

Abstract
Background
The time course and extent of recovery after revascularization of viable dysfunctional myocardium is variable. While fibrosis is a major determinant, myocyte structural and molecular remodeling may also play important roles.
Objective
This study sought to determine whether persistent myocyte loss and/or irreversibility of protein changes that develop in hibernating myocardium have an impact on functional recovery in the absence of infarction.
Methods
Swine instrumented with a chronic left anterior descending artery (LAD) stenosis to produce hibernating myocardium underwent percutaneous revascularization with serial functional recovery evaluated for 1 month (n = 12). Myocardial tissue was evaluated to assess myocyte size, nuclear density, and proliferation indexes in comparison to normal animals and nonrevascularized controls. Proteomic analysis by 2-dimensional differential in-gel electrophoresis (2D-DIGE) was used to determine the reversibility of molecular adaptations of hibernating myocytes.
Results
At 3 months, physiological features of hibernating myocardium were confirmed, with depressed LAD wall thickening and no significant infarction. Revascularization normalized LAD flow reserve, with no immediate change in LAD wall thickening. Regional LAD wall thickening slowly improved, but remained depressed 1 month post-percutaneous coronary intervention (PCI). Surprisingly, revascularization was associated with histological evidence of myocytes reentering the growth phase of the cell cycle and increased cKit+ cells. Myocyte nuclear density returned to normal, while regional myocyte hypertrophy regressed. Proteomic analysis demonstrated heterogeneous effects of revascularization. Up-regulated stress and cytoskeletal proteins normalized, while reduced contractile and metabolic proteins persisted.
Conclusions
Delayed recovery of hibernating myocardium in the absence of scar may reflect persistent reductions in contractile and metabolic proteins. While revascularization appears to stimulate myocyte proliferation, the persistence of small immature myocytes may also contribute to delayed functional recovery.
Keywords: Coronary Blood Flow, Myocyte Regeneration, Proteomics
Introduction
Hibernating myocardium is characterized by viable, dysfunctional myocardium that develops as an adaptive response to chronic repetitive ischemia from a flow-limiting stenosis. It arises from a severe impairment in coronary flow reserve (1,2), which leads to myocyte apoptosis with regional myocyte loss, compensatory cellular hypertrophy (3), reduced myocardial oxygen consumption (4), regional down-regulation of enzymes involved in oxidative metabolism, and up-regulation of stress proteins that allow the heart to adapt and prevent infarction (5). In some patients, these adaptations are incomplete and progressive fibrosis and myocyte loss develops. In others, these adaptations ultimately minimize stress-induced ischemia, which prevents further myocyte death, but at the expense of chronic regional contractile dysfunction.
While contractile dysfunction in hibernating myocardium can improve after revascularization, complete recovery is infrequent (<25% of patients)(6,7) and 1 in 4 dysfunctional segments without fibrosis (determined by magnetic resonance imaging [MRI]) fail to improve (8). The time course of functional recovery is also variable. Different studies demonstrate rapid recovery (9), recovery within several weeks (10–12), or delayed improvement requiring 1 year (13–15). This protracted course contrasts with the complete normalization of function within hours or days following acute stunning and short-term hibernation (16). The roles of myocyte loss (3) and myocardial protein changes (5,17) in the delayed response to revascularization are unclear. We performed percutaneous revascularization of swine with chronic hibernating myocardium to determine the initial time course of functional recovery and the cellular mechanisms contributing to persistent dysfunction in the absence of scar. We demonstrate that revascularization of hibernating myocardium stimulates myocyte proliferation. Nevertheless, functional improvement is delayed and incomplete, with persistent reductions in metabolic and contractile proteins 1 month after revascularization.
Methods
Experimental procedures and protocols conformed to institutional guidelines for the care and use of animals in research and were approved by the University at Buffalo Institutional Animal Care and Use Committee.
Coronary Artery Instrumentation
Juvenile farm-bred pigs (8 to 10 kg) were chronically instrumented with a left anterior descending coronary artery (LAD) stenosis to produce hibernating myocardium using a modification of previously published techniques (1). Fasted pigs pre-medicated with tiletamine 50 mg/ml and zolazepam 50 mg/ml (tiletamine)/ketamine (100 mg/ml)(0.037 ml/kg intramuscular [IM]), cefazolin (0.5 g intravenous [IV]) and gentamicin (40 mg IV) were intubated and anesthetized with isoflurane (1 to 2%). A thoracotomy was performed in the fourth left intercostal space. The proximal LAD was instrumented with a short piece of expandable 1.5 mm internal diameter silicone tubing with a longitudinal slit for vessel insertion (18). It was secured with circumferential sutures that could be expanded using standard angioplasty balloon inflation pressures. The chest was closed, intercostal nerves were infiltrated with 2% lidocaine, and the pneumothorax was evacuated. Postoperative antibiotics and analgesics (butorphanol 0.025 mg/kg) were administered as needed for pain.
Serial Physiological Studies and Percutaneous Intervention
Pigs with hibernating myocardium undergoing revascularization (n = 12) were compared to nonrevascularized animals (n = 12) and normal shams that underwent stent insertion (n = 10). Animals underwent baseline physiological studies at ~3 months post-instrumentation in the closed-chest sedated state (Telazol/xylazine IM and propofol 2 to 5 mg·kg−1·min−1 IV). Coronary angiography was performed with a 5-F multipurpose catheter and nonionic contrast to quantify stenosis severity and angiographic collaterals. Myocardial perfusion was measured using fluorescent microspheres at rest and after adenosine vasodilation (0.9 mg/kg per min; with coinfusion of phenylephrine to prevent hypotension)(19). Regional function was measured using off-axis M-mode echocardiography (GE Vivid7)(19).
Percutaneous intervention (PCI) was performed 89 ± 3 days after placement of the LAD stenosis. Five days before the procedure, animals undergoing PCI were pre-treated with clopidogrel (300 mg orally) followed by daily aspirin (325 mg) and clopidogrel (75 mg). After heparin (5,000 U IV) a 6-F guiding catheter was inserted and a 0.014 guide wire advanced into the LAD. If total LAD occlusion was present, pre-dilation was performed with a low-profile balloon. Subsequently, percutaneous revascularization was performed with a bare-metal stent (Multi-Link Zeta, Abbott Vascular, Santa Clara, California) placed in most animals (n = 9) to prevent recoil of the silastic stenosis.
Flow and function were reassessed 2 h after revascularization. To define the time-course of functional recovery, animals were brought back for serial echocardiography at 1 day, 3 days, 1 week, and 1 month. Angiography and flow measurements were repeated at 1 month, after which animals were euthanized under general anesthesia 72 h later. Hearts were stained with triphenyltetrazolium chloride (TTC) to exclude infarction and samples were obtained for histology.
Proteomic Profiling
Samples for proteomics taken from the LAD subendocardium were immediately flash-frozen at −80°C. We used 2-dimensional differential in-gel electrophoresis (2D-DIGE) to assess protein expression, using published techniques (5,17), as described in the Online Supplement. Total protein extracted from hibernating swine that were revascularized (n = 12) and nonrevascularized (n = 12) were compared to a pooled sham control sample (5). Samples were labeled with CyDye DIGE Fluor, run on 2D gels, scanned, and imported into DeCyder v6.5 software (GE Healthcare) for gel matching and analysis. Spot identification was performed by in-gel trypsin digestion, MALDI-TOF mass spectrometry (Mascot search engine), and LC/MS (Bioworks software and SEQUEST). A heat map was created to compare proteins identified as dysregulated in hibernating myocardium in animals with and without revascularization (R package version 2.13.0.(20)). Selected enzyme activities were assayed as described (5).
Myocyte Nuclear Density and Morphometry
Myocyte nuclear density was quantified using hematoxylin and eosin-stained sections (3). Periodic acid-Schiff (PAS)-stained sections were used to quantify myocyte diameter (100 longitudinal myocytes per region) in revascularized (n = 12), nonrevascularized (n = 8), and sham normal (n = 6) samples (19). Myocytes were included regardless of size, as long as myofilaments could be identified surrounding the nucleus.
Immunohistochemical Assessment of Myocyte Proliferation
Paraffin-fixed tissue sections (~4 μm thickness) were incubated with anti-Ki67 (mouse monoclonal antibody, clone MIB-1, 1:200; Dako, Carpinteria, California,) or anti-phospho-histone-H3 (rabbit polyclonal antibody,1:1000; Upstate Biotech, Lake Placid, New York) and costained with anti-cTnI (rabbit polyclonal antibody, 1:200; Santa Cruz Biotechnology, Dallas, Texas,) to confirm colocalization in myocytes (19,21). Myocardial cKit+/CD45− cells (both antibodies 1:200 dilution; AbDSerotec, Raleigh, North Carolina,) and cKit+/GATA4+ cells (Santa Cruz Biotechnology, 1:200 dilution; Dalla, Texas) were quantified in frozen sections (19,21), post-treated with FITC conjugated anti-mouse and TexasRed-conjugated anti-rabbit antibody (Dako). Nuclei were stained with TO-PRO-3 (Molecular Probes, Life Technologies, Grand Island, New York) or DAPI (4′,6-diamidino-2-phenylindole; Vectashield, Vector Laboratories, Burlingame, California). Image acquisition was performed with a confocal microscope (Bio-Rad MRC 1024, Hercules, California) and an ApoTome-equipped AxioImager (Zeiss, Thornwood, New York). Results represent means of 363 ± 25 fields (57 ± 4 mm2).
Statistics
Data are expressed as means ± SEMs (SigmaStat 3.0, SPSS, Systat Software Inc., San Jose, California). Within-group comparisons over time and comparisons between hibernating and normally-perfused remote regions were assessed using paired Student t tests. Between-group differences were assessed using a 2-way Analysis of Variance (ANOVA) and the post-hoc Holm-Sidak test. An unpaired Student t test was used for statistical analysis of proteomics data and a 1-way ANOVA was used for analysis of enzyme activity data.
Results
Temporal Functional Improvement After Revascularization
Serial angiographic images from an animal with hibernating myocardium are displayed in Figure 1. Revascularized and nonrevascularized animals exhibited slight differences in stenosis severity (93 ± 2% versus 99 ± 1%, p < 0.05), but similar reductions in subendocardial LAD flow reserve (2.0 ± 0.5 vs. 2.1 ± 1.3, p = 0.93) and LAD wall thickening (LADΔWT 2.9 ± 0.3 vs. 3.5 ± 0.3 mm, p = 0.17). Restenosis 1 month after PCI was insignificant (23% ± 5%). TTC staining showed <1% infarction and LAD connective tissue was similar (9.8 ± 1.3% vs. 7.9 ± 0.9% after PCI, p = 0.28).
Figure 1. Selected Angiographic Images From an Animal With Hibernating Myocardium.
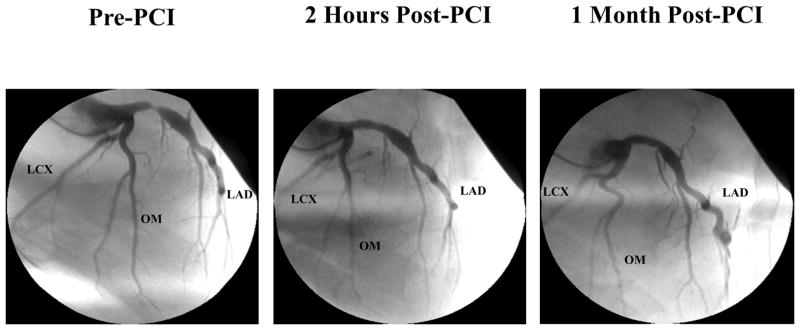
The pre-PCI image demonstrates a severe proximal LAD stenosis. The 2-h post-PCI image demonstrates a widely patent LAD with no significant restenosis after 1 month. LAD = left anterior descending artery; PCI = percutaneous coronary intervention.
Hemodynamics and measurements of regional wall thickening are summarized in Table 1. Indices of global function were normal at all time points and are summarized in Online Table 1. Despite immediate normalization of LAD flow reserve (Figure 2), wall thickening initially remained unchanged (LADΔWT 2.9 ± 0.4 vs. 2.9 ± 0.3 mm, p = 0.77). Regional LAD function gradually increased in the first week following revascularization and became significant after 1 week (LADΔWT 2.9 ± 0.3 to 4.2 ± 0.4 mm; p < 0.05, Figure 3). There was little additional improvement thereafter and, despite complete revascularization, LAD dysfunction persisted at 1 month (LADΔWT 4.6 ± 0.4 vs. 6.3 ± 0.3 mm in remote; p < 0.05).
Table 1.
Hemodynamics in Revascularized and Nonrevascularized Animals with Hibernating Myocardium.
| Heart Rate (beats/min) | LV Systolic Pressure (mm Hg) | LVEDP (mm Hg) | LV dP/dtMax (mm Hg/s) | LV dP/dtMin (mm Hg/s); | Regional Wall Thickening | ||||
|---|---|---|---|---|---|---|---|---|---|
| WT (%) | ΔWT (mm) | ||||||||
| LAD | Remote | LAD | Remote | ||||||
| Revascularized | |||||||||
| Baseline | 108 ± 4 | 108 ± 14 | 28 ± 3 | 2549 ± 165 | −2467 ± 148 | 38.6 ± 4.0‡ | 92.2 ± 7.5 | 2.9 ± 0.3‡ | 5.8 ± 0.3 |
| 2 h Post-PCI | 105 ± 6 | 113 ± 6 | 30 ± 3 | 1988 ± 89* | −2247 ± 125 | 35.2 ± 4.6‡ | 86.6 ± 7.2 | 2.9 ± 0.4‡ | 5.7 ± 0.4 |
| 1 Month Post-PCI | 101 ± 2 | 128 ± 5 | 28 ± 3 | 2439 ± 136† | −2514 ± 96 | 58.6 ± 4.9*‡ | 86.9 ± 6.9 | 4.6 ± 0.4*‡ | 6.3 ± 0.3 |
| Nonrevascularized | |||||||||
| Baseline | 107 ± 5 | 121 ± 4 | 23 ± 2 | 2284 ± 168 | −2225 ± 154 | 34.0±2.1‡ | 80.6 ± 8.4 | 3.5 ± 0.3‡ | 6.1 ± 0.4 |
| 1 Month | 103 ± 5 | 120 ± 4 | 23 ± 2 | 2073 ± 69 | −2184 ± 139 | 26.5 ± 3.2‡ | 76.9 ± 5.4 | 2.8 ± 0.3‡ | 7.2 ± 0.5 |
Values are mean ± SEM; Baseline measurements were made 3 months after surgical placement of a LAD stenosis.
LV = left ventricular; LVEDP = left ventricular end diastolic pressure; PCI = percutaneous coronary intervention; WT = wall thickening;
p < 0.05 vs. baseline;
p < 0.05 vs. 2 h post-PCI;
p < 0.05 LAD vs. remote
Figure 2. Coronary Flow Reserve and Regional Wall Thickening in Hibernating Myocardium Before and Immediately After PCI.

LAD subendocardial flow reserve (A) and wall thickening (B) were reduced in hibernating myocardium (pre-PCI). Flow reserve normalized 2 h later (post-PCI), but LAD wall thickening remained depressed. Abbreviations as in Figure 1.
Figure 3. Delayed Time Course of Functional Improvement After PCI.

Regional wall thickening was reduced at rest (pre-PCI). There was no immediate effect of revascularization, but a delayed improvement in function became evident after 1 week. No further functional improvement occurred at 1 month and LAD wall thickening remained depressed. Abbreviations as in Figure 1.
The Effects of Revascularization on the Hibernating Myocardium Proteome
Table 2 summarizes the effects of revascularization on proteomic adaptations in hibernating myocardium (5,17,22) and proteins correlating with increases in flow and function after revascularization are summarized in the heat map (Figure 4). Figure 5 summarizes average changes in selected proteins from each category (proteins italicized in Table 2). In general, reductions in contractile proteins and metabolic enzymes persisted after revascularization. NADH dehydrogenase (42 and 30 kDa subunits), ATP synthase (F1 alpha and beta chains), pyruvate dehydrogenase (E1 alpha, dihydrolipoamide dehydrogenase), and isocitrate dehydrogenase remained down-regulated, while long-chain acyl-CoA dehydrogenase decreased. The 51 kDa subunit of NADH dehydrogenase, flavoprotein subunit of complex II, cytochrome Bc1 core protein-1, pyruvate dehydrogenase (E1β and E2), malate dehydrogenase, and medium-chain acyl-CoA dehydrogenase increased, but did not completely normalize. Cytochrome C oxidase and citrate synthase activities, which were reduced in hibernating myocardium, corroborated the heterogeneous effects of revascularization on metabolic enzymes (Figure 6). Despite variable effects of revascularization on metabolic enzymes, alleviation of repetitive ischemia by PCI normalized cytosolic stress and structural proteins (Figure 4, Figure 5). Stress proteins including αβ-crystalline, GRP-78, and HSP-20, as well as structural proteins, such as vimentin and desmin, were no longer up-regulated. Revascularization had limited effects on contractile proteins, with most that were reduced in hibernating myocardium remaining so after revascularization. This failure to reverse chronic reductions in contractile and metabolic proteins may contribute to persistent dysfunction and delayed functional recovery of hibernating myocardium.
Table 2.
Proteomic Profiling in Swine With Hibernating Myocardium With and Without. Revascularization*
| Protein Name | n | Average Ratio
|
|
|---|---|---|---|
| Hibernating LAD/sham | Revascularized LAD/sham | ||
| Citric Acid Cycle Proteins | |||
| Pyruvate dehydrogenase E1 α subunit | 12 | 0.68† | 0.74 |
| Pyruvate dehydrogenase E1 β subunit | 12 | 0.60† | 0.77†‡ |
| Dihydrolipoamide acetyltransferase (PDH E2) | 12 | 0.74† | 0.85†‡ |
| Dihydrolipoamide dehydrogenase (PDH E3) | 12 | 0.86 | 0.87 |
| Dihydrolipoamide dehydrogenase (PDH E3) | 12 | 0.77† | 0.79 |
| Aconitase | 10 | 1.28† | 1.05‡ |
| Aconitase | 12 | 1.06 | 1.05 |
| Aconitase | 12 | 0.86 | 0.98 |
| Aconitase | 9 | 0.95 | 0.81 |
| Aconitase | 11 | 1.00 | 1.18 |
| NAD+ isocitrate dehydrogenase (α subunit) | 12 | 0.69† | 0.75† |
| NAD+ isocitrate dehydrogenase (α subunit) | 12 | 0.72† | 0.83†‡ |
| Dihydrolipoamide succinyltransferase | 12 | 0.87 | 0.96 |
| Dihydrolipoamide succinyltransferase | 11 | 0.97 | 1.02 |
| Malate dehydrogenase (mitochondrial) | 11 | 0.77 | 0.91‡ |
| Malate dehydrogenase (mitochondrial) | 9 | 0.85 | 0.91 |
| Malate dehydrogenase (mitochondrial) | 12 | 0.84 | 0.91 |
| Electron Transport Chain and ATP Synthesis Proteins | |||
| NADH dehydrogenase (Complex I) 75 kDa subunit | 12 | 0.86 | 0.84 |
| NADH dehydrogenase (Complex I) 75 kDa subunit | 11 | 0.72 | 0.76 |
| NADH dehydrogenase (Complex I) 51 kDa subunit | 12 | 0.82 | 0.89 |
| NADH dehydrogenase (Complex I) 51 kDa subunit | 12 | 0.67† | 0.79†‡ |
| NADH dehydrogenase (Complex I) 30 kDa subunit | 10 | 0.61† | 0.62† |
| NADH dehydrogenase (Complex I) 24 kDa subunit | 11 | 0.78 | 0.88 |
| Flavoprotein subunit of Complex II | 12 | 0.58† | 0.74‡ |
| Cytochrome Bc1 core protein I (complex III) | 12 | 0.69† | 0.95‡ |
| ATP synthase F1 α chain | 7 | 0.87 | 1.00 |
| ATP synthase F1 α chain | 7 | 0.91 | 0.94 |
| ATP synthase F1 α chain | 12 | 0.86 | 0.89 |
| ATP synthase F1 α chain | 12 | 0.71† | 0.81 |
| ATP synthase F1 β chain | 12 | 1.20 | 0.91‡ |
| ATP synthase F1 β chain | 12 | 0.84 | 0.77† |
| Fatty Acid Oxidation | |||
| Medium-chain acyl-CoA dehydrogenase (MCAD) | 12 | 0.88 | 0.80 |
| Long-chain acyl-CoA dehydrogenase (LCAD) | 12 | 0.84 | 0.81‡ |
| Other Mitochondrial Matrix Proteins | |||
| 60 kDa heat shock protein, mitochondrial | 12 | 0.95 | 0.94 |
| Aspartate aminotransferase, mitochondrial | 8 | 1.07 | 1.05 |
| Creatine kinase, mitochondrial | 6 | 1.01 | 0.96 |
| Creatine kinase, mitochondrial | 12 | 0.80 | 0.78 |
| Isovaleryl-CoA dehydrogenase | 12 | 0.78 | 0.82 |
| Mitochondrial stress-70 protein (GRP 75) | 12 | 0.92 | 0.98 |
| Protein Name | n | Average Ratio | |
|---|---|---|---|
| Hibernating LAD/sham | Revascularized LAD/sham | ||
| NADP+ isocitrate dehydrogenase (47 kDa subunit) | 11 | 0.58† | 0.77‡ |
| NADP+ isocitrate dehydrogenase (47 kDa subunit) | 11 | 0.62 | 0.76‡ |
| Succinyl-CoA 3-ketoacid-CoA transferase 1 | 12 | 0.69† | 0.78† |
| Translation elongation factor EF-Tu | 12 | 0.76† | 0.84 |
| Other Mitochondrial Membrane Proteins | |||
| Mitofilin | 10 | 1.07 | 1.23 |
| Mitofilin | 12 | 0.78† | 0.95 |
| Prohibitin | 3 | 0.83 | 0.95 |
| Voltage-dependent anion channel 2 (VDAC2) | 12 | 0.70† | 0.96‡ |
| Voltage-dependent anion channel 1 (VDAC1) | 8 | 0.85 | 0.93 |
| Voltage-dependent anion channel 1 (VDAC1) | 12 | 0.87 | 0.97 |
| Glycolytic Proteins | |||
| Fructose biphosphate aldolase A | 7 | 0.61 | 0.78 |
| Fructose biphosphate aldolase A | 11 | 0.67 | 0.87‡ |
| Triosephosphate isomerase | 12 | 0.67† | 0.73† |
| Triosephosphate isomerase | 12 | 0.99 | 0.83†‡ |
| GAPDH | 11 | 1.58 | 1.19‡ |
| GAPDH | 12 | 1.27 | 0.93‡ |
| GAPDH | 12 | 0.96 | 0.77‡ |
| GAPDH | 8 | 1.05 | 0.87‡ |
| Phosphoglycerate kinase 1 | 6 | 0.67† | 0.90‡ |
| Phosphoglycerate mutase 2 M isozyme | 12 | 0.70† | 0.71† |
| Enolase 3 | 12 | 0.94 | 0.90 |
| α Enolase | 11 | 0.98 | 1.09 |
| β Enolase | 12 | 0.84 | 0.86 |
| Muscle specific phosphopyruvate hydratase (enolase) | 12 | 0.81† | 0.77† |
| Pyruvate kinase | 12 | 0.66† | 0.86 |
| Anaerobic Metabolism Proteins | |||
| L-lactate dehydrogenase B chain | 6 | 0.64† | 0.68† |
| L-lactate dehydrogenase B chain | 12 | 0.68† | 0.89‡ |
| Contractile Proteins | |||
| Actin, alpha-cardiac | 12 | 0.83 | 0.89 |
| Actin, alpha-cardiac | 12 | 1.20 | 1.01‡ |
| Actin, alpha-cardiac | 3 | 0.80 | 0.71 |
| Actin, alpha-cardiac | 11 | 1.03 | 0.86 |
| Actin, alpha-cardiac | 12 | 0.65† | 0.86‡ |
| Actin, alpha-cardiac | 11 | 0.45† | 0.57†‡ |
| Actin, alpha-cardiac | 8 | 0.58† | 0.78‡ |
| Actin, alpha-cardiac | 10 | 0.92 | 0.77†‡ |
| Myosin heavy chain α (cardiac) | 11 | 0.99 | 0.92 |
| Myosin heavy chain α (cardiac) | 10 | 0.66† | 1.29 |
| Myosin heavy chain β | 8 | 0.69 | 0.95 |
| Myosin heavy chain β | 6 | 0.77 | 0.88 |
| Myosin heavy chain β | 6 | 0.92 | 1.08 |
| Myosin heavy chain β | 3 | 1.20 | 0.84 |
| Protein Name | n | Average Ratio | |
|---|---|---|---|
| Hibernating LAD/sham | Revascularized LAD/sham | ||
| Myosin heavy chain β | 12 | 0.94 | 0.95 |
| Myosin heavy chain β | 9 | 0.87 | 1.36 |
| Myosin heavy chain β | 4 | 1.46 | 1.11 |
| Myosin heavy chain β | 3 | 1.14 | 1.05 |
| Myosin heavy chain β | 9 | 1.23 | 1.07 |
| Myosin light chain 3 | 11 | 0.65† | 0.71† |
| Myosin light chain 3 | 11 | 0.60† | 0.67† |
| Myosin light chain 3 | 11 | 0.83† | 0.74† |
| Troponin T, cardiac | 6 | 0.69 | 0.86‡ |
| Troponin T, cardiac | 12 | 0.70† | 0.74† |
| Troponin T (cardiac isoform type 2) | 12 | 0.67† | 0.71† |
| Troponin T, cardiac | 12 | 0.73† | 0.60*‡ |
| Troponin T cardiac | 12 | 0.74† | 0.64*‡ |
| Tropomysin α chain | 11 | 0.82 | 0.81 |
| Tropomysin β chain | 12 | 0.61† | 0.63† |
| Structural/Cytoskeletal Proteins | |||
| Actin-interacting protein | 12 | 0.79 | 0.97‡ |
| Desmin | 12 | 1.23 | 1.21 |
| Desmin | 11 | 1.14 | 0.87‡ |
| Desmin | 12 | 1.34† | 0.90‡ |
| Desmin | 10 | 0.85 | 1.44 |
| Desmin | 12 | 1.37† | 0.93‡ |
| Gelsolin precursor | 11 | 1.42 | 1.21 |
| Vimentin | 12 | 1.54† | 1.10‡ |
| Vinculin | 12 | 1.14 | 1.09 |
| Stress Proteins | |||
| αB crystallin | 12 | 1.50† | 0.90‡ |
| αB crystallin | 12 | 0.90 | 0.70†‡ |
| Annexin 2 | 5 | 1.24 | 1.29 |
| Annexin 2 | 12 | 1.05 | 1.04 |
| GRP78 | 12 | 1.29 | 0.99‡ |
| GRP78 | 12 | 1.22† | 1.17 |
| HSP70 (70 Kda protein 1) | 12 | 1.11 | 0.99‡ |
| HSP70 (70 Kda protein 1) | 12 | 0.83 | 0.96‡ |
| HSP60 | 12 | 0.95 | 0.94 |
| HSP27 | 12 | 1.39 | 0.99 |
| HSP27 | 12 | 1.07 | 0.91 |
| HSP27 | 4 | 1.07 | 0.85 |
| HSP20 β-6 | 12 | 1.69† | 0.73† |
| T-complex protein 1 (chaperonin) | 12 | 0.88 | 0.87 |
| Antioxidant Proteins | |||
| Peroxiredoxin-2 | 12 | 0.90 | 0.87 |
| Peroxiredoxin-6 | 12 | 0.73† | 0.75† |
| Other Cytoplasmic Proteins | |||
| Aconitase 1 (Fe regulatory protein 1) | 11 | 0.97 | 1.06 |
| Protein Name | n | Average Ratio | |
|---|---|---|---|
| Hibernating LAD/sham | Revascularized LAD/sham | ||
| Aminoacylase-1 | 5 | 0.55† | 0.68‡ |
| Aspartate aminotransferase (cytoplasmic) | 12 | 0.52† | 0.64† |
| Creatine kinase, M chain (cytoplasmic) | 11 | 0.84 | 0.69†‡ |
| Creatine kinase, M chain (cytoplasmic) | 12 | 0.73† | 0.71† |
| Creatine kinase, M chain (cytoplasmic) | 12 | 0.56† | 0.65† |
| Cytosolic aminopeptidase | 12 | 1.00 | 0.97 |
| Dihydropyrimidinase-related protein 2 (DRP-2) | 12 | 0.87† | 0.83† |
| Malate dehydrogenase (cytoplasmic) | 12 | 0.64† | 0.69† |
| Myoglobin | 8 | 0.62 | 0.61 |
| Myoglobin | 10 | 0.43† | 0.36† |
| Transitional endoplasmic reticulum ATPase | 11 | 0.99 | 1.04 |
| Endoplasmic Reticulum Proteins | |||
| Protein disulfide isomerase A3 | 12 | 1.04 | 1.06 |
Bolded and italicized proteins are depicted in Figure 5.
p < 0.05 vs. sham
p < 0.05 vs. hibernating
Figure 4. Heat Map Demonstrating Differential Expression of Proteins Correlating With Increased Flow and/or Function From Animals With Hibernating Myocardium.

Following revascularization and the alleviation of ischemia, up-regulated (red) stress and cytoskeletal proteins and glycolytic enzymes generally normalized (yellow). In contrast, many mitochondrial and contractile proteins remained depressed (green) 4 weeks after revascularization.
Figure 5. Summary of Selected Proteins from 2D-DIGE.

(A) Mitochondrial proteins were reduced in animals with hibernating myocardium, with little improvement after revascularization. (B) Revascularization had heterogeneous effects on cytosolic proteins. Increases in stress and structural proteins tended to normalize after alleviation of ischemia. In contrast, contractile proteins such as troponin T remained persistently down-regulated 4 weeks after revascularization. 2D-DIGE = 2-dimensional differential in-gel electrophoresis.
Figure 6. Variable Effects of Revascularization on Enzymatic Activity.

Cytochrome c oxidase activity was reduced in hibernating myocardium (n = 9) and remained unchanged following revascularization (n = 12). In contrast, citrate synthase activity normalized following PCI. Abbreviations as in Figure 1.
Revascularization Stimulates Myocyte Proliferation in Hibernating Myocardium
We evaluated the effects of revascularization on myocyte number (nuclear density) and cell diameter (Figure 7). Nonrevascularized animals exhibited regionally-reduced nuclear density and increased myocyte diameters, as we previously reported in hibernating myocardium (3). In contrast, we found a prominent increase in LAD nuclear density (998 ± 52 to 1,406 ± 103 myocyte nuclei/mm2, p < 0.05) 1 month after revascularization, with smaller-diameter cardiomyocytes (10.3 ± 0.3 vs.15.7 ± 0.5 μm, p < 0.05). Despite these prominent changes, LAD end-diastolic wall thickness was unchanged after revascularization (7.8 ± 0.4 to 8.2 ± 0.6, p = 0.63). The relative increase in end-diastolic wall thickness of 4.7 ± 4.1 % was similar to the 6.0 ± 2.4% change observed in nonrevascularized animals (p = 0.28).
Figure 7. Effect of PCI on Myocyte Diameter and Nuclear Density in Hibernating Myocardium.

Untreated animals exhibited cardiomyocyte hypertrophy and a reduction in myocyte nuclear density. Four weeks after revascularization, myocyte nuclear density increased and myocyte diameter decreased, suggesting new myocyte formation. Abbreviations as in Figure 1.
To explain the increased myocyte nuclear density, we evaluated the effects of revascularization on indexes of myocyte proliferation. Ki-67, a marker of cell cycling, and the mitotic marker, phospho-histone-H3 (pHH3), were no different in LAD myocytes from sham and nonrevascularized animals. In contrast, Ki67 increased from 410 ± 82 to 2,109 ± 400 nuclei/106 myocyte nuclei after revascularization (Figure 8A). Likewise, myocyte pHH3 increased from 9 ± 5 to 350 ± 50 nuclei/106 myocyte nuclei (Figure 8B). Resident cardiac stem cells (LAD c-Kit+/CD45− cells, Figure 8C) were rare in shams (20 ± 11 cells/106 myocyte nuclei, n = 6), but tended to be higher in hibernating myocardium (178 ± 59 cells/106 myocyte nuclei, n = 7, p = 0.052), with significant increases 1 month after revascularization (401 ± 43 cells/106 myocyte nuclei, n = 6, p < 0.05). Progenitor cells committed to a cardiac lineage (cKit+/GATA4+) were not detected in shams, but increased in hibernating myocardium (15 ± 10 cells/106 myocyte nuclei, p < 0.05 vs. sham) and 1 month after revascularization (43 ± 20 cells/106 myocyte nuclei, p < 0.05 vs. sham; p = 0.38 vs. hibernating myocardium). The increased proliferative indexes, reductions in myocyte size, and increased myocyte nuclear density, collectively support the notion that revascularization stimulated new myocyte formation in hibernating myocardium.
Figure 8. Revascularization of Hibernating Myocardium Increases cKit+ Cells, Ki67+ Myocytes and pHH3+ Myocytes.

(A) Confocal images of Ki67+ staining (green) in a myocyte (cardiac troponin I; red) and nonmyocyte after revascularization. Nuclei are stained with DAPI (blue). Revascularization significantly increased Ki67+ myocytes consistent with increased DNA synthesis. (B) Confocal images depicting a pHH3+ nucleus (green), indicating a myocyte undergoing mitosis. Revascularization increased pHH3+ myocytes as compared to nonrevascularized animals with hibernating myocardium and sham controls. (C) Confocal images of a cKit+ cell between cardiomyocytes. Myocardial cKit+ cells increased after revascularization. DAPI = 4′,6-diamidino-2-phenylindole; pHH3 = phosphorylated histone H3.
Discussion
Despite complete revascularization of chronic hibernating myocardium and the absence of scar, there is no immediate improvement in regional LAD wall thickening. While it improved 1 week after revascularization, there was little subsequent improvement, and it remained depressed 1 month later. Tissue analysis at this time point demonstrated variable effects of revascularization on the proteome. Up-regulated stress and cytoskeletal proteins normalized, but reductions in contractile, metabolic, and mitochondrial proteins persisted. Unexpectedly, revascularization stimulated myocyte proliferation. These results suggest that the prolonged time course required for functional improvement in some patients with viable dysfunctional myocardium reflects heterogeneous effects of revascularization on the myocardial proteome, as well as the potentially time-dependent maturation of small, newly-formed cardiomyocytes.
Time Course of Functional Recovery Following Revascularization
Patients with viable dysfunctional myocardium display considerable variability in the magnitude and time course of functional recovery after revascularization (13,14). This is in part due to variable degrees of fibrosis and subendocardial infarction, but even completely viable myocardium exhibits variability. This likely reflects the underlying mechanisms of dysfunction, which arise from reversible ischemia (acute stunning, chronic stunning, and chronic hibernating myocardium)(23). The spectrum of viable dysfunctional myocardium leads to heterogeneity in the extent of myocyte loss (3,24) and variability in chronic molecular adaptations induced in response to reversible ischemia (5,17). We previously demonstrated that similar degrees of chronic contractile dysfunction from a chronic stenosis result in variable degrees of myocyte loss and proteomic remodeling (17,24). These variations are related to physiological stenosis severity, with more profound abnormalities developing in hibernating myocardium where coronary flow reserve becomes critically impaired.
Our results in swine with chronic hibernating myocardium exclude infarction as a covariate and demonstrate several phases of functional recovery (Figure 3). Despite complete restoration of perfusion, there was no immediate change in function, nor did it increase in the first several days after revascularization. A significant, but delayed, increase in LADΔWT was observed 1 week later. This early time course is reminiscent of the normalization seen following short-term hibernation (16,23). No further functional improvement occurred up to 1 month later. This delay in functional recovery contrasts with prior animal models (18,25,26), where dysfunction was not as chronic and resting function normalized early (within 1 to 2 weeks after revascularization). Thus, the present findings are more compatible with clinical observations following revascularization in chronic ischemic cardiomyopathy, where function rarely normalizes and frequently requires 3 to 12 months to reach a new steady state (13).
Incomplete Proteomic Remodeling after Revascularization
We previously demonstrated a spectrum of proteomic abnormalities in viable dysfunctional myocardium, which progress as stenosis severity increases (17). Proteomic changes in nonrevascularized animals with hibernating myocardium were characterized by down-regulation of multiple mitochondrial proteins, including enzymes that are entry points to oxidative metabolism (e.g., pyruvate dehydrogenase and acyl-CoA-dehydrogenase), down-regulation of contractile proteins, and up-regulation of stress and cytoskeletal proteins, as previously described (5,27). Other investigators have shown that up-regulated stress and cytoskeletal proteins normalize 1 month after revascularization, accompanied by reductions in interstitial connective tissue (26,28). In contrast, revascularization had only modest effects on metabolic enzymes and reductions in contractile proteins persisted. We previously demonstrated that regional metabolic proteins and mitochondrial oxygen consumption progressively declines as stenosis severity increases in viable dysfunctional myocardium (5,17,22,29). Consonant with our findings, Kelly et al. reported persistent reductions in the mitochondrial proteome before and after surgical revascularization of hibernating myocardium. Nevertheless, resting contractile function normalized 1 month post-revascularization (25). In their study, attenuation of the flow and functional responses to dobutamine persisted, suggesting that mitochondrial dysfunction continued to limit oxygen consumption during beta-adrenergic stimulation (30,31). This attenuated response could also reflect persistent regional sympathetic denervation and reduced beta-adrenergic responsiveness, despite revascularization (18,32,33).
Stimulation of Myocyte Regeneration by Revascularization
An unexpected finding was the prominent reverse myocyte remodeling induced by restoring perfusion to hibernating myocardium. This has not been evaluated in patients because it would require serial left ventricular (LV) biopsies after revascularization. Consonant with previous studies, nonrevascularized animals with hibernating myocardium had reductions in myocyte nuclear density and compensatory cellular hypertrophy that maintained near-normal regional wall thickness in the face of apoptosis (3,21). Revascularization increased myocyte nuclear density and reduced cellular hypertrophy, despite LAD wall thickness remaining unchanged. Myocyte proliferation after revascularization was suggested by immunohistochemistry of myocytes in the synthetic phase of the cell cycle using Ki-67 and increases in the myocyte mitotic marker, phospho-histone-H3. Previous studies in this model have shown that myocyte apoptosis returns to low values (~0.003%) during the time period in which experiments were performed in the present study (19). Thus, proliferation indexes were 1 to 2 orders of magnitude higher than myocyte apoptosis at this time, resulting in a net increase in myocyte number. These findings suggest that de novo myocyte formation with delayed maturation may contribute to delayed functional recovery. The observation that revascularization stimulates new myocyte formation is also reinforced by recent clinical observations. First, some patients with reversible dysynergic myocardium have regional wall thinning that reverses after revascularization (34). While originally thought to indicate irreversibility from infarction, some of these regions develop profound increases in wall thickness and systolic function after revascularization.
Revascularization might plausibly have stimulated endogenous myocyte proliferation. Secondly, D’Amario recently isolated cardiac stem cells (CSC) from biopsies obtained from patients undergoing coronary bypass surgery and demonstrated that CSC proliferation and function in vitro correlates with late functional improvement after revascularization (35). Increased telomere length and reduced population doubling time in c-Kit+ CSCs predicted increased wall thickness, increased LV mass, and an increase in LV ejection fraction 12 months after revascularization. Our results are compatible with these clinical observations, but a limitation relates to our inability to determine whether the new myocytes arose from the increase in c-Kit+ cardiac stem cells or from the proliferation of existing myocytes; experimental evidence is available to support (36) and to refute (37) cKit+ cells as a source of new myocytes. Elucidating the answer will require sophisticated approaches to study endogenous myocyte fate, which currently are largely confined to transgenic animal studies. Regardless of their source, our findings suggest that new myocyte formation and/or maturation may delay functional recovery after revascularization.
Methodological Limitations
Farm-bred pigs require 3 to 4 months to develop hibernating myocardium and, due to their size, could only be studied for 1 month after revascularization in our facility. While providing new insight into the factors responsible for persistent dysfunction, we cannot assess long-term functional recovery, proteomic remodeling, or myocyte proliferation. Thus, it is uncertain whether regional function would completely recover over a longer follow-up period and whether changes in protein expression would normalize. Mini-swine (31) may facilitate longer-term studies of coronary revascularization. Likewise, young healthy animals may have more plasticity in myocyte regeneration than humans with disease, which could also significantly impact the extent of functional recovery and remodeling.
Our results demonstrate that revascularization elicits a significant increase in myocyte number and reciprocal reduction in myocyte size. While consistent with new myocyte formation, it is unclear if these myocytes would continue to increase in size and ultimately develop into functionally mature cardiomyocytes over a longer follow-up period. In this case, the delayed time course of new myocyte maturation may contribute to the delayed recovery of contractility after PCI. However, if the myocytes remain small and immature, their long-term contribution to contractile function may be limited (38,39). Future studies with a longer follow-up period are necessary to definitively address this question.
Finally, proteomics involves the parallel analysis of multiple proteins on a 2-dimensional gel, which can lead to false discovery of differential protein expression via Type I errors (40). These can be minimized using large samples, as employed in our study, which reduces the impact of biological and experimental variability. Nearly all protein changes in nonrevascularized animals were previously reported in independent experiments using the same experimental approach in this model, and in an expanded protein analysis using label-free quantification with LC/MS (22).
Clinical Implications
Our study demonstrates that, even in the absence of infarction, the delayed improvement in LV function after revascularization of hibernating myocardium is associated with an incomplete reversal of the molecular phenotype and the stimulation of new cardiomyocytes originally lost via regional apoptosis from reversible ischemia. Thus, adjunctive treatments, such as intracoronary stem cells (21) and growth factors (19), may accelerate functional recovery following revascularization of high-risk patients with heart failure, as well as those in whom complete revascularization is not technically feasible. Studies evaluating hybrid strategies to treat viable dysfunctional myocardium will be needed to test these hypotheses.
Supplementary Material
Figure 9. Central Illustration. Revascularization of Hibernating Myocardium.

Delay in the functional recovery of hibernating myocardium is partly related to stimulating new myocyte formation with myocyte cellular hypertrophy replaced by small new myocytes (upper panels). Immunohistochemistry in the lower panel shows that revascularization increases proliferating myocytes (pHH3 and Ki67 stains), as well as cKit positive cardiac stem cells. pHH3= phosphorylated histone H3.
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE
In patients with ischemic cardiomyopathy, improvement in ventricular function after revascularization may be delayed as a consequence of chronic cellular protein adaptations to repetitive ischemia and immature cardiomyocyte formation, even in zones of viable myocardium.
TRANSLATIONAL OUTLOOK
Beyond alleviating ischemia through revascularization, the efficacy of hybrid interventions that concurrently accelerate endogenous myocyte formation to enhance functional recovery of hibernating myocardium warrants further study.
Acknowledgments
The authors thank Anne Coe, Deana Gretka, Elaine Granica, and Amy Johnson for technical assistance. This project was supported by NHLBI HL-55324 and HL-61610, and by the Albert and Elizabeth Rekate Fund.
Abbreviations
- LAD
left anterior descending artery
- LADΔWT
LAD wall thickening (end-systolic to end-diastolic wall thickness)
- LV
left ventricular
- MRI
magnetic resonance imaging
- PCI
percutaneous coronary intervention
- pHH3
phosphorylated histone H3
- TTC
triphenyltetrazolium chloride
- 2D-DIGE
2-dimensional differential in-gel electrophoresis
Footnotes
Disclosures: The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fallavollita JA, Perry BJ, Canty JM., Jr 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: Evidence for transmural variations in chronic hibernating myocardium. Circulation. 1997;95:1900–9. doi: 10.1161/01.cir.95.7.1900. [DOI] [PubMed] [Google Scholar]
- 2.Vanoverschelde JLJ, Wijns W, Depre C, et al. Mechanisms of chronic regional postischemic dysfunction in humans. New insights from the study of noninfarcted collateral-dependent myocardium. Circulation. 1993;87:1513–23. doi: 10.1161/01.cir.87.5.1513. [DOI] [PubMed] [Google Scholar]
- 3.Lim H, Fallavollita JA, Hard R, et al. Profound apoptosis-mediated regional myocyte loss and compensatory hypertrophy in pigs with hibernating myocardium. Circulation. 1999;100:2380–6. doi: 10.1161/01.cir.100.23.2380. [DOI] [PubMed] [Google Scholar]
- 4.Fallavollita JA, Malm BJ, Canty JM., Jr Hibernating myocardium retains metabolic and contractile reserve despite regional reductions in flow, function, and oxygen consumption at rest. Circ Res. 2003;92:48–55. doi: 10.1161/01.res.0000049104.57549.03. [DOI] [PubMed] [Google Scholar]
- 5.Page B, Young R, Iyer V, et al. Persistent regional downregulation in mitochondrial enzymes and upregulation of stress proteins in swine with chronic hibernating myocardium. Circ Res. 2008;102:103–12. doi: 10.1161/CIRCRESAHA.107.155895. [DOI] [PubMed] [Google Scholar]
- 6.Dilsizian V, Bonow RO. Current diagnostic techniques of assessing myocardial viability in patients with hibernating and stunned myocardium. Circulation. 1993;87:1–20. doi: 10.1161/01.cir.87.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Haas F, Augustin N, Holper K, et al. Time course and extent of improvement of dysfunctioning myocardium in patients with coronary artery disease and severely depressed left ventricular function after revascularization: correlation with positron emission tomographic findings. J Am Coll Cardiol. 2000;36:1927–34. doi: 10.1016/s0735-1097(00)00968-2. [DOI] [PubMed] [Google Scholar]
- 8.Kim RJ, Wu E, Rafael A, et al. The use of contrast-enhanced magnetic resonance imaging to identify reversible myocardial dysfunction. N Engl J Med. 2000;343:1445–53. doi: 10.1056/NEJM200011163432003. [DOI] [PubMed] [Google Scholar]
- 9.Topol EJ, Weiss JL, Guzman PA, et al. Immediate improvement of dysfunctional myocardial segments after coronary revascularization: Detection by intraoperative transesophageal echocardiography. J Am Coll Cardiol. 1984;4:1123–34. doi: 10.1016/s0735-1097(84)80131-x. [DOI] [PubMed] [Google Scholar]
- 10.Ceconi C, La Canna G, Alfieri O, et al. Revascularization of hibernating myocardium: rate of metabolic and functional recovery and occurrence of oxidative stress. Eur Heart J. 2002;23:1877–85. doi: 10.1053/euhj.2002.3364. [DOI] [PubMed] [Google Scholar]
- 11.Shivalkar B, Maes A, Borgers M, et al. Only hibernating myocardium invariably shows early recovery after coronary revascularization. Circulation. 1996;94:308–15. doi: 10.1161/01.cir.94.3.308. [DOI] [PubMed] [Google Scholar]
- 12.Afridi I, Qureshi U, Kopelen HA, et al. Serial changes in response of hibernating myocardium to inotropic stimulation after revascularization: a dobutamine echocardiographic study. J Am Coll Cardiol. 1997;30:1233–40. doi: 10.1016/s0735-1097(97)00308-2. [DOI] [PubMed] [Google Scholar]
- 13.Bax JJ, Visser FC, Poldermans D, et al. Time course of functional recovery of stunned and hibernating segments after surgical revascularization. Circulation. 2001;104:I314–8. doi: 10.1161/hc37t1.094853. [DOI] [PubMed] [Google Scholar]
- 14.Haas F, Jennen L, Heinzmann U, et al. Ischemically compromised myocardium displays different time-courses of functional recovery: correlation with morphological alterations? Eur J Cardiothorac Surg. 2001;20:290–8. doi: 10.1016/s1010-7940(01)00781-3. [DOI] [PubMed] [Google Scholar]
- 15.Bondarenko O, Beek AM, Twisk JWR, et al. Time course of functional recovery after revascularization of hibernating myocardium: a contrast-enhanced cardiovascular magnetic resonance study. Eur Heart J. 2008;29:2000–5. doi: 10.1093/eurheartj/ehn266. [DOI] [PubMed] [Google Scholar]
- 16.Heusch G, Schulz R, Rahimtoola SH. Myocardial hibernation: a delicate balance. Am J Physiol Heart Circ Physiol. 2005;288:H984–99. doi: 10.1152/ajpheart.01109.2004. [DOI] [PubMed] [Google Scholar]
- 17.Page BJ, Young RF, Suzuki G, et al. The physiological significance of a coronary stenosis differentially affects contractility and mitochondrial function in viable chronically dysfunctional myocardium. Basic Res Cardiol. 2013;108:354. doi: 10.1007/s00395-013-0354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shivalkar B, Flameng W, Szilard M, et al. Repeated stunning precedes myocardial hibernation in progressive multiple coronary artery obstruction. J Am Coll Cardiol. 1999;34:2126–36. doi: 10.1016/s0735-1097(99)00467-2. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki G, Lee TC, Fallavollita JA, et al. Adenoviral gene transfer of FGF-5 to hibernating myocardium improves function and stimulates myocytes to hypertrophy and reenter the cell cycle. Circ Res. 2005;96:767–75. doi: 10.1161/01.RES.0000162099.01268.d1. [DOI] [PubMed] [Google Scholar]
- 20.Warnes Gregory R, Bolker Ben, Bonebakker Lodewijk, et al. [Accessed December 8, 2014.];g-plots: Various R programming tools for plotting data v2.15.0. 2014 Dec; Available at: http://cran.r-project.org/web/packages/gplots/index.html.
- 21.Suzuki G, Iyer V, Lee TC, et al. Autologous mesenchymal stem cells mobilize cKit+ and CD133+ bone marrow progenitor cells and improve regional function in hibernating myocardium. Circ Res. 2011;109:1044–54. doi: 10.1161/CIRCRESAHA.111.245969. [DOI] [PubMed] [Google Scholar]
- 22.Qu J, Young R, Page BJ, et al. A reproducible ion current-based approach for 24-plex comparison of the tissue proteomes of hibernating versus normal myocardium in swine models. J Proteome Res. 2014;13:2571–84. doi: 10.1021/pr5000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canty JM, Jr, Suzuki G. Myocardial perfusion and contraction in acute ischemia and chronic ischemic heart disease. J Mol Cell Cardiol. 2012;52:822–31. doi: 10.1016/j.yjmcc.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fallavollita JA, Lim H, Canty JM., Jr Myocyte apoptosis and reduced SR gene expression precede the transition from chronically stunned to hibernating myocardium. J Mol Cell Cardiol. 2001;33:1937–44. doi: 10.1006/jmcc.2001.1457. [DOI] [PubMed] [Google Scholar]
- 25.Kelly RF, Cabrera JA, Ziemba EA, et al. Continued depression of maximal oxygen consumption and mitochondrial proteomic expression despite successful coronary artery bypass grafting in a swine model of hibernation. J Thorac Cardiovasc Surg. 2011;141:261–8. doi: 10.1016/j.jtcvs.2010.08.061. [DOI] [PubMed] [Google Scholar]
- 26.Verheyen F, Racz R, Borgers M, et al. Chronic hibernating myocardium in sheep can occur without degenerating events and is reversed after revascularization. Cardiovasc Pathol. 2014;23:160–8. doi: 10.1016/j.carpath.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 27.McFalls EO, Sluiter W, Schoonderwoerd K, et al. Mitochondrial adaptations within chronically ischemic swine myocardium. J Mol Cell Cardiol. 2006;41:980–8. doi: 10.1016/j.yjmcc.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Dewald O, Frangogiannis NG, Zoerlein M, et al. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci U S A. 2003;100:2700–5. doi: 10.1073/pnas.0438035100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu Q, Suzuki G, Young RF, et al. Reductions in mitochondrial O2 consumption and preservation of high-energy phosphate levels after simulated ischemia in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol. 2009;297:H223–32. doi: 10.1152/ajpheart.00992.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McFalls EO, Kelly RF, Hu Q, et al. The energetic state within hibernating myocardium is normal during dobutamine despite inhibition of ATP-dependent potassium channel opening with glibenclamide. Am J Physiol Heart Circ Physiol. 2007;293:H2945–51. doi: 10.1152/ajpheart.00012.2007. [DOI] [PubMed] [Google Scholar]
- 31.Jameel MN, Li Q, Mansoor A, et al. Long-term preservation of myocardial energetic in chronic hibernating myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H836–44. doi: 10.1152/ajpheart.00540.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer V, Canty JM., Jr Regional desensitization of beta-adrenergic receptor signaling in swine with chronic hibernating myocardium. Circ Res. 2005;97:789–95. doi: 10.1161/01.RES.0000184675.80217.9e. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez SF, Ovchinnikov V, Canty JM, Jr, et al. Hibernating myocardium results in partial sympathetic denervation and nerve sprouting. Am J Physiol Heart Circ Physiol. 2013;304:H318–27. doi: 10.1152/ajpheart.00810.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah DJ, Kim HW, James O, et al. Prevalence of regional myocardial thinning and relationship with myocardial scarring in patients with coronary artery disease. JAMA. 2013;309:909–18. doi: 10.1001/jama.2013.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D’Amario D, Leone AM, Iaconelli A, et al. Growth properties of cardiac stem cells are a novel biomarker of patients’ outcome after coronary bypass surgery. Circulation. 2014;129:157–72. doi: 10.1161/CIRCULATIONAHA.113.006591. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 37.van Berlo JH, Kanisicak O, Maillet M, et al. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–41. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Wilson RM, Kubo H, et al. Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ Res. 2007;100:536–44. doi: 10.1161/01.RES.0000259560.39234.99. [DOI] [PubMed] [Google Scholar]
- 39.Dawn B, Guo Y, Rezazadeh A, et al. Postinfarct cytokine therapy regenerates cardiac tissue and improves left ventricular function. Circ Res. 2006;98:1098–105. doi: 10.1161/01.RES.0000218454.76784.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen X, Young R, Canty JM, et al. Quantitative proteomics in cardiovascular research: global and targeted strategies. Proteomics Clin Appl. 2014;8:488–505. doi: 10.1002/prca.201400014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.