

Abstract
Cultivated cochineal (Dactylopius coccus) produces carminic acid, a valuable red dye used to color textiles, cosmetics, and food. Extant native D. coccus is largely restricted to two populations in the Mexican and the Andean highlands, although the insect's ultimate center of domestication remains unclear. Moreover, due to Mexican D. coccus cultivation's near demise during the 19th century, the genetic diversity of current cochineal stock is unknown. Through genomic sequencing, we identified two divergent D. coccus populations in highland Mexico: one unique to Mexico and another that was more closely related to extant Andean cochineal. Relic diversity is preserved in the crops of small-scale Mexican cochineal farmers. Conversely, larger-scale commercial producers are cultivating the Andean-like cochineal, which may reflect clandestine 20th century importation.
Keywords: Cochineal, genomics, Mexico, Peru, phylogeography
Introduction
Domesticated cochineal (Dactylopius coccus) is a New-World scale insect cultivated for carminic acid, a potent scarlet dye used to color textiles, cosmetics, and food (Chávez-Moreno et al. 2009). With the use of mordants and adjuncts, carminic acid dyes produce colors ranging from pinks to deep purples and black (Phipps 2010). From the conquest of the Aztec Empire by the Spanish until the advent of laboratory-synthesized colorants in the 19th century, cochineal dye was the preeminent source of scarlet coloring. Cochineal was one of the primary exports from New Spain (after gold and silver) and played a critical role in the highland Mexican economy, where commercial production was centered (Chávez-Moreno et al. 2009). Cochineal dye's monetary value was so high that its production was a Spanish state secret and pre-Columbian codices describing its use were destroyed to prevent piracy. After the development of artificial red dyes, cochineal production nearly disappeared, including from highland Mexico. Since the 1970s, cochineal production has started to resurge due to the discovery of carcinogenic and hazardous properties of many synthesized dyes (Chávez-Moreno et al. 2009).
Cochineal insects (Dactylopius spp.) are endemic American phytophagous scale insects of the monogeneric family Dactylopiidae. Ten species are currently recognized (Van Dam and May 2012), although highly divergent biotypes within individual species have been identified, suggesting possible cryptic speciation (Mathenge et al. 2009). Four wild species are endemic to north and central Mexico (D. confusus,D. gracilipilus,D. opuntiae, and D. tomentosus), while an additional five wild species (D. austrinus,D. ceylonicus,D. confertus,D. salmianus, and D. zimmermanni) are endemic to South America (Rodríguez et al. 2001; Chávez-Moreno et al. 2009; Van Dam and May 2012). As an antimicrobial and antipredatory defense mechanism, all cochineal insects (both wild and cultivated species) synthesize the anthraquinone carminic acid. Of the Dactylopius species, domesticated D. coccus produces the most carminic acid (∽20% of dry body weight) (Wouters and Verhecken 1989; Chávez-Moreno et al. 2009). Additionally, D. coccus lacks the protective waxy coating that the wild forms possess, making it more susceptible to both weather fluctuations and predation (Chávez-Moreno et al. 2009).
Cochineal insects are obligate parasites of cacti (primarily Opuntia spp.), with individual Dactylopius species/biotypes preferring different host cactus species. D. coccus can survive on a wide range of host cactus species. While cultivated insects are primarily raised on domesticated nopal (Opuntia ficus-indica), D. coccus can also parasitize Nopalea cochenillifera and numerous Opuntia species including O. atropes,O. crassa,O. fuliginosa,O. hyptiacantha,O. jaliscana,O. megacantha,O. pilifera,O. robusta,O. streptacantha,O. tomentosa, and O. undulata (Rodríguez et al. 2001; Chávez-Moreno et al. 2011). D. coccus competes with other Dactylopius species for these hosts across its range, although some other species also parasitize cactus species not utilized by D. coccus (e.g., Cylindropuntia spp.) (Chávez-Moreno et al. 2011).
The geographic origin of domesticated cochineal is debated (Fig.1). “Native” populations are located in highland Mexico (centered in Oaxaca state, but also found in Puebla, Tlaxcala, and the Valley of Mexico) and in the Andes of southern Peru (Chávez-Moreno et al. 2009; de Ávila Blomberg 2005; Rodríguez et al. 2001). Feral populations have also been reported in neighboring Chile. Coccidoculture was successfully introduced to Spain, the Canary Islands, Argentina, Guatemala, and South Africa during the 19th and 20th centuries. This disjunct distribution is unexpected as cochineal species have limited dispersion capability: female cochineals are sessile, attaching themselves to the host plant immediately after hatching, while males are winged, but die quickly after fertilizing females, surviving only approximately three days in their adult form (de Ávila Blomberg 2005). Furthermore, although its host Opuntia species can thrive in multiple ecological zones, D. coccus is limited to arid and semi-arid habitats (Chávez-Moreno et al. 2009).
Figure 1.
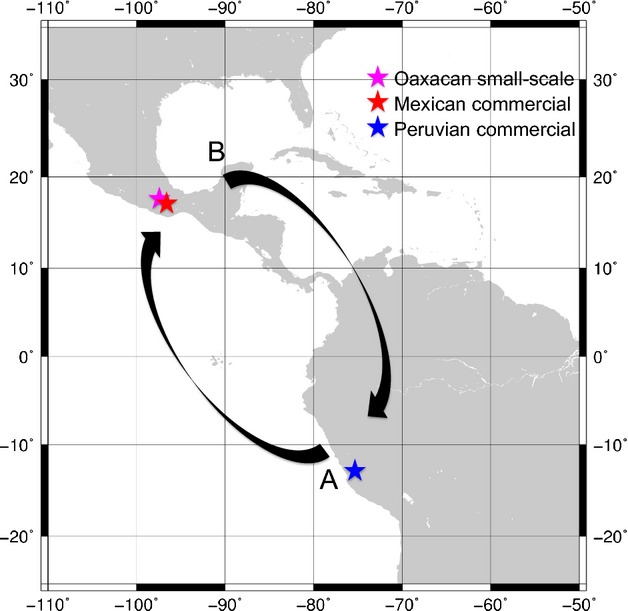
Map depicting the competing Dactylopius coccus origin hypotheses: (A) D. coccus originated in Peru and subsequently spread to Mexico; (B) D. coccus evolved in Mexico and was later introduced to Peru, possibly after domestication. D. coccus sampling locations for the genomic analyses are also shown.
D. coccus's dispersed geographical pattern raises the question of whether the current day distribution is natural or the result of deliberate introduction of the insects in prehistory. The earliest known cochineal-dyed textiles were discovered in Paracas, Peru (10th to 12th century AD), but the first evidence of cochineal farming was found in Mexican Toltec (10th century AD) sites (Rodríguez et al. 2001; Chávez-Moreno et al. 2009). Based on a phylogenetic analysis of morphological characters, Rodríguez et al. (2001) argued for a South American origin. Additionally, Mexican D. coccus is reliant on human propagation and protection for survival, while Andean insects survive ferally (Ramírez-Puebla et al. 2010). Conversely, de Ávila Blomberg (2005) argued that the presence of eight species that prey on domesticated cochineal in Mexico, as opposed to only one extant species in the Andes, indicates a Mexican origin. Genetic evidence is lacking: before this project, only 58 short DNA sequences (<800 bp each) were available for the entire Dactylopius genus.
Although genetic analyses could clarify the history of domesticated cochineal, they require phylogenetically informative variation to exist in extant populations. Whether extant Mexican cochineal exhibits such variation is unclear. While Oaxaca, Mexico was once the center of cochineal production, the Oaxacan cochineal industry nearly disappeared during the 19th century (Chávez-Moreno et al. 2009). Cochineal crops were deliberately destroyed during the Mexican War of Independence. The industry never recovered due to the competition from foreign production and the development of synthetic dyes. This bottleneck may have greatly reduced the level of diversity. Furthermore, Mexican populations may have become introgressed with Peruvian stocks during the 20th century (Chávez-Moreno et al. 2009). After the destruction of the Oaxacan cochineal industry, the center of production shifted to Peru. As the majority of Mexican D. coccus crops had gone extinct, some Mexican farmers may have been forced to obtain Peruvian stocks to start production. Trade of D. coccus stocks with the Canary Islands has also been noted in Mexico (Chávez-Moreno et al. 2009), although this is less likely to obscure phylogeographic information since the Canary Island population was introduced from Mexico around 1825 A.D. (Piña Luján 1980). Here, we assess the level of extant diversity of Mexican D. coccus through analysis of mitochondrial genetic markers and de novo whole-genomic sequencing.
Materials and Methods
Cochineal sample collection
Grana (dried female cochineal used for dye production) and fresh D. coccus females were obtained from small-scale farmers and large-scale commercial vendors in Mexico, Chile, and Peru (Table1). As large-scale commercial vendors may conglomerate crops from different farmers in each year, we tested multiple crop years from several producers (Table1). We also obtained historic grana of unknown provenance from the Peabody Museum of Archaeology and Ethnology (Harvard University) to evaluate whether extinct diversity might be preserved in historic specimens. Additionally, we collected wild female cochineal (Dactylopius spp.) by hand in Oaxaca, Mexico, for comparison with the cultivated species.
Table 1.
Single-insect samples collected and analyzed for mitochondrial markers. The geographic and/or commercial source of the material as well as year of collection is given for each sample. Also noted is whether the sample was obtained from a small-scale cochineal farmer (“Small-scale”), a large-scale commercial vendor (“Commercial”), or wild-caught (“Wild”). “Sample Type” states whether the sample was derived from grana or fresh insects. The total sample size and the number of sequenced cytochrome c oxidase I (cox1) and 12S rRNA mitochondrial genes are also given
| Sample | Source | Year | Cultivation type | Sample type | N | cox1 | 12S rRNA |
|---|---|---|---|---|---|---|---|
| Oaxaca1 | Oaxaca, Mexico | 2012 | Small-scale | Fresh | 20 | 14 | 0 |
| Oaxaca2 | Oaxaca, Mexico | 2010 | Small-scale | Grana | 5 | 0 | 0 |
| Oaxaca3 | Oaxaca, Mexico | 2010 | Small-scale | Grana | 5 | 4 | 0 |
| Oaxaca4 | Oaxaca, Mexico | 2010 | Small-scale | Grana | 5 | 0 | 0 |
| Oaxaca5 | Oaxaca, Mexico | 2010 | Small-scale | Grana | 5 | 0 | 0 |
| Mexico1 | Mexico (textile store “Teotitlan,” Oaxaca) | 2011 | Commercial | Grana | 10 | 10 | 10 |
| Mexico2 | Mexico (textile store “Teotitlan,” Oaxaca) | 2012 | Commercial | Grana | 5 | 4 | 0 |
| Peru1 | Peru (wildcolours.org.uk) | 2011 | Commercial | Grana | 10 | 10 | 10 |
| Peru2 | Peru (aurorasilk.com) | 2011 | Commercial | Grana | 10 | 5 | 10 |
| Peru3 | Peru (La Tierra Dye Co.) | 2011 | Commercial | Grana | 10 | 10 | 0 |
| Peru4 | Peru (aurorasilk.com) | 2012 | Commercial | Grana | 10 | 10 | 0 |
| Chile1 | Chile (aurorasilk.com) | 2011 | Commercial | Grana | 10 | 0 | 0 |
| Chile2 | Chile (aurorasilk.com) | 2012 | Commercial | Grana | 10 | 1 | 0 |
| Museum | No provenance (Peabody Museum) | Unknown | Unknown | Grana | 10 | 0 | 0 |
| OaxacaWild | Oaxaca, Mexico | 2012 | Wild | Fresh | 41 | 25 | 0 |
Mitochondrial marker analyses
DNA was extracted from 166 single insects using the PowerSoil kit (MO BIO, Carlsbad, California, USA) and the QIAamp® DNA Mini Kit (Qiagen, Valencia, California, USA) according to manufacturer's instructions. The dataset included 40 insects cultivated by small-scale Oaxacan farmers, 75 from large-scale commercial producers (15 Mexican, 40 Peruvian, and 20 Chilean), 10 historic grana samples without provenance, and 41 wild Dactylopius from Oaxaca (Table1). The mitochondrial cytochrome c oxidase I (cox1) and 12S rRNA genes were amplified by the polymerase chain reaction and dideoxy-terminator sequenced (Appendix). The 12S rRNA experiments were omitted for most individuals as we found only three single-nucleotide polymorphisms in an initial subset of 30 individuals (10 Mexican and 20 Peruvian grana from commercial vendors), and the results were in agreement with the more informative cox1 results (Table1; Fig.2). The obtained sequences were compared with 11 cox1 (representing D. opuntiae [n = 1] and D. tomentosus [n = 10]) and seven 12S rRNA sequences (including D. opuntiae [n = 3], Mexican D. coccus [n = 1], D. confusus [n = 1], D. ceylonicus [n = 1], and D. tomentosus [n = 1]) obtained from GenBank. While this sample is not representative of the entire Dactylopius genus, it includes all publicly available data for these genes.
Figure 2.
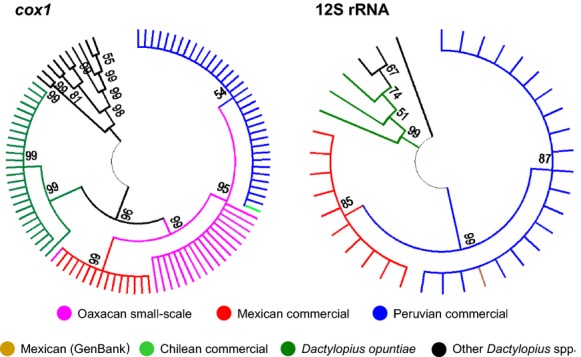
Condensed maximum-likelihood trees of Dactylopius coccus cytochrome c oxidase I (cox1) and 12S rRNA mitochondrial genes. Topology robustness was tested with 100 bootstrap replicates.
Whole-genome sequencing library construction
Due to the discovery of limited mitochondrial variation, we conducted whole-genomic sequencing on Dactylopius coccus to better understand domesticated cochineal phylogenies. Three bulk extracts representing cochineal raised by Oaxacan small-scale farmers or sold by Mexican and Peruvian commercial vendors were subjected to Pool-Seq (Schlötterer et al. 2014; Fig.1). Bulk DNA was extracted from 50 individuals each (Schlötterer et al. 2014; Appendix). Sequencing libraries were prepared from the bulk extracts using the PrepX Illumina Kit (IntegenX, Pleasanton, California, USA) and NEXTflex™ DNA Barcodes (Bioo Scientific, Austin, Texas, USA) on the Apollo 324 robotic platform (IntegenX). Paired-end 150-bp sequences were generated on one-quarter of an Illumina HiSeq 2500 lane. A total of 5.2–5.5 million paired sequences were obtained per library.
Identification and phylogenetic analysis of genomic sequence variants
A draft Dactylopius coccus genome assembly was constructed using JR-Assembler 1.02 (Chu et al. 2013; Table2). The final assembly was 18.6 Mbp long with an N50 of 378,999 bp (Table2). The quality-controlled merged sequence reads were aligned against the D. coccus assembly using BWA 0.7.5 (Li and Durbin 2009, 2010) in order to identify sequence variants. A total of 1.99 Gbp of reads (106.8× mean depth) were aligned to the assembly. Analysis of the assembly using BEDTools 2.17.0 (Quinlan and Hall 2010), however, showed significant variation in coverage across the genome and between samples (per sample mean depth ± standard deviation: 9.4× ± 78.7×, 43.3× ± 26.9×, and 54.0× ± 34.0× for the Oaxacan small-scale farm, Mexican commercial, and Peruvian commercial samples, respectively). Genotypes were called using SAMtools 0.1.19 (Li et al. 2009).
Table 2.
Dactylopius coccus genome assembly statistics
| Assembly length | 18,613,147 bp | Mean sequencing depth | 106.8× |
| N50 | 378,999 bp | L50 count | 12 |
| No. scaffolds | 1499 | Mean scaffold length | 12,417 bp |
| Maximum scaffold length | 1,388,629 bp | Minimum scaffold length | 200 bp |
| Genome %A | 20.89% | Genome %T | 20.97% |
| Genome %G | 29.02% | Genome %C | 29.12% |
Selection on the Dactylopius coccus genome
To determine whether the cochineal genome was undergoing detectable natural or artificial selection, we predicted genic sequences using GeneMark-ES 2.3c (Borodovsky and Lomsadze 2011). The ratio of nonsynonymous to synonymous (N/S) SNPs was calculated using SnpEff 3.6a (Cingolani et al. 2012). Tajima's D was calculated using 500-bp windows with VCFtools 1.0.9 (Danecek et al. 2011).
Results
Mitochondrial DNA analyses
The grana accessions' DNA preservation varied, probably due to different procedures used for preparation (e.g., boiling and air drying). We were unable to obtain sequences for all individuals due to the variation in DNA preservation. We obtained 68 cox1 (18 from Oaxacan small-scale farmers, 14 from commercial Mexican vendors, 35 from commercial Peruvian vendors, and 1 from Chilean commercial vendors) and 30 12S rRNA Dactylopius coccus sequences (10 Mexican and 20 Peruvian insects from commercial vendors) (Table1; Fig.2). We sequenced 25 wild Oaxacan cochineal cox1 genes. All the Oaxacan wild cochineal we collected clustered with Dactylopius opuntiae (Fig.2).
We observed nine credible substitutions in 1003 bp of D. coccus mitochondrial DNA (0.90% divergence): six substitutions in 559 bp of cox1 sequence (1.1% divergence) and three substitutions in 454 bp of 12S rRNA (0.66% divergence). We identified three cox1 and two 12S rRNA D. coccus haplotypes (Fig.2). Peruvian commercial cochineal cox1 sequences differed by one substitution from the Oaxacan small-scale farm insect specimens. A third divergent cox1 haplotype (an additional five substitutions) was found in Mexican commercial samples. The Chilean sample clustered with the Peruvian commercial grana. The 12S rRNA tree resolved the same two major clades (Peruvian commercial/Oaxacan small-scale farm insects versus Mexican commercial cochineal).
Genomic SNP phylogenetic analyses
A total of 11,517 genomic variants (including 10,598 polymorphic single-nucleotide polymorphisms [SNPs]) were identified in the three D. coccus pools. To account for sequencing errors, collapsed repetitive regions and apparent variants deriving from D. coccus-like environmental contaminants, we refined the SNP dataset by requiring that each site be sequenced a minimum depth of 5× per pool (15× total depth) and a maximum of 100× per pool (300× total depth). The refined SNP dataset included 82 high-confidence polymorphic SNPs (135× mean total sequencing depth). Both the raw and filtered SNP datasets were analyzed by principal component (PCA) and identity-by-state relatedness analyses using SNPRelate 0.9.12 (Zheng et al. 2012; Fig.3). While SNPRelate was designed to analyze individuals, no similar software is yet available for Pool-Seq data. To corroborate the SNPRelate results, we calculated genomic differentiation (mean FST) of the informative sites using PoPoolation2 1.201 (Kofler et al. 2011) using the same SNP filtering criteria as in the SNPRelate analyses. Additionally, SNP-sharing analysis was performed on the raw SNP dataset using VCFtools 1.0.9 (Danecek et al. 2011).
Figure 3.
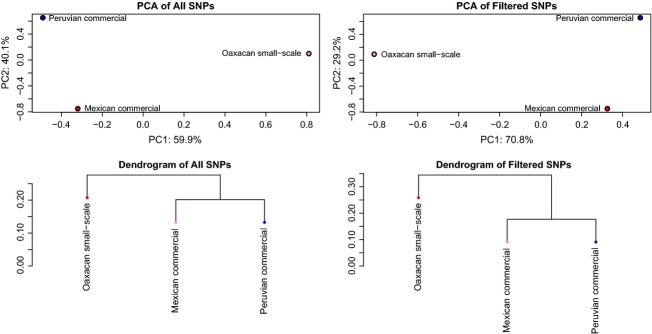
Relatedness between Oaxacan small-scale farm, Mexican commercial, and Peruvian commercial cochineal bulk samples. Principle component analysis (top row) separates the Oaxacan small-scale farm insects from the commercial specimens, with the first principle component explaining the majority of the variation (59.9% and 70.8% in the unfiltered and filtered SNP datasets, respectively). Identity-by-state analysis (bottom row) of these SNP datasets produces dendrograms with congruent topology.
All genomic SNP analyses had congruent results (Fig.3; Table3). The first principal component separated the Oaxacan small-scale farm sample from the Mexican and Peruvian commercial vendor specimens. Similarly, in the identity-by-state relatedness analyses, the Mexican and Peruvian commercial samples form a clade, with the Oaxacan small-scale farm sample being more distantly related (Fig.3). Genomic differentiation analysis also separated the Oaxacan small-scale farm sample from the two commercial samples (Table3). Additionally, the commercial samples from Mexico and Peru share more SNPs with each other than either do with the Oaxacan small-scale farm sample (Fig.4). These results indicate that the Mexican and Peruvian commercial samples are more closely related to each other than they are to Oaxacan small-scale farm cochineal. Notably, both the genomic differentiation and the SNP-sharing analyses show that the Oaxacan small-scale farm sample is slightly closer related to the Mexican commercial cochineal than to the Peruvian cochineal (Table3; Fig.4). Unfortunately, we are unable to ascertain precise ages of these genomic clades as we have no paleontological calibration point and the most closely related sequenced genome, the pea aphid (Acyrthosiphon pisum), is too divergent to align against the D. coccus draft genome sequence (International Aphid Genomics Consortium 2010).
Table 3.
Genomic differentiation between the three cochineal bulk samples. Values are listed as mean FST ± standard deviation
| Oaxacan small-scale | Mexican commercial | Peruvian commercial | |
|---|---|---|---|
| Oaxacan small-scale | 0.0842 ± 0.0901 | 0.1097 ± 0.0897 | |
| Mexican commercial | 0.0842 ± 0.0901 | 0.0096 ± 0.0058 | |
| Peruvian commercial | 0.1097 ± 0.0897 | 0.0096 ± 0.0058 |
Figure 4.
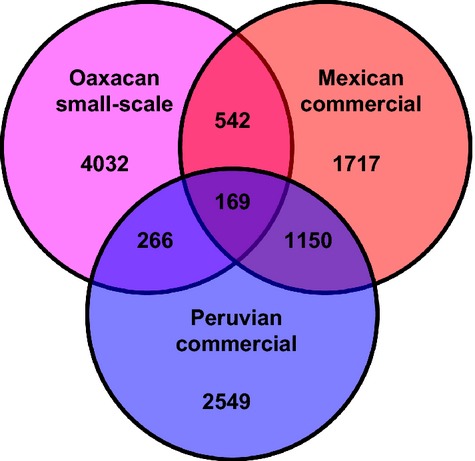
Venn diagram depicting numbers of genomic SNPs unique to and shared between each bulk Dactylopius coccus sample.
Selection on the Dactylopius coccus genome
GeneMark-ES predicted 8003 genes. A total of 4245 SNPs were located in putative exonic regions, of which 3028 were nonsynonymous and 1217 were synonymous substitutions (combined N/S for all samples = 2.49). Although the N/S ratio was greater than one for all bulk samples (1.96, 2.73, and 2.73 for the Oaxacan small-scale farm, Mexican commercial, and Peruvian commercial samples, respectively), Tajima's D found no strong evidence of selection on the cochineal genome (mean absolute value of D ± standard deviation: 0.0560 ± 0.260). Furthermore, there was no difference in selection effect between genic (0.0600 ± 0.271) and nongenic (0.0552 ± 0.258) regions of the genome (Student's t-test, P = 0.2151), which suggests that the high N/S ratios are not associated with selection.
Discussion
We find no effect on the mitochondrial DNA diversity that can be attributed solely to human management. Nevertheless, the cox1 and 12S rRNA mitochondrial diversity is limited (three and two haplotypes, respectively) with one Mexican haplotype diverging from the other two, suggesting some form of bottleneck in the past. Nonfunctionally constrained mitochondrial markers (such as the control region) may be more variable. While it is tempting to attribute the observed bottleneck to human management, a more likely explanation is cytoplasmic incompatibility due to Wolbachia infection, a process that can produce false phylogeographic signal in arthropod phylogenetic trees (Hurst and Jiggins 2005). Dactylopius host numerous endosymbionts (Ramírez-Puebla et al. 2010), including the Alphaproteobacterium Wolbachia (Pankewitz et al. 2007). We detected Wolbachia sequences in both the single-marker and genomic analyses (Appendix). Furthermore, we found only one mitochondrial haplotype in the wild Oaxacan cochineal (D. opuntiae), suggesting that limited mitochondrial diversity is common across Dactylopius species.
Similarly, we found no conclusive evidence that the cochineal genome is under strong natural or artificial selection. Nevertheless, we observed only one D. coccus genomic sequence variant every ∽1600 nucleotides, which suggests a relatively slow mutation rate for insects (for comparison, Drosophila simulans has a SNP every ∽40 bases) (Begun et al. 2007; Hu et al. 2013). Further research is required to determine whether the slow mutation rate reflects selection.
The genomic phylogeny suggests that extant Mexican D. coccus derive from at least two source populations. One of these populations appears to be Mexican in origin, while the other is more closely related to Peruvian cochineal. Moreover, the distinctiveness between the “Mexican” and “Peruvian” clades suggests long-term isolation between the populations, which does not support the hypothesis of continuous and extensive trading of cochineal stocks during the pre-Columbian era as has been proposed previously (Chávez-Moreno et al. 2009). This observation supports contentions by local Mexican cochineal farmers that Peruvian stock may have been recently imported into Oaxaca with the renewed interest in cochineal production. However, our genomic differentiation and SNP-sharing results suggest that Mexican commercial cochineal may also have some local Mexican ancestry, even if it primarily derives from recently imported Peruvian stock.
Notably, the mitochondrial and genomic phylogenies are incongruent. The cox1 tree clusters the Peruvian grana and Mexican fresh insect accessions, but the genomic SNP data indicate that the two grana samples form a clade. Wolbachia infection is a likely cause of the discrepancy between the mitochondrial DNA and the genomic variant phylogenies (Hurst and Jiggins 2005). Alternatively, this incongruence could reflect recent introgression (Zakharov et al. 2009), which would be consistent with recent importation of South American cochineal into Mexico.
Further genomic research is required to establish D. coccus's domestication center(s) with confidence. Our cochineal dataset does not permit us to identify the ultimate source population. Additionally, although Wolbachia strains exhibit phylogenetic and phylogeographic patterning (Russell et al. 2009), we were unable to pinpoint the source location of D. coccus through sequencing and analysis of its Wolbachia endosymbiont (Appendix). Our results, however, show that phylogenetically informative variation survives in the crops of Oaxacan small-scale cochineal farmers. Nevertheless, future analyses will need to carefully control for the effects of recent clandestine Peruvian introgression into Mexican stocks.
Acknowledgments
The Wenner-Gren Foundation (to NT and NRG), the David Rockefeller Center for Latin American Studies (to NT), Harvard University's Department of Human Evolutionary Biology, and the Science of the Human Past Initiative supported this research. Alejandro de Ávila Blomberg (Jardín Etnobotánico de Oaxaca) and Eric Chávez Santiago (Museo Textil de Oaxaca) kindly provided specimens. Hannah Koon, Linda Reynard, and the Instituto Nacional de Antropología e Historia assisted in sample collection.
Appendix
Single-marker Analyses
Based on the available cochineal and scale insect phylogenetic literature, we analyzed the mitochondrial cytochrome c oxidase I (cox1) and 12S rRNA genes and the nuclear 18S rRNA and elongation factor 1α (EF1α) genes (C.W. Mathenge, P. Holford, R. Spooner-Hart, G.A.C. Beattie, Unpublished data; Morse and Normark 2006; Ramírez-Puebla et al. 2010). DNA targets were amplified by PCR on an MJ Research PTC-200 DNA engine thermocycler. Each 25 μL reaction contained 1× BIOLASE Diamond mix (Bioline, Taunton, Massachusetts, USA), 0.2–0.4 μmol/L each primer and 3–5 μL DNA. 12S and 18S reactions also contained 10 ng BSA. Primers are listed in TableA1. Final cox1, 12S rRNA, and 18S rRNA thermocycling programs consisted of an initial denaturation step at 95°C for 5 min (cox1, 12S rRNA) or 12 min (18S rRNA), 45 cycles of denaturation at 94°C for 30 sec, 50°C for 30 sec and 72°C for 45 sec, and a final extension step at 72°C for 10 min. The EF1α thermocycling program consisted of an initial denaturation step at 95°C for 4 min, 45 cycles of denaturation at 94°C for 30 sec, 50–55°C for 1 min and 72°C for 1 min, and a final extension step at 72°C for 4 min. PCR products were assessed by agarose gel electrophoresis. EF1α reactions produced multiple bands; therefore, the expected fragment of ∽1150 bp was isolated from the gel and purified using the QIAquick® Gel Extraction Kit (Qiagen). PCR products were treated with ExoSAP-IT™ (GE Healthcare) and then sequenced in both directions on an ABI 3730xl (Applied Biosystems) sequencer.
Table A1.
PCR primers used to amplify Dactylopius coccus genetic markers
| Marker | Forward primer | Reverse primer | Reference |
|---|---|---|---|
| 12S rRNA | 5′-AAGAGTGACGGGCRATTTGTACATA-3′ | 5′-GTGCCAGCAGTWGCGGTTA-3′ | Ramírez-Puebla et al. (2010) |
| 18S rRNA | 5′-CTGGTTGATCCTGCCAGTAG-3′ | 5′-CCGCGGCTGCTGGCACCAGA-3′ | Ramírez-Puebla et al. (2010) |
| cox1 | 5′-TCCGRATAGAACTWATAAAYACYAA-3′ | 5′-TAAACTTCAGGGTGACCAAAAAATCA-3′ | C.W. Mathenge, P. Holford, R. Spooner-Hart, G.A.C. Beattie, Unpublished data |
| EF1α | 5′-GATGCTCCGGGACAYAGA-3′ | 5′-ATGTGAGCGGTGTGGCAATCCAA-3′ | Morse & Normark (2006) |
Some initial cox1 PCRs using the forward primer 5′-GGTCAACAAATCATAAAGATATTGG-3′ amplified sequences matching Wolbachia (Folmer et al. 1994). These contaminants were discarded. We observed no variation in the nuclear markers (13 EF1α and seven 18S rRNA sequences) consistent with their relatively slow rates of mutation. Therefore, these markers were not considered further. For the final cox1 and 12S rRNA datasets, condensed maximum-likelihood trees were constructed with 100 bootstrap replicates under a Hasegawa–Kishino–Yano (Hasegawa et al. 1985) substitution model with invariant sites and a gamma distribution (four gamma categories) for substitutions in MEGA 6.06 (Tamura et al. 2013).
Whole-genome Sequencing
Bulk extracts were constructed including 50 fresh insects or grana each. Fresh insects were digested using proteinase K in buffer ATL from the QIAamp® DNA Mini Kit (Qiagen) and ethanol-precipitated on site in Mexico. The precipitated DNA was transported dry back to the laboratory at Harvard where it was resuspended and purified using Econo-Pac 10DG columns (Bio-Rad). Grana were digested using buffers ATL (with proteinase K) and AL from the QIAamp® kit. Digested grana bulk extracts were vacuum-filtered and concentrated using Vivaspin® 15 30-kDa MWCO columns. Extracts were then exchanged into PCR-grade water and fractionated using Econo-Pac 10DG columns in order to separate the DNA from carminic acid. DNA-rich fractions were collected and purified using the QIAquick PCR Purification Kit (Qiagen).
Bulk extracts were sheared to ∽200 bp average length using a S220 Focused-Ultrasonicator (Covaris, Inc., Woburn, Massachusetts, USA). DNA-sequencing libraries were constructed using the PrepX Illumina Kit (IntegenX) and NEXTflex™ DNA Barcodes (Bioo Scientific) on the Apollo 324 robotic platform (IntegenX) according to the manufacturer's instructions. Libraries were quality-controlled via analysis on an Agilent 2100 using a high-sensitivity DNA chip and quantified using the KAPA Library Quantification Kit – Illumina/Universal (KAPA Biosystems) and a Qubit® Fluorometer. A total of 13 PCR cycles using the NEXTflex™ kit enriched the indexed libraries to sequenceable concentrations. PCR-enriched libraries were requantified and pooled in equimolar ratios. Paired-end 150-bp sequences were generated on one-quarter of an Illumina HiSeq 2500 lane.
After demultiplexing using CASAVA 1.8.2, mate-paired sequences were merged using PANDAseq 2.4.0 (Masella et al. 2012). Adapter artifacts were removed using TagDust 1.12 (Lassman et al. 2009). PCR duplicates were removed using CD-HIT 4.6 (Li et al. 2012). Final datasets were quality-controlled using FastQC 1.32 (Andrews n.d.).
A Dactylopius coccus genome assembly was constructed using JR-Assembler 1.02 from the original unpaired reads (Chu et al. 2013). The final four base pairs of each read were removed to improve sequence quality as recommended by Chu and colleagues (Chu et al. 2013). JR-Assembler uses complete reads to assemble the genome sequence via seed extension, which improves assembly of large genomes in comparison with de Bruijn graph assemblers such as SOAPdenovo2 (Luo et al. 2012) and ABySS (Simpson et al. 2009). We found that de Bruijn graph assemblers (SOAPdenovo2 and ABySS) produced unsatisfactory D. coccus assemblies, probably due to the relatively low sequencing depth and presence of repetitive regions. The original reads were aligned very poorly against the SOAPdenovo2 and ABySS assemblies, possibly due to misassemblies after chopping the reads into k-mers. Moreover, analysis of the sequence datasets using KmerGenie 1.5658 (Chikhi and Medvedev 2014) found no optimal k-mer solution.
The assembly was aligned against the GenBank nonredundant nucleotide database using MegaBLAST 2.2.27+ and the National Center for Biotechnology Information contamination screen (Zhang et al. 2000; ). The MegaBLAST results were analyzed in MEGAN 4.70.4 (Huson et al. 2011). Contigs and scaffolds matching contaminants (e.g., Proteobacteria) were removed from the assembly. Genome assembly statistics were calculated using the assemblathon_stats.pl script from the Assemblathon 2 competition (Bradnam et al. 2013). Genome completeness was evaluated using the Core Eukaryotic Gene (CEGs) approach implemented in CEGMA (Parra et al. 2009). The assembly included 47 of 248 complete CEGs (19%) with an additional 53 partial CEGs (21%). As a final test of assembly quality, the known mitochondrial cox1 and 12S rRNA sequences were identified in the assembly. JR-Assembler had correctly assembled these sequences and placed them in the same scaffold.
Phylogeographic Analysis of the Dactylopius coccus Strain of Wolbachia Genome
Wolbachia strains exhibit phylogenetic and phylogeographic patterning (Russell et al. 2009). We therefore assembled and analyzed the genome of the Dactylopius coccus strain of Wolbachia (strain “wCoc”) in order to pinpoint the ultimate geographic source of D. coccus. The merged cochineal reads were aligned against two complete Wolbachia genomes (strains wMel and wPip) (Wu et al. 2004; Klasson et al. 2008) using BWA 0.7.4 (Li and Durbin 2009, 2010). Aligned reads were removed from the sequence pools using a custom script. These reads were used to de novo assemble wCoc using SOAPdenovo2 (127 bp k-mer length, 32 bp minimum mapped read length) (Luo et al. 2012). The wCoc genome was then iteratively aligned against the remaining merged reads, the newly aligned sequences were removed from the datasets, and then, the wCoc genome was reassembled including the newly removed sequences. This process was repeated until no more reads aligned against the draft genome (two iterations). The final wCoc genome sequence totaled 1.13 Mb with an N50 of 1387 bp (TableA2). Previously sequenced Wolbachia genomes range in length between 1.0 and 1.5 Mb, indicating that we have sequenced ∽75–100% of the wCoc genome.
Table A2.
Wolbachia strain “wCoc” genome assembly statistics
| Assembly length | 1,125,157 bp | Mean sequencing depth | 42.9× |
| N50 | 1387 bp | L50 count | 208 |
| No. scaffolds | 1065 | Mean scaffold length | 1056 bp |
| Maximum scaffold length: | 16,603 bp | Minimum scaffold length | 183 bp |
| Genome %A | 33.02% | Genome %T | 32.71% |
| Genome %G | 17.18% | Genome %C | 17.09% |
Wolbachia strains are classified primarily by the ftsZ gene (Lo et al. 2002). After identification of this gene in wCoc, we aligned it against 797 ∽428-bp partial Wolbachia ftsZ sequences obtained from GenBank. The analyzed region corresponded to neighboring positions 624,163–624,590 of the wPip genome (Klasson et al. 2008; GenBank accession AM999887.1). A condensed maximum-likelihood tree (100 bootstrap replicates) was then constructed in MEGA 6.06 (Tamura et al. 2013) under a Tamura–Nei (Tamura and Nei 1993) substitution model with invariant sites and a gamma distribution for substitution rates (four gamma categories). wCoc fell in clade B with most other insects (Fig.A1). We found little phylogeographic patterning, although it clustered with strains hosted by other scale insects including Kerria lacca and Bemisia tabaci.
Figure A1.
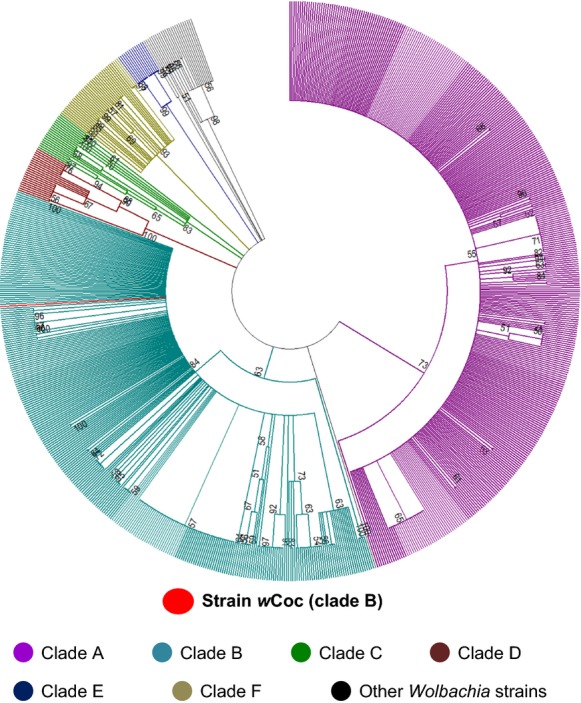
Condensed maximum-likelihood tree of 797 partial Wolbachia ftsZ genes. The Wolbachia endosymbiont of Dactylopius coccus (strain “wCoc”) falls in clade B. Clade nomenclature follows Lo et al. 2002. The tree was constructed under a Tamura–Nei substitution model with invariant sites and a gamma distribution for substitution rates (four gamma categories) and tested with 100 bootstrap replicates. Only clades supported by at least 50 replicates are noted.
Data Accessibility
Sanger sequences have been deposited in GenBank (accessions KJ701865–KJ702008). Genome assemblies and associated sequence reads have been deposited in the BioProject archive (accession PRJNA244295).
Conflict of Interest
None declared.
References
- Andrews S. FastQC: a quality control tool for high throughput sequence data www.bioinformatics.babraham.ac.uk/projects/fastqc/ )
- de Ávila Blomberg A, et al. El insecto humanizado: biología y mexicanidad en los textos de Alzate y sus contemporáneos acerca de la grana. In: Sánchez Silva C, de Ávila Blomberg A, editors. La Grana y el Nopal en los Textos de Alzate. Mexico City: CONACULTA; 2005. pp. 29–73. [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh Y-P, Hahn MW, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky M. Lomsadze A. Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr. Prot. Bioinformat. 2011;Chapter 4:4.6.1–4.6.10. doi: 10.1002/0471250953.bi0406s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience. 2013;2:10. doi: 10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez-Moreno CK, Tecante A. Casas A. The Opuntia (Cactaceae) and Dactylopius (Hemiptera: Dactylopiidae) in Mexico: a historical perspective of use, interaction and distribution. Biodivers. Conserv. 2009;18:3337–3355. [Google Scholar]
- Chávez-Moreno CK, Tecante A, Casas A. Claps LE. Distribution and habitat in Mexico of Dactylopius Costa (Hemiptera; Dactylopiidae) and their cacti hosts (Cactaceae: Opuntioideae. Neotrop. Entomol. 2011;40:62–71. doi: 10.1590/s1519-566x2011000100009. [DOI] [PubMed] [Google Scholar]
- Chikhi R. Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30:31–37. doi: 10.1093/bioinformatics/btt310. [DOI] [PubMed] [Google Scholar]
- Chu T-C, Lu C-H, Liu T, Lee GC, Li W-H. Shih A C-C. Assembler for de novo assembly of large genomes. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E3417–E3424. doi: 10.1073/pnas.1314090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118iso-2; iso-3. Fly. 2012;6:1–13. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo M A, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer O, Black M, Hoeh W, Lutz R. Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech. 1994;5:304–313. [PubMed] [Google Scholar]
- Hasegawa M, Kishino H. Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hu TT, Eisen MB, Thornton KR. Andolfatto P. A second-generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Res. 2013;23:89–98. doi: 10.1101/gr.141689.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst GDD. Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proceed. Royal Soc. B: Biol. Sci. 2005;272:1525–1534. doi: 10.1098/rspb.2005.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh H-J, Weber N. Schuster S C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21:1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010;8:e1000313. doi: 10.1371/journal.pbio.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, Walker T, Sebaihia M, Sanders MJ, Quail MA, Lord A, et al. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol. Biol. Evol. 2008;25:1877–1887. doi: 10.1093/molbev/msn133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Pandey RV. Schlötterer C. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq) Bioinformatics. 2011;27:3435–3436. doi: 10.1093/bioinformatics/btr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassman T, Hayashizaki Y. Daub CO. TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Durbin R. Fast and accurate long read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hansaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;16:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Fu L, Niu B, Wu S. Wooley J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinformat. 2012;13:656–668. doi: 10.1093/bib/bbs035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo N, Casiraghi M, Salati E, Bazzocchi C. Bandi C. How many Wolbachia supergroups exist? Mol. Biol. Evol. 2002;19:341–346. doi: 10.1093/oxfordjournals.molbev.a004087. [DOI] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masella AP, Bartram AK, Truszkowski JM, Brown DG. Neufeld JD. PANDAseq: PAired-eND assembler for Illumina sequences. BMC Bioinformat. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathenge CW, Holford P, Hoffmann JR, Spooner-Hart R. Beattie GAC. Distinguishing suitable biotypes of Dactylopius tomentosus (Hemiptera: Dactylopiidae) for biological control of Cylindropuntia fulgida var. fulgida (Caryophyllales: Cactaceae) in South Africa. Bull. Entomol. Res. 2009;99:619–627. doi: 10.1017/S0007485309006671. [DOI] [PubMed] [Google Scholar]
- Morse GE. Normark BB. A molecular phylogenetic study of armoured scale insects (Hemiptera: Diaspididae) Syst. Entomol. 2006;31:338–349. [Google Scholar]
- Pankewitz F, Zöllmer A, Hilker M. Gräser Y. Presence of Wolbachia in insect eggs containing antimicrobially active anthraquinones. Microb. Ecol. 2007;54:713–721. doi: 10.1007/s00248-007-9230-5. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T. Korf I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009;37:289–297. doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps E. Cochineal red: the art history of a color. New York: Metropolitan Museum of Art; 2010. [Google Scholar]
- Piña Luján I. Dispersión de dos especies del género Opuntia en las Islas Canarias. Cactaceas y Suculentas Mexicanas. 1980;25:3–6. [Google Scholar]
- Quinlan AR. Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Puebla ST, Rosenblueth M, Chávez-Moreno CK, Catanho Pereira de Lyra MC, Tecante A. Mart E. Molecular phylogeny of the genus Dactylopius (Hemiptera: Dactylopiidae) and identification of the symbiotic bacteria. Environ. Entomol. 2010;39:1178–1183. doi: 10.1603/EN10037. [DOI] [PubMed] [Google Scholar]
- Rodríguez LC, Méndez MA. Niemeyer HM. Direction of dispersion of cochineal (Dactylopius coccus Costa) within the Americas. Antiquity. 2001;75:73–77. [Google Scholar]
- Russell JA, Goldman-Huertas B, Moreau CS, Baldo L, Stahlhut J K, Werren JH, et al. Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution. 2009;63:624–640. doi: 10.1111/j.1558-5646.2008.00579.x. [DOI] [PubMed] [Google Scholar]
- Schlötterer C, Tobler R, Kofler R. Nolte V. Sequencing pools of individuals — mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014;15:749–763. doi: 10.1038/nrg3803. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM. Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. Nei S. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A. Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam AR. May B. A new species of Dactylopius Costa (Dactylopius gracilipilus sp. nov.) (Hemiptera: Coccoidea: Dactylopiidae) from the Chihuahuan Desert, Texas, U.S.A. Zootaxa. 2012;3573:33–39. [Google Scholar]
- Wouters J. Verhecken A. The coccid insect dyes: HPLC and computerized diode-array analysis of dyed yarns. Stud. Conserv. 1989;34:189–200. [Google Scholar]
- Wu M, Sun LV, Vamathevan J, Deboy R, Brownlie JC, McGraw EA, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2:327–341. doi: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov EV, Lobo NF, Nowak C. Hellmann JJ. Introgression as a likely cause of mtDNA paraphyly in two allopatric skippers (Lepidoptera: Hesperiidae) Heredity. 2009;102:590–599. doi: 10.1038/hdy.2009.26. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L. Miller W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C. Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sanger sequences have been deposited in GenBank (accessions KJ701865–KJ702008). Genome assemblies and associated sequence reads have been deposited in the BioProject archive (accession PRJNA244295).