

Abstract
Stimulator of interferon genes (STING) is an important regulator of the innate immune response to cytoplasmic DNA. However, regulation of STING itself is largely unknown. Here, we show that STING transcription is induced by innate immune activators, such as cyclic dinucleotides (CDNs), through an IFNAR1- and STAT1-dependent pathway. We also identify a STAT1 binding site in the STING promoter that contributes to the activation of STING transcription. Furthermore, we show that induction of STING mediates the positive feedback regulation of CDN-triggered IFN-I. Thus, our study demonstrates that STING is an interferon-stimulated gene (ISG) and its induction is crucial for the IFN-I positive feedback loop.
Keywords: CDN, cGAMP, IFN, ISG, STING
Introduction
Stimulator of interferon genes (STING, also named MITA, MYPS, or ERIS) plays a critical role in sensing pathogenic nucleic acid as an adapter protein or as a DNA sensor 1, 2, 3, 4, 5, 6, 7. STING not only can directly complex with DNA such as cyclic dinucleotides (CDNs) and pathogen-related ssDNA and dsDNA 6, 7, 8, but also can interact with other DNA sensors such as gamma-interferon-inducible protein16 (IFI16) and DEAD (Asp-Glu-Ala-Asp) box polypeptide 41 (DDX41) 9, 10. Following activation by pathogenic DNAs or its upstream DNA sensors, STING translocates with TANK-binding kinase 1 (TBK1) to perinuclear endosomes, leading to the activation of interferon regulatory factors (IRFs) and NF-κB, which triggers the expression of type I interferon (IFN-I) and other immune response genes 1, 2, 5. STING has emerged as central for DNA-induced IFN-I activation through the STING–TBK1–IRF3 signaling axis 1, 2, 3, 4, 5, 6, 7, 8, 9, 10. Although STING-dependent IFN-I pathway has been extensively studied, it is largely unknown how STING is regulated, particularly at the transcriptional level.
Rapid and robust induction of IFN-I is a critical event during viral and bacterial infections 11, 12. The initial production of IFN-I is further regulated by a positive feedback loop that is based on the ability of IFNβ and IFNα4 to induce numerous IFN-stimulated genes (ISGs). For example, induction of cytosolic RNA and DNA sensors such as RIG-I, MDA5, and IFI16 enhances the induction of IFN-I by sensing more pathogen-derived nucleic acids 12, 13, 14. In addition, induction of transcription factor (TF) IRF7 drives numerous of IFNα genes expression and initiates IFN-I positive feedback through the well-established IRF3–IFNβ–IRF7–IFNα/β signaling axis 15, 16, 17. Both the first wave induction and the subsequent positive feedback regulation of IFN-I are important for host innate immune responses. Several CDNs that can trigger IFN-I have been identified in recent studies 6, 7, 18. Cyclic diguanosine monophosphate (c-di-GMP) and cyclic diadenosine monophosphate (c-di-AMP) are two key secondary messengers with essential roles in regulating bacterial metabolism, motility, and virulence 19, 20. Cyclic GMP–AMP (cGAMP) is a metazoan endogenous second messenger derived from cytoplasmic DNA via the synthase activity of cyclic GMP–AMP synthase (cGAS) 6, 21. All these CDNs produced by bacteria and cellular cGAS could trigger IFN-I production through the STING-dependent pathway 6, 7, 18. However, it is unclear whether the positive feedback loop is required for CDNs-triggered IFN-I production.
Here, our data indicate that STING expression could be induced by IFN-I via a STAT1 binding site in its promoter region; positive feedback regulation loop is required for optimal production of CDNs-triggered IFN-I. Furthermore, we show that induction of STING by the first wave of IFN-I plays a role in the subsequent positive feedback regulation of CDNs-triggered IFN-I production, and IRF7-dependent IFNα production further amplifies STING induction at the late stage.
Results and Discussion
STING is an ISG
By analyzing the gene expression profile of the IFN-I- and IFN-II-stimulated bone marrow-derived macrophages (BMMs) 14, we found that STING mRNA expression was significantly upregulated in IFNα-treated BMMs, as the well-known IFN-inducible TF IRF7 (Fig1A). However, other genes in the downstream of STING signaling pathway, such as Tbk1, Irf3, Ifnar1, and Ifnar2, could not be induced by IFNα (Fig1A and Supplementary Fig S1A). Consistent with the STING mRNA expression data, STING protein level was significantly elevated in IFNα-treated BMMs (Fig1B). STING also could be induced by IFNα in human macrophages and dendritic cells (DCs) (Fig1C and D). To further determine whether STING is an ISG, we transfected polyI:C and polydA:dT, the mimics of pathogen-derived nucleic acid which trigger large amount of IFN-I production in immune cells, into different BMMs and compared the STING expression. Higher STING mRNA was detected in WT BMMs than Ifnar1−/− BMMs activated by either polyI:C or polydA:dT (Fig1E). In addition, polyI:C triggered more STING transcripts in WT BMMs than Trif−/− BMMs, while polydA:dT triggered similar amount of STING mRNA in WT and Trif−/− BMMs (Fig1F). Given that polyI:C-triggered IFN-I is mainly TRIF dependent while polydA:dT activates IFN-I in a TRIF-independent manner 22, our data here suggest that induction of STING by polyI:C or polydA:dT requires IFN-I production and its downstream signaling. Furthermore, we found that cGAMP induced STING expression in a dose-dependent manner in WT BMMs but not in Ifnar1−/− cells (Fig1G). Consistently, STING protein was induced by cGAMP in WT BMMs but not in Ifnar1−/− BMMs (Fig1H). Interestingly, there is no upregulation of STING mRNA in LPS-stimulated WT BMMs, though LPS also could trigger IFN-I production via TRIF-dependent pathway (Fig1I). LPS induced STING mRNA in Myd88−/− BMMs in a time-dependent manner (Fig1J), which implied that LPS could suppress STING expression through the MyD88-dependent pathway and induce STING expression through the TRIF-dependent pathway. Furthermore, by analyzing the STING mRNA level in LPS-activated WT and Trif−/− BMMs, we found that STING expression was suppressed by LPS after 4 h stimulation and recovered after 12 h stimulation in WT BMMs, while STING mRNA was suppressed at both time points examined in Trif−/− BMMs (Fig1J and Supplementary Fig S1B). The TLR2 ligand, Pam3CSK4, activates MyD88 pathway but does not trigger IFN-I production 23. STING expression was suppressed in Pam3CSK4-activated WT BMMs but not in Myd88−/− BMMs (Supplementary Fig S1C), which further suggested that STING could be suppressed by MyD88-dependent signaling. Taken together, we have demonstrated that STING transcripts could be induced by IFN-I and most of the ligands which could trigger IFN-I in BMMs, therefore demonstrating that STING is an ISG. LPS plays dual roles in controlling STING expression. In LPS-activated BMMs, TRIF-dependent signaling induces STING expression by triggering IFN-I production, while MyD88-dependent pathway suppresses STING expression via an unknown mechanism.
Figure 1.

- BMMs were treated with 62.5 U/ml IFNα for 2.5 h. RNA was extracted and gene expression profile was detected by Affymetrix 430.2 Chip. Sting, Irf3, and Irf7mRNA levels are shown as probe intensity from microarray.
- BMMs were treated with 100 U/ml IFNα for the indicated time, and the protein level of Sting in these cells was detected by WB. α-tubulin are shown as a loading control.
- Human macrophage (C) and dendritic cells (D) were treated with 100 U/ml recombinant human IFNα for the indicated time, and STING mRNA level in these cells was detected by qPCR and normalized to RPL32.
- WT and Ifnar1−/− (E) or Trif−/− (F) BMMs were transfected with 1 μg/ml polyI:C or polydA:dT for 4 h, and StingmRNA level in these cells was detected by qPCR and normalized to Rpl32.
- WT and Ifnar1−/− BMMs were transfected with the indicated amount of cGAMP for 4 h, and StingmRNA from these cells was detected by qPCR and normalized to Rpl32.
- WT and Ifnar1−/− BMMs were transfected with 3 μg/ml cGAMP for the indicated times, Sting protein level was detected by WB, and α-tubulin are shown as a loading control.
- WT and Myd88−/− BMMs were stimulated with 100 ng/ml LPS for 4 h or 12 h, and supernatant IFNβ was measured by ELIAS (I). StingmRNA from these cells was detected by qPCR and normalized to Rpl32 (J).
Data information: Data of (A, C–G, I, J) are from three independent experiments (mean ± s.e.m.), *P < 0.05, **P < 0.01 (Student's t-test). Data of (B) and (H) are from one representative experiment. Similar results were obtained in three independent experiments.
Several studies have described the posttranslational modifications of STING. Phosphorylation of STING on Ser358 by TBK1 is required for STING-mediated activation of IRF3 5. However, phosphorylation of STING at S366 by UNC-51-like kinase (ULK1) suppressed IRF3 activation 24. K63-linked ubiquitination of STING by TRIM56 and TRIM32 induced STING dimerization, which is a prerequisite for recruitment of TBK1 and subsequent induction of IFN-I 25, 26. However, K48-linked ubiquitination of STING by RNF5 mediates degradation of STING 27. Besides these posttranslational regulations of STING, our results here have added a new layer regulation of STING at transcription level.
A STAT1 binding site is critical for induction of STING by IFN-I
To determine how STING is induced by IFN-I, we analyzed the potential TF binding sites in STING 5'-UTR region. Mouse Sting locates in chromosome 18 and is encoded by the negative strand of DNA (Fig2A). Among all the predicted TF binding sites around the Sting transcription start site (TSS), there are two potential STAT1 binding sites accounting for induction of STING by IFN-I. Both sites got high matrix score in the TRANSFAC software 28, 29, and the sequences of the two STAT1 binding sites are conserved in mouse, rat, and human STING (Fig2B). To verify Stat1 binding at the Sting promoter, we downloaded and processed Stat1 ChIP-seq (chromatin immunoprecipitation sequencing) data in BMMs 30, 31. Interestingly, we found a significant Stat1 binding peak around the Stat1#1 region of the Sting promoter in the BMMs treated with IFNβ and IFNγ (Fig2C). In addition, we observed the dynamic change of the Stat1 binding in this region in BMMs treated with IFNγ for different time points (Supplementary Fig S2A). We further verified the ChIP-Seq data by ChIP-qPCR assay and detected significantly higher Stat1 binding in IFNα- or IFNγ-treated BMMs comparing with the untreated BMMs (Fig2D). Three reporter constructs were made to verify the potential function of the two STAT1 binding sites (Fig2E). IFNα activated both WT and Δ#2 luciferase reporters, but not the Δ#2-mut#1 reporter in RAW264.7 cells (Fig2F), which suggested that Stat1#1 binding site was a major site of STING regulated by IFN-I. IPS1, TBK1, and IRF1 trigger IFN-I production in HEK293T cells 32, 33. Consistent with the results from RAW264.7 cells, all of the IPS1, TBK1, and IRF1 could activate WT and Δ#2 luciferase reporters, but not the Δ#2-mut#1 reporter in HEK293T cells (Fig2G). Furthermore, IFNα treatment or STAT1 transfection plus IFNα treatment could significantly activates WT and Δ#2 luciferase reporters, but not the Δ#2-mut#1 reporter in HEK293T cells (Fig2H). Considering that STAT1 is a common TF activated by both IFN-I and IFN-II, we checked the STING mRNA in IFNγ-stimulated BMMs. IFNγ stimulation also significantly induced STING mRNA in BMMs (Supplementary Fig S2B) and THP1 cells (Supplementary Fig S2C), which further suggested that activation of STAT1 was required for induction of STING. Our data therefore suggest that both IFN-I and IFN-II can induce STING expression at transcription level through a STAT1 binding site in the STING promoter.
Figure 2.

- The chromosome location of Sting and its nearby genes. The diagram was modified from NCBI gene (Gene ID: 72512).
- The potential Stat1 binding sites in the promoter of Sting. Transcription factor binding site prediction was performed by TRANSFAC. The location of the Stat1 binding sites and the matrix score are shown. The conservation comparison of the predicted Stat1 binding sites between human, rat, and mouse was according to the sequence from Ensembl Genome Browser. TSS, transcription start site; CDS, coding DNA sequence.
- BMMs were treated with 100 U/ml IFNβ and IFNγ for 6 h, Stat1 ChIP-Seq data were analyzed, and the Stat1 binding region in Sting promoter is shown. The Stat1 ChIP-Seq raw data were downloaded from GEO (accession no. GSE33913).
- BMMs were treated with 100 U/ml IFNα or IFNγ for 2 h, and the binding of Sting promoter region with Stat1 (or IgG) was detected by ChIP-qPCR. The data are shown as fold change of Stat1/IgG.
- The sequence of the mutated Stat1#1 binding site and the schematic diagram of the Sting promoter reporter plasmids.
- Control vector (pGL4.2) and indicated Sting promoter reporter constructs expressing firefly luciferase were transfected into RAW264.7 cells by nucleofection system. pRL-TK-luc vector expressing Renilla luciferase was co-transfected as a control for transfection efficiency. Data are shown as the relative luciferase activity.
- Flag, IPS1, TBK1, or IRF1 was co-transfected with the indicated Sting promoter reporter constructs and pRL-TK-luc vector into HEK293T cells. Data are shown as the relative luciferase activity.
- Flag or STAT1 was co-transfected with the indicated Sting promoter reporter constructs and pRL-TK-luc vector into HEK293T cells for 12 h, and these cells were treated with control or 500 U/ml IFNα for another 12 h. Firefly and Renilla luciferase activities were measured and relative luciferase activity are shown.
Data information: Data of (D) are from three independent experiments (mean ± s.e.m.), **P < 0.01 (Student's t-test). Data of (F–H) are from one representative experiment (mean ± s.d., n = 6), *P < 0.05, **P < 0.01 (Student's t-test). Similar results were obtained in three independent experiments.
Activation of IFNAR by IFN-I leads to the formation of STAT1–STAT2–IRF9 (known as ISGF3, IFN-stimulated gene factor 3) complexes and STAT1–STAT1 homodimers, which bind to IFN-stimulated response elements (ISREs) and IFNγ-activated site (GAS) elements, respectively. Activation of IFNγ receptor (IFNGR) by IFNγ only triggers dimerization of STAT1 and induces the genes with GAS elements in their promoters 34. “TTC/ANNNG/TAA” is the typical GAS motif 34. The Stat1#1 site in the Sting promoter harbors the core sequence “TTCGGGGAA”, a GAS motif. This motif gives the reason for why STING could be induced by both IFN-I and IFN-II. Considering that IFNγ treatment only leads to dimerization of STAT1, while IFNα or IFNβ treatment results in two STAT1-containing complexes, it is reasonable that IFNγ induces slightly more STING transcripts and triggers more STAT1 binding in the STING promoter region than IFN-I, as the results shown in Fig2D and Supplementary Fig S2C.
Optimal production of CDNs-triggered IFN-I requires IFNAR signaling
In response to cytosolic DNA, host cells produce cGAMP to trigger IFN-I production utilizing the STING–TBK1–IRF3 signaling axis 6. Interestingly, more induction of IFNβ and IFNα4 transcripts was detected in cGAMP-activated WT BMMs than Ifnar1−/− BMMs (Fig3A and B). Consistently, higher supernatant IFNβ and IFNα were detected in cGAMP-activated WT BMMs than Ifnar1−/− BMMs (Fig3C and D). Similar studies in J2 virus-immortalized macrophage cell line (J2-BMMs) supported that cGAMP-induced IFN-I transcripts and protein production were positively regulated by IFNAR1 and STAT1 (Fig3E–H), which suggested that optimal production of cGAMP-triggered IFN-I requires IFNAR signaling.
Figure 3.

- WT or Ifnar1−/−BMMs were transfected with the indicated amount of cGAMP for 4 h (A, B) or 16 h (C, D). Ifnb (A) and Ifna4 (B) mRNA levels from BMMs transfected with cGAMP for 4 h were detected by qPCR. IFNβ (C) and IFNα (D) in the supernatant of BMMs transfected with cGAMP for 16 h were measured by ELISA.
- WT, Ifnar1−/−, and Stat1−/− J2-BMMs were transfected with 3 μg/ml cGAMP for 4 h (E, F) or 16 h (G, H). Ifnb (E) and Ifna4 (F) mRNA levels from BMMs transfected with cGAMP for 4 h were detected by qPCR. IFNβ (G) and IFNα (H) in the supernatant of BMMs transfected with cGAMP for 16 h were measured by ELISA.
- BMMs were pretreated with 250 U/ml or 1,000 U/ml IFNα for 2 h, and the cells were transfected with 3 μg/ml cGAMP for 4 h. Ifnb (I) and Ifna4 (J) from these BMMs were measured by qPCR.
- BMMs were pretreated with 250 U/ml or 1,000 U/ml IFNα for 2 h, and the cells were transfected with 0.5 or 2 μg/ml cGAMP for 16 h; IFNβ in the supernatant was measured by ELISA.
Data information: **P < 0.01 (Student's t-test). Data are from three independent experiments (mean ± s.e.m.).
To further determine the requirement of IFNAR signaling in cGAMP-triggered IFN-I induction, BMMs were pretreated with IFNα and then activated by cGAMP. Priming of BMMs with IFNα significantly enhanced cGAMP-mediated induction of IFNβ and IFNα4 transcripts in a dose-dependent manner (Fig3I and J). Similarly, significantly higher IFNβ protein was produced after IFNα pretreatment, both by low-dose and high-dose cGAMP-transfected BMMs (Fig3K).
Bacterial CDNs such as c-di-AMP and c-di-GMP are also able to elicit a host IFN-I innate immune response 7, 18. Transfection with c-di-AMP and c-di-GMP triggers IFNβ mRNA expression in a dose-dependent manner (Supplementary Fig S3A and B). Induction of IFNβ transcript and protein by either c-di-AMP or c-di-GMP was impaired in Ifnar1−/− BMMs and J2-BMMs (Supplementary Fig S3). Taken together, these data indicated that optimal production of both endogenous and bacterial CDNs-triggered IFN-I requires IFNAR signaling, which suggested that the positive feedback loop played a role in the CDNs-triggered IFN-I. Given that the induction of cytosolic RNA and DNA sensors such as RIG-I, MDA5, and IFI16 by IFN-I positively regulates IFN-I production by sensing more pathogen nucleic acids 12, 13, 14, we hypothesized that the induction of STING could increase the host's ability of sensing CDNs and positively regulate CDNs-triggered IFN-I production.
IRF7-dependent IFNα mediates the late stage of STING induction in cGAMP-activated BMMs
IRF7 is a well-known ISG that mediates IFN-I positive feedback loop mainly by inducing IFNα during viral infection 15, 16, 17. Therefore, we tested whether induction of STING by cGAMP requires IRF7 and whether the classical IRF3–IFNβ–IRF7–IFNα/β signaling axis plays a role in the positive feedback loop of cGAMP-triggered IFN-I production. Firstly, we found that IRF3 expression determined basal and inducible level of IRF7 in untreated and cGAMP-activated J2-BMMs (Fig4A), which supported that IRF7 was an ISG and induction of IRF7 was IRF3 dependent in cGAMP-activated macrophages. To test whether IRF7 is required for cGAMP-triggered IFN-I induction, we compared the induction of IFNβ and IFNα4 in WT, Irf3−/−, and Irf7−/− J2-BMMs triggered by cGAMP. Significantly attenuated induction of IFNβ mRNA was observed in Irf3−/− but not in Irf7−/− J2-BMMs activated by transfection of cGAMP for 4 h, while IFNα4 mRNA induction was impaired in both Irf3−/− and Irf7−/− J2-BMMs (Fig4B and C). Significantly less supernatant IFNβ and IFNα were detected in Irf3−/− and Irf7−/− J2-BMMs activated by transfection of cGAMP for 16 h, although the downregulation of IFNβ was not as dramatic as IFNα in Irf7−/− J2-BMMs (Fig4D and E). Consistent with the IFN-I production results, induction of STING was impaired in Irf3−/− J2-BMMs activated by transfection of cGAMP for 4 h and 16 h, while modest but significant downregulation of STING induction was observed in Irf7−/− J2-BMMs activated by transfection of cGAMP for 16 h (Fig4F). These data indicate that IRF7 is critical for cGAMP-triggered IFNα production and modestly regulates IFNβ production at the late stage. However, IRF7 is dispensable for early stage of IFNβ induction in cGAMP-activated macrophages. Classic IFN-I positive feedback loop via IRF3–IFNβ–IRF7–IFNα/β signaling plays a role in cGAMP-triggered total IFN-I production. However, considering that induction of STING is IFNAR dependent but IRF7 independent at the early stage of cGAMP transfection, induction of DNA sensors such as STING by the first wave production of IFN-I is an alternative mechanism for IFN-I positive feedback, particularly at the early stage.
Figure 4.

- WT, Irf3−/−, or Irf7−/− J2-BMMs were transfected with 3 μg/ml cGAMP for 4 h. Irf7 (A), Ifnb (B), and Ifna4 (C) mRNA levels in these cells were detected by qPCR and normalized to Rpl32.
- WT, Irf3−/−, or Irf7−/− J2-BMMs were transfected with 3 μg/ml cGAMP for 16 h, and IFNβ (D) and IFNα (E) in the supernatant of these cells were measured by ELISA.
- WT, Irf3−/−, or Irf7−/− J2-BMMs were transfected with 3 μg/ml cGAMP for the indicated times, and StingmRNA level in these cells was detected by qPCR and normalized to Rpl32.
Data information: Data are from three independent experiments (mean ± s.e.m.), *P < 0.05, **P < 0.01 (Student's t-test).
Overexpression of STING abolishes the difference of CDNs-triggered IFN-I production between WT and Ifnar1−/− BMMs
To determine whether the induction of STING by IFN-I plays a role in the positive feedback loop of CDNs-triggered IFN-I production, we overexpressed mouse Sting in both WT and Ifnar1−/− J2-BMMs by lentiviral gene transduction to get a similar level of Sting expression in these cells. Sting mRNA was elevated dramatically after transducing with Sting-overexpressing lentiviruses, and the expression levels of Sting were similar between Sting-overexpressing WT and Ifnar1−/− J2-BMMs (Fig4A). In addition, the Sting protein level is comparable between J2-BMMs transduced with LV-Sting and BMMs triggered by cGAMP (Supplementary Fig S4A). Considering the almost-saturated Sting expression in these Sting-overexpressing cells, CDNs-triggered IFN-I may not affect the STING expression in these cells. We found less difference of cGAMP-triggered IFNβ production between Sting-overexpressing WT and Ifnar1−/− J2-BMMs, comparing to LV-Empty transduced WT and Ifnar1−/− J2-BMMs (Fig4B and C). Overexpression of Sting rescues the impaired cGAMP-triggered IFNβ production but not the defect of cGAMP-triggered IFNα production in Irf7−/− macrophages (Supplementary Fig S4B–D). These results indicated that overexpression of Sting rescued the defect of IFNβ production in Ifnar1−/− macrophages and reduced the difference of CDNs-triggered IFNβ production between WT and Ifnar1−/− J2-BMMs, which suggested that induction of Sting by IFN-I contributed to the positive feedback loop of IFN-I production. Classical IRF3–IFNβ–IRF7–IFNα/β signaling pathway plays a role at the late stage of STING induction by triggering IFNα production, which accounts for how IRF7 and STING work together to play roles in type I IFN positive feedback loop (Fig5D).
Figure 5.

- WT and Ifnar1−/− J2-BMMs were transduced with empty or Sting-expressing lentivirus (LV-Empty or LV-Sting) for 3 days. StingmRNA was detected by qPCR.
- WT and Ifnar1−/− J2-BMMs were transduced with LV-Empty or LV-Sting for 3 days; then, the medium was replaced and the cells were left untreated or transfected with 3 μg/ml cGAMP for another 16 h. IFNβ in the supernatant of these cells was measured by ELISA.
- J2-BMMs were treated and induction of IFNβ was measured as described in (B). IFN-β/(IFN-β)WT in percent was calculated and compared for LV-Empty- and LV-STING-transduced cGAMP-activated cells.
- Induction of STING plays a role in the positive feedback loop of CDNs-triggered IFN-I production. Viral DNA such as HSV and HIV DNA is recognized and converted into cGAMP by cGAS. Bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis can produce the other CDNs, for example, c-di-AMP and c-di-GMP. These CDNs are recognized by STING and trigger the STING–TBK1–IRF3 signaling axis to produce the first wave of IFN-I. Initial production of IFN-I induces the IFN-stimulated gene, STING, via the IFNAR signaling, which further amplifies the CDNs recognition and the IFN-I production, as shown in step (1). In addition, induction of transcription factor IRF7 by first wave IFN-I drives the production of multiple IFNα, which mediates the further induction of STING and IFNα at the later stage, as shown in step (2).
Data information: Data of (A–C) are from three independent experiments (mean ± s.e.m.), **P < 0.01 (Student's t-test).
In summary, our study not only has demonstrated a novel mechanism by which STING is induced by IFN-I via a STAT1 binding site in the promoter region of STING, but also suggested a role of the induction of STING in IFN-I positive feedback regulation loop. Although we have described a new layer regulation of STING at transcription level other than the previous reported regulation at posttranslational level 5, 24, 25, 26, 27, it seems that the regulation of STING during innate immune responses is much more complicated than we think. LPS positively regulates STING expression via TRIF-dependent signaling while negatively suppresses STING expression through MyD88-dependent pathway. Our results indicate that cGAMP induces STING mRNA and protein in macrophage by triggering IFN-I production, while cGAMP destabilizes STING protein by triggering ULK1 phosphorylation in primary MEF and hTERT-BJ1 cells 24. These data suggested that STING is fine-regulated during host innate immune responses. Activation of STING by CDNs results in the induction of numerous genes that suppress pathogen replication and facilitate adaptive immunity 1, 2, 3, 4, 5. However, persistent transcription of innate immune genes causes inflammatory disorders and autoimmune diseases 35, 36, 37. Thus, STING expression should be tightly regulated at multiple levels to maintain functional host innate immune responses and homeostasis.
IRF3–IFNβ–IRF7–IFNα/β signaling axis is a well-established loop for IFN-I positive feedback during viral infections. In our study, we show that induction of STING is IRF3 dependent but IRF7 independent in cGAMP-activated BMMs at the early stage, which suggest that STING-dependent IFN-I positive feedback loop is independent of the classical IRF7-dependent IFN-I positive feedback pathway, particularly at early stage. It is interesting to investigate whether other IFN-inducible DNA sensors and RNA sensors such as cGAS and RIG-I mediate IFN-I positive feedback independent of IRF3–IFNβ–IRF7–IFNα/β axis or not. Our study here focuses on the positive regulation of STING expression, and we suggest that this positive regulation leads to more induction of CDNs-triggered IFN-I. It is interesting that STING-dependent IFN-I production facilitates host clearance of VSV and HSV-1 infections while inhibits cell-mediated immunity to Listeria monocytogenes infection 1, 2, 5, 38. However, we have no evidence to demonstrate the roles of STING-dependent type I IFN positive feedback loop in host innate immune responses against certain pathogens, and also the roles of this loop in inflammatory disorders and autoimmune diseases. Further experiments are required for fully understanding the physiological or pathophysiological significance of the mechanism proposed by this study.
Materials and Methods
Mice and reagents
Wild-type C57BL/6 (6–8 weeks of age) and age-matched Ifnar1−/−, Stat1−/−, MyD88−/−, and Trif−/− male mice were purchased from Jackson Laboratory. Irf3−/− mice were from Dr. Tadatsugu Taniguchi's laboratory (University of Tokyo). All mice experiments were performed in accordance with guidelines from the University of California, Los Angeles, Institutional Animal Care and Use Committee. TLR2 ligand Pam3CSK4, cGAMP, c-di-AMP, c-di-GMP, polyI:C, and polydA:dT were purchased from InvivoGen (San Diego, CA). TLR4 ligand LPS (Escherichia coli 0111:B4) and anti-α-tubulin antibody were from Sigma-Aldrich (St. Louis, MO). Anti-STING antibody (#3337) was from Cell Signaling Technology (Danvers, MA). Recombinant human and mouse IFNα was from PBL interferon source (Piscataway, NJ), and recombinant mouse IFNγ was from R&D systems (Minneapolis, MN).
Cell culture and activation
HEK293T, RAW264.7, and THP-1 cell lines were obtained from American Type Culture Collection (Manassas, VA). HEK293T and RAW264.7 cells were maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin. THP-1 cells were cultured in RPMI1640 supplemented with 10% FBS and 1% penicillin/streptomycin. For human macrophage and DC differentiation, CD14+ human PBMCs purified from whole blood (obtained from heath donors, with informed consent, UCLA I.R.B. #92-10-591-31) were cultured for 7 days with 800 U/ml GM-CSF (Immunex) and 500 U/ml IL-4 (Peprotech) for DC differentiation, or with 50 ng/ml M-CSF (R&D Systems) for macrophage differentiation. Cytokines were replenished at day 3, and the media was replaced on day 7 prior to activation of the cells. For mice bone marrow-derived macrophage (BMM) differentiation, bone marrow was harvested from wild-type or indicated knockout mice and differentiated in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 10 ng/ml of M-CSF for 7 days. The media was replaced on day 3 and day 6, and the cells were used for experiments as BMMs on day 7. For J2 virus-immortalized macrophages (J2-BMMs), a cell line transformed by retrovirus expressing v-raf and c-myc was established (called GG2EE) and grown in RPMI1640 (10 mM HEPES pH 7.8, 10% FBS, 1% penicillin/streptomycin). The supernatant containing J2 viruses was harvested and filtered through 0.22-μM filter. Bone marrow cells were infected with the J2 virus and immortalized as described previously 39, 40. Femur and tibia from Irf7−/− mice (8 weeks, male, C57BL/6 background) were overnight-shipped from Michael S. Diamond's laboratory (Washington University). Irf7−/− bone marrow cells were differentiated into BMMs and immortalized to Irf7−/− J2-BMMs. To activate BMMs or J2-BMMs, 100 ng/ml LPS was added into culture medium, or an indicated amount of cGAMP, c-di-AMP, c-di-GMP, polyI:C, or polydA:dT was transfected into cells by Lipofectamine 2000 (Life Technologies). The ratio of transfection reagent to ligands was 2.5 (μl/μg). Detailed Lipofectamine 2000 transfection protocol was followed as described in the manufacturer's instructions.
RNA isolation and quantitative PCR (qPCR)
Total RNA was extracted with TRIzol reagent (Life Technologies) according to the manufacturer's instructions. 1 μg RNA from each sample was reverse-transcribed by using iScript One-Step RT-PCR Kit with SYBR Green (Bio-Rad). Real-time quantitative RT–PCR analysis was performed by using SensiFAST SYBR & Fluorescein Kit (Bioline) and the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). The relative mRNA expression level of genes was normalized to the internal control ribosomal protein gene Rpl32 by using 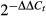 cycle threshold method 41. The primer sequences for qPCR were from primer bank 42, and they are available upon request.
cycle threshold method 41. The primer sequences for qPCR were from primer bank 42, and they are available upon request.
Microarray
On day 7, the wild-type BMMs were stimulated with 62.5 U/ml IFNα or 1 U/ml IFNγ for 2.5 h, and the total RNA was extracted for microarray analyses. Microarrays were done on the Affymetrix 430.2 Chip (University of California, Los Angeles Genotyping and Sequencing Core). The microarray data analysis was performed using GeneSpring software (Agilent Technologies) 14. The microarray data have been deposited in the Gene Expression Omnibus (GEO) database (accession no. GSE35825).
ChIP-Seq data analysis and Stat1 ChIP
Stat1 ChIP-Seq raw data for BMMs were downloaded from the GEO (accession no. GSE33913). BMMs cells differentiation and activation were described previously 30. Sequenced reads were aligned to mouse genome (mm9) allowing up to two mismatches using Bowtie 43. The data were processed as previously described 44. For peak calling, mouse genome was divided into 100-bp windows. A P-value for Poisson distribution of enriched ChIPed DNA over input DNA for each window was calculated. Significant peaks were defined as the windows with significant P-value less than 10−3 and with two neighboring windows at the same significance. Stat1 ChIP assays were performed according to the protocol of the ChIP assay kit (Magna ChIP™ HiSens kit, catalog No. 17-10460, Millipore). Anti-STAT1 antibody (sc-592x) from Santa Cruz Biotechnology was used for ChIP assay. Primer pair specific for the STING promoter Stat1#1 region was 5′-TTGGCTATCTGGACCTGGAC-3′ (forward) and 5′-AGCACTCTTTTCGGGGAAAT-3′ (reverse). Sting promoter region in both input and immunoprecipitated genomic DNA was detected by qPCR. The percentage of Sting promoter region immunoprecipitated by anti-STAT1 or its isotype IgG relative to input DNA was calculated, and the data are shown as the enrichment of the Sting promoter region by STAT1.
ELISA and immunoblot
IFNα and IFNβ in cell culture supernatant were quantified with VeriKine Mouse Interferon Alpha and Beta ELISA Kit (PBL interferon source), respectively. The ELISAs were performed according to the manufacturer's instructions. For immunoblot analysis, cells were collected in Triton lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 5% glycerol) containing complete protease inhibitors (Roche). Protein concentrations of the extracts were measured with a BCA assay (Thermo Scientific) and equalized with the lysis buffer. Equal amounts of the extracts were loaded and subjected to SDS–PAGE, transferred onto PVDF membranes (Millipore), and then blotted with enhanced chemiluminescence (Pierce) or Odyssey Imaging Systems (LI-COR Biosciences).
STING promoter reporter and dual-luciferase reporter assay
The potential transcription factor (TF) binding sites in the mouse Sting promoter region were predicted by TRANFAC 28, 29. The conservation of the TF binding sites among the mammalian species was analyzed by UCSC genome browser (http://genome.ucsc.edu/). Different length of Sting promoters were amplified from C57BL/6 genome DNA and subcloned into the pGL4.20 [luc2/Puro] vector (Promega, Madison, WI) to generate wild-type Sting promoter reporter (WT-luc) and Stat1#2-deficient Sting promoter reporter (Δ#2-luc). The Stat1#1 region of the Δ#2-luc reporter was mutated to generate Δ#2-luc-mut#1 reporter construct via QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). The indicated STING promoter reporter construct was co-transfected with Renilla luciferase reporter (pRL-TK-luc) into RAW264.7 cells by Amaxa Cell Line Nucleofector Kit V (Lonza). 12 h posttransfection, the medium was replaced and the cells were stimulated with 500 U/ml IFNα for another 12 h; then, the cells were lysed by passive lysis buffer, and the firefly luciferase activity of the STING promoter reporters was measured and normalized by Renilla luciferase activity according to the manufacturer's instructions of the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). HEK293T cells were co-transfected with indicated vectors (Flag, TBK1, IPS1, IRF1, or STAT1), STING promoter reporter constructs, and pRL-TK-luc according the manufacturer's instructions of Jet-PEI (Polyplus-transfection). 24 h posttransfection (in some experiments, cells were transfected for 12 h and treated with 500 U/ml IFNα for another 12 h), the cells were lysed and the relative luciferase activity was measured as in RAW264.7 cells.
Lentivirus packaging and lentiviral transduction
Full-length mouse Sting gene was cloned into the lentiviral vector pCDF1-CMV-MCS2-EF1-copGFP (CD111B-1; System Bioscience) to make the expression constructs LV-Sting. LV-Empty or LV-Sting vector was co-transfected into HEK293T cells with the pPACKF1 Packaging plasmids mix (LV100A-1, System Bioscience). Control or Sting-overexpressing lentiviruses were produced, and the WT, Ifnar1−/−, or Irf7−/− J2-BMM cells were transduced by these lentiviruses according to the user's manual (System Bioscience) and the previous study 45.
Acknowledgments
We thank Kislay Parvatiyar and Amir Ali Ghaffari for their helpful discussions and Dr. Tadatsugu Taniguchi (University of Tokyo) and Dr. Michael S. Diamond (Washington University) for sharing Irf3−/− and Irf7−/− mice. We appreciate Neda Arora for her excellent technical support, Aiping Wu for his bioinformatics instructions, and Jing Zhu for editing the manuscript. This work was funded by NIH RO1AI078389 and AI069120 grants. We apologize for works not cited in this paper owing to limited space.
Author contributions
FM and GC designed the study. FM, BL, YY, SSI, and MS performed the experiments and analyzed the data. FM wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure Legends
Review Process File
References
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe T, Harashima A, Xia T, Konno H, Konno K, Morales A, Ahn J, Gutman D, Barber GN. STING recognition of cytoplasmic DNA instigates cellular defense. Mol Cell. 2013;50:5–15. doi: 10.1016/j.molcel.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med. 2012;209:1969–1983. doi: 10.1084/jem.20121960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA. 2012;109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- Pesavento C, Hengge R. Bacterial nucleotide-based second messengers. Curr Opin Microbiol. 2009;12:170–176. doi: 10.1016/j.mib.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity. 2010;33:765–776. doi: 10.1016/j.immuni.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hu MM, Wang YY, Shu HB. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem. 2012;287:28646–28655. doi: 10.1074/jbc.M112.362608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, Wang YY, Zhang XL, Shu HB. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407. doi: 10.1016/j.immuni.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Qian Z, Cai YD, Li Y. Automatic transcription factor classifier based on functional domain composition. Biochem Biophys Res Commun. 2006;347:141–144. doi: 10.1016/j.bbrc.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SL, Friedman BA, Schmid S, Gertz J, Myers RM, Tenoever BR, Maniatis T. IkappaB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc Natl Acad Sci USA. 2011;108:21170–21175. doi: 10.1073/pnas.1119137109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M, Jr, Barber GN, Stetson DB. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer KA, Durack J, Portnoy DA. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 2014;10:e1003861. doi: 10.1371/journal.ppat.1003861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleroni AV, Varesio L, Wright RB, Brunda MJ. Tumoricidal alveolar macrophage and tumor infiltrating macrophage cell lines. Int J Cancer. 1991;49:296–302. doi: 10.1002/ijc.2910490226. [DOI] [PubMed] [Google Scholar]
- Ma F, Liu SY, Razani B, Arora N, Li B, Kagechika H, Tontonoz P, Nunez V, Ricote M, Cheng G. Retinoid X receptor alpha attenuates host antiviral response by suppressing type I interferon. Nat Commun. 2014;5:5494. doi: 10.1038/ncomms6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Su T, Ferrari R, Li JY, Kurdistani SK. A unique epigenetic signature is associated with active DNA replication loci in human embryonic stem cells. Epigenetics. 2014;9:257–267. doi: 10.4161/epi.26870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure Legends
Review Process File