.2740-2748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun. 2006;74:5391–6. doi: 10.1128/IAI.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao M, Yang M, Baranov E, Wang X, Penman S, Moossa AR, et al. Spatial-temporal imaging of bacterial infection and antibiotic response in intact animals. Proc Natl Acad Sci U S A. 2001;98:9814–8. doi: 10.1073/pnas.161275798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Demidova TN, Gad F, Zahra T, Francis KP, Hamblin MR. Monitoring photodynamic therapy of localized infections by bioluminescence imaging of genetically engineered bacteria. J Photochem Photobiol B. 2005;81(1):15–25. doi: 10.1016/j.jphotobiol.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niska JA, Shahbazian JH, Ramos RI, Pribaz JR, Billi F, Francis KP, et al. Daptomycin and tigecycline have broader effective dose ranges than vancomycin as prophylaxis against a Staphylococcus aureus surgical implant infection in mice. Antimicrob Agents Chemother. 2012;56(5):2590–7. doi: 10.1128/AAC.06291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niska JA, Shahbazian JH, Ramos RI, Francis KP, Bernthal NM, Miller LS. Vancomycin-rifampin combination therapy has enhanced efficacy against an experimental Staphylococcus aureus prosthetic joint infection. Antimicrob Agents Chemother. 2013;57(10):5080–6. doi: 10.1128/AAC.00702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meighen EA. Genetics of bacterial bioluminescence. Annu Rev Genet. 1994;28:117–39. doi: 10.1146/annurev.ge.28.120194.001001. [DOI] [PubMed] [Google Scholar]
- 22.Cronin M, Sleator RD, Hill C, Fitzgerald GF, van Sinderen D. Development of a luciferase-based reporter system to monitor Bifidobacterium breve UCC2003 persistence in mice. BMC Microbiol. 2008;8:161. doi: 10.1186/1471-2180-8-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cronin M, Akin AR, Collins SA, Meganck J, Kim JB, Baban CK, et al. High resolution in vivo bioluminescent imaging for the study of bacterial tumour targeting. PLoS One. 2012;7(1):e30940. doi: 10.1371/journal.pone.0030940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis KP, Joh D, Bellinger-Kawahara C, Hawkinson MJ, Purchio TF, Contag PR. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect Immun. 2000;68:3594–35600. doi: 10.1128/IAI.68.6.3594-3600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Warawa JM, Long D, Rosenke R, Gardner D, Gherardini FC. Bioluminescent diagnostic imaging to characterize altered respiratory tract colonization by the Burkholderia pseudomallei capsule mutant. Front Microbiol. 2011;2:133. doi: 10.3389/fmicb.2011.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen H, Gold SE, Tamaki SJ, Keen NT. Construction of Tn7-lux system for gene expression studies in Gram-negative bacteria. Gene. 1992;122:27–34. doi: 10.1016/0378-1119(92)90028-N. [DOI] [PubMed] [Google Scholar]
- 27.Riedel CU, Monk IR, Casey PG, Morrissey D, O’Sullivan GC, Tangney M, et al. Improved luciferase tagging system for Listeria monocytogenes allows real-time monitoring in vivo and in vitro. Appl Env Microbiol. 2007;73(9):3091–4. doi: 10.1128/AEM.02940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owen SJ, Batzloff M, Chehrehasa F, Meedeniya A, Casart Y, Logue CA, et al. Nasal-associated lymphoid tissue and olfactory epithelium as portals of entry for Burkholderia pseudomallei in murine melioidosis. J Infect Dis. 2009;199(12):1761–70. doi: 10.1086/599210. [DOI] [PubMed] [Google Scholar]
- 29.Massey S, Johnston K, Mott TM, Judy BM, Kvitko BH, Schweizer HP, et al. In vivo bioluminescence imaging of Burkholderia mallei respiratory infection and treatment in the mouse model. Front Microbiol. 2011;2:174. doi: 10.3389/fmicb.2011.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damron FH, McKenney ES, Barbier M, Liechtie GW, Schweizer HP, Goldberg JB. Construction of mobilizable mini-Tn7 vectors for bioluminescent detection and single copy promoter lux reporter analysis in Gram-negative bacteria. Appl Env Microbiol. 2013;79:4149–53. doi: 10.1128/AEM.00640-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Boer HA, Comstock LJ, Vasser M. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci U S A. 1983;80:21–5. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levesque C, Brassard S, Lapointe J, Roy P. Diversity and relative strength of tandem promoters for the antibiotic resistance genes of several integrons. Gene. 1994;142:49–54. doi: 10.1016/0378-1119(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 33.Deshazer D, Woods DE. Broad-host-range cloning and cassette vectors based on the R388 trimethoprim resistance gene. Biotechniques. 1996;20:762–4. doi: 10.2144/96205bm05. [DOI] [PubMed] [Google Scholar]
- 34.Reznikoff WS. The Tn5 transposon. Annu Rev Microbiol. 1993;47:945–63. doi: 10.1146/annurev.mi.47.100193.004501. [DOI] [PubMed] [Google Scholar]
- 35.Peters JE, Craig NL. Tn7: smarter than we thought. Nat Rev Mol Cell Biol. 2001;2:806–14. doi: 10.1038/35099006. [DOI] [PubMed] [Google Scholar]
- 36.de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in Gram-negative bacteria. J Bacteriol. 1990;172:6568–72. doi: 10.1128/jb.172.11.6568-6572.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2:443–8. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 38.Choi K-H, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protocols. 2006;1:153–61. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 39.McKenzie GJ, Craig NL. Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol. 2006;6:39. doi: 10.1186/1471-2180-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar A, Dalton C, Cortez-Cordova J, Schweizer HP. Mini-Tn7 vectors as genetic tools for single copy gene cloning in Acinetobacter baumannii. J Microbiol Methods. 2010;82:296–300. doi: 10.1016/j.mimet.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 41.Crepin S, Harel J, Dozois CM. Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Env Microbiol. 2012;78(17):6001–8. doi: 10.1128/AEM.00986-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi K-H, DeShazer D, Schweizer HP. mini-Tn7 insertion in bacteria with multiple glmS-linked attTn7 sites: example Burkholderia mallei ATCC 23344. Nat Protocols. 2006;1:162–9. doi: 10.1038/nprot.2006.25. [DOI] [PubMed] [Google Scholar]
- 43.Choi K-H, Mima T, Casart Y, Rholl D, Kumar A, Beacham IR, et al. Genetic tools for select agent compliant manipulation of Burkholderia pseudomallei. Appl Env Microbiol. 2008;74:1064–75. doi: 10.1128/AEM.02430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi K-H, Schweizer HP. mini-Tn7 insertion in bacteria with secondary, non-glmS-linked attTn7 sites: example Proteus mirabilis HI4320. Nat Protocols. 2006;1:170–8. doi: 10.1038/nprot.2006.26. [DOI] [PubMed] [Google Scholar]
- 45.Parks AR, Peters JE. Tn7 elements: engendering diversity from chromosomes to episomes. Plasmid. 2009;61(1):1–14. doi: 10.1016/j.plasmid.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herrero M, De Lorenzo V, Timmis KN. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in Gram-negative bacteria. J Bacteriol. 1990;172:6557–67. doi: 10.1128/jb.172.11.6557-6567.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimada T, Kori A, Ishihama A. Involvement of the ribose operon repressor RbsR in regulation of purine nucleotide synthesis in Escherichia coli. FEMS Microbiol Lett. 2013;344(2):159–65. doi: 10.1111/1574-6968.12172. [DOI] [PubMed] [Google Scholar]
- 48.Katzen F. Gateway recombinational cloning: a biological operating system. Expert Opin Drug Discov. 2007;2:571–89. doi: 10.1517/17460441.2.4.571. [DOI] [PubMed] [Google Scholar]
- 49.Liss L. New M13 host: DH5αF’ competent cells. Focus. 1987;9:13. [Google Scholar]
- 50.Kvitko BH, Bruckbauer S, Prucha J, McMillan I, Breland EJ, Lehman S, et al. A simple method for construction of pir+ Enterobacterial hosts for maintenance of R6K replicon plasmids. BMC Res Notes. 2012;5:157. doi: 10.1186/1756-0500-5-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopez CM, Rholl DA, Trunck LA, Schweizer HP. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol. 2009;75(20):6496–503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Propst KL, Mima T, Choi KH, Dow SW, Schweizer HP. A Burkholderia pseudomallei ΔpurM mutant is avirulent in immune competent and immune deficient animals: candidate strain for exclusion from Select Agent lists. Infect Immun. 2010;78:3136–43. doi: 10.1128/IAI.01313-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehtinen J, Jarvinen S, Virta M, Lilius EM. Real-time monitoring of antimicrobial activity with the multiparameter microplate assay. J Microbiol Methods. 2006;66(3):381–9. doi: 10.1016/j.mimet.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 54.Kong Y, Akin AR, Francis KP, Zhang N, Troy TL, Xie H, et al. Whole-body imaging of infection using fluorescence. Curr Prot Microbiol. 2011;Chapter 2:Unit 2C 3. doi: 10.1002/9780471729259.mc02c03s21. [DOI] [PubMed] [Google Scholar]
- 55.Sambrook J, Russell DW. Molecular Cloning. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 56.Kvitko BH, McMillan IA, Schweizer HP. An improved method for oriT-directed cloning and functionalization of large bacterial genomic regions. Appl Env Microbiol. 2013;79:4869–78. doi: 10.1128/AEM.00994-13. [DOI] [PMC free article] [PubMed] [Google Scholar]