

Abstract
The long-term consequences of traumatic brain injury (TBI) are closely associated with the development of histopathological deficits. Notably, TBI may predispose long-term survivors to age-related neurodegenerative diseases, such as Parkinson's disease (PD), which is characterized by a gradual degeneration of the nigrostriatal dopaminergic neurons. However, preclinical studies on the pathophysiological changes in substantia nigra (SN) after chronic TBI are lacking. In the present in vivo study, we examined the pathological link between PD-associated dopaminergic neuronal loss and chronic TBI. Sixty days post-TBI, rats were euthanized and brain tissues harvested. Immunostaining was performed using tyrosine hydroxylase (TH), an enzyme required for the synthesis of dopamine in neurons, α-synuclein, a presynaptic protein that plays a role in synaptic vesicle recycling, and major histocompatibility complex II (MHCII), a protein found in antigen presenting cells such as inflammatory microglia cells, all key players in PD pathology. Unbiased stereology analyses revealed significant decrease of TH-positive expression in the surviving dopaminergic neurons of the SN pars compacta (SNpc) relative to sham control. In parallel, increased α-synuclein accumulation was detected in the ipsilateral SN compared to the contralateral SN in TBI animals or sham control. In addition, exacerbation of MHCII+ cells was recognized in the SN and cerebral peduncle ipsilateral to injury relative to contralateral side and sham control. These results suggest α-synuclein as a pathological link between chronic effects of TBI and PD symptoms as evidenced by significant overexpression and abnormal accumulation of α-synuclein in inflammation-infiltrated SN of rats exposed to chronic TBI. J. Cell. Physiol. 230: 1024–1032, 2015. © 2014 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
Traumatic brain injury (TBI), defined as a jolt to the head resulting in brain damage, accounts for approximately 52,000 deaths a year in the United States, and an estimated 1.7 million people including military personnel and athletes suffer from TBI annually (Fabrizio and Keltner, 2010; Faul et al., 2010). TBI is now recognized as a disease process than an event. It is often accompanied by primary necrotic injury during the acute and sub-acute state, and secondary injury during the chronic state; the latter has been associated with apoptosis, oxidative stress, neuro-inflammation, abnormal protein aggregation, edema, and progressive neuronal cell death (Werner and Engelhard, 2007; Yu et al., 2009 Glover et al., 2012).
The chronic consequences of TBI are closely associated with the development of chronic neuro-inflammation, sensory-motor, and cognitive problems years after the initial insult (Ettenhofer and Abeles, 2009; Ozen and Fernandes, 2012; Acosta et al., 2013). We and others have shown that chronic TBI causes secondary injuries which are closely associated with the development of neurodegenerative disorders such as dementia pugilistica (DP), Alzheimer's disease (AD) and Parkinson's disease (PD) among other pathologies (Saing et al., 2012; Acosta et al., 2013; Xiong et al., 2013). Furthermore, clinical studies have shown that previously concussed athletes, boxers, and ex-military personnel are at a higher risk of developing idiopathic Parkinsonism or PD- like syndrome, of which symptoms develop several years after the primary injury of TBI (Prins et al., 2013; Kokjohn et al., 2012; Karantzoulis and Randolph, 2013; Smith, 2013; Smith et al., 2013; Chauhan, 2014; Levin and Bhardwaj, 2014). Among the many pathological mechanisms implicated in PD and/or PD-like pathology, two major hallmarks have been recognized, namely, progressive degeneration of dopaminergic neurons, and the overexpression and inclusion of α-synuclein as Lewy bodies in the substantia nigra (SN) of the brain (Wan and Chung, 2012; Ulusoy and Di Monte, 2013). Dopaminergic neuronal degeneration causes remarkable decreases in tyrosine hydroxylase (TH) expression, the rate-limiting enzyme for dopamine, and consequently lowering the nigrostriatal dopamine levels and leading to neurotransmission impairments and eventually to PD symptoms (Chaudhuri et al., 2006; Jankovic, 2008). α-Synuclein is a presynaptic protein that is activated through a number of processes such as phosphorylation and plays a role in synaptic vesicle recycling. Regardless of the etiology of PD, all genetic and cellular modifications eventually lead to overexpression of total α-synuclein whereby accumulation of α-synuclein has been demonstrated to cause neuronal damage (Goedert, 2001; Wakabayashi et al., 2007; Klein and Westenberger, 2012; Shahaduzzaman et al., 2013; Rodrigues et al., 2014)
The presence of aberrant overexpression of α-synuclein can disrupt the cell membrane of dopaminergic neurons (Conway et al., 2000b,2000a). However, evidence of its neurotoxicity in an in vivo model of chronic brain injury is still limited. Here, we provided evidence that α-synuclein may serve as a pathological link between chronic effects of TBI and PD symptoms as evidenced by significant overexpression and abnormal accumulation of α-synuclein in the SN of rats exposed to chronic TBI.
Methods
Subjects
Experimental procedures were approved by the University of South Florida Institutional Animal Care and Use Committee (IACUC). All male rats were housed under normal conditions (20 °C, 50% relative humidity, and a 12-h light/dark cycle). The light/dark cycle was not reversed. Necessary precautions were taken to reduce pain and suffering of animals throughout the study. Analgesic agents were administered to the animal during the surgery and/or once a day for the next 3 days after surgery to relieve pain and distress. Animals were closely checked daily throughout the 2 months study period, with daily weights recorded for the first week post-surgery, then twice weekly thereafter throughout this 2 months study. All studies were performed by personnel blinded to the treatment condition.
Surgical procedures
Ten-week old Sprague–Dawley rats (n = 12) were exposed to either sham or to TBI surgery. TBI surgery was done using a controlled cortical impactor (CCI; Pittsburgh Precision Instruments, Inc, Pittsburgh, PA). Deep anesthesia was achieved using 1–2% isoflurane in nitrous oxide/oxygen (69/30%) delivered via a nose mask. All animals were secured in a stereotaxic frame (David Kopf Instruments, Tujunga, CA). TBI injury surgeries consisted of animals subjected to scalp incision to expose the skull, and craniectomy. An electric drill was used to perform the craniectomy of about 2.5 mm radius with coordinates calculated from +0.2 anterior and −0.2 mm lateral right from bregma (Paxinos and Watson, 2005). Subsequently, the brain was impacted at the fronto-parietal cortex with a velocity of 6.0 m/s reaching a depth of 1.0 mm below the dura matter layer and the impactor remained in the brain for 150 msec. The impactor rod was angled 15° degrees vertically to maintain a perpendicular position in reference to the tangential plane of the brain curvature at the impact surface. A linear variable displacement transducer (Macrosensors, Pennsauken, NJ), which was connected to the impactor, measured the velocity and duration to verify consistency. The analgesic Ketoprofen (Ketofen 100 mg/m, Fort Dodge Animal Health) was administered subcutaneously once a day for 3 days post-operatively to minimize pain and discomfort at a dose of 5 mg/kg, and at a concentration of 1.25 mg/0.1 ml per rat weighting approximately 250 grams. Sham surgeries consisted of animals subjected to scalp incision and craniectomy only. Of note, the animals did not develop overt behavioral abnormalities, although based on our experience with this TBI model, when these injured animals are challenged with sensitive motor and cognitive tests, they exhibit task-dependent deficits. For the present study, however, our focus is on the histopathology thus we did not subject the animals to behavioral tests.
Histology
Under deep anesthesia, the rats were sacrificed at 8 weeks after TBI surgery. Approximately 200 ml of ice cold phosphate buffer saline (PBS) followed by 200 ml of 4% paraformaldehyde in PBS were used for brain perfusion through the ascending aorta. After perfusion, brains were removed and post-fixed in the same fixative for 24 h and stored at 4 °C, next brains transferred to 30% sucrose in phosphate buffer (PB) and stored at 4 °C for 1 week. Thereafter, they were frozen at −24 °C, mounted onto a 40 mm specimen disk using embedding medium. Coronal sectioning was carried out at a thickness of 40 μm by cryostat.
Immunohistochemistry of substantia nigra
Staining for MHCII activated microglia cells (via OX6), tyrosine hydroxylase (TH), and total α-synuclein was conducted on every 6th coronal section throughout the entire SN (coordinates −4.36 to −6.4 anterior posterior) (Paxinos and Watson, 2005). In all animals, sections were anatomically matched. Series of eight sections per rat were processed for staining. For TH, and α-synuclein stainings; normal goat serum was used, and for OX6 horse serum was used. Eight free-floating coronal sections (40 μm) were washed three times in 0.1 M phosphate-buffered saline (PBS) to clean the section from the cryoprotectant. Afterwards, all sections were incubated in 0.3% hydrogen peroxide (H2O2) solution for 20 min and washed three times with PBS for 10 min each wash. Next, all sections were incubated in blocking solution for 1 h using PBS supplemented with 3% normal serum and 0.2% Triton X-100. Sections were then incubated overnight at 4 °C with rabbit anti-rat TH (1:100 tyrosine hydroxylase; Millipore; AB152) or mouse anti-rat α-synuclein (C terminal amino acids 111–131 of α-synuclein; 1:300 abcam; AB15530) (anti α-synuclein antibody LB 509; Abcam; AB27766; immunoreactivity to epitodes in amino acids 115–122 of alpha synuclein) or goat anti-OX6 (major histocompatibility complex or MHC class II; 1:750 BD), antibody markers in PBS supplemented with 3% normal serum and 0.1% triton X-100. Sections were then washed three times with PBS and incubated in biotinylated goat anti-rabbit secondary antibody (1:300; Vector Laboratories, Burlingame, CA) for the TH staining, and goat anti-mouse secondary antibody (1:200; Vector Laboratories, Burlingame, CA) for α-synuclein staining and horse anti-goat secondary antibody (1:200; Vector Laboratories Burlingame, CA) for MHCII (activated microglia) staining in PBS supplemented with normal serum, and 0.1% Triton X-100 for 1 h. Next, the sections were incubated for 60 min in avidin–biotin substrate (ABC kit, Vector Laboratories, Burlingame, CA) and washed three times with PBS for 10 min each wash. All sections were then incubated for 1 min in 3, 30-diaminobenzidine (DAB) solution (Vector Laboratories) and washed three times with PBS for 10 min each wash. Sections were mounted onto glass slides, set to dry for 24 h and dehydrated in ascending ethanol concentration (70%, 95%, and 100%) for 2 min each and one time in xylenes for 2 min of clarification, then cover-slipped using mounting medium.
Immunohistochemistry of cerebral peduncle
Staining for MHCII activated microglia cells (via OX6) was conducted on every 6th coronal section throughout the entire posterior cerebral peduncle (coordinates −5.52 to −6.4 anterior posterior) (Paxinos and Watson, 2005). In all animals, sections were anatomically matched. Series of six sections per rat were processed for MHCII staining. Six free-floating coronal sections (40 μm) were washed three times in 0.1 M phosphate-buffered saline (PBS) to clean the section from the cryoprotectant. Afterwards, all sections were incubated in 2% hydrogen peroxide (H2O2) solution for 20 min and washed three times with PBS for 10 min each wash. Next, all sections were incubated in blocking solution for 1 h using PBS supplemented with 5% normal serum and 0.1% Triton X-100. Sections were then incubated overnight at 4 °C with goat anti-OX6 (major histocompatibility complex or MHC class II; 1:750 BD), antibody markers in PBS supplemented with 3% normal serum and 0.1% triton X-100. Sections were then washed three times with PBS and incubated in biotinylated horse anti-goat secondary antibody (1:200; Vector Laboratories, Burlingame, CA) in PBS supplemented with normal horse serum, and 0.1% Triton X-100 for 1 h. Next, the sections were incubated for 60 min in avidin–biotin substrate (ABC kit, Vector Laboratories, Burlingame, CA) and washed three times with PBS for 10 min each wash. All sections were then incubated for 1 min in 3, 30-diaminobenzidine (DAB) with methal enhancer (Vector Laboratories) and washed three times with PBS for 10 min each wash. Sections were then mounted onto glass slides, dehydrated in ascending ethanol concentration (70%, 95%, and 100%) for 2 min each and 2 min in xylenes, then cover-slipped using mounting medium.
Immunofluorescent staining
Immunofluorescent staining for tyrosine hydroxylase (TH) and total α-synuclein was conducted on every 6th coronal section throughout the entire SN (coordinates −4.36 to −6.4 anterior posterior) (Paxinos and Watson, 2005). In all animals, sections were anatomically matched. Series of eight sections per rat were processed for staining. For TH, and α-synuclein stainings; normal goat serum was used. Eight free-floating coronal sections (40 μm) were washed three times in 0.1 M phosphate-buffered saline (PBS) to clean the section from the cryoprotectant. Afterwards, all sections were incubated in 2% hydrogen peroxide (H2O2) solution for 20 min and washed three times with PBS for 10 min each wash. Next, all sections were incubated in blocking solution for 1 h using PBS supplemented with 3% normal goat serum and 0.1% Triton X-100. Sections were then incubated overnight at 4 °C with rabbit anti-rat TH (1:100 tyrosine hydroxylase; Millipore; AB152) or mouse anti-rat α-synuclein (anti α-synuclein antibody LB 509; Abcam; AB27766; immunoreactivity to epitodes in amino acids 115–122 of alpha synuclein), antibody markers in PBS supplemented with 5% normal serum and 0.1% triton X-100. Sections were washed three times for 10 min in PBS and then incubated in PBS supplemented with 3% normal goat serum and 0.1% Triton X-100 containing corresponding secondary antibodies, goat anti-rabbit IgG-Alexa 488 (green) (1:500; Invitrogen) and goat anti-mouse igG-Alexa 594 (red) (1:750; Invitrogen), for 90 min. Finally, sections were washed five times for 10 min in PBS, then processed for Hoechst (1:200; bisBenzimideH 33258 trihydrochloride, Sigma) for 30 min, washed in PBS three times for 10 min each wash, and cover-slipped with Fluoromount (Sigma; F4680). Sections were analyzed in independent channels with an Olympus FV1000 laser scanning confocal microscope equipped with Fluoview SV1000 imaging software. SN sections were examined in independent channels with an Olympus FV1000 laser scanning confocal microscope equipped with Fluoview SV1000 imaging software. Control studies included exclusion of primary antibodies substituted with 3% normal goat serum in PBS. No immunoreactivity was observed in these controls.
The characterization of α-synuclein within the SNpc was done on both DAB and fluorescent staining. We used DAB staining couple with unbiased stereology (optical fractionator probe) to count every cell expressing α-synuclein within the SNpc of both ipsilateral and contralateral side. Subsequently, we analyzed the density per mm2 of α-synuclein expression within neurons and along neurites couple with its colocalization with TH positive cells using immunofluorescent techniques as seen in the Results section. Density per mm2 was quantified following the morphometric method explained elsewhere (Amstrong, 2003; Amstrong et al., 2014).
Stereological analysis
Unbiased stereology was performed on brain sections immunostained with TH, α-synuclein, and OX6. Sets of 1/6 section, ∼8 systematically random sections, of about 240 μm apart, were taken from the brain spanning AP – 4.36 mm to AP – 6.4 mm in all 12 rats. Of note, section thicknesses were confirmed as being between 20.0–21.0 μm after dehydration, and this did not statistically differ between groups. Expression of TH, and of α-synuclein in dopaminergic neurons of the substantia nigra pars compacta (SNpc) were visualized by staining with TH and α-synuclein, respectively. Positive stains were analyzed with a Nikon Eclipse 600 microscope and quantified using Stereo Investigator software, version 10 (MicroBrightField, Colchester, VT). The estimated number of TH-positive cells and of α-synuclein was examined using the optical fractionator estimator probe of the unbiased stereological cell technique revealing the total estimated number of TH in the SNpc. TH-positive cells and α-synuclein were counted within the SNpc, in both hemispheres (ipsilateral and contralateral), using the optical fractionator probe of unbiased stereological cell counting technique. MHCII (OX6) positive cells were examined using the Cavalieri estimator probe of the unbiased stereological cell technique (Mayhew, 1991) revealing the volume of MHCII+ in the SN and cerebral peduncle (CP) in both hemispheres (ipsilateral and contralateral). All samplings were optimized to count at least 300 cells per animal with error coefficients less than 0.07. The Cavalieri estimator was executed using a point grid spaced equally both across and down directions. The grid space used was 100 μm in order to cover the entire SN which represented our region of interest (ROI) (Mayhew et al., 1991). Each counting frame (140 × 140 μm2 for TH, and α-synuclein) was placed at an intersection of the lines forming a virtual grid (175 × 175 μm2), which was randomly generated and placed by the software within the outlined structure.
Morphometric method
In the ipsilateral SNpc, the density of α-synuclein in the dopaminergic neurons was measured. A rectangular contour was used and the location of random plots within the SNpc was determined by a counting sample frame of 250 × 50 μm2 (Amstrong, 2003; Amstrong et al., 2014) in order to minimize bias. A total of six counting plots were generated randomly covering the rectangular area per section and the number of α-synuclein positive cells per unit area (density) was quantified throughout the entire ipsilateral SNpc (Amstrong, 2003; Amstrong et al., 2014).
Statistical analysis
For data analyses, contralateral and ipsilateral matching brain regions were used providing two sets of raw data per treatment condition (TBI vs. sham control). Mean estimated numbers of TH, α-synuclein, and OX6-positive cells were used for data analyses using one-way analysis of variance (ANOVA) for group comparisons, followed by pairwise comparisons using post hoc Bonferonni test. Pearson's correlation analysis was performed to identify the correlation between α-synuclein and TH expressions. All data are presented as the mean values ± SEM. Statistical significance was set at P < 0.05 for all analyses.
Results
We investigated whether chronic TBI was associated with dopaminergic neuronal loss and mediated by α-synuclein overexpression. Immunohistochemical data revealed downregulation of TH-positive cells accompanied by increased accumulation of α-synuclein in the SNpc of TBI animals in the long-term (i.e., 60 days post-TBI). The reduction in TH-positive cells inversely correlated with upregulation of α-synuclein-positive cells in the SNpc of TBI animals. Moreover, an elevated inflammatory response, as evidence by infiltration of MHCII+ activated cells, occurred in tandem with the depletion of TH-positive cells and the surge of α-synuclein-positive cells in the SNpc, which extended also to the CP of TBI animals.
Downregulation of TH-positive cells in the SNpc of chronic TBI rats
The estimated number of TH-positive dopaminergic neurons at 60 days post-TBI was quantified using stereological techniques. An ANOVA revealed a significant effect of TBI on the estimated number of TH-positive dopaminergic neurons in the SNpc of rats subjected to chronic TBI (SNpc, F3,20 = 11.79; P < 0.0001). Post hoc Bonferroni's analysis revealed that significantly fewer TH-positive cells were found in the SNpc ipsilateral to TBI compared to the contralateral side (P < 0.05) or sham injury side (P's < 0.05). (Figs. 1A–D).
Figure 1.
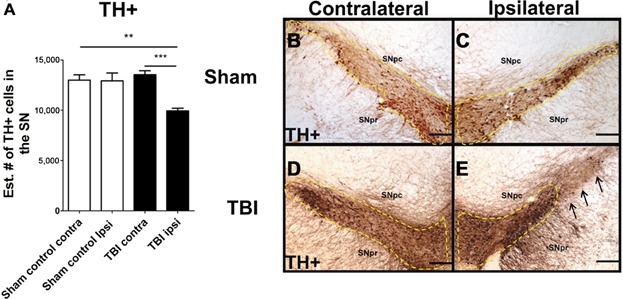
Downregulation of TH-positive dopaminergic neurons in the SNpc in chronic TBI. Arrows indicate downregulation of TH-positive dopaminergic neurons in TBI ipsilateral SNpc. Quantification of TH immunostaining reflects mean estimated number of TH-positive dopaminergic cells in the SNpc (A). SNpc, F3,20 = 11.79; P < 0.0001. Photomicrographs correspond to representative SN in coronal sections immunostained with TH antibody. (B) Sham contralateral SNpc, (C) Sham ipsilateral SNpc, (D) TBI contralateral SNpc, (E) TBI ipsilateral SNpc. Scale bar = 100 μm. Significance at P's < 0.05.
Overexpression of α-synuclein-positive cells in the SNpc of chronic TBI rats
The estimated number of α-synuclein-positive dopaminergic neurons at 60 days post-TBI was quantified using stereological techniques. An ANOVA revealed a significant effect of TBI on the estimated number of α-synuclein-positive dopaminergic neurons in the SNpc of rats subjected to chronic TBI (F3,20 = 17.32; P < 0.0001). Post hoc comparisons revealed that significantly more α-synuclein-positive cells were found in the SNpc ipsilateral to TBI relative to contralateral side (P < 0.05) or sham injury side (P's < 0.05) (Figs. 2A–D), at 60 days after the initial TBI.
Figure 2.

Overexpression of α-synuclein-positive cells in the SNpc in chronic TBI rats. Upregulation of α-synuclein-positive cells in the ipsilateral side of SNpc of chronic TBI rats relative to contralateral side and sham control. Arrows indicate positive expression of α-synuclein-positive cells in TBI ipsilateral SNpc. Quantification of α-synuclein immunostaining reflects mean estimated number of α-synuclein-positive cells in the SNpc (A). SNpc, F3,20 = 17.32; P < 0.0001. Photomicrographs correspond to representative SN in coronal sections immunostained with α-synuclein antibody. (B) Sham contralateral SNpc, (C) Sham ipsilateral SNpc, (D) magnification of sham ipsilateral SNpc, (E) TBI contralateral SNpc, (F) TBI ipsilateral SNpc, (G) magnification of TBI ipsilateral SNpc. Arrows indicate expression of α-synuclein in cells in all groups within the SNpc (Figs. 2B–E). Scale bar for B, C, E, F = 500 μm and D, G =0.1 μm. Significance at P's < 0.05.
The density of α-synuclein in dopaminergic neurons was quantified in the contralateral and ipsilateral side of the entire SNpc of sham control and chronic TBI rats (Fig. 3). An ANOVA revealed a significant effect of TBI on the density of α-synuclein in the ipsilateral SNpc of rats subjected to chronic TBI (SNpc α-synuclein density, F3,20 = 15.37; P < 0.0001 (Figs. 3A and B). Pairwise comparison revealed that chronic TBI resulted in a significant upregulation of α-synuclein in dopaminergic neurons (P < 0.05) in the ipsilateral SNpc compared to contralateral side or either side sham injury side (P's < 0.05). α-Synuclein expression in dopaminergic neurons was found to be abundant and scattered around the entire SN compared to the contralateral side and sham control (Fig. 3B-I). It was found that there was a significant threefold expression of α-synuclein scattered around the soma and neurites of TH-positive and negative dopaminergic neurons ipsilateral to injury relative to the contralateral side or either sham injury side. Fluorescent confocal photomicrographs of ipsilateral SNpc of chronic TBI and sham controls demonstrated significant positive expression of α-synuclein in dopaminergic cells ipsilateral to injury in the chronic TBI brain 60 days post-TBI (Fig. 3B–I). Positive α-synuclein staining was found to be adjacent to the nucleus in the cytoplasm and along neurites of dopaminergic neurons in the SNpc (Fig. 3E–I).
Figure 3.

Quantification of α-synuclein density in dopaminergic neurons in the SNpc of chronic TBI rat. Results revealed upregulation of α-synuclein as measured by α-synuclein density in TH-positive dopaminergic neurons in the ipsilateral side of SNpc of chronic TBI rats relative to contralateral side and sham control. (A) Arrows denote positive expression of α-synuclein detected in the cytoplasm of the soma and neurites. Quantification of total α-synuclein immunostaining reflects the density of α-synuclein-positive expression in the cytoplasm and neurites in the SNpc (A). F3,20 = 15.37; P < 0.0001. Confocal photomicrographs of positive expression of TH (green) (B–F), α-synuclein (red) (C–G), and Hoechst (blue) (D–H) within the SNpc dopaminergic neurons of sham control rats and TBI rats at 60 days post-TBI. (E) Colocalization of TH, α-synuclein, and Hoechst shows minimum expression of α-synuclein in dopaminergic neurons of the SNpc contralateral side and sham control rats. (I) Colocalization of TH, α-synuclein and Hoechst shows positive expression of α-synuclein in the soma of dopaminergic neurons (yellow arrows) and along dendrites and axonal projections (white arrows) of the ipsilateral SNpc of 60 days post-TBI. Scale bar: 50 μm. Significance at P's < 0.05.
Correlation between α-synuclein-positive and TH-positive dopaminergic neurons in the SNpc
For each rat, the number of α-synuclein-positive neurons and the number of TH-positive neurons were plotted. Pearson's correlation analysis revealed negative correlation between the number of α-synuclein-expressing neurons and the number of TH-positive neurons (Pearson r = −0.9115, R2 = −0.8381, P < 0.01). These results indicate that overexpression of α-synuclein negatively affects TH-positive neuron expression in dopaminergic neurons of the SNpc (Fig. 4A).
Figure 4.
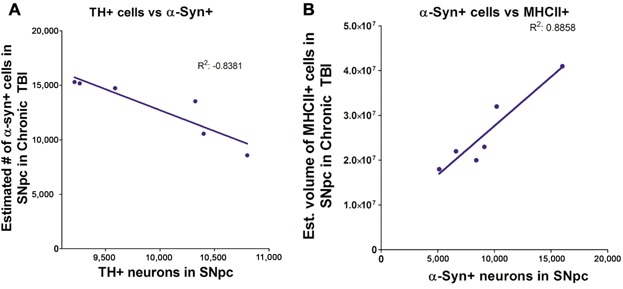
Correlation of TH-positive expression vs. α-synuclein overexpression and MHCII+ cells vs. α-synuclein. Mean estimated number of α-synuclein expressing neurons negatively correlates (A) with the number of TH-positive neurons in the SNpc ipsilateral to TBI (Pearson r = −0.9115, R2 = −0.8381, P < 0.01). Mean estimated number of α-synuclein expressing neurons positively correlates (B) with the volume of MHCII+ cells in the SNpc ipsilateral to TBI (Pearson r = 0.9412, R2 = 0.8858, P < 0.01). Significance at P's < 0.05.
Correlation between α-synuclein-positive and MHCII+ cells in the SNpc
For each rat, the number of α-synuclein-positive neurons and the volume of MHCII activated cells were plotted. Pearson's correlation analysis revealed positive correlation between the number of α-synuclein-expressing neurons and the volume of MHCII activated cells (Pearson r = 0.9412, R2 = 0.8858, P < 0.01). These results indicate that overexpression of α-synuclein was associated with increased migration of MHCII+ cells in the ipsilateral SNpc (Fig. 4B).
Upregulation of MHCll-positive activated microglia cells in SN and CP in chronic TBI
The volume of MHCII-positive cells was examined in the contralateral and ipsilateral side of the entire SN and CP of sham control and chronic TBI rats (Fig. 5). An ANOVA revealed a significant robust effect of TBI on the volume of MCHII-positive cells within the SN and CP of rats subjected to chronic TBI (SN, F3,2 = 7.951; P < 0.001; CP, F3,2 = 16.18; P < 0.0001). For the SN area, post hoc Bonferroni's analysis revealed that chronic TBI resulted in a significant increase in the volume of MHCII-positive cells ipsilateral to TBI compared to the contralateral side (P's < 0.05) or either sham injury side (P's < 0.05) (Figs. 5A and C). For the CP area, post hoc Bonferroni's analysis revealed that chronic TBI also resulted in a significant elevation in the volume of MHCII-positive cells ipsilateral to TBI compared to the contralateral side (P < 0.05) or either sham injury side (P's < 0.05) (Figs. 5B and C). All data are represented as mean values ± SEM.
Figure 5.
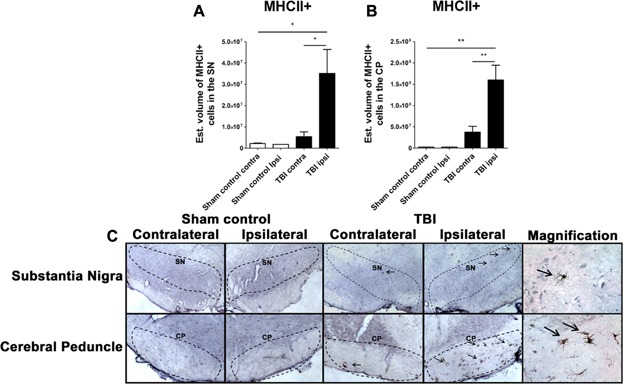
Upregulation of MHCII+ microgia cells in the SN and CP in chronic TBI rats. Quantification of MHCII immunostaining reflects mean estimated volume of MHCII+ activated microglia cells in the SN and CP in the contralateral and ipsilateral side (A, B). SN, F3,2 = 7.951; P < 0.001; CP, F3,2 = 16.18; P < 0.0001. Arrows indicate MHCII+ activated microglia cells ipsilateral SN and CP from TBI injury. Photomicrographs correspond to representative SN in coronal sections immunostained with MHCII/OX6 antibody. (C) Top part left: SN, sham control (contralateral, ipsilateral, and magnification); top part right: SN, TBI (contralateral, ipsilateral, and magnification); bottom part left: CP, sham control (contralateral, ipsilateral, and magnification); bottom part right: CP, TBI (contralateral, ipsilateral, and magnification); scale bar = 100 μm, magnification = 1 μm. Significance at P's < 0.05.
Discussion
In the present study, we found significant downregulation of TH-expressing dopaminergic neurons, a threefold increase in expression of α-synuclein in the ipsilateral SNpc coupled with robust upregulation of MHCII+ inflammatory cells in the ipsilateral SN and CP of rats subjected to chronic TBI. Moreover, a close examination revealed the presence of overexpressed α-synuclein adjacent to the nucleus in the cytoplasm and along neurites of dopaminergic neurons of the SNpc ipsilateral to injury in TBI rats. Furthermore, correlational analysis showed that the increase of α-synuclein-positive dopaminergic neurons negatively correlated with the decrease in the expression of TH-positive neurons in the ipsilateral SNpc and positively correlated with the increase in the volume of MHCII+ cells in the ipsilateral SNpc. These findings indicate that increased upregulation of activated microglia cells, and aberrant overexpression of α-synuclein in the SN may alter the expression of TH-positive dopaminergic neurons in the SNpc, implicating α-synuclein overexpression coincident with inflammation in the SN as key hallmarks of chronic TBI posing as the likely pathological link between TBI and PD and/or PD-like pathology (Fig. 6).
Figure 6.
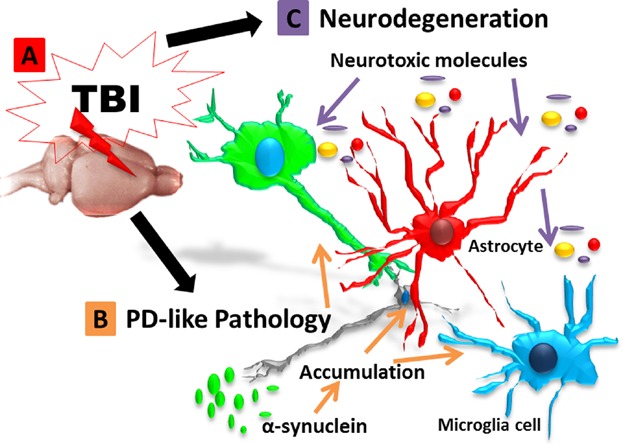
Schematic rendition of the pathological link between TBI and PD via α-synuclein. Overexpression of α-synuclein serves as a pathological link between TBI and PD-associated pathology. After TBI exposure (A), aberrant accumulation of α-synuclein is detected in neurons, microglial cells, and astrocytes (B), leading to propagation of the neurodegeneration (C). The resulting cascade of cell death events contributes to the progressive neurodegeneration associated with increased risk to develop synucleinopathies, such as Parkinson's disease.
The present results are in accordance with those reported in animal and clinical research whereby TBI was found to induce overexpression of α-synuclein and PD-like symptoms in humans and animal models of TBI (Bower et al., 2003; Goldman et al., 2006; Wong and Hazrati, 2013). Accordingly, studies in animal models of TBI have shown that several pathological mechanisms after the primary injury appear to potentiate the acute and chronic inflammation as well as overexpression of oxidized, phosphorylated, and/or nitrated forms of α-synuclein (Cheng et al., 2012; Protter et al., 2012; Stefanis, 2012; Abdul-Muneer et al., 2013). In a recent animal study, it was demonstrated that TBI alone or in combination with pesticide agents can induce a progressive degeneration of dopaminergic neurons along with upregulation of microglia activation during acute and chronic times points (Hutson et al., 2011). Furthermore, clinical data from infants showed a fivefold increase in α-synuclein expression in the cerebrospinal fluid during sub-acute and acute TBI stages, followed by a late progressive 10 fold increase one week after TBI (Su et al., 2010b). Along this line, neuronal injury has been shown to induce overexpression and accumulation of total α-synuclein in human cerebrospinal fluid and correlated with secondary neuronal cell death, exacerbated inflammation, downregulation of TH expression, and the development of neuropathological symptoms following severe TBI (Yu et al., 2004; Uryu et al., 2007; Siebert et al., 2010; Mondello et al., 2013). Some clinical and pre-clinical studies, however, reported acute levels of α-synuclein overexpression in the cerebral spinal fluid after neuronal cell death. While these findings suggest transient and short-lived aggregation due to primary neuronal cell death immediately after TBI (Uryu et al., 2007; Siebert et al., 2010; Su et al., 2010a; Stefanis, 2012; Mondello et al., 2013), progressive neuronal death, apoptosis, and neuro-inflammation characterize the TBI brain during chronic stages, suggesting long-lived progressive neuropathological mechanisms in chronic TBI that could instigate further aberrant α-synuclein secretion (Yu et al., 2009; Acosta et al., 2013; Tajiri et al., 2014).
In an in vitro study, TH mRNA and protein expressions were significantly decreased in MES23.5 rat dopaminergic cells transfected with wild-type human α-synuclein cDNA constructs (Yu et al., 2004). Interestingly, blocking α-synuclein potentiates the release of striatal dopamine (Abeliovich et al., 2000). The mechanism by which chronic TBI-induced overexpression of α-synuclein triggers dopaminergic cell death may initially involve neuro-inflammation followed by apoptosis (Emmanouilidou et al., 2010; Danzer et al., 2011; Wagner et al., 2011). After TBI, α-synuclein accumulation in specific regions of the brain is associated with increased neuro-inflammation that precedes apoptosis and impaired neurotransmission (Abeliovich et al., 2000; Chandra et al., 2004; Auluck et al., 2010; Burre et al., 2010; Nemani et al., 2010; Wagner et al., 2011; Gaugler et al., 2012; Sato et al., 2013). Taken together, the findings from the present study as well as the previous reports demonstrate that α-synuclein functions as a neuro-inflammatory initiator that causes apoptosis and dysfunctional neurotransmission in dopaminergic neurons. Interestingly, trauma-induced compression in the dorsolateral midbrain and cerebral peduncle has been associated with PD pathology in the clinic (Matsuda et al., 2003; Jannetta et al., 2011), suggesting the involvement of the dopaminergic system such as substantia nigra and central tegmental area in the evolution of PD-like symptoms after TBI.
In conclusion, the present study provides insights into the potential pathophysiological response of the brain after chronic TBI. Specifically, we found that α-synuclein overexpression may serve as the pathological link between TBI and the development of PD-like pathology. Our data indicate that α-synuclein overexpression and upregulated microglial activation altered TH-expression of dopaminergic neurons in the SN, providing neurodegenerative pathology in chronic TBI. These findings warrant further investigations into the clinical relevance of the aberrant upregulation of α-synuclein as a potential prognostic biomarker for the progressive secondary cell death of inflammation, which may evolve into a neurodegenerative process (i.e., PD symptoms) in chronic TBI. In addition, that α-synuclein may serve as the pathological link between TBI and PD opens new avenues for the development of treatment interventions designed to reduce α-synuclein load in order to prevent the manifestation of PD-like symptoms in chronic TBI patients.
Literature Cited
- Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Grimmig B, Diamond DM, Sanberg PR, Bickford PC, Kaneko Y, Borlongan CV. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PloS One. 2013;8:e53376. doi: 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amstrong RA. Quantifying the pathology of neurodegerative disorders: Quantitative measurements, sampling strategies and data analysis. Histopathology. 2003;42:521–529. doi: 10.1046/j.1365-2559.2003.01601.x. [DOI] [PubMed] [Google Scholar]
- Amstrong RA, Kotzbauer PT, Perlmutter JS, Cambell MC, Hurth KM, Schmidt RE, Cairns NJ. A quantitative study of α-synuclein pathology in fifteen cases of dementia associated with Parkinson disease. J Neural Transm. 2014;121:171–181. doi: 10.1007/s00702-013-1084-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, Caraveo G, Lindquist S. Alpha-Synuclein: Membrane interactions and toxicity in Parkinson's disease. Ann Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Bower JH, Maraganore DM, Peterson BJ, McDonnell SK, Ahlskog JE, Rocca WA. Head trauma preceding PD: A case-control study. Neurology. 2003;60:1610–1615. doi: 10.1212/01.wnl.0000068008.78394.2c. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha synuclein promotes SNARE complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proce Natl Acad Sci USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson's disease: Diagnosis and management. Lancet Neurol. 2006;5:235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- Chauhan NB. Chronic neurodegenerative consequences of traumatic brain injury. Restorative Neurol Neurosci. 2014;32:337–365. doi: 10.3233/RNN-130354. [DOI] [PubMed] [Google Scholar]
- Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167:699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT., Jr Accelerated oligomerization by Parkinson's disease linked alpha-synuclein mutants. Ann NY Acad Sci. 2000b;7:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000a;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J: Off Publ Fed Am Soc Exp Biol. 2011;25:326–336. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettenhofer KS, Abeles N. The significant of mild traumatic brain injury to cognition and self-reported sympons in long term recovery from injury. J Clin Exp Neuropsychol. 2009;31:363–372. doi: 10.1080/13803390802175270. [DOI] [PubMed] [Google Scholar]
- Fabrizio KS, Keltner NL. Traumatic brain injury in operation enduring freedom/operation iraqi freedom: A primer. Nursing Clinics North America. 2010;45:569–580. doi: 10.1016/j.cnur.2010.06.003. vi. [DOI] [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: Emergency department visits, hospitalizations and deaths. Atlanta (GA): Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun JC, Schneggenburger R, Knott GW, Aebischer P, Schneider BL. Nigrostriatal overabundance of alpha-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW. Head injury and Parkinson's disease risk in twins. Ann Neurol. 2006;60:65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Glover LE, Tajiri N, Lau T, Kaneko Y, van Loveren H, Borlongan CV. Immediate, but not delayed, microsurgical skull reconstruction exacerbates brain damage in experimental traumatic brain injury model. PloS One. 2012;7:e33646. doi: 10.1371/journal.pone.0033646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–1801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J. Parkinson's disease: Clinical features and diagnosis. J Neurol, Neurosurg Psychiatr. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jannetta PJ, Whiting DM, Fletcher LH, Hobbs JK, Brillman J, Quigley M, Fukui M, Williams R. Parkinson's disease: An inquiry into the etiology and treatment. Neurol Int. 2011;3:e7. doi: 10.4081/ni.2011.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantzoulis S, Randolph C. Modern chronic traumatic encephalopathy in retired athletes: What is the evidence. Neuropsychol Rev. 2013;23:350–360. doi: 10.1007/s11065-013-9243-4. [DOI] [PubMed] [Google Scholar]
- Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harbor Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokjohn TA, Maarouf CL, Daugs ID, Hunter JM, Whiteside CM, Malek-Ahmadi M, Rodriguez E, Kalback W, Jacobson SA, Sabbagh MN, Beach TG, Roher AE. Neurochemical profile of dementia pugilistica. J Neurotrauma. 2013;30:981–997. doi: 10.1089/neu.2012.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin B, Bhardwaj A. Chronic traumatic encephalopathy: A critical appraisal. Neurocrit Care. 2014;20:334–344. doi: 10.1007/s12028-013-9931-1. [DOI] [PubMed] [Google Scholar]
- Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: Report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol. 2003;74:1571–1573. doi: 10.1136/jnnp.74.11.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayhew TM. The new stereological methods for interpreting functional morphology from slices of cells and organs. Exp Physiol. 1991;76:639–665. doi: 10.1113/expphysiol.1991.sp003533. [DOI] [PubMed] [Google Scholar]
- Mondello S, Buki A, Italiano D, Jeromin A. Alpha-synuclein in CSF of patients with severe traumatic brain injury. Neurology. 2013;80:1662–1668. doi: 10.1212/WNL.0b013e3182904d43. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen LJ, Fernandes MA. Slowing down after a mild traumatic brain injury: A strategy to improve cognitive task performance. Arch Clin Neuropsychol. 2012;27:85–100. doi: 10.1093/arclin/acr087. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 5th ed. San Diego, CA: Academic Press; 2005. [Google Scholar]
- Prins M, Greco T, Alexander D, Giza CC. The pathophysiology of traumatic brain injury at a glance. Dis Models Mech. 2013;6:1307–1315. doi: 10.1242/dmm.011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter D, Lang C, Cooper AA. Alpha-synuclein and mitochondrial dysfunction: A pathogenic partnership in Parkinson's disease. Parkinson's Dis. 2012;2012:829207. doi: 10.1155/2012/829207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues TM, Jeronimo-Santos A, Outeiro TF, Sebastiao AM, Diogenes MJ. Challenges and promises in the development of neurotrophic factor-based therapies for Parkinson's disease. Drugs Aging. 2014;31:239–236. doi: 10.1007/s40266-014-0160-x. [DOI] [PubMed] [Google Scholar]
- Saing T, Dick M, Nelson P, Kim RC, Cribbs DH, Head E. Frontal cortex neuropathy in dementia pugilistica. J Neurotrauma. 2012;29:1054–1070. doi: 10.1089/neu.2011.1957. DOI: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Kato T, Arawaka S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson's disease: A review of in vivo models. Rev Neurosci. 2013;24:115–123. doi: 10.1515/revneuro-2012-0071. [DOI] [PubMed] [Google Scholar]
- Siebert H, Kahle PJ, Kramer ML, Isik T, Schlüter OM, Schulz-Schaeffer WJ, Brück W. Over-expression of alpha-synuclein in the nervous system enhances axonal degeneration after peripheral nerve lesion in a transgenic mouse strain. J Neurochem. 2010;114:1007–1018. doi: 10.1111/j.1471-4159.2010.06832.x. [DOI] [PubMed] [Google Scholar]
- Shahaduzzaman Md, Acosta S, Bickford PC, Borlongan CV. α-Synuclein is a pathological link and therapeutic target for Parkinson's disease and traumatic brain injury. Med Hypotheses. 2013;81:675–680. doi: 10.1016/j.mehy.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Smith C. Review: The long-term consequences of microglial activation following acute traumatic brain injury. Neuropathol Appl Neurobiol. 2013;39:35–44. doi: 10.1111/nan.12006. [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: Substrates of dementia. Nat Rev Neurol. 2013;9:211–221. doi: 10.1038/nrneurol.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L. Alpha-synuclein in Parkinson's disease. Cold Spring Harbor Perspect Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LJ, Auluck PK, Outeiro TF, Yeger-Lotem E, Kritzer JA, Tardiff DF, Strathearn KE, Liu F, Cao S, Hamamichi S, Hill KJ, Caldwell KA, Bell GW, Fraenkel E, Cooper AA, Caldwell GA, McCaffery JM, Rochet JC, Lindquist S. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Dis Models Mech. 2010a;3:194–208. doi: 10.1242/dmm.004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su E, Bell MJ, Wisniewski SR, Adelson PD, Janesko-Feldman KL, Salonia R, Clark RS, Kochanek PM, Kagan VE, Bayır H. Alpha-synuclein levels are elevated in cerebrospinal fluid following traumatic brain injury in infants and children: the effect of therapeutic hypothermia. Dev Neurosci. 2010b;32:385–395. doi: 10.1159/000321342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri N, Acosta SA, Shahaduzzaman M, Ishikawa H, Shinozuka K, Pabon M, Hernandez-Ontiveros D, Kim DW, Metcalf C, Staples M, Dailey T, Vasconcellos J, Franyuti G, Gould L, Patel N, Cooper D, Kaneko Y, Borlongan CV, Bickford PC. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34:313–326. doi: 10.1523/JNEUROSCI.2425-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy A, Di Monte DA. Alpha-synuclein elevation in human neurodegenerative diseases: Experimental, pathogenetic, and therapeutic implications. Mol Neurobiol. 2013;47:484–494. doi: 10.1007/s12035-012-8329-y. [DOI] [PubMed] [Google Scholar]
- Uryu K1, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Amin KB, Niyonkuru C, Postal BA, McCullough EH, Ozawa H, Dixon CE, Bayir H, Clark RS, Kochanek PM, Fabio A. CSF Bcl-2 and cytochrome C temporal profiles in outcome prediction for adults with severe TBI. J Cereb Blood Flow Metab. 2011;31:1886–1896. doi: 10.1038/jcbfm.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27:494–506. doi: 10.1111/j.1440-1789.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- Wan OW, Chung KK. The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson's disease. PLoS One. 2012;7:e38545. doi: 10.1371/journal.pone.0038545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Wong JC, Hazrati LN. Parkinson's disease, parkinsonism, and traumatic brain injury. Crit Rev Clin Lab Sci. 2013;50:103–106. doi: 10.3109/10408363.2013.844678. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Kaneko Y, Bae E, Stahl CE, Wang Y, van Loveren H, Sanberg PR, Borlongan CV. Severity of controlled cortical impact traumatic brain injury in rats and mice dictates degree of behavioral deficits. Brain Res. 2009;1287:157–163. doi: 10.1016/j.brainres.2009.06.067. [DOI] [PubMed] [Google Scholar]
- Yu S, Zuo X, Li Y, Zhang C, Zhou M, Zhang YA, Uéda K, Chan P. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci Lett. 2004;367:34–39. doi: 10.1016/j.neulet.2004.05.118. [DOI] [PubMed] [Google Scholar]