

Abstract
Background
Surgery is entering a new phase with the revolution in genomic technology. Cheap, mass access to next‐generation sequencing is now allowing the analysis of entire human genomes at the DNA and RNA level. These data sets are being used increasingly to identify the molecular differences that underlie common surgical diseases, and enable them to be stratified for patient benefit.
Methods
This article reviews the recent developments in the molecular biology of colorectal, oesophagogastric and breast cancer.
Results
The review specifically covers developments in genetic predisposition, next‐generation sequencing studies, biomarkers for stratification, prognosis and treatment, and other 'omics technologies such as metabolomics and proteomics.
Conclusion
There are unique opportunities over the next decade to change the management of surgical disease radically, using these technologies. The directions that this may take are highlighted, including future advances such as the 100 000 Genomes Project.
Short abstract
May individualize cancer treatment
Introduction
The field of molecular biology has undergone rapid advancement in the past 5 years, with exciting consequences for the diagnosis, treatment and follow‐up of surgical patients.
A series of enabling technologies and projects have expanded the knowledge of how basic molecular biology can assist in the management of surgical disease. The first, and most important, was the Human Genome Project, established in 1990 by the US National Institutes of Health and the UK Sanger Centre1. This established the reference human genome by carrying out sequencing of multiple fragments of a reference human genome using the dye‐terminator technique described by Sanger and colleagues2. A consequence of this technology is that the project took 10 years to produce a single genome and cost over US $3 billion to complete.
The development of microarray technology and next‐generation sequencing (NGS) within the past 5 years has led to a step‐change in the implementation of genomic technologies; before this, the bulk of genetic research was carried out on DNA microarrays.
Genome‐wide association studies
DNA microarrays are available from a variety of manufacturers (Illumina, Affymetrix and Agilent) and consist of silicon or glass slides with oligonucleotides complementary to the DNA sequence being studied, which are annealed to their surface. This allows cheap, mass production of microarrays that can be used for large population‐based studies. Typically these microarrays have between 500 000 and 1·5 million genomic markers, usually single‐nucleotide polymorphisms (SNPs). SNPs are single‐nucleotide changes within a gene that lead to protein change and subsequent change in the function of that gene. When scanned with a laser, each individual oligonucleotide fluoresces a specific colour, depending on the bound oligonucleotide fragment (Figs 1 and 2).
Figure 1.
Image of scanned oligonucleotide array (from Wikimedia Commons)
Figure 2.

Affymetrix microarray chip from Wikimedia Commons
A variety of projects have been undertaken using DNA microarrays, typically taking the form of the genome‐wide association study (GWAS). These are usually case–control studies with cases enriched for the disease of interest. SNPs of interest are identified and taken forward to validation in larger cohorts, giving insights into the disease process being studied. Examples of GWASs include the COGENT (COlorectal cancer GENeTics) Consortium, and the Wellcome Trust Case Control Consortium 1/2 (WTCCC 1/2) examining colorectal cancer, Crohn's disease, diabetes, ischaemic heart disease, and several other common diseases and pathologies.
Next‐generation sequencing
The most widely used NGS technology, sequencing by synthesis (Illumina, San Diego, California, USA) allows entire human genomes to be sequenced within 24 h at low cost, with the sub $1000 genome barrier (X‐prize) being achieved earlier this year. Other innovative technologies include single molecular real‐time sequencing (PacBio® SMRT™; Pacific Biosciences, Menlo Park, California, USA), semiconductor sequencing (Ion Torrent™; Life Technologies, Paisley, UK) and nanopore sequencing (Oxford Nanopore, Oxford, UK). Until recently, NGS was limited to small studies on a few samples owing to cost constraints, but because of the rapid fall in price‐per‐sample (Fig. 3), large studies are now in progress. The Cancer Genome Atlas (TCGA) project is sequencing the cancer genome of 33 different cancer types in the US population; the 100 000 Genomes Project in the UK is undertaking to sequence 50 000 cancer genomes and 50 000 rare disease genomes over the next 5 years.
Figure 3.
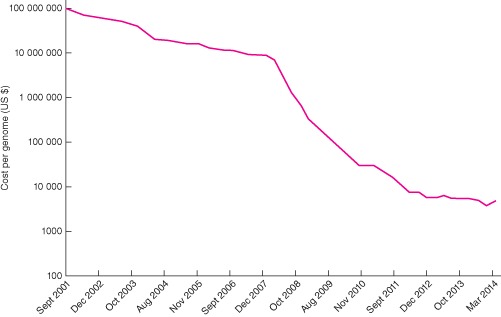
Cost in US dollars per genome sequenced in 2001–2014, at 5‐month intervals (data from http://www.genome.gov/sequencingcosts/)
To carry out NGS, an inherently massively parallel technique, several steps are required. All technologies require capture of DNA or RNA into sequencing libraries. Illumina Solexa™ then uses this captured DNA/RNA to generate clusters, which are amplified. Cluster amplification is the process whereby target DNA is immobilized on to spatially separated template sites, allowing sequencing reactions to occur in parallel. Sequencing is then carried out on the clusters present on the glass slide. The slide is portioned into eight channels, enabling independent samples to be run simultaneously. Typically, reads of between 75 and 100 base pairs are possible, and the nucleotide incorporation cycle is repeated until sufficient depth is covered for all targeted regions. Data generated are then aligned with a reference sequence, and variants are called after comparing the sequencing data to a reference. This allows a tumour specimen to be compared with its paired normal tissue sample.
A critical difference in analysis of human cancers is the concept of germline and somatic mutations. Germline mutations are the constitutive DNA that the patient is born with, and variation within the germline confers an increased (or decreased) risk of cancer. Somatic mutations occur as a consequence of tumour development, although they may initiate tumour development by occurring spontaneously as a result of external factors such as ionizing radiation or carcinogens.
Microarray and sequencing technologies also allow analysis of other types of genetic information, such as DNA methylation (epigenetics), which acts as a switch in the regulation of gene expression. NGS also allows analysis of gene expression by NGS of mRNA (RNA‐seq) and other genetic modifications, such as sequencing of chromatin‐immunoprecipitated DNA (ChIP‐seq). Non‐coding small RNAs that may affect gene function, such as long non‐coding RNA (lncRNA), small interfering RNA (siRNA) and small nucleolar RNA (snoRNA), can also be analysed by NGS.
Other 'omics technologies
Other technologies are emerging as potential methods for the downstream analysis and stratification of patient samples in surgical disease. Two examples of these technologies are metabolomics and proteomics. Metabolomics uses either nuclear magnetic resonance or mass spectrometry to ascertain the presence of metabolites in surgical specimens. The patterns and relative abundances of the metabolites observed can give clues as to the underlying biological processes at work in the tissues studied3.
Proteomics uses mass spectrometry to understand the structure of proteins. Several methods exist to allow proteins to be studied in the ionized form without fragmentation or damage including matrix‐assisted laser desorption ionization (MALDI)4 and electrospray ionization5. Because proteins are modified after they are produced by transcription (post‐translational modification), study of both the protein and RNA involved in tissues allows a fuller appreciation of the changes that may be occurring in a particular disease.
These technologies have allowed advances in understanding of the initiation and progression of multiple surgical diseases, and in adjuvant therapies for surgical disease such as chemotherapy and radiotherapy. They also allow the possibility of population screening of asymptomatic carriers.
Lower gastrointestinal tract: colorectal cancer and inflammatory bowel disease
Genetic predisposition
The predominant surgically relevant disease types studied in the lower gastrointestinal (GI) tract have been colorectal cancer, Crohn's disease and ulcerative colitis. The COGENT Consortium6 has undertaken multiple GWASs of patients with colorectal cancer associated with a strong family history or extreme phenotype (such as young age of onset), identifying ten SNPs associated with colorectal cancer at population‐wide significance. Although inheritance of one disease association SNP confers a population risk of odds ratio approximately 1·05 (Table 1, taken from Tenesa and Dunlop7), inheritance of multiple SNPs (more than 10) confers a cumulative threefold risk of cancer. Other successes from the GWAS/NGS approach have been identification of the GREM‐associated duplication in hereditary mixed polyposis syndrome8 and mutations in the DNA polymerase genes POLE and POLD as a cause of hereditary polyposis9. Identification of these mutations allows familial testing, enhanced surveillance and reduction in the risk of developing colorectal cancer.
Table 1.
Frequency of identified single‐nucleotide polymorphisms in colorectal cancer, and their effect sizes (from Tenesa and Dunlop7)
| Gene/locus | Chromosome | SNP | Effect size (odds ratio) | Allele frequency | Population attributable risk (%) |
|---|---|---|---|---|---|
| – | 8q24 | rs6983267 | 1·21 (1·15, 1·27) | 0·51 | 9·7 |
| GREM1 | 15q13 | rs4779584 | 1·26 (1·19, 1·34) | 0·18 | 4·5 |
| SMAD7 | 18q21 | rs4939827 | 1·18 (1·12, 1·23) | 0·52 | 8·6 |
| – | 11q23 | rs3802842 | 1·12 (1·07, 1·17) | 0·29 | 3·4 |
| EIF3H | 8q23 | rs16892766 | 1·25 (1·19, 1·32) | 0·07 | 1·7 |
| — | 10p14 | rs10795668 | 1·12 (1·10, 1·16) | 0·67 | 7·4 |
| BMP4 | 14q21 | rs4444235 | 1·11 (1·08, 1·15) | 0·46 | 4·8 |
| CDH1 | 16q22 | rs9929218 | 1·10 (1·06, 1·12) | 0·71 | 6·6 |
| RHPN2 | 19q13 | rs10411210 | 1·15 (1·10, 1·20) | 0·90 | 11·9 |
| BMP2 | 20q12 | rs961253 | 1·12 (1·08, 1·16) | 0·35 | 4·0 |
Values in parentheses are 95 per cent c.i. SNP, single‐nucleotide polymorphism.
In Crohn's disease, multiple large‐population GWAS studies10 have been undertaken identifying multiple SNPs of predisposition, suggesting that Crohn's disease has a strong heritable component. In total, more than 73 SNPs have been identified, with the strongest association in the NOD2 gene, which plays an important role in immunity. In total, these loci make up about 20 per cent of the observed inheritability of Crohn's disease.
Comparatively less research has been undertaken in germline susceptibility to ulcerative colitis; several large population GWAS studies11, 12, 13 have demonstrated over 30 associated SNPs. These SNPs are in a variety of genes, but are associated predominantly with immune system and immunity‐related genes. In addition, approximately 50 per cent of identified loci overlap with those of Crohn's disease.
Genomic analysis of colorectal cancer
The colorectal cancer TCGA project14 has carried out exome sequencing (sequencing of the protein coding regions of the genome), RNA‐seq, genome‐wide methylation analysis and protein expression (via reverse‐phase protein arrays; RPPAs) of, at the time of writing, 461 colorectal tumours. This group has confirmed recurrent driver mutations in APC, TP53, SMAD4, PIK3CA and KRAS, but also found novel therapeutic targets in ARID1A, SOX9 and FAM123B. Another study15 observed gene fusions (merging of two genes, which causes abnormal function) in R‐spondin. The mutations observed in ARID1A are particularly exciting as they present a potential therapeutic target16. These data sets provide a wealth of information about colorectal cancer, and linkage to a clinical data set provides opportunities for future biomarker studies.
Recent work has examined the role of integration of multiple 'omics data sets to produce classifiers of disease17, also known as endotypes. These are based on mutation, expression and immunological data sets. The Colorectal Cancer Subtyping Consortium found four distinct Colorectal cancer Molecular Subtypes (CMSs) (Table 2). The classifiers identified provide insight into the biology of the distinct types. CMS1 consisted of microsatellite‐unstable, immunologically active tumours occurring mainly on the right side in the elderly, whereas CMS2 (the most frequent endotype) consisted of chromosomally unstable, microsatellite‐stable tumours. Further study of these classifiers may permit finer stratification, allowing precisely targeted therapy.
Table 2.
The Colorectal Cancer Subtyping Consortium classification of colorectal cancer
| Classifier | Frequency (%) | Characteristics |
|---|---|---|
| CMS1 | 14 | MSI, immune pathway activation/expression, right‐side tumours, older age at diagnosis, females, hypermutation, BRAF mutation, intermediate survival |
| CMS2 | 41 | High CIN, MSS, strong Wnt/Myc pathway activation, left‐side tumours, TP53 mutation, EGFR amplification/overexpression, better survival |
| CMS3 | 8 | Low CIN, moderate Wnt/Myc pathway activation, KRAS mutation, PIK3CA mutation, IGFBP2 overexpression, intermediate survival |
| CMS4 | 20 | CIN/MSI heterogeneous, mesenchymal/TGF‐β activation, younger age at diagnosis, NOTCH3/VEGFR2 overexpression, worse survival |
CMS, Colorectal cancer Molecular Subtype; MSI, microsatellite instability; CIN, chromosomal instability; MSS, microsatellite stable; TGF, transforming growth factor.
Screening biomarkers
A wide variety of biomarkers have been examined18 in colorectal cancer, as both markers of screening and of prognosis. The ideal biomarker would be easily detectable in either stool or blood, cheap and highly accurate. Unfortunately, no current biomarker fits these criteria precisely owing to the molecular heterogeneity associated with colorectal cancer. The most promising markers seem to be associated with abnormal DNA methylation. For example, in colorectal cancer, differential methylation of the septin 9 gene has been shown to have 72 per cent sensitivity and 90 per cent specificity for the detection of malignancy. However, many biomarker studies across all cancer types are plagued by poor study design, insufficient power and non‐hypothesis‐driven marker selection. Currently, a well designed, UK‐based trial of methylated biomarkers, the ENDCaP‐C study (Enhanced Neoplasia Detection and Cancer Prevention in Chronic Colitis) is under way19, examining their value in the detection of dysplasia in a screened population of patients with ulcerative colitis.
Another rich field of developing interest in colorectal cancer is sequencing of the microbial genomes that exist within the colon. Experimental murine models seem to indicate that the microbiome within the colon alters the risk of colorectal cancer by modulating inflammation20. This has also been demonstrated to be the case in patients with inflammatory bowel disease, for both ulcerative colitis and Crohn's disease21.
Metabolomic techniques also show promise in acting as screening biomarkers. Mirnezami and co‐workers22 undertook high‐resolution magic‐angle spinning nuclear magnetic resonance spectroscopy in 44 tumour–normal pairs, finding cancer‐specific metabolite patterns allowing differentiation of cancer from normal tissues, in addition to finding changes in the metabolome as the tumour progressed. A number of proteomic studies have also been carried out in colorectal cancer23, although these all suffer from lack of validation, and the variety of different markers identified undoubtedly reflects the varying populations from which they were sampled.
Targeted therapies, prognostic and predictive biomarkers
The discovery that the epidermal growth factor (EGF) pathway in colorectal cancer was sensitive to inhibition by anti‐EGF receptor (EGFR) monoclonal antibodies (mAbs) led to the rapid development of panitumumab and cetuximab. However, it was found in initial trials that the antibodies seemed to have no clinical effect against colorectal cancer; although a proportion of patients seemed to benefit from therapy, the majority did not24. It was found subsequently that mutations in the EGFR pathway genes (KRAS, BRAF, NRAS and PIK3CA) conferred resistance to anti‐EGFR mAbs due to hyperactivation of the pathway independent of the EGFR25. The CRYSTAL trial26 compared patients with a KRAS mutation and those without, finding a clear response and survival benefit for anti‐EGFR mAbs in patients without mutation. A number of other pathway‐specific inhibitors exist for colorectal cancer, including the antivascular endothelial growth factor (VEGF) mAb bevacizumab, MEK inhibitors that target EGF pathway mutated cancers and cancer vaccines. The FOCUS4 trial27 is currently recruiting patients for a molecularly stratified trial of metastatic colorectal cancer therapy: patients are selected for a specific therapy when they possess a mutation specific to that cancer. This raises the intriguing possibility of molecular‐targeted therapy for primary, non‐metastatic tumours as neoadjuvant therapy before surgery.
A commercially available test exists for prediction of recurrence and benefit for 5‐fluorouracil chemotherapy (Oncotype DX®; Genomic Health, Redwood City, California, USA), based on a multigene panel of RNA expression from formalin‐fixed paraffin‐embedded tissues28. This was developed by screening 761 candidate genes against a large cohort of 1851 patients undergoing surgery for colorectal cancer, with or without adjuvant 5‐fluorouracil therapy. Validation was carried out on the QUASAR study29, which could stratify prognosis, but did not correlate with benefit from chemotherapy.
Another intriguing possibility is the use of immune‐based stratification to estimate colorectal cancer prognosis. Lal et al.30 used the TCGA expression data set to identify four different immune classifiers based on the expression of immune system‐related genes. Immunogenicity is thought to be related to survival, as tumours that are more visible to the immune system are more likely to undergo destruction by the immune system. One of the mechanisms that may occur in highly mutated tumours, such as tumours with a POLE mutation or those with microsatellite instability, is where the large number of mutations causes a variety of frameshift mutations. These frameshift mutations drive the production of neoantigens31 caused by the alternative splicing of multiple genes, which increases the visibility of the tumour to the immune system.
Upper gastrointestinal tract: oesophagogastric cancer
Genetic predisposition
Research into predisposition to gastro‐oesophageal cancer is complicated by the fact that it can arise in two histologically different epithelial types: squamous cell carcinoma (SCC) and adenocarcinoma. A significant proportion of the risk is likely to comprise lifestyle factors such as smoking, gastro‐oesophageal reflux and diet.
In adenocarcinoma, a GWAS of the premalignant stage of oesophageal cancer32, Barrett's oesophagus, demonstrated associations between the major histocompatibility locus and a gene associated with oesophageal development (FOXF1). It was also found that the predisposition to Barrett's oesophagus was made up of multiple common variants of small effect, rather than a single genetic driver. A further GWAS of oesophageal adenocarcinoma32 demonstrated associations with transcription factors (CRTC1, FOXP1, BARX1). In light of these results it is difficult to highlight SNPs that may act as markers for increased disease risk or act as molecular targets for therapy.
Several GWAS studies have been undertaken, predominantly in Chinese populations at high risk of oesophageal SCC33, 34. They highlighted SNPs in the riboflavin transporter C20orf54, and a cell growth and differentiation gene, PLCE1. Riboflavin deficiency was identified before this study as a risk factor35 for oesophageal SCC. The PLCE1 variant was further found to interact specifically with tobacco smoke exposure36.
Genomic analysis of oesophagogastric cancer
Both the Broad Institute (Cambridge, Massachusetts, USA) and the OCCAMS (Oesophageal Cancer Clinical and Molecular Stratification) Consortium have studied oesophageal adenocarcinoma. The Broad Institute project37 undertook whole‐exome and whole‐genome sequencing in 149 tumour–normal pairs, verifying previously identified mutations in TP53, CDKN2A, SMAD4, ARID1A and PIK3CA. Previously unidentified mutations in SPG20, TLR4, ELMO1 and DOCK2 were also found, and a possible role for the RAC1 pathway (a modulator of epithelial–mesenchymal transition) was identified.
The OCCAMS Consortium38 examined whole‐genome sequencing of oesophageal adenocarcinoma, as well as targeted sequencing of never‐dysplastic Barrett's oesophagus (which did not progress to malignancy) and high‐grade dysplasia. They found that TP53 was the dominant mutation seen in adenocarcinoma, in over 80 per cent of samples, but that these mutations were also present in biopsies from never‐dysplastic patients, contrary to what was expected based on the known oncological progression of these lesions. The only stage‐specific mutations seen in high‐grade dysplasia and adenocarcinoma were in TP53 and SMAD4.
Whole‐genome and whole‐exome sequencing of oesophageal SCC has also been performed39. Whole‐genome sequencing of 17 tumour–normal pairs and whole‐exome sequencing in a further 71 tumour–normal pairs identified recurrent mutations in TP53, RB1, CDKN2A, PIK3CA, NOTCH1 and NFE2L2, as well as ADAM29 and FAM135B. The genomic landscape of oesophageal SCC was significantly different from that of oesophageal adenocarcinoma, highlighting the different therapeutic strategies that are needed in this disease.
Biomarkers of predisposition/sensitivity and screening
Given the above OCCAMS findings, it is difficult to use TP53 mutation as a biomarker of adenocarcinoma, as it has also been identified in biopsies from never‐dysplastic patients, who should not progress to adenocarcinoma. As a precursor to the OCCAMS study38, the same group undertook combined‐array CGH (comparative genomic hybridization, a type of microarray analysis looking at chromosomal abnormalities) in tumour samples and gene expression via microarray40. They found a pattern of copy number alterations and associated expression change that could identify poor‐prognosis oesophageal adenocarcinoma. The OCCAMS group also examined gene expression changes in adenocarcinoma using RNA microarrays41, finding a four‐gene expression panel of DCK, PAPSS2, SIRT2 and TRIM44 that were independently predictive of survival.
There have been a number of attempts at developing methylated biomarkers in oesophageal adenocarcinoma, both as screening biomarkers and to identify high‐risk Barrett's oesophagus. The genes studied include CDKN2A, vimentin, P14ARF, CDX2, SOCS1/3, SFRP1/2/4/5 and WIF1 42, 43, 44. Unfortunately no consistent marker can be identified that successfully differentiates adenocarcinoma from normal oesophagus and high‐grade Barrett's dysplasia. These types of focused biomarker will become increasingly important to stratify therapy.
Attempts have been made using proteomics45 to distinguish oesophagogastric cancer from benign disease, to allow screening. MALDI mass spectrometry was used to compare the differences between oesophageal cancer and normal mucosa, and gastric cancer and normal mucosa. It was found that, although there were clear differences between cancer and normal tissue, there was a wide variety of changes that varied between different tumours, making determination of a specific biomarker difficult.
Targeted therapies
Despite the recent advances in discovery science in gastro‐oesophageal cancer, few therapeutic targets currently exist. A small proportion of cancers overexpress the human EGFR 2 (HER2) protein46, and clinical trials of trastuzumab, a mAb against the HER–receptor complex are ongoing. The ToGA (Trastuzumab for Gastric Cancer) study47 investigated the addition of trastuzumab to standard chemotherapy, and demonstrated a small survival benefit in HER2‐positive cancer. Targeting by anti‐VEGF therapy is also undergoing clinical trials48; however, results have been mixed with no improvement in overall survival, but improvements in response rates and progression‐free survival.
Breast cancer
Genetic predisposition
The role of inherited variability in breast cancer has been investigated extensively. A very strong signal for variants in the fibroblast growth factor receptor 2 gene (FGFR2) have been found in GWAS studies across multiple populations49, 50, 51. Carriers of the two low‐risk alleles at FGFR2 (frequency 38 per cent of the population) have a relative risk of breast cancer of 0·83 compared with the general population52; carriers of one high‐risk and one low‐risk allele (47 per cent) have a relative risk of 1·05; and carriers of two high‐risk alleles (14 per cent) have a relative risk of 1·2653. FGFR2 mutations are of particular interest as they may represent a therapeutic target in breast cancer54.
A recent GWAS55 in oestrogen receptor (ER)‐negative breast cancer demonstrated four variants that reached genome‐wide significance in MDM4, LGR6, FTO and a SNP within the 2p24·1 region. These SNPs were present only in ER‐negative breast cancers, in contrast to the findings in a combined ER‐positive/ER‐negative GWAS56. This found variants located with PTHLH, known to have a role in breast development, and NRIP1, a co‐factor of the ER.
Next‐generation sequencing
A number of NGS projects have examined the mutation spectrum in breast cancer. The most comprehensive of these is from the TCGA project57, which carried out exome sequencing, RNA‐seq, methylation array analysis and RPPA of 463 patients. Recurrent mutations were found in TP53, PIK3CA and GATA3 at frequencies of over 10 per cent, reinforcing their role as driver mutations in breast cancer, as well as mutations in several dozen genes previously identified in breast cancer. Study of the role of expression subtypes in breast cancer demonstrated four separate subtypes (luminal A/B, basal and HER2E), with the mutational burden and spectrum varying in each. The HER2E subtype demonstrated a relatively low mutational frequency, whereas the luminal A subtype demonstrated large numbers of significantly mutated genes, the most frequent mutation being in PIK3CA.
In common with colorectal cancer, methylation array analysis of breast cancer revealed a hypermethylator phenotype, associated with the luminal B expression subtype, and a hypomethylated phenotype associated with a basal expression subtype and comparatively higher frequency of TP53 mutation. Copy number analysis was also performed, with the previously identified amplifications in HER2 and EGFR being identified, as well as novel amplifications in PIK3CA and FOXA1 and deletions in RB1 and PTEN. A striking finding throughout the study was the detection of genetic heterogeneity both within tumours and between samples, highlighting the diverse nature of this disease.
Shah and colleagues58 examined the mutational spectrum in triple‐negative breast cancer (ER/progesterone receptor/HER‐negative), again finding that TP53 and PIK3CA mutations were clonally dominant, but also finding a wide variety of mutation spectra, including tumours with few driving mutations and tumours with extremely complex mutational spectra (the hypermutated phenotype). A separate study59 identified recurrent mutations in the transcription factor genes CBFB and RUNX1, as well as a gene fusion seen only in triple‐negative breast cancers, the MAGI3–AKT3 fusion transcript. An exome sequencing study60 of 100 breast cancers at the Wellcome Trust Sanger Institute (Cambridge, UK) identified more than 40 driver mutations in a breast cancer cohort, including genes now known to be important therapeutic targets, such as AKT1/2, ARID1B, CASP8 and MAP3K1.
NGS also has applications in the monitoring of metastatic disease or in recurrence. Dawson et al.61 used a combination of targeted NGS, digital PCR and whole‐genome sequencing to examine DNA circulating in the bloodstream that is shed from metastatic tumours. They found that increasing amounts of circulating DNA correlated with poorer overall survival, but also that, by comparing mutations in the primary tumour with the cell‐free DNA obtained, recurrence of the primary tumour could be detected by the presence of the same somatic mutations in the circulating DNA. This technology has applications for the detection of recurrence in multiple tumour types across the disease spectrum.
Biomarkers of predisposition/sensitivity and screening
One‐step nucleic acid amplification (OSNA) is an example of the use of genetic technologies to stratify patients. It works by the detection of raised copy number of the cytokeratin 19 gene (CK19) as a surrogate marker of the presence of breast cancer within sentinel lymph nodes62, and is used as a proxy marker for axillary lymph node positivity in breast cancer. OSNA has been approved as a technological solution to determine sentinel lymph node positivity by the UK National Institute for Health and Care Excellence (NICE), and has been shown to be equivalent to both radioisotope‐ and dye‐based technologies for mapping lymph nodes63.
Another recent development is the DNA damage response assay for the prediction of response to anthracycline/cyclophosphamide‐based chemotherapy in breast cancer64. The study examined RNA expression in patients with a DNA damage response deficiency and developed a 44‐gene RNA expression panel that could predict response to chemotherapy in sporadic breast cancer.
Targeted therapies
A rich variety of targeted therapies exist for breast cancer, based on the underlying biology of the tumour, gained by the extensive molecular research carried out on breast cancer. Aromatase inhibitors (such as anastrazole) and targeted ER modulators (such as tamoxifen) have been used extensively for many years, based on the observation that a proportion of breast cancers are ER‐positive. Identification of the overexpression of HER2 in breast cancer has allowed targeting with both mAbs (trastuzumab) and small‐molecule tyrosine kinase inhibitors such as lapatanib, with appreciable survival benefits.
More recently, several novel pathways have been identified as being dysregulated in breast cancer: the insulin‐like growth factor (IGF) 1 pathway; the BRCA‐associated double‐strand break repair protein, poly(ADP‐ribose) polymerase (PARP) 1; and the phosphatidylinositol‐3‐kinase (PIK3)–Akt–mammalian target of rapamycin (mTOR) pathway. Immunohistochemistry of IGF‐1 has revealed that it is overexpressed in more than 50 per cent of breast cancers, and a mAb (cixutumumab) that targets these pathways is currently in phase 1 studies65.
PARP inhibitors were identified from work targeting BRCA mutant breast cancer66 as a therapeutic strategy to treat patients with BRCA mutations. A small molecular PARP inhibitor, olaparib, has been used to treat germline BRCA mutant breast cancer67, with a clinical trial (the OlympiAD trial) currently under way. PARP inhibitors also show promise in triple‐negative breast cancer, as studies have shown that a proportion of these women possess BRCA mutations68, 69 and may respond to PARP inhibitors. mTOR pathway inhibitors, such as everolimus, should theoretically be of benefit in breast cancer as a result of the dysregulation of this pathway demonstrated by molecular studies, although initial results have been disappointing70.
The future
The union of basic biological research with surgery has allowed the field of translational surgical biology to develop, utilizing modern molecular technologies to stratify surgical disease based on therapeutic and outcome response.
A number of possibilities exist for future research. First, whole‐genome studies using NGS technologies of prospectively collected samples will allow identification of biomarkers for response (such as radiotherapy in rectal cancer) and treatment (for instance, molecularly targeted therapies). An example of current clinical issues that could be answered using this technology is the response of rectal cancer to radiotherapy. Around 5–10 per cent of patients with advanced rectal cancer given preoperative radiotherapy have a complete response71. Research is currently under way to understand what makes these tumours particularly sensitive (the so‐called extreme responders) to radiation, and it is likely that a highly focused approach using NGS will identify responsible pathways.
Stratification of premalignant lesions is also an important focus of research. In colorectal, upper GI and breast cancer, few biomarkers exist to predict which premalignant lesions will progress to invasive cancer. Typically less than 1 per cent of patients with Barrett's oesophagus will progress to invasive oesophageal cancer, but no markers reliably predict this. The use of retrospective cohorts of patients with progressive Barrett's oesophagus and NGS analysis might identify genetic markers.
The concept of disease classifiers will play an increasingly important role and, as more NGS data sets become available, the granularity of classifiers will improve to the extent whereby a more precise understanding of the biology underlying a tumour will be available. This will be enhanced by the integration of multiplatform (NGS, metabolomics, proteomics) data into these classifiers, and will allow tailoring of therapy to the underlying disease.
The availability of NGS data sets for the surgical population will be enhanced by the commissioning of the UK 100 000 Genomes Project72. This exciting project will carry out whole‐genome sequencing of 50 000 tumour–normal pairs from patients with a range of cancers, but concentrating primarily on colorectal, breast, lung and prostate cancer. The data made available by this project will allow a highly detailed examination of the drivers in these cancer types; this is the largest project of its type in the world.
Although the technological advances are numerous, they are not without their own challenges. The recent identification of intratumoral heterogeneity, long hypothesized but only recently identified definitively in renal cell cancer73 and in preneoplastic lesions74, provides unique challenges for stratification. It is likely that multiple subclones of tumour exist within a primary tumour, each with differing characteristics. These will dictate prognosis and response to therapy, and further investigation is needed to understand the extent and consequences of this phenomenon.
Technological leaps in molecular biology will enable selection of the right therapy for the right patient at the right time, and further build on the surgical and anaesthetic improvements achieved in the past century to provide maximal benefit for the patient.
Acknowledgements
A.D.B. is supported by a Wellcome Trust Post‐Doctoral Fellowship for Clinician Scientists (102732/Z/13/Z). A.D.B. undertakes collaborative research with Illumina UK into colorectal cancer; sequencing was provided free of charge by Illumina UK as part of the research project.
Disclosure: The authors declare no other conflict of interest.
Click here to listen to the author discuss the contents of this article.
References
- 1. Dulbecco R. A turning point in cancer research: sequencing the human genome. Science 1986; 231: 1055–1056. [DOI] [PubMed] [Google Scholar]
- 2. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain‐terminating inhibitors. Proc Natl Acad Sci U S A 1977; 74: 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patti GJ, Yanes O, Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol 2012; 13: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lattová E, Chen VC, Varma S, Bezabeh T, Perreault H. Matrix‐assisted laser desorption/ionization on‐target method for the investigation of oligosaccharides and glycosylation sites in glycopeptides and glycoproteins. Rapid Commun Mass Spectrom 2007; 21: 1644–1650. [DOI] [PubMed] [Google Scholar]
- 5. Ho CS, Lam CW, Chan MH, Cheung RC, Law LK, Lit LC et al Electrospray ionisation mass spectrometry: principles and clinical applications. Clin Biochem Rev 2003; 24: 3–12. [PMC free article] [PubMed] [Google Scholar]
- 6. Tomlinson IP, Dunlop M, Campbell H, Zanke B, Gallinger S, Hudson T et al COGENT (COlorectal cancer GENeTics): an international consortium to study the role of polymorphic variation on the risk of colorectal cancer. Br J Cancer 2010; 102: 447–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome‐wide association studies. Nat Rev Genet 2009; 10: 353–358. [DOI] [PubMed] [Google Scholar]
- 8. Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR et al Hereditary mixed polyposis syndrome is caused by a 40‐kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 2012; 44: 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P et al Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 4ac5: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franke A, McGovern DP, Barrett JC, Wang K, Radford‐Smith GL, Ahmad T et al Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet 2010; 42: 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. UK IBD Genetics Consortium , Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA et al Genome‐wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 2009; 41: 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franke A, Balschun T, Sina C, Ellinghaus D, Hasler R, Mayr G et al Genome‐wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL). Nat Genet 2010; 42: 292–294. [DOI] [PubMed] [Google Scholar]
- 13. McGovern DP, Gardet A, Torkvist L, Goyette P, Essers J, Taylor KD et al Genome‐wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet 2010; 42: 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB et al Recurrent R‐spondin fusions in colon cancer. Nature 2012; 488: 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sayegh J, Cao J, Zou MR, Morales A, Blair LP, Norcia M et al Identification of small molecule inhibitors of Jumonji AT‐rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J Biol Chem 2013; 288: 9408–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dienstmann R, Guinney J, Delorenzi M, De Reynies A, Roepman P, Sadanandam A et al Colorectal Cancer Subtyping Consortium (CRCSC) identification of a consensus of molecular subtypes. J Clin Oncol 2014; 32(Suppl): 5s (Abstract). [Google Scholar]
- 18. Newton KF, Newman W, Hill J. Review of biomarkers in colorectal cancer. Colorectal Dis 2012; 14: 3–17. [DOI] [PubMed] [Google Scholar]
- 19. National Institute for Health Research . EME ‐ 11/100/29: Enhanced Neoplasia Detection and Cancer Prevention in Chronic Colitis (ENDCaP‐C). http://www.nets.nihr.ac.uk/projects/eme/1110029 [accessed 1 October 2014]. [Google Scholar]
- 20. Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY et al The gut microbiome modulates colon tumorigenesis. mBio 2013; 4: e00692–e00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV et al Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mirnezami R, Jiménez B, Li JV, Kinross JM, Veselkov K, Goldin RD et al Rapid diagnosis and staging of colorectal cancer via high‐resolution magic angle spinning nuclear magnetic resonance (HR‐MAS NMR) spectroscopy of intact tissue biopsies. Ann Surg 2014; 259: 1138–1149. [DOI] [PubMed] [Google Scholar]
- 23. de Wit M, Fijneman RJ, Verheul HM, Meijer GA, Jimenez CR. Proteomics in colorectal cancer translational research: biomarker discovery for clinical applications. Clin Biochem 2013; 46: 466–479. [DOI] [PubMed] [Google Scholar]
- 24. Markman B, Javier Ramos F, Capdevila J, Tabernero J. EGFR and KRAS in colorectal cancer. Adv Clin Chem 2010; 51: 71–119. [DOI] [PubMed] [Google Scholar]
- 25. Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF et al KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006; 66: 3992–3995. [DOI] [PubMed] [Google Scholar]
- 26. Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A et al Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360: 1408–1417. [DOI] [PubMed] [Google Scholar]
- 27. Kaplan R, Maughan T, Crook A, Fisher D, Wilson R, Brown L et al Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol 2013; 31: 4562–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Connell MJ, Lavery I, Yothers G, Paik S, Clark‐Langone KM, Lopatin M et al Relationship between tumor gene expression and recurrence in four independent studies of patients with stage II/III colon cancer treated with surgery alone or surgery plus adjuvant fluorouracil plus leucovorin. J Clin Oncol 2010; 28: 3937–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gray RG, Quirke P, Handley K, Lopatin M, Magill L, Baehner FL et al Validation study of a quantitative multigene reverse transcriptase–polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011; 29: 4611–4619. [DOI] [PubMed] [Google Scholar]
- 30.Lal N, Beggs AD, Willcox BE, Middleton GW. An immunogenomic stratification of colorectal cancer: implications for development of targeted immunotherapy. Oncoimmunology 2014; (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maletzki C, Schmidt F, Dirks WG, Schmitt M, Linnebacher M. Frameshift‐derived neoantigens constitute immunotherapeutic targets for patients with microsatellite‐instable haematological malignancies: frameshift peptides for treating MSI + blood cancers. Eur J Cancer 2013; 49: 2587–2595. [DOI] [PubMed] [Google Scholar]
- 32. Su Z, Gay LJ, Strange A, Palles C, Band G, Whiteman DC et al; Esophageal Adenocarcinoma Genetics Consortium; Wellcome Trust Case Control Consortium 2. Common variants at the MHC locus and at chromosome 16q24·1 predispose to Barrett's esophagus. Nature Genet 2012; 44: 1131–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang LD, Zhou FY, Li XM, Sun LD, Song X, Jin Y et al Genome‐wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nat Genet 2010; 42: 759–763. [DOI] [PubMed] [Google Scholar]
- 34. Wu C, Wang Z, Song X, Feng XS, Abnet CC, He J et al Joint analysis of three genome‐wide association studies of esophageal squamous cell carcinoma in Chinese populations. Nat Genet 2014; 46: 1001–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He Y, Ye L, Shan B, Song G, Meng F, Wang S. Effect of riboflavin‐fortified salt nutrition intervention on esophageal squamous cell carcinoma in a high incidence area, China. Asian Pac J Cancer Prev 2009; 10: 619–622. [PubMed] [Google Scholar]
- 36. Duan F, Xie W, Cui L, Wang P, Song C, Qu H et al Novel functional variants locus in PLCE1 and susceptibility to esophageal squamous cell carcinoma: based on published genome‐wide association studies in a central Chinese population. Cancer Epidemiol 2013; 37: 647–652. [DOI] [PubMed] [Google Scholar]
- 37. Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C et al Exome and whole‐genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013; 45: 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weaver JM, Ross‐Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M et al Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet 2014; 46: 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song Y, Li L, Ou Y, Gao Z, Li E, Li X et al Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014; 509: 91–95. [DOI] [PubMed] [Google Scholar]
- 40. Goh XY, Rees JR, Paterson AL, Chin SF, Marioni JC, Save V et al Integrative analysis of array‐comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut 2011; 60: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 41. Peters CJ, Rees JR, Hardwick RH, Hardwick JS, Vowler SL, Ong CA et al; Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Study Group. A 4‐gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 2010; 139: 1995–2004.e15. [DOI] [PubMed] [Google Scholar]
- 42. Bian YS, Osterheld MC, Fontolliet C, Bosman FT, Benhattar J. p16 inactivation by methylation of the CDKN2A promoter occurs early during neoplastic progression in Barrett's esophagus. Gastroenterology 2002; 122: 1113–1121. [DOI] [PubMed] [Google Scholar]
- 43. Moinova H, Leidner RS, Ravi L, Lutterbaugh J, Barnholtz‐Sloan JS, Chen Y et al Aberrant vimentin methylation is characteristic of upper gastrointestinal pathologies. Cancer Epidemiol Biomarkers Prev 2012; 21: 594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zou H, Molina JR, Harrington JJ, Osborn NK, Klatt KK, Romero Y et al Aberrant methylation of secreted frizzled‐related protein genes in esophageal adenocarcinoma and Barrett's esophagus. Int J Cancer 2005; 116: 584–591. [DOI] [PubMed] [Google Scholar]
- 45. Singhal R, Carrigan JB, Wei W, Taniere P, Hejmadi RK, Forde C. MALDI profiles of proteins and lipids for the rapid characterisation of upper GI‐tract cancers. J Proteomics 2013; 80C: 207–215. [DOI] [PubMed] [Google Scholar]
- 46. Hu Y, Bandla S, Godfrey TE, Tan D, Luketich JD, Pennathur A et al HER2 amplification, overexpression and score criteria in esophageal adenocarcinoma. Mod Pathol 2011; 24: 899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A et al Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010; 376: 687–697. [DOI] [PubMed] [Google Scholar]
- 48. Ku GY, Ilson DH. Emerging tyrosine kinase inhibitors for esophageal cancer. Expert Opin Emerg Drugs 2013; 18: 219–230. [DOI] [PubMed] [Google Scholar]
- 49. Elgazzar S, Zembutsu H, Takahashi A, Kubo M, Aki F, Hirata K et al A genome‐wide association study identifies a genetic variant in the SIAH2 locus associated with hormonal receptor‐positive breast cancer in Japanese. J Hum Genet 2012; 57: 766–771. [DOI] [PubMed] [Google Scholar]
- 50. Fletcher O, Johnson N, Orr N, Hosking FJ, Gibson LJ, Walker K et al Novel breast cancer susceptibility locus at 9q31·2: results of a genome‐wide association study. J Natl Cancer Inst 2011; 103: 425–435. [DOI] [PubMed] [Google Scholar]
- 51. Rinella ES, Shao Y, Yackowski L, Pramanik S, Oratz R, Schnabel F et al Genetic variants associated with breast cancer risk for Ashkenazi Jewish women with strong family histories but no identifiable BRCA1/2 mutation. Hum Genet 2013; 132: 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beggs AD, Hodgson SV. Genomics and breast cancer: the different levels of inherited susceptibility. Eur J Hum Genet 2009; 17: 855–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pharoah PD, Antoniou AC, Easton DF, Ponder BA. Polygenes, risk prediction, and targeted prevention of breast cancer. N Engl J Med 2008; 358: 2796–2803. [DOI] [PubMed] [Google Scholar]
- 54. Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C et al Drug‐sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A 2008; 105: 8713–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Garcia‐Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN et al Genome‐wide association studies identify four ER negative‐specific breast cancer risk loci. Nat Genet 2013; 45: 392–398, 398.e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ghoussaini M, Fletcher O, Michailidou K, Turnbull C, Schmidt MK, Dicks E et al Genome‐wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet 2012; 44: 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Network Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y et al The clonal and mutational evolution spectrum of primary triple‐negative breast cancers. Nature 2012; 486: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Banerji S, Cibulskis K, Rangel‐Escareno C, Brown KK, Carter SL, Frederick AM et al Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012; 486: 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC et al The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486: 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF et al Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013; 368: 1199–1209. [DOI] [PubMed] [Google Scholar]
- 62. Pegolo E, Puppin C, Gerometta A, Damante G, Puglisi F, Di Loreto C. One‐step nucleic acid amplification (OSNA) for intraoperative evaluation of sentinel lymph node status in breast cancer: a comparative study between CK19 protein expression and CK19 mRNA level in primary tumors and lymph node metastasis. Virchows Arch 2013; 463: 7–15. [DOI] [PubMed] [Google Scholar]
- 63. Klingler S, Marchal F, Rauch P, Kenouchi O, Chretien AS, Genin P et al Using one‐step nucleic acid amplification (OSNA) for intraoperative detection of lymph node metastasis in breast cancer patients avoids second surgery and accelerates initiation of adjuvant therapy. Ann Oncol 2013; 24: 2305–2309. [DOI] [PubMed] [Google Scholar]
- 64. Mulligan JM, Hill LA, Deharo S, Irwin G, Boyle D, Keating KE et al Identification and validation of an anthracycline/cyclophosphamide‐based chemotherapy response assay in breast cancer. J Natl Cancer Inst 2014; 106: djt335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ma CX, Suman VJ, Goetz M, Haluska P, Moynihan T, Nanda R et al A phase I trial of the IGF‐1R antibody cixutumumab in combination with temsirolimus in patients with metastatic breast cancer. Breast Cancer Res Treat 2013; 139: 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB et al Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–921. [DOI] [PubMed] [Google Scholar]
- 67. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN et al Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet 2010; 376: 235–244. [DOI] [PubMed] [Google Scholar]
- 68. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K et al Olaparib in patients with recurrent high‐grade serous or poorly differentiated ovarian carcinoma or triple‐negative breast cancer: a phase 2, multicentre, open‐label, non‐randomised study. Lancet Oncol 2011; 12: 852–861. [DOI] [PubMed] [Google Scholar]
- 69. Greenup R, Buchanan A, Lorizio W, Rhoads K, Chan S, Leedom T et al Prevalence of BRCA mutations among women with triple‐negative breast cancer (TNBC) in a genetic counseling cohort. Ann Surg Oncol 2013; 20: 3254–3258. [DOI] [PubMed] [Google Scholar]
- 70. Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y et al Everolimus plus exemestane for hormone‐receptor‐positive, human epidermal growth factor receptor‐2‐negative advanced breast cancer: overall survival results from BOLERO‐2. Ann Oncol 2014; [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Habr‐Gama A, Perez R, Proscurshim I, Gama‐Rodrigues J. Complete clinical response after neoadjuvant chemoradiation for distal rectal cancer. Surg Oncol Clin N Am 2010; 19: 829–845. [DOI] [PubMed] [Google Scholar]
- 72. Genomics England . The 100 000 Genomes Project. http://www.genomicsengland.co.uk/the‐100000‐genomes‐project/ [accessed 1 October 2014]. [Google Scholar]
- 73. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Beggs AD, Domingo E, Abulafi M, Hodgson SV, Tomlinson IP. A study of genomic instability in early preneoplastic colonic lesions. Oncogene 2013; 32: 5333–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]