

Abstract
Background
Pomacea canaliculata is an important invasive species worldwide. However, little is known about the molecular mechanisms behind species displacement, adaptational abilities, and pesticide resistance, partly because of the lack of genomic information that is available for this species. Here, the transcriptome sequences for the invasive golden apple snail P. canaliculata and the native mudsnail Cipangopaludina cahayensis were obtained by next-generation-sequencing and used to compare genomic divergence and identify molecular markers.
Results
More than 46 million high quality sequencing reads were generated from P. canaliculata and C. cahayensis using Illumina paired-end sequencing technology. Our analysis indicated that 11,312 unigenes from P. canaliculata and C. cahayensis showed significant similarities to known proteins families, among which a total of 4,320 specific protein families were identified. KEGG pathway enrichment was analyzed for the unique unigenes with 17 pathways (p-value < 10−5) in P. canaliculata relating predominantly to lysosomes and vitamin digestion and absorption, and with 12 identified in C. cahayensis, including cancer and toxoplasmosis pathways, respectively. Our analysis also indicated that the comparatively high number of P450 genes in the P. canaliculata transcriptome may be associated with the pesticide resistance in this species. Additionally, 16,717 simple sequence repeats derived from expressed sequence tags (EST-SSRs) were identified from the 14,722 unigenes in P. canaliculata and 100 of them were examined by PCR, revealing a species-specific molecular marker that could distinguish between the morphologically similar P. canaliculata and C. cahayensis snails.
Conclusions
Here, we present the genomic resources of P. canaliculata and C. cahayensis. Differentially expressed genes in the transcriptome of P. canaliculata compared with C. cahayensis corresponded to critical metabolic pathways, and genes specifically related to environmental stress response were detected. The CYP4 family of P450 cytochromes that may be important factors in pesticide metabolism in P. canaliculata was identified. Overall, these findings will provide valuable genetic data for the further characterization of the molecular mechanisms that support the invasive and adaptive abilities of P. canaliculata.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-015-0175-2) contains supplementary material, which is available to authorized users.
Keywords: Biological invasion, Pomacea canaliculata, Cipangopaludina cahayensis, EST-SSR, Transcriptome
Background
Biologically invasive species are one of the major threats to global biodiversity, and they can cause substantial economic losses as well as pose a public health risk [1-8]. The golden apple snail (Pomacea canaliculata) is native to South America and is beginning to emerge worldwide, among others China. It has become a highly damaging invasive species, affecting agriculture and fisheries, as well as pubilc heatlth [9-14]. The snail was first introduced to Zhongshan (Guangdong Province, China) as a human food source or aquarium pet [15]. It adapted quickly and is now found at least 11 provinces in southern China [16]. Currently, P. canaliculata has invaded local habitats, including rice fields and ponds, causing severe crop damage and substantial ecological destruction such as the destruction of aquatic product resources [9,17,18] and the displacement of the native mudsnail Cipangopaludina cahayensis. In addition, P. canaliculata serves as a major intermediate host for the nematode Angiostrongylus cantonensis, which has led to the emergence of human eosinophilic meningitis in China [16,19].
Genetic divergence between the alien and native species may play an important role in the highly adaptive nature of P. canaliculata. However, few genomic resources are available for P. canaliculata and C. cahayensis, and this lack of information has hindered the understanding of possible molecular mechanisms [20]. Previous studies using mitochondrial DNA have provided insights into the continental expansion and molecular phylogeny of P. canaliculata [12,13,18,21-24], but any genomic factors pertaining to competition and displacement are still unknown.
Recently, next generation sequencing technologies have revolutionized the fields of genomics and transcriptomics, providing an opportunity for the rapid and cost-effective generation of genome-scale data [25]. These technologies have been applied successfully in many invasive species, including Bemisia tabaci [26,27], Anguillicola crassus [28], Aedes aegypti [29] and Mytilus galloprovincialis [30]. In the present study, we sequenced and assembled the transcriptome of the native C. cahayensis from mainland China and the invasive P. canaliculata using de novo sequence assembly. Transcriptome divergence between the native and invasive species was examined to identify important candidate genes related to competitiveness, resistance to environmental stress, and invasive potential. This approach enabled the prediction of expressed sequence tag-simple sequence repeat (EST-SSR) markers to facilitate gene mapping and genetic variation analysis in P. canaliculata.
Result and discussion
Sequencing data and de novo assembly
Using Illumina paired-end sequencing technology, the transcriptome sequencing produced 65,198,546 reads with a total length of 6.5 Gb for C. cahayensis, which generated 161,941 contigs and 151,518 unigenes (Table 1). For P. canaliculata, 94,808,488 reads were obtained, and 94,518 contigs and 76,082 unigenes were generated (Table 1). Using the SOAP de novo assembly program, high quality reads were assembled into 160,256 contigs longer than 200 bp, with a mean length of 1,080 bp and a N50 of 1,004 bp for the native C. cahayensis. For P. canaliculata, 94,518 contigs longer than 200 bp, with a mean length of 916 bp and a N50 of 1,854 bp were generated. In C. cahayensis, the lengths of 104,713 (65.34%) of the contigs ranged from 200 to 500 bp, 28,918 (18.04%) contigs ranged from 500 to 1,000 bp, and 15,191 (9.50%) contigs ranged from 1000 to 2,000 bp; the remaining contigs were longer than 2,000 bp (Figure 1). In P. canaliculata, the lengths of 41,544 (43.95%) of the contigs ranged from 200 to 500 bp, 19,289 (20.41%) contigs ranged from 500 to 1000 bp, and 17,619 (18.16%) contigs ranged from 1000 to 2,000 bp; the remaining contigs were longer than 2,000 bp. The related data were submitted to the NCBI data under accession numbers: SRA191276 (P. canaliculata) and SRA192725 (C. cahayensis).
Table 1.
Transcriptome summary for indigenous Cipangopaludina cahayensis and Pomacea canaliculata
| Cipangopaludina cahayensis | Pomacea canaliculata | |
|---|---|---|
| Total number of reads | 65,198,546 | 94,808,488 |
| Total base pair (bp) | 6,519,854,600 | 3,507,914,056 |
| Total number of contigs | 161,941 | 94,518 |
| Mean length of contigs (bp) | 1,080 | 916 |
| Total number of Unigenes | 151,518 | 76,082 |
| Mean length of Unigenes (bp) | 1,004 | 1,854 |
Figure 1.
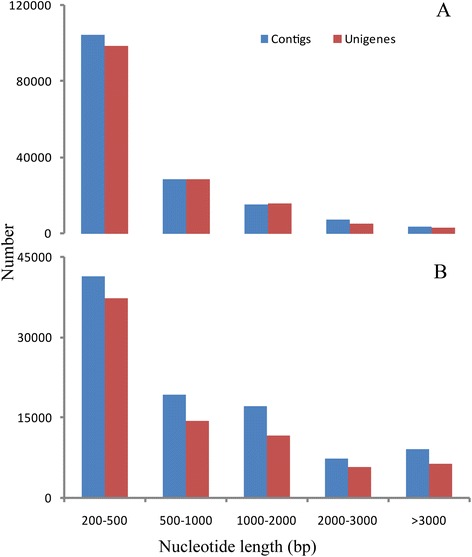
Assessment of transcriptome assembly quality of Cipangopaludina cahayensis (A) and Pomacea canaliculata (B).
Functional annotation
To annotate the C. cahayensis and P. canaliculata sequences, searches were conducted against the NCBI non-redundant protein (Nr) database, the Swiss-Prot protein database, Cluster of Orthologous Groups (COG), and Kyoto Encyclopedia of Genes and Genomes (KEGG) database using BLASTX (E-value ≤ 1 × 10−5). The alignment results were used to predict unigene transcriptional orientations and coding regions. Gene ontology (GO) terms were assigned to the annotated sequences and 14,864 sequences from C. cahayensis and 56,300 sequences from P. canaliculata were categorized into the three GO categories, biological process, cellular component, and molecular function (Figure 2). We found that the distribution and percentages of the assigned gene functions were similar in both species. In the biological process category, death (22.1%) was prominent, while in the molecular function category, cell (30%–31%) and cell part (30%–31%) were prominently represented. In the cellular component category, binding (47.8%–49%) was predominant, followed by catalytic activity (36%). Overall, the transcriptome sequencing yielded a great number of unique genes in the two species, in agreement with similar results reported in other species [20]. Several differences were noted between the two species, with more genes noted in P. canaliculata (56,300 genes) compared with in C. cahayensis (14,864 genes). Furthermore, the percentage of genes annotated as metabolic process/pigmentation under the biological process category was higher in P. canaliculata (15.7%/7.46%) compared with C. cahayensis (7.93%/1.6%), implying a possible relation to various environmental stressors. Moreover, the percentage of genes annotated as metallochaperone activity and translation regulator activity under the cellular component category was much higher in P. canaliculata compared with C. cahayensis. These results indicated that P. canaliculata might contain additional genes that are able to confer high competitiveness or strong resistance to envrionmental stress compared to C. cahayensis.
Figure 2.
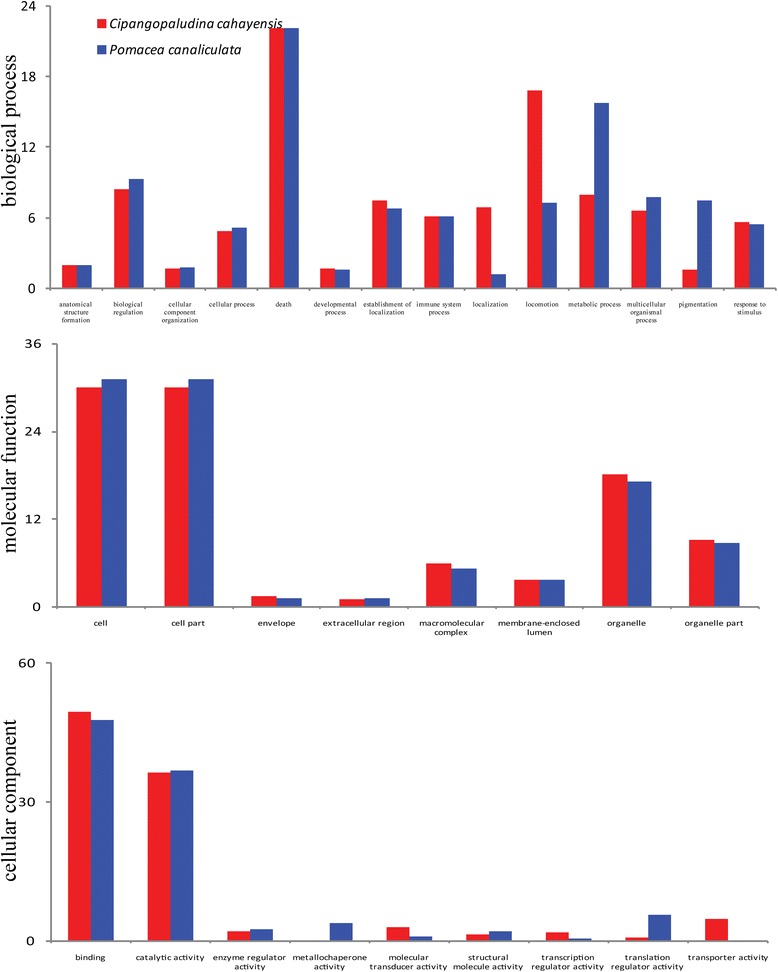
Comparing functional annotations of contigs between Cipangopaludina cahayensis (red) and invasive Pomacea canaliculata (blue) transcriptome. The distribution of gene ontology (GO) terms is given for each of each of the three main GO categories (biological process, molecular function, and cellular component).
Furthermore, all of the C. cahayensis and P. canaliculata unigenes were subjected to functional prediction and classification using the COG database. The unigenes were assigned to 25 COG categories (Figure 3), among which “general function prediction” represented the largest group (4,081 (17.9%) genes for C. cahayensis; 4,346 (19%) genes for P. canaliculata). For C. cahayensis, the next most represented category was translation, ribosomal structure and biogenesis (1915 (8.41%) genes), while for P. canaliculata, replication, recombination and repair (1,883 (8.23%) genes,) was the next most represented category.
Figure 3.
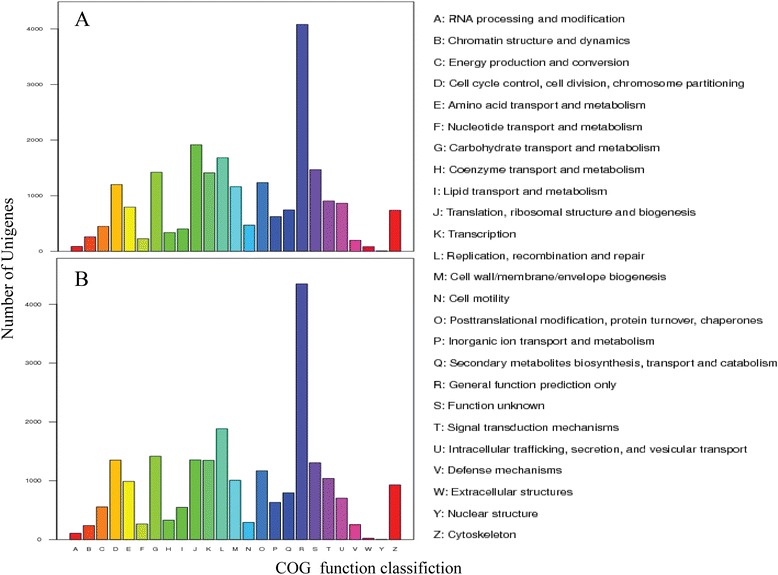
Clusters of orthologous group (COG) classifications for Cipangopaludina cahayensis (A) and Pomacea canaliculata (B) transcriptome. All unigenes were aligned to COG database to predict and classify possible functions.
To identify differentially regulated biological pathways between C. cahayensis and P. canaliculata, the annotated unigenes were mapped to reference pathways in the KEGG database [31]. We found that 13,351 C. cahayensis unigenes mapped to 276 pathways and 13,808 P. canaliculata genes mapped to 240 pathways, with different pathway associations between the two species. In C. cahayensis, the largest number of genes included cancer (577 (4.32%) genes; pathway: ko05200), focal adhesion (496 (3.72%) genes; pathway: ko04510), ubiquitin mediated proteolysis (427 (3.2%) genes; pathway: ko04120), and Huntington’s disease (333 (2.49%) genes; pathway: ko05016). In P. canaliculata, the predominant pathways were metabolic (2241 (16.23%) genes; pathway: ko01100), cancer (530 (3.84%) genes; pathway: ko05200), focal adhesion (415 (3.01%) genes; pathway: ko04510) and Huntington’s disease (348 (2.52%) genes; pathway: ko05016). Collectively, these transcriptome sequences and pathway annotations provide an essential resource for further screening and expression analysis of candidate genes related to the invasive abilities of P. canaliculata.
Analysis of protein families and genes
A total of 15,632 protein families were identified based on sequence similarities (Figure 4); 13,490 families for C. cahayensis and 13,453 families for P. canaliculata. When the transcriptomes of the two species were compared, a total of 11,312 protein families were found to be conserved between the C. cahayensis and P. canaliculata transcriptomes, and 2142 and 2178 families for P. canaliculata and C. cahayensis, respectively, were found to be differentially expressed. Some of the differentially expressed proteins may be responsible for the unique features of each of these species. An enriched analysis of the GO terms assigned to the 11,312 conserved protein families, identified 12 protein families that were significantly enriched (Table 2), including RNA transport (380 (2.6%) genes), spliceosome (383 (2.62%) genes), and endoplasmic reticulum protein processing (358 (2.45%) genes), which are related to protein transportation and metabolism. The finding that GO terms related to protein transportation and metabolism were enriched is inconsistent with the results reported for other invasive species such as Bemisia tabaci [32], possibly suggesting the critical roles of these pathways in these two species. We identified a total of 12 protein families (p-value < 10−5) encoded by the differentially expressed genes in C. cahayensis (Table 3), including those assigned to pathways pertaining to cancer (97 (6.92%) genes), toxoplasmosis (87 (6.21%) genes), and apoptosis (71 (5.06%) genes). In P. canaliculata, we identified a total of 17 protein families (p-value < 10−5) encoded by the differentially expressed genes, including those assigned to pathways pertaining to lysosomes (84 (4.02%) genes), vitamin digestion and absorption (71 (3.4%) genes), ECM-receptor interaction (57 (2.73%) genes), and metabolism of xenobiotics by cytochrome P450 (49 (2.35%) genes). We used reads per kilobase per million mapped reads (RPKM) to analyze the expression levels of P. canaliculata genes and identified 20 annotated genes with very high expression levels (RPKM > 2000), which were predicted to be involved in cell and protein structure (ferritin [Swiss-Prot: C7TNT3] and augerpeptide hhe53 [Swiss-Prot: P0CI21]) and ribosomes (60S ribosomal proteins and 40S ribosomal protein S8) (Table 4).
Figure 4.
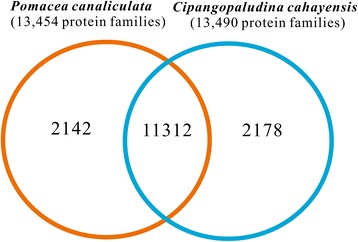
Protein families from the transcriptomes of Cipangopaludina cahayensis and Pomacea canaliculata. Protein families were identified for all the translated genes of the two transcriptomes using Blastp and a Markov Cluster algorithm (MCL), with the total number of protein families belonging to each category listed in the figure for the 11,312 protein families belonging to the two transcriptomes.
Table 2.
Statistically common enriched Gene Ontology (GO) terms between Cipangopaludina cahayensis and Pomacea canaliculata for the 11,312 protein families
| KO term | No. of DEGs | No. of genes | p-value | Pathways |
|---|---|---|---|---|
| ko03050 | 87 ((0.6%)) | 104 (0.38%) | 1.177307e-10 | proteasome |
| ko04141 | 358 (2.45%) | 533 (1.96%) | 1.450501e-10 | protein processing in endoplasmic reticulum |
| ko03013 | 380 (2.6%) | 571 (2.1%) | 2.417814e-10 | RNA transport |
| ko00190 | 222 (1.52%) | 315 (1.16%) | 7.983256e-10 | oxidative phosphorylation |
| ko00020 | 111 (0.76%) | 148 (0.54%) | 7.634897e-08 | citrate cycle (TCA cycle) |
| ko00010 | 143 (0.98%) | 199 (0.73%) | 1.168776e-07 | Glycolysis/Gluconeogenesis |
| ko04130 | 65 (0.45%) | 80 (0.29%) | 2.460867e-07 | SNARE interactions in vesicular transport |
| ko00280 | 131 (0.9%) | 183 (0.67%) | 5.546992e-07 | Valine, leucine and isoleucine degradation |
| ko03040 | 383 (2.62%) | 609 (2.24%) | 2.553937e-06 | spliceosome |
| ko00030 | 69 (0.47%) | 89 (0.33%) | 2.854414e-06 | pentose phosphate pathway |
| ko04910 | 287 (1.97%) | 453 (1.67%) | 1.953777e-05 | insulin signaling pathway |
| ko04380 | 155 (1.06%) | 232 (0.85%) | 3.401093e-05 | osteoclast differentiation |
Table 3.
Statistically unique protein families in Cipangopaludina cahayensis and Pomacea canaliculata
| KO term | No. of DEGs* | No. of genes** | p-value | Pathway |
|---|---|---|---|---|
| Cipangopaludina cahayensis | ||||
| ko05145 | 87 (6.21%) | 316 (2.37%) | 8.262866e-18 | toxoplasmosis |
| ko04210 | 71 (5.06%) | 251 (1.88%) | 2.041735e-15 | apoptosis |
| ko05222 | 64 (4.56%) | 264 (1.98%) | 8.751653e-11 | small cell lung cancer |
| ko05144 | 23 (1.64%) | 55 (0.41%) | 1.741389e-09 | malaria |
| ko04621 | 47 (3.35%) | 190 (1.42%) | 1.417618e-08 | NOD-like receptor signaling pathway |
| ko05014 | 29 (2.07%) | 102 (0.76%) | 3.722068e-07 | amyotrophic lateral sclerosis (ALS) |
| ko05200 | 97 (6.92%) | 577 (4.32%) | 1.527724e-06 | pathways in cancer |
| ko05146 | 39 (2.78%) | 170 (1.27%) | 1.852183e-06 | amoebiasis |
| ko00590 | 29 (2.07%) | 119 (0.89%) | 1.106722e-05 | arachidonic acid metabolism |
| ko05416 | 35 (2.5%) | 158 (1.18%) | 1.392634e-05 | viral myocarditis |
| ko05210 | 27 (1.93%) | 116 (0.87%) | 5.248618e-05 | colorectal cancer |
| ko05323 | 20 (1.43%) | 76 (0.57%) | 7.901248e-05 | rheumatoid arthritis |
| Pomacea canaliculata | ||||
| ko00940 | 22 (1.05%) | 23 (0.17%) | 1.61937e-17 | Phenylpropanoid biosynthesis |
| ko04977 | 71 (3.4%) | 172 (1.25%) | 7.427869e-17 | vitamin digestion and absorption |
| ko00140 | 52 (2.49%) | 120 (0.87%) | 1.004048e-13 | Steroid hormone biosynthesis |
| ko04512 | 57 (2.73%) | 158 (1.14%) | 5.811497e-11 | ECM-receptor interaction |
| ko00830 | 49 (2.35%) | 126 (0.91%) | 6.057395e-11 | Retinol metabolism |
| ko00980 | 49 (2.35%) | 129 (0.93%) | 1.610139e-10 | Metabolism of xenobiotics by cytochrome P450 |
| ko00130 | 24 (1.15%) | 41 (0.3%) | 2.008634e-10 | Ubiquinone and other terpenoid-quinone biosynthesis |
| ko00591 | 45 (2.15%) | 116 (0.84%) | 4.002728e-10 | linoleic acid metabolism |
| ko00533 | 22 (1.05%) | 38 (0.28%) | 1.543998e-09 | glycosaminoglycan biosynthesis - keratan sulfate |
| ko00740 | 21 (1.01%) | 37 (0.27%) | 5.989671e-09 | riboflavin metabolism |
| ko00360 | 26 (1.24%) | 54 (0.39%) | 1.007722e-08 | Phenylalanine metabolism |
| ko00982 | 47 (2.25%) | 142 (1.03%) | 6.201926e-08 | drug metabolism-cytochrome P450 |
| ko00627 | 41 (1.96%) | 119 (0.86%) | 1.229762e-07 | aminobenzoate degradation |
| ko04142 | 84 (4.02%) | 321 (2.32%) | 1.553304e-07 | lysosome |
| ko00590 | 45 (2.15%) | 138 (1%) | 1.921892e-07 | arachidonic acid metabolism |
| ko00983 | 47 (2.25%) | 147 (1.06%) | 2.012819e-07 | drug metabolism-other enzymes |
| ko02020 | 16 (0.77%) | 31 (0.22%) | 2.208469e-06 | two-component system |
*The number of differentially expressed genes (DEGs) that belong to a KEGG pathway.
**The total number of orthologous genes that belong to a KEGG pathway.
Table 4.
Highly expressed genes in the transcriptome of Pomacea canaliculata
| Gene ID | Number of reads* | RPKM** | Swissprot annotation | E-value |
|---|---|---|---|---|
| Unigene0034597 | 370469 | 7963.2 | Ferritin | 2.00E-79 |
| Unigene0070417 | 178665 | 6508.7 | Temptin | 4.00E-22 |
| Unigene0099572 | 1758831 | 6234.6 | Auger peptide hhe53 | 1.00E-11 |
| Unigene0095431 | 309172 | 5711.9 | Cysteine-rich secretory protein Mr30 | 6.00E-50 |
| Unigene0102121 | 48348 | 5217.9 | Polyubiquitin | 7.00E-39 |
| Unigene0069599 | 333123 | 4748.7 | Elongation factor 1-alpha, somatic form | 0 |
| Unigene0122375 | 169714 | 4112.1 | Fibrinogen C domain-containing protein 1-B | 1.00E-53 |
| Unigene0115512 | 780688 | 4335.5 | Paramyosin | 0 |
| Unigene0087254 | 227698 | 4289.3 | Metalloproteinase inhibitor 3 | 7.00E-07 |
| Unigene0114631 | 361080 | 4118.2 | Actin, adductor muscle | 0 |
| Unigene0069690 | 284495 | 4020.1 | Tubulin beta chain | 0 |
| Unigene0121686 | 60035 | 3534.1 | 60S ribosomal protein L36 | 2.00E-31 |
| Unigene0006316 | 82422 | 2973.3 | 40S ribosomal protein S8 | 1.00E-89 |
| Unigene0102783 | 157410 | 2712.7 | 60S ribosomal protein L5 | 1.00E-123 |
| Unigene0033792 | 48918 | 2226.8 | 60S ribosomal protein L24 | 4.00E-57 |
| Unigene0099167 | 208322 | 2530.9 | Myosin, essential light chain, adductor muscle | 1.00E-47 |
| Unigene0083872 | 75749 | 2346.9 | 60S ribosomal protein L44 | 3.00E-47 |
| Unigene0099241 | 78798 | 2247.8 | 60S ribosomal protein L7a | 2.00E-113 |
| Unigene0034297 | 55506 | 2045.0 | 60S ribosomal protein L23a | 1.00E-57 |
| Unigene0123696 | 49155 | 2011.4 | Ubiquitin-60S ribosomal protein L40 | 7.00E-68 |
*The total number of reads mapped to each gene.
**Gene expression levels were determined by calculating the number of reads for each gene and then normalizing to RPKM.
P. canaliculata has become an important pest in China and has exhibitied resistance to pesticides such as metaldehyde and niclosamide ethanolamine salt [33-35]; however, the molecular mechanisms underlying this resistance are still unclear. To detect unique resistance-related sequences, the unigenes were edited manually to remove redundant and overlying short sequences and the edited sequences were then used to identify genes encoding proteins related to the metabolism of pesticides. We identified P450 cytochromes (CYPs), a major family of enzymes involved in detoxification and metabolism, as potential major detoxification component proteins [36-38]. Previous studies have reported a correlation between increased exposure to metabolic neurotoxic pesticides and over-expression of P450 genes in many pest species [39-46]. In our study, 210 P450-related sequences were identified in P. canaliculata and only 159 were found in C. cahayensis, indicating that the number of P450 genes may be one of the contributory factors to pesticides resistance in P. canaliculata. While the number of P450 genes detected is not necessarily related to gene expression levels, an increased gene number of genes may increase metabolic enzyme detoxification activity, and contribute to the development of a progressive resistance in P. canaliculata. These findings will enhance the understanding of pesticide metabolism and help in the development of effective treatments for invasive species. To investigate the relationship between the P450 sequences from both species a phylogenetic tree was constructed using the neighbor joining (NJ) method in conjunction with bit-score values. Sixty of the sequences showed high homology and were classified into the CYP2, CYP3, and CYP4 families based on their similarity to sequences in the Nr database. These sequences clustered into three clades in the phylogentic tree that corresponded to the same three P450 families (Figure 5). We found a high concentration of P. canaliculata genes in the CYP4 family, possibly implying that these genes played important roles in the metabolism of pesticides in this invasive species. While these finding are insightful, they need to be examined further using RACE technology and RT-PCR before they can be accepted.
Figure 5.
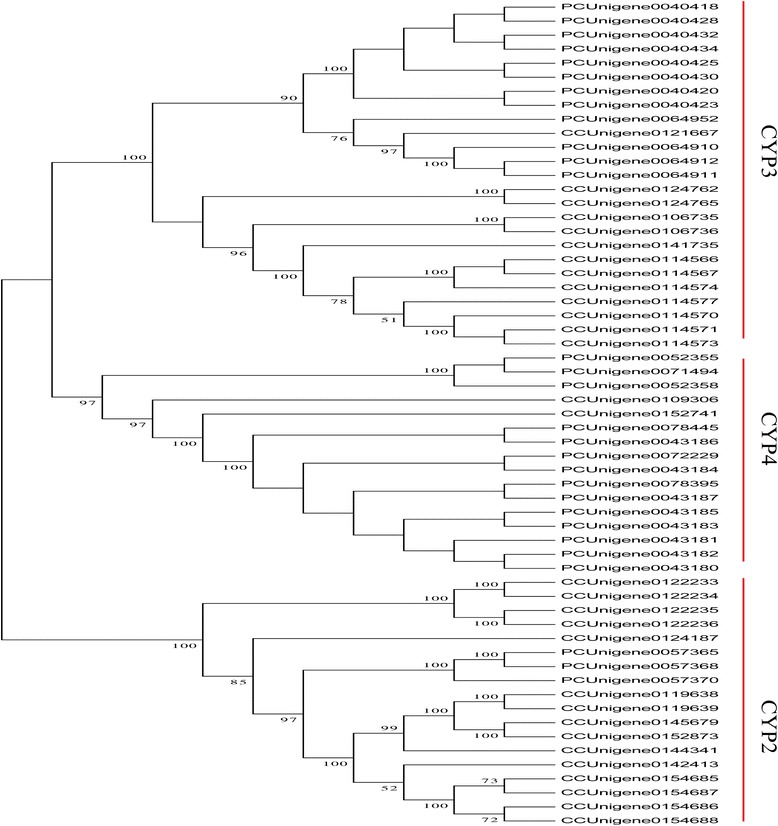
Neighbor-joining phylogenetic analysis of cytochrome P450 from Cipangopaludina cahayensis (CC) and Pomacea canaliculata (PC). CYP represent cytochrome P450.
Detection of intraspecific genetic variation
EST-SSRs serve as effective molecular markers for genetic mapping, comparative genomics and population genetic analysis in many invasive species. Characterization of EST-SSRs may enable breakthroughs in the detection of cryptic species, aid in defining the number and location of establishment events, and help trace the routes of alien species as they spread into new regions [47-51]. Compared with traditional methods, EST-SSRs are more transferable and advantageous than random genomic SSRs, enabling improved genetic studies related to population genetics [52]. Unitl now, only a few SSRs have been identified in P. canaliculata [20,53], which has hampered marker applications in this species. To further understand the invasive and adaptive mechanism in P. canaliculata, six P. canaliculata samples were collected from three. invasive regions/habitats in mainland China and examined for polymorphisms. A total of 16,717 potential SSRs were identified. As shown in Table 5, the di-nucleotide repeats were the most abundant (10,554, 63.1%), followed by tri-(4,480, 26.8%), tetra- (1,021, 6.10%), hexa-(341, 2.0%), and penta-nucleotide (321, 1.9%) repeats. The most abundant repeat combination was AG/CT (40.4%), followed by AT/AT (18.3%), AAG/CTT (7.8%), AAT/ATT (4.7%), AC/GT (4.0%) and ATC/ATG (3.4%) (Figure 6A). Based on the SSR-containing sequences, 8,428 SSR primers were developed and 100 SSRs (Additional file 1: Table S1) were selected to design EST-SSR primers based on the information (name and longer length of gene identified). Of the 100 SSRs examined by PCR amplification, 26 (26.0%) PCR products exhibited more than one band, which may have resulted from high heterozygosity, while the others SSRs generated bands of the expected length. In total, 143 amplicons were detected from the 100 primer pairs. The number of amplicons per primer pair ranged from one to three, with an average of 1.43 (Figure 6B). To estimate EST-SSR marker novelty, the amplicons were evaluated against previously reported P. canaliculata markers [20,53]. We found that the 100 EST-SSR markers had not been reported previously. Thus, other EST-SSR primers can be designed from the 8,428 identified EST-SSR to contribute further to the characterization of the invasive and adaptive processes. P. canaliculata and C. cahayensis have very similar morphological features, especially at the immature stages, which makes early identification difficult. Therefore, a molecular means for the identification and characterization of these two species is essential. Using the P. canaliculata SSR primers, we identified a unique amplicon (FSLssr64; Additional file 1: Table S1) that was present in P. canaliculata but absent in C. cahayensis (Figure 6C). Thus, FSLssr64 could serve as a species-specific molecular marker to distinguish these two species and aid in the prevention and detection of invasive P. canaliculata in different regions.
Table 5.
Summary of EST-SSRs identified in the Pomacea canaliculata transcriptome
| Searching item | Numbers |
|---|---|
| Total number of Unigene examined | 135,121 |
| Total size of examined Unigene (bp) | 117,356,620 |
| Total number of identified SSRs | 16,717 |
| Number of Unigene containing SSR | 14,722 |
| Number of Unigene containing more than 1 SSR | 1,748 |
| Number of SSRs present in compound formation | 753 |
| Di-nucleotide | 10,554 (63.1%) |
| Tri-nucleotide | 4,480 (26.8%) |
| Tetra-nucleotide | 1,021 (6.10%) |
| Penta-nucleotide | 321 (1.9%) |
| Hexa-nucleotide | 341 (2.0%) |
Figure 6.
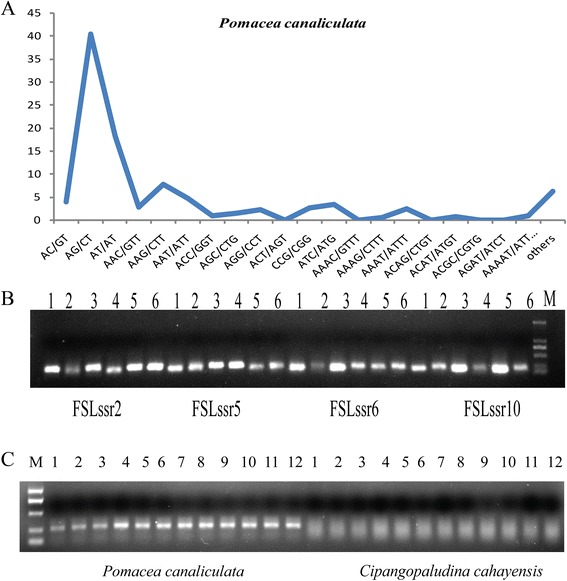
Frequencies and polymorphisms of classified SSR repeat types and molecular characterization of Pomacea canaliculata. (A): The graph shows the frequency of each repeat motif classified, considering the sum of the frequencies for complementary sequences (for example, the sum of frequencies for the dinucleotides AC and its complementary GT). (B) Polymorphism and validation of a subset of the microsatellite primer pairs for six P. canaliculata samples by agarose-gel profiling. 1–6 represent GZ1, GZ2, HN1, HN2, SG1, and SG2, respectively. (C) The SSR primer (FSLssr64) for species-specific identification between P. canaliculata and C. cahayensis.
Conclusions
The transcriptomes of the invasive golden apple snail (P. canaliculata) and the native mudsnail (C. cahayensis) were characterized using the Illumina next-generation sequencing technique. This allowed the identification of a number of the differentially expressed genes, some of which were found to be related specifically to environmental stress; for example, the CYP4 family of cytochrome P450s. These findings can contribute to a better understanding of pesticide metabolism and will provide valuable genetic data to facilitate future studies towards understanding the successful invasive and adaptive mechanism of P. canaliculata. In addition, the 16,717 EST-SSRs predicted in this study should provide a solid genetic basis for molecular markers development and aid in ecological studies pertaining to genetic variation in P. canaliculata.
Methods
Ethics statement
This study was approved by the Animal Care and Use committee of Aquatic Invasive Risk Assessment Center, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences.
Sample collection, RNA extraction, and next generation sequencing
P. canaliculata (20–25 mm shell length; 25.23 ± 0.34 g; 10 individuals) and C.cahayensis (20.4–23.2 mm shell length; 22.43 ± 0.46 g; 10 individuals) were collected without the use of chemicals and grown in the Aquatic Invasive Risk Assessment Center, Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China. Tissues samples from the foot, muscle, liver, and kidney were rinsed separately with water pretreated by diethyl pyrocarbonate to cleanse the samples and inactivate RNases [32]. Total RNA of each sample was extracted using a Trizol Kit (Promega) according to the manufacturer’s instructions. RNA quality was assessed using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and RNase-free agarose gel electrophoresis, with the total RNA concentration measured using a 2100 Bioanalyzer. Equal amounts of RNA from each sampled tissue were combined for subsequent experiments and RNA purity was assessed at absorbance ratios of OD260/280 and OD260/230. RNA integrity was confirmed by 1% agarose gel electrophoresis.
De novo assembly and gene annotation of Illumina reads
Transcriptome de novo assembly was carried out with the short-read assembly program Trinity [54]. The Trinity program has three independent modules: Inchworm, Chrysalis, and Butterfly. Inchworm assembled the RNA sequencing data into unique transcripts that we called Inchworm contigs; Chrysalis clustered the Inchworm contigs, then constructed complete de Bruijn graphs for each cluster and partitioned the full read set among these disjoint graphs; and Butterfly processed the individual graphs in parallel, tracing the paths based on reads and pair-end information, ultimately reporting full-length transcripts for alternatively spliced isoforms. After assembly, the TIGR Gene Indices clustering tools (TGICL) [55] were used to cluster and remove redundant transcripts. The remaining sequences after TGICL clustering were defined as unigenes. BLASTX searches (E-value < 10−5) were conducted to screen the unigenes against the Nr database (http://www.ncbi.nlm.nih.gov/), Swiss-Prot protein database (http://www.expasy. ch/sprot/), the KEGG pathway database (http://www.genome.jp/kegg/), and COG database (http://www.ncbi.nlm.nih.gov/cog/). High scoring alignments were used to determine the unigene sequence direction. When alignment results varied between databases, the results from the Nr database were preferentially selected, followed by the Swiss-Prot, KEGG and COG databases. When a unigene sequence did not match entries in any of these databases, ESTScan was used to predict the coding regions and determine sequence directionality.
Functional annotation and differential expression analysis of unigenes
Unigene sequences were aligned to the protein databases (listed above) using BLASTX (E-value < 10−5) and to the nucleotide sequence database Nt (E-value < 10−5) using BLASTN to obtain both protein and functional annotation information. Based on the annotations in the protein databases, Blast2GO [56] was used to obtain GO annotations for the aligned unigene sequences and the Web Gene Ontology Annotation Plot (WEGO) software [57] was used to establish GO functional classifications for all unigenes. The unigenes were aligned to the COG database to predict and classify possible functions and the KEGG database was used [31] to obtain pathway annotations (E-value threshold 10−5). RPKM was used to calculate unigene expression levels, which eliminated the influence of gene length and sequencing level on the estimation of gene expression
Analysis of genes related to pesticide
BLASTX searches against the Nr database (E-value < 10−5) were used to detect genes related to pesticide resistance. Sequences that returned redundant BLAST results or showed high sequence homology were eliminated and presumed to be allelic variants or different parts of the same gene. Thirty P450 gene sequences (Additional file 1: Table S1) with a range of bit-score values were identified and aligned using MUSCLE [58], and their phylogenetic relationships and genotype classifications were determined using MEGA 5 software [59]. The NJ method [60] was used to create phylogenetic trees, with positions containing alignment gaps or missing data eliminated via pairwise deletion. Tree branch strength was evaluated via a bootstrap analysis of 1000 replication trees.
Development and detection of EST-SSR markers
MIcroSAtellite (MISA) (http://pgrc.ipk-gate-rsleben.de/misa/) was used for microsatellite mining. SSRs were considered to contain motifs of two to six nucleotides and a minimum of five contiguous repeat units. Based on the MISA results, Primer 6.0 was used with the default settings to design primer pairs that would generate PCR products ranging from 100 to 280 bp in length. A total of 100 pairs of primers were designed (Additional file 1: Table S1) and validated by PCR in six P. canaliculata samples, including Guangzhou 1–2 (GZ1-2), Hainan1-2 (HN1-2), and Shaoguang1-2 (SG1-2) that were collected from three major invasive regions in Guangdong Province, China. PCR amplification was carried out as follows: an initial denaturation at 94°C for 4 min; 33 cycles of 94°C for 1 min (denaturation), 50°C for 30 s (annealing), and 72°C for 45 s (extension); followed by a final extension at 72°C for 8 min. The PCR products were analyzed by electrophoresis on a 8.0% non-denaturing polyacrylamide gel and silver stained.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31300468, u1131006) and Agricultural Biological Resources Protection and Utilization Project (2130108).
Additional file
60 P450-related gene sequences for phylogenetic tree in Cipangopaludina cahayensis and Pomacea canaliculata.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XDM designed the study, analyzed the data, and drafted the whole manuscript. GYH, PX, and HMS extracted the RNA, analyzed the bioinformatics data and participated in the manuscript revision. HMS extracted the RNA and helped draft the manuscript. DL, DEG and MX collected samples, assisted with data analysis. JRL and JEZ analyzed the data and helped draft the manuscript. YCH co-designed the experiments and obtained the funds. All authors read and approved the final manuscript.
Contributor Information
Xidong Mu, Email: muxd1019@163.com.
Guangyuan Hou, Email: hougy@cafs.ac.cn.
Hongmei Song, Email: shm1227@126.com.
Peng Xu, Email: xupeng@cafs.ac.cn.
Du Luo, Email: luodu2012@163.com.
Dangen Gu, Email: gudangen@163.com.
Meng Xu, Email: xm0557@126.com.
Jianren Luo, Email: olfishlo@163.com.
Jiaen Zhang, Email: jeanzh@scau.edu.cn.
Yinchan Hu, Email: huyc22@163.com.
References
- 1.Pimentel DS, McNair J, Janecka J, Wightman J, Simmonds C, O’Connell C, Wong E, Russel L, Zern J, Aquino T, Tsomondo T. Economic and environmental threats of alien plant, animal, and microbe invasions. Agr Ecosyst Environ. 2001;84:1–20. doi: 10.1016/S0167-8809(00)00178-X. [DOI] [Google Scholar]
- 2.Pimental D, Zuniga R, Morrison D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol Econ. 2005;52:273–288. doi: 10.1016/j.ecolecon.2004.10.002. [DOI] [Google Scholar]
- 3.Simberloff D. The politics of assessing risk for biological invasions: the USA as a case study. Trends Ecol Evol. 2005;20:216–222. doi: 10.1016/j.tree.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy TA, Naeem S, Howe KM, Knops JMH, Tilman D, Reich P. Biodiversity as a barrier to ecological invasion. Nature. 2002;417:636–638. doi: 10.1038/nature00776. [DOI] [PubMed] [Google Scholar]
- 5.Allendorf FW, Lundquist LL. Introduction: population biology, evolution, and control of invasive species. Conserv Biol. 2003;17:24–30. doi: 10.1046/j.1523-1739.2003.02365.x. [DOI] [Google Scholar]
- 6.Lowe MR, Wu W, Peterson MS, Brown-Peterson NJ, Slack WT, Schofield PJ. Survival, growth and reproduction of non-native Nile tilapia II: fundamental niche projections and invasion potential in the northern Gulf of Mexico. PLoS One. 2012;7:e41580. doi: 10.1371/journal.pone.0041580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carey MP, Wahl DH. Native fish diversity alters the effects of an invasive species on food webs. Ecology. 2010;91:2965–2974. doi: 10.1890/09-1213.1. [DOI] [PubMed] [Google Scholar]
- 8.Ma YZ. Exotic invasion losses an annual 200$ plus billion. Econ weekly. 2009;21:43–45. [Google Scholar]
- 9.Carlsson NOL, Bronmark C, Hansson L. Invading herbivory: the golden apple snail alters ecosystem functioning in Asian wetlands. Ecology. 2004;85:1575–1580. doi: 10.1890/03-3146. [DOI] [Google Scholar]
- 10.Kwong KL, Robert KY, Qiu JW. The potential of the invasive snail Pomacea canaliculata as a predator of various life-stages of five species of freshwater snails. Malacologia. 2009;51:343–356. doi: 10.4002/040.051.0208. [DOI] [Google Scholar]
- 11.Kwong KL, Dudgeon D, Wong PK, Qiu JW. Secondary production and diet of an invasive snail in freshwater wetlands: implications for resource utilization and competition. Biol Invasions. 2010;12:1153–1164. doi: 10.1007/s10530-009-9537-x. [DOI] [Google Scholar]
- 12.Rawlings TA, Hayes KA, Cowie RH, Collins TM. The identity, distribution, and impacts of non-native apple snails in the continental United States. BMC Evol Biol. 2007;7:97. doi: 10.1186/1471-2148-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayes KA, Cowie RH, Jørgensen A, Schultheiß R, Albrecht C, Thiengo SC. Molluscan models in evolutionary biology: apple snails (Gastropoda: Ampullariidae) as a system for addressing fundamental questions. Am Malacol Bull. 2009;27:47–58. doi: 10.4003/006.027.0204. [DOI] [Google Scholar]
- 14.Fang L, Wong PK, Lin L, Lan C, Qiu JW. Impact of invasive apple snails in Hong Kong on wetland macrophytes, nutrients, phytoplankton and filamentous algae. Freshwater Biol. 2010;55:1191–1204. doi: 10.1111/j.1365-2427.2009.02343.x. [DOI] [Google Scholar]
- 15.Yang YX, Hu YC, Li XH, Wang XJ, Mu XD, Song HM, et al. Historical invasion, expansion process and harm investigation of Pomacea canaliculata in China. Chinese Agri Sci Bull. 2010;26:245–250. [Google Scholar]
- 16.Lv S, Zhang Y, Liu HX, Hu L, Yang K, Steinmann P, et al. Invasive snails and an emerging infectious disease: results from the first national survey on Angiostrongylus cantonensis in China. PLoS Neglect Trop D. 2009;3:e368. doi: 10.1371/journal.pntd.0000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu YF, Li F, Li YF, Liu WH, Zeng QG, Su WJ. The distribution situation, damage and early warning on diffusion risk of golden apple snail, Pomacea canaliculata lamark in Human. Acta Hydro Sinica. 2011;35:1067–1071. [Google Scholar]
- 18.Carlsson NOL, Lacoursiere JO. Herbivory on aquatic vascular plants by the introduced golden apple snail (Pomacea canaliculata) in Lao PDR. Biol Invasions. 2005;7:233–241. doi: 10.1007/s10530-004-0741-4. [DOI] [Google Scholar]
- 19.Lv S, Zhang Y, Liu HX, Hu L, Liu Q, Wei FR, et al. Phylogenetic evidence for multiple and secondary introductions of invasive snails: Pomacea species in the People’s Republic of China. Divers Distrib. 2013;19:147–156. doi: 10.1111/j.1472-4642.2012.00924.x. [DOI] [Google Scholar]
- 20.Sun J, Wang M, Wang H, Zhang H, Zhang X, Thiyagarajan V, et al. De novo assembly of the transcriptome of an invasive snail and its multiple ecological applications. Mol Ecol Resour. 2012;12:1133–1144. doi: 10.1111/1755-0998.12014. [DOI] [PubMed] [Google Scholar]
- 21.Hayes KA, Joshi RC, Thiengo SC, Cowie RH. Out of South America: multiple origins of non-native apple snails in Asia. Divers Distrib. 2008;14:701–712. doi: 10.1111/j.1472-4642.2008.00483.x. [DOI] [Google Scholar]
- 22.Hayes KA, Cowie RH, Thiengo SC. A global phylogeny of apple snails: Gondwanan origin, generic relationships, and the influence of outgroup choice (Caenogastropoda: Ampullariidae) Biol J Linn Soc. 2009;98:61–76. doi: 10.1111/j.1095-8312.2009.01246.x. [DOI] [Google Scholar]
- 23.Hayes KA, Cowie RH, Thiengo SC, Strong EE. Comparing apples with apples: clarifying the identities of two highly invasive neotropical Ampullariidae (Caenogastropoda) Zool J Linn Soc. 2012;166:723–753. doi: 10.1111/j.1096-3642.2012.00867.x. [DOI] [Google Scholar]
- 24.Matsukura K, Okuda M, Cazzaniga NG, Wada T. Genetic exchange between two freshwater apple snails, Pomacea canaliculata and Pomacea maculata invading East and Southeast Asia. Biol Invasions. 2013;15:2039–2048. doi: 10.1007/s10530-013-0431-1. [DOI] [Google Scholar]
- 25.Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genom Hum G. 2009;10:135–151. doi: 10.1146/annurev-genom-082908-145957. [DOI] [PubMed] [Google Scholar]
- 26.Wang XW, Luan JB, Li JM, Su YL, Xia J, Liu SS. Transcriptome analysis and comparison reveal divergence between two invasive whitefly cryptic species. BMC Genomics. 2011;12:458. doi: 10.1186/1471-2164-12-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YL, Wang YJ, Luan JB, Yan GH, Liu SS, Wang XW. Analysis of the transcriptional differences between indigenous and invasive whiteflies reveals possible mechanisms of whitefly invasion. PLoS One. 2013;8:e62176. doi: 10.1371/journal.pone.0062176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heitlinger E, Bridgett S, Montazam A, Taraschewski H, Blaxter M. The transcriptome of the invasive eel swim bladder nematode parasite Anguillicola crassus. BMC Genomics. 2013;14:87. doi: 10.1186/1471-2164-14-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akbari OS, Antoshechkin I, Amrhein H, Williams B, Diloreto R, Sandler J, et al. he developmental transcriptome of the mosquito Aedes Aegypti, an invasive species and major arbovirus vector. G3. 2013;3:1493–1509. doi: 10.1534/g3.113.006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lockwood BL, Sanders JG, Somero GN. Transcriptomic responses to heat stress in invasive and native blue mussels (genus Mytilus): molecular correlates of invasive success. J Exp Biol. 2010;213:3548–3558. doi: 10.1242/jeb.046094. [DOI] [PubMed] [Google Scholar]
- 31.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XW, Zhao QY, Luan JB, Wang YJ, Yan GH, Liu SS. Analysis of a native whitefly transcriptome and its sequence divergence with two invasive whitefly species. BMC Genomics. 2012;13:529. doi: 10.1186/1471-2164-13-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh FL, Lai PS, Huang DJ. Preliminary studies of molluscicide resistance of apple snails, Pomacea canaliculata in Taiwan. Bull Malacol. 2010;34:49–62. [Google Scholar]
- 34.Liu XM. Study on distinctions of pesticides resistance and its physiology biochemistry in different populations of Pomacea canaliculata. 1st ed. Nanning: Guangxi University Press; 2011
- 35.He MQ, Luo MZ, Zhang JE, Song CX. Damage status and control counter measure of Pomacea canaliculata in Guangdong. Guizhou Agr Sci. 2011;39:100–103. [Google Scholar]
- 36.Tijet N, Helvig C, Feyereisen R. The cytochrome P450 gene superfamily in Drosophila melanogaster: annotation, intron-exon organization and phylogeny. Gene. 2001;262:189–198. doi: 10.1016/S0378-1119(00)00533-3. [DOI] [PubMed] [Google Scholar]
- 37.Daborn P, Yen J, Bogwitz M, Le Goff G, Feil E, Jeffers S, et al. A single P450 allele associated with insecticide resistance in Drosophila. Science. 2002;297:2253–2256. doi: 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- 38.Puinean AM, Foster SP, Oliphant L, Denholm I, Field LM, Millar NS, et al. Amplification of a cytochrome P450 gene is associated with resistance to neonicotinoid insecticides in the aphid Myzus persicae. PLoS Genet. 2010;6:e1000999. doi: 10.1371/journal.pgen.1000999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58:737–747. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bogwitz MR, Chung H, Magoc L, Rigby S, Wong W, O’Keefe M, et al. CYP12A4 confers lufenuron resistance in anatural population of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2005;102:12807–12812. doi: 10.1073/pnas.0503709102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joußen N, Heckel DG, Haas M, Schuphan I, Schmidt B. Metabolism of imidacloprid and DDT by P450 CYP6G1 expressed in cell cultures of Nicotiana tabacum suggests detoxification of these insecticides in Cyp6g1-overexpressing strains of Drosophila melanogaster, leading to resistance. Pest Manag Sci. 2008;64:65–73. doi: 10.1002/ps.1472. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin WS, Marko PB, Nelson DR. The cytochrome P450 (CYP) gene superfamily in Daphnia pulex. BMC Genomics. 2009;10:169. doi: 10.1186/1471-2164-10-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones RT, Bakker SE, Stone D, Shuttleworth SN, Boundy S, McCart C, et al. Homology modelling of Drosophila cytochrome P450 enzymes associated with insecticide resistance. Pest Manag Sci. 2010;66:1106–1115. doi: 10.1002/ps.1986. [DOI] [PubMed] [Google Scholar]
- 44.Zhu F, Parthasarathy R, Bai H, Woithe K, Kaussmann M, Nauen R, et al. A brain-specific cytochrome P450 responsible for the majority of deltamethrin resistance in the Qtc279 strain of Tribolium castaneum. Proc Natl Acad Sci U S A. 2010;107:8557–8562. doi: 10.1073/pnas.1000059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bass C, Carvalho R, Oliphant L, Puinean A, Field L, Nauen R, et al. Overexpression of a cytochrome P450 monooxygenase, CYP6ER1, is associated with resistance to imidacloprid in the brown planthopper, Nilaparvata lugens. Insect Mol Biol. 2011;20:763–773. doi: 10.1111/j.1365-2583.2011.01105.x. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell SN, Stevenson BJ, Müller P, Wilding CS, Egyir-Yawson A, Field SG, et al. Identification and validation of a gene causing cross-resistance between insecticide classes in Anopheles gambiae from Ghana. Proc Natl Acad Sci U S A. 2012;109:6147–6152. doi: 10.1073/pnas.1203452109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herborg LM, Weetman D, Oosterhout C, Hanfling B. Genetic population structure and contemporary dispersal patterns of a recent European invader, the Chinese mitten crab, Eriocheir sinensis. Mol Ecol. 2007;16:231–242. doi: 10.1111/j.1365-294X.2006.03133.x. [DOI] [PubMed] [Google Scholar]
- 48.Darling JA, Bagley MJ, Roman J, Tepolt CK, Geller JB. Genetic patterns across multiple introductions of the globally invasive crab genus Carcinus. Mol Ecol. 2008;17:4992–5007. doi: 10.1111/j.1365-294X.2008.03978.x. [DOI] [PubMed] [Google Scholar]
- 49.Cao LL, Zhou LZ, Zhang BW. Genetic patterns of an invasive Procambarus clarkii population in the three river basins of Anhui Province. Biodiver Sci. 2010;18:398–407. [Google Scholar]
- 50.Yue GH, Li JL, Bai ZY, Wang CM, Feng F. Genetic diversity and population structure of the invasive alien red swamp crayfish. Biol Invasions. 2012;12:2697–2706. doi: 10.1007/s10530-009-9675-1. [DOI] [Google Scholar]
- 51.Zhan A, Perepelizin PV, Ghabooli S, Paolucci E, Sylvester F, Sardiña P, et al. Scale-dependent post-establishment spread and genetic diversity in an invading mollusc inSouth America. Divers Distrib. 2012;18:1042–1055. doi: 10.1111/j.1472-4642.2012.00894.x. [DOI] [Google Scholar]
- 52.Zhang L, Yan HF, Wu W, Yu H, Ge XJ. Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii) BMC Genomics. 2013;14:329. doi: 10.1186/1471-2164-14-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L, Xu HG, Li H, Wu J, Ding H, Liu Y. Isolation and characterization of sixteen polymorphic microsatellite loci in the golden apple snail Pomacea canaliculata. Int J Mol Sci. 2011;12:5993–5998. doi: 10.3390/ijms12095993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pertea G, Huang XQ, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 56.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 57.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]