

Abstract
Oncostatin M (OSM) is an interleukin-6-like inflammatory cytokine reported to play a role in a number of pathological processes including cancer. Full-length OSM is expressed as a 26 kDa protein that can be proteolytically processed into 24 kDa and 22 kDa forms via removal of C-terminal peptides. In this study, we examined both the ability of OSM to bind to the extracellular matrix (ECM) and the activity of immobilized OSM on human breast carcinoma cells. OSM was observed to bind to ECM proteins collagen types I and XI, laminin, and fibronectin in a pH-dependent fashion, suggesting a role for electrostatic bonds that involves charged amino acids of both the ECM and OSM. The C-terminal extensions of 24 kDa and 26 kDa OSM, which contains six and thirteen basic amino acids, respectively, enhanced electrostatic binding to ECM at pH 6.5–7.5 when compared to 22 kDa OSM. The highest levels of OSM binding to ECM, though, were observed at acidic pH 5.5, where all forms of OSM bound to ECM proteins to a similar extent. This indicates additional electrostatic binding properties independent of the OSM C-terminal extensions. The reducing agent dithiothreitol also inhibited the binding of OSM to ECM suggesting a role for disulfide bonds in OSM immobilization. OSM immobilized to ECM was protected from cleavage by tumor-associated proteases and maintained activity following incubation at acidic pH for extended periods of time. Importantly, immobilized OSM remained biologically active and was able to induce and sustain the phosphorylation of STAT3 in T47D and ZR-75-1 human breast cancer cells over prolonged periods, as well as increase levels of STAT1 and STAT3 protein expression. Immobilized OSM also induced epithelial-mesenchymal transition-associated morphological changes in T47D cells. Taken together, these data indicate that OSM binds to ECM in a bioactive state that may have important implications for the development of chronic inflammation and tumor metastasis.
Keywords: Cytokines, extracellular matrix, collagens, cancer, metastasis, inflammation, Col11a1, oncostatin M, OSM, electrostatic binding, arthritis
1. INTRODUCTION
Oncostatin M (OSM) is a member of the interleukin-6 (IL-6) cytokine family, which signals cancer cells primarily through STAT3 to promote a metastatic phenotype via effects on tumor cell proliferation, attachment to substratum, migration, invasion, metastasis and the epithelial-to-mesenchymal transition (EMT) (1–8). OSM is expressed as a 26 kDa MW protein (full-length) that can be proteolytically processed into 24 kDa OSM via proteolytic cleavage of eighteen C-terminal amino acids, as well as a 22 kDa form via removal of an additional thirteen C-terminal amino acids by a trypsin-like protease (9). OSM with MWs of 22 kDa and 26 kDa are reported in rat testis that differ in MW due to OSM glycosylation, and are suggested to have different functions in spermatogenesis (10). OSM is reported to induce acute inflammation upon subcutaneous injection into mice (11) and can regulate inflammation via the induction of both proteases and protease inhibitors (12, 13). OSM regulates proteolysis and extracellular matrix (ECM) turnover via the induction of cysteine and aspartic proteases, as well as metalloproteinases (3, 14, 15). In addition to its role in the inflammatory process associated with tumor progression, OSM drives other pathological conditions involving chronic inflammation including arthritis (16–20), Alzheimer’s disease (21–23) and atherosclerosis (24). The importance of inflammation in cancer is well established, since patients suffering from chronic inflammatory conditions such as pancreatitis, inflammatory bowel disease, prostatitis, and chronic obstructive pulmonary disease are more prone to develop cancer in affected tissues (25–28). In addition, a link between arthritis-induced inflammation and secondary metastasis associated with breast cancer has recently been established (29, 30).
IL-6 family members, including OSM, and the JAK/STAT pathway have recently emerged as key players in cancer-associated inflammation (4, 27, 31–34). Most inflammatory signals affect tumor progression by activating STAT3 and NF-κB (32). OSM signaling through STAT3 is involved in the regulation of both systemic and local inflammatory responses in a variety of tumors (33, 35), shaping the tissue microenvironment into one that is pro-tumorigenic and inflammatory (36). Interestingly, unphosphorylated STAT3 is also reported to activate cell signaling associated with inflammation (37, 38).
The ECM is a complex and dynamic material that provides structural support for cells (39, 40). The ECM also functions in ways beyond structural support and is important in phenomena as diverse as developmental patterning, stem cell niches, genetic diseases, and cancer (39, 40). ECM proteins regulate paracrine, autocrine, juxtacrine, and matricrine signaling in cells (41) and numerous growth factors and cytokines promote expression of ECM proteins (39, 40). OSM is reported to enhance fibronectin expression in T47D breast tumor cells, as well as regulate ECM deposition in MCF-7 breast tumor cells (8). OSM also stimulates excessive ECM production and accumulation in connective tissue disease in vivo (42), and in vitro in human dermal fibroblasts, pancreatic stellate cells, and lung fibroblasts (42–46). Additionally, the ECM acts as a reservoir of biologically active molecules and as a storage site for growth factors, cytokines, and proteases (39, 47, 48), regulating their distribution, activation, and presentation to cells (41). Recently, the invasiveness and malignant properties of ovarian tumors were reported to be enhanced by collagen type XI expression (49).
Many interactions between ECM proteins and cytokines are highly specific. Variations in localization of specific ECM protein isoforms, the extent of ECM protein glycosylation and the type of glycosaminoglycan groups present on proteoglycan molecules all work together to determine the distribution of specific cytokines within the ECM, providing an additional layer of regulation for cytokine activity (41). As solid phase ligands, ECM proteins integrate complex, multivalent signals to cells in a spatially and temporally patterned fashion (40). The ability of particular cytokines to bind and be immobilized by ECM proteins can accentuate their action by: 1) promoting the accumulation of cytokines in a particular microenvironment and to increase their encounter with target cells; 2) activating cytokines by inducing conformational changes in the bound cytokine; 3) promoting conformation-dependent association of cytokines and their cell-surface receptors, as well as the assembly of the appropriate protein complexes necessary to elicit signal transduction; and 4) protecting cytokines from both physiological and chemical degradation (41).
Limited studies have previously indicated an interaction between OSM and specific ECM molecules including collagen type I (50) and fibromodulin (51). OSM possesses charged clusters of basic amino acids within its C-terminal region, which are proposed to enhance binding to ECM components (51). In addition, OSM contains five cysteine residues, three of which are localized to its surface and are reported to mediate immobilization to gold nano-clusters (52). In this study, the interactions between OSM and the ECM molecules collagen types I and XI, fibronectin, and laminin were examined, and their binding properties investigated for potential involvement in chronic inflammation and tumor metastasis. Our data indicate that OSM binds to ECM proteins in a bioactive conformation, which may play an important role in the development of an inflammatory, pro-tumorigenic environment.
2. EXPERIMENTAL PROCEDURES
2.1 Materials
The following reagents were obtained from commercial sources: full-length 26 kDa (227 amino acids) OSM (Invitrogen, Life Technologies, Grand Island, NY); 22 kDa (196 amino acids) OSM and neutralizing antibody (R&D Systems, Minneapolis, MN); 24 kDa (209 amino acids) OSM (Peprotech, Oak Park, CA); antibody to OSM employed in immunoblots (Santa Cruz Biotechnologies, CA); collagen type I, laminin and fibronectin, ECM-coated 6-well plates (Collaborative Biomedical Products, Bedford, MA); protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO); Cleland’s reagent (DTT) (Sigma, St. Louis, MS) and RPMI-1640 growth medium (Hyclone, Logan, UT). Recombinant collagen α1(XI) fragment was synthesized in our laboratory as previously reported (53).
2.2 Co-culture experiments
Neutrophils were isolated from whole blood employing Polymorphprep™ solution (Sigma, St. Louis, MO). Neutrophils were added to tumor cells at a ratio of 10:1 and cultured in RPMI, 10% FBS, with penicillin and streptomycin.
2.3 Immunohistochemistry
Formalin fixed, paraffin-embedded tumor specimens were obtained from the archives of the Department of Veterans Affairs Medical Center, Boise ID. Following deparaffinization in xylenes and rehydration in a graded ethanol series, the sections were washed with 0.1 M Tris-HCl, 0.1 M NaCl, pH 7.5 and incubated with hydrogen peroxide in methanol to neutralize endogenous peroxidase activity. Sections were incubated with proteinase K (Sigma, St. Louis, MO), followed by microwaving for 2.5 min in Antigen Unmasking Solution (Vector Labs, Burlingame, CA). Slides were then washed and incubated in 20 % FBS (Sigma, St. Louis MO) to reduce non-specific binding. Two distinct OSM-directed monoclonal antibodies (a kind gift from Dr. Phil Wallace, Bristol-Myer Squibb Pharmaceutical Research Institute, Seattle, WA) were previously characterized (54, 55). This OSM antibody cocktail was added (20.0 μg/mL in 0.05 % FBS) and slides were incubated overnight at 4°C. The tumor sections were incubated with biotinylated anti-rabbit/anti-mouse secondary antibody (Dako Corp., Carpinterio, CA) for 20 min, followed by incubation with streptavidin/horseradish peroxidase (Dako Corp.) for 20 min. The slides were then incubated with substrate (3-amino-9-ethylcarbazole in N,N-dimethylformamide (Dako, Corp.) for up to 10 min and counterstained with hematoxylin (Buffalo Supply, Louisville, CO) for 1 min. After counterstaining, the tissue sections were prepared with Aqua-Mount (Lerner Laboratories, Pittsburg, PA).
2.4 Binding of OSM to ECM components in vitro
Tissue culture plates were coated with ECM preparations (100 μl of 0.2 mg/mL in PBS, room temp) for 60 min. Excess ECM solution was then decanted and plates were air dried in a cell culture hood and stored at 4°C. Wells were rinsed with serum-free RPMI-1640 culture media (SFM) for 15 min and then incubated for 15 min with SFM at the particular pH to be tested. Fifty-seventy nanograms of 22 kDa, 24 kDa or 26 kDa OSM was added to each well in 0.5 mL SFM, incubated for 3 h at 37°C, pH 5.5, 6.5 or 7.5, and then washed 4 times for 10 minutes with 1 mL of culture medium of the corresponding pH. SFM was employed in the binding experiments to examine the binding of OSM in the ionic milieu associated with a physiological environment and to make this model relevant to tissue culture. After washing the samples, 80 μl of boiling lysis solution (1.0 % SDS; 1 mM sodium vanadate and 10 mM Tris-HCl, pH 7.4) was applied to recover the OSM bound to ECM, followed by a 10 μl lysis solution rinse that was pooled with the corresponding samples. Experiments were also conducted with extended washing steps up to 48 h to validate the properties of immobilized OSM.
2.5 Binding analysis by surface plasmon resonance
Surface plasmon resonance (SPR) measurements were performed at 25°C using a Reichert SR7000 instrument in-line with a Surveyor LC pump and autosampler. OSM was immobilized to the surface of a PEG-10% carboxyl mixed self-assembled monolayer surface on gold sensor chip. Briefly, the chip surface was activated with a 0.4 mM N-ethyl-N-(3-diethylaminopropyl) carbodiimide and 0.1 mM N-hydroxysuccinimide in 0.5 M MES buffer followed by the injection of OSM at a concentration of 20 μg/ml in 10 mM sodium acetate buffer (pH 4.5). Unreacted N-hydroxysuccinimide ester groups were blocked with 1 M ethanolamine hydrochloride. ECM samples were prepared in phosphate buffered saline (PBS) with 0.05% Tween-20 (pH 7.4), which was filtered through a 0.45 μm filter prior to use. All samples consisted of 100 μl injections at a flow rate of 50 μl/min. After an association phase, dissociation phase was initiated and monitored in running buffer without the presence of ECM analyte for 180 s. The surface was regenerated with a 15 s injection of 250 mM NaCl in PBS. The association and dissociation phases were followed in real-time by monitoring changes in signal expressed in refractive index units and the data displayed as refractive index units versus time. Each concentration was run in triplicate for each analyte injected across the chip surfaces. Blank samples were included in the experiment to control for background signal and subtracted during post-processing. Data were fit to an association-then-dissociation kinetic binding model with GraphPad Prism version 5.0 d for Mac OS X (GraphPad Software, La Jolla California USA, www.graphpad.com).
2.6 Immunoblot procedure
Nitrocellulose was incubated in Superblock (Pierce, Thermo Fisher Scientific Inc., Rockford, IL) for 1 h, rinsed in Tris-buffered saline with 0.05 % Tween-20 (TTBS) and incubated with primary antibody (1 μg/ml) overnight at 5°C. Blots were then washed with TTBS 3X followed by incubation with secondary antibody (1/3000) in TTBS for 1–1.5 h at room temperature. Blots were then washed 3X in TTBS and developed using ECL substrates (Pierce, Thermo Fisher Scientific Inc., Rockford, IL).
2.7 Assessment of the bioactivity of ECM-bound and soluble OSM
Bioactivity was assessed by adding tumor cells to culture plates containing OSM immobilized to ECM proteins. Prior to the addition of tumor cells to ECM-bound OSM, one set of sample wells was treated with an OSM neutralizing antibody directed against the receptor binding site of OSM (3 μg/ml) for 60 min. T47D or ZR-75-1 tumor cells were plated in each well to approximately 75% confluence. Following incubations ranging from 2 h-5 days, tumor cell samples with ECM-bound OSM were harvested with 100 μl of boiling lysis buffer and 3 μl of protease inhibitor cocktail. Phospho-STAT3, STAT3 and STAT1 levels induced by immobilized OSM bound were assessed by immunoblot and compared to the levels of OSM in wells that received neutralizing antibody, or control wells that did not receive OSM.
2.8 Protein array analysis of STAT1 and STAT3 expression
ZR-75-1 cells were treated with OSM (20 ng/ml) for 48 h in confluent 75 cm2 culture flasks. Control- and OSM-treated breast tumor cells were then harvested with boiling lysis solution. Samples were submitted to BD Transduction Labs for a BD Powerblot protein array analysis that included STAT1 and STAT3 proteins. Samples were processed in duplicate by immunoblot analysis and quantitated by densitometry via the ratio of protein expression in OSM treated samples versus control samples.
2.9 OSM Circle assay
To study the effects of immobilized OSM on the properties and morphology of breast tumor cells, as well as study the properties of OSM bound to ECM for prolonged incubation periods the ‘OSM circle’ assay was developed. OSM (50 ng: 100μg/ml stock) was mixed 1:2 (vol/vol) with complete RPMI and spotted (20 μl total) into the middle of a culture well (OSM circle), incubated for 3h (37°C, pH7.5) and then washed extensively to remove any OSM not bound to substratum. T47D cells were either added after washing, or following an extended incubation of immobilized OSM (up to 8 days) in complete RPMI with additional washing. The effect of immobilized OSM on the morphology of cells within the ‘OSM circle’ was compared to cells outside the ‘OSM circle’ for up to 28 days utilizing phase-contrast microscopy.
2.10 Electrostatic and disulfide bond-dependent interaction between OSM and ECM
Truncated 22 kDa OSM (196 amino acids), 24 kDa OSM (209 amino acids), and 26 kDa full-length OSM (227 amino acids) were bound to ECM (as described above) at pH 7.5, 6.5 and 5.5 for 2–3 h at 37°C; harvested with boiling lysis buffer (100 μl) and then assessed by immunoblot. To study the role of disulfide bonds in OSM immobilization to ECM, 1 mM Cleland’s reagent (dithiothreitol; DTT) was included in the binding assay.
2.11 The proteolytic processing of soluble versus immobilized OSM by tumor secreted proteases
Full-length 26 kDa OSM was bound to ECM and extensively washed. Tumor conditioned media (CM) was generated by incubating a confluent 75 cm2 culture flask of breast tumor cells in 10 mL of SFM for 24h. The CM was collected and clarified by brief centrifugation. In the immobilized OSM processing assay 0.5 ml of CM or SFM was added to wells and following 24h incubation (room temp, pH7.5), 70 μl of boiling lysis buffer was added along with 3 μl of protease inhibitor cocktail. OSM band intensities were examined by immunoblot analysis. The processing of immobilized OSM was also compared to the processing of soluble OSM. The soluble OSM processing assay consisted of 50 ng of 26 kDa OSM in 35 μl of CM or SFM (room temp, pH7.5). The assay was stopped after 24h by the addition of SDS-PAGE sample buffer. Samples were examined via immunoblot.
3. RESULTS
3.1 OSM is associated with ECM in an in vitro co-culture model and in human tumor tissue
Neutrophils are a primary source of OSM within a tumor (6, 17, 56). We have previously established that, while both neutrophils and breast cancer cells alone express relatively low levels of OSM, 24-hour co-culture of these two cell types results in tumor cell-promoted neutrophil OSM expression and secretion (6). In the current study, OSM expression was observed within human neutrophils after co-culturing with MDA-MB-231 breast cancer cells for 24 hours (Fig. 1A). Interestingly, after co-culture for 48 hours, OSM was detected in the neutrophils, as well as in the pericellular matrix surrounding the breast cancer cells (Fig. 1B). This experiment indicated that co-culture of neutrophils and tumor cells resulted in a reservoir of OSM associated with the ECM. Furthermore, OSM was detected within the ECM in several human tumors of different histological origin, representing a general phenomenon that is not restricted to breast cancer cells alone. A colorectal tumor with a high number of infiltrating neutrophils expressing OSM is shown in Figure 1C. A high magnification image (100×) indicated that OSM was also associated with the tumor ECM (Fig. 1D). Association of OSM with tumor ECM was also observed in an ovarian carcinoma, as depicted in serial sections of the same tumor (Fig. 1E and F). OSM was also localized around an orifice not associated with endothelial cells (arrow in Fig. 1F). This type of tumor structure may be produced by neutrophils creating a neutrophil canal (57), which can be observed in an ovarian carcinoma containing OSM-expressing neutrophils (Fig. 1G). Together, these results demonstrated that OSM is associated with the ECM of tumor cells both in vitro and in vivo.
Figure 1. In vitro and in vivo evidence that OSM binds to ECM.
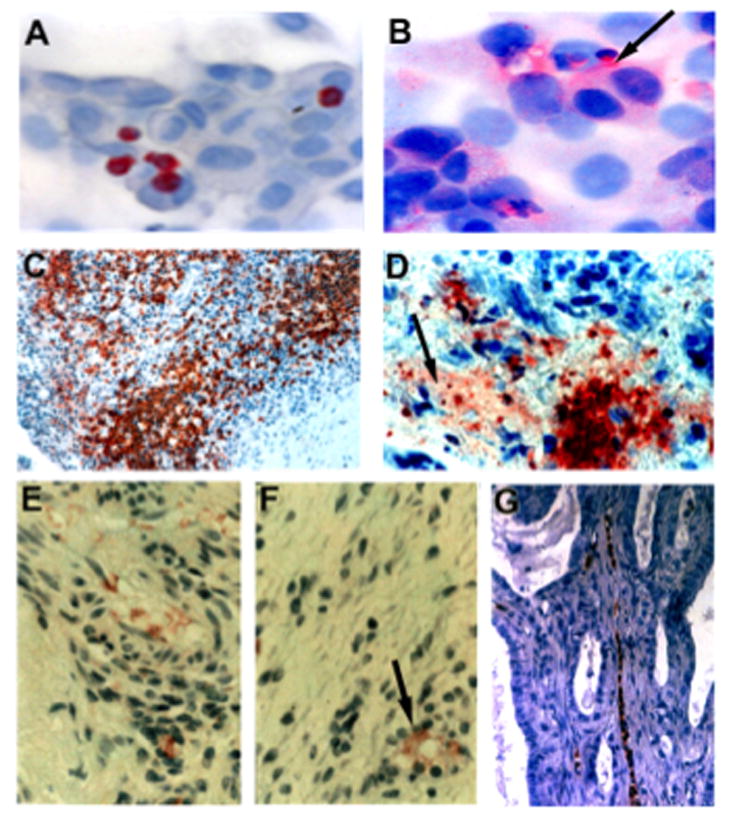
(A) OSM-expressing neutrophils were co-cultured with MDA-MB-231 breast cancer cells (10:1 ratio neutrophils to tumor cells), and no association of OSM with tumor cell ECM was apparent at 24 h. (B) Following 48 h incubation with neutrophils, OSM immobilized to the ECM of tumor cells was observed (arrow). (C–D) Low and high magnification photomicrographs of OSM expressing neutrophils in a colorectal tumor section. The colon cancer cells were negative for OSM expression, although a large population of OSM-expressing neutrophils that had infiltrated into the tumor was apparent (C). Upon high magnification, OSM associated with tumor ECM was observed (D; arrow). (E–F) OSM associated with the ECM of serial sections of an ovarian tumor. The similar patterns of OSM localized to the ECM of serial sections provided evidence against artifactual staining. (F) A circular orifice surrounded by OSM staining was apparent (arrow), which may represent a neutrophil canal. (G) An ovarian tumor section, which contains a neutrophil canal with OSM-expressing neutrophils.
3.2 OSM binds electrostatically to ECM via charged amino acids in OSM and ECM proteins with a further potential role for disulfide bonds
Having established a physiological relevancy for ECM-immobilized OSM, we next examined the ability of full-length 26 kDa OSM (227 amino acids) versus 22 kDa C-terminal truncated OSM (196 amino acids) to bind to the ECM molecules collagen types I and XI, laminin and fibronectin. OSM binding to ECM at physiological pH (7.5) was compared to more acidic pH, including pH 6.5 and pH 5.5, which is characteristic of an inflammatory microenvironment (58, 59). A pH dependent binding of OSM to each ECM protein was observed, which suggested an electrostatic binding mechanism (Fig. 2A). Full length OSM (26 kDa) was observed to bind to ECM proteins to a greater extent than 22 kDa OSM at pH 7.5 and 6.5, as assessed by band intensities in the immunoblot (Fig. 2A). The most significant difference between the binding of 26 kDa OSM versus 22 kDa OSM to ECM at pH 6.5 and 7.5 was observed for recombinant collagen type XI at neutral pH, where full-length OSM readily bound to collagen type XI, whereas very little 22 kDa OSM bound (Fig. 2A). This experiment indicated that the C-terminal extension of full-length OSM enhanced binding to ECM at physiological pH. At acidic pH 5.5, full-length and 22 kDa OSM bound equally well to ECM proteins except for collagen type XI (Fig. 2A). The highest overall levels of OSM binding to ECM were also observed at acidic pH, which was apparent from the intensities of the OSM bands (Fig. 2A). These observations indicated electrostatic OSM binding properties at pH 5.5 independent of the C-terminal extension of full length OSM.
Figure 2. OSM binds ECM proteins via electrostatic bonds: additional evidence for covalent disulfide bonds.
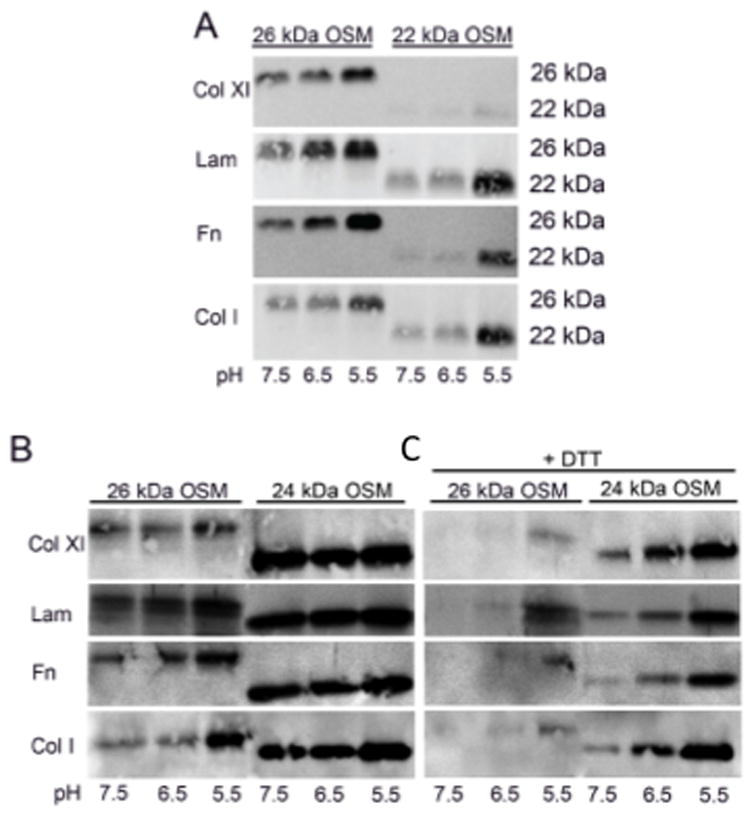
(A) Full-length 26 kDa OSM and mature 22 kDa OSM were bound to ECM in a 3 h incubation, 37°C in SFM at pH 5.5, 6.5 and 7.5. OSM was observed to bind to all ECM proteins in a pH-dependent fashion with greater 26 kDa OSM binding observed at pH 6.5–7.5. (B) The binding of full-length OSM compared to 24 kDa OSM. 24 kDa OSM was observed to bind to each ECM protein to a greater extent than 26 kDa OSM at pH 6.5–7.5, with more similar levels of binding apparent at pH 5.5. (C) The binding of OSM to ECM in the presence or absence of DTT (1 mM) inhibited, or partially inhibited the binding of OSM to each of the ECM proteins tested.
A role for pairs or doublets of basic amino acids in OSM binding to ECM molecules such as fibromodulin and osteoadherin has been reported previously (51). Full-length OSM has five basic amino acid doublets clustered in its C-terminus compared to one pair for 22 kDa OSM (Table 1). To further examine the nature of OSM binding to ECM, the binding of 26 kDa OSM was compared to the binding of 24 kDa OSM. Six of the thirteen amino acids in the C-terminal extension of 24 kDa OSM are charged 197-HSPHQALRKGVRR-209 (Table 1). 24 kDa OSM was observed to bind collagen types I and XI, fibronectin, and laminin to a greater extent than 26 kDa OSM at pH 6.5 and 7.5 (Fig. 2B). At pH 5.5, similar levels of OSM binding to ECM proteins were observed. A pH dependent binding was apparent for all ECM proteins except for 24 kDa OSM binding to collagen type XI, which tended to bind at similar high levels. This set of OSM binding experiments demonstrated that OSM MW variants were associated with different ECM binding properties that were regulated by pH, which was indicative of electrostatic binding. These experiments also indicated that particular basic amino acids, or doublets, as well the position and accessibility of these basic residues to ECM binding sites may be important determinants of OSM binding to ECM. In addition, at pH 5.5 changes in the properties of OSM binding sites, as well as ECM binding sites may affect OSM binding to ECM.
Table 1.
Charged amino acids in C-terminus of OSM MW species, IL-6 and LIF
| OSM 196 amino acids, 22 kDa | R182,K186,E189,R193,R195,R196 |
| OSM 209 amino acids, 24 kDa | ------------------------------------------H197,H200,R204,K205,R208,R209 |
| OSM 227 amino acids, 26 kDa | ---------------------------------------------------------------------------------------R211,R214,K215,K217,R218,R222,R227 |
| IL-6 184 amino acids, 21 kDa | D160,H164,R168,K171,E172,R179,R182 |
| LIF 180 amino acids, 20 kDa | D149,K153,D154,K158,K159,K160,K168,K1170 |
Since OSM contains three cysteine residues at its surface (60), experiments were conducted to determine whether disulfide bonds were also involved in the immobilization of OSM to ECM. Full-length 26 kDa and 24 kDa OSM were incubated with the disulfide bond-reducing reagent dithiothreitol (DTT; Cleland’s reagent; 1 mM) and the effects of DTT on OSM binding to ECM were then assessed by immunoblot. The binding properties of 22 kDa OSM were not examined due to the low levels of ECM binding observed in Figure 2A. In these experiments, DTT inhibited or reduced the binding of OSM to collagens type I and XI, laminin and fibronectin at pHs 7.5, 6.5 and 5.5 (Fig. 2B and C), suggesting a change in OSM conformation due to DTT dissociation of disulfide bonds. The effect of DTT appeared to be greater on the binding of 26 kDa versus 24 kDa OSM for each ECM protein examined. This experiment suggested a potential role for disulfide bonds in OSM immobilization to ECM, involving cysteine residues in OSM.
To examine OSM binding to ECM from a different perspective, OSM molecules were immobilized to a PEG-sensor chip and the binding of ECM was examined (pH 7.4) by surface plasmon resonance measurements (Fig. 3, Table 2). Binding was observed between OSM and collagen type XI, fibronectin, and laminin.
Figure 3. Binding kinetics assessed by surface plasmon resonance.
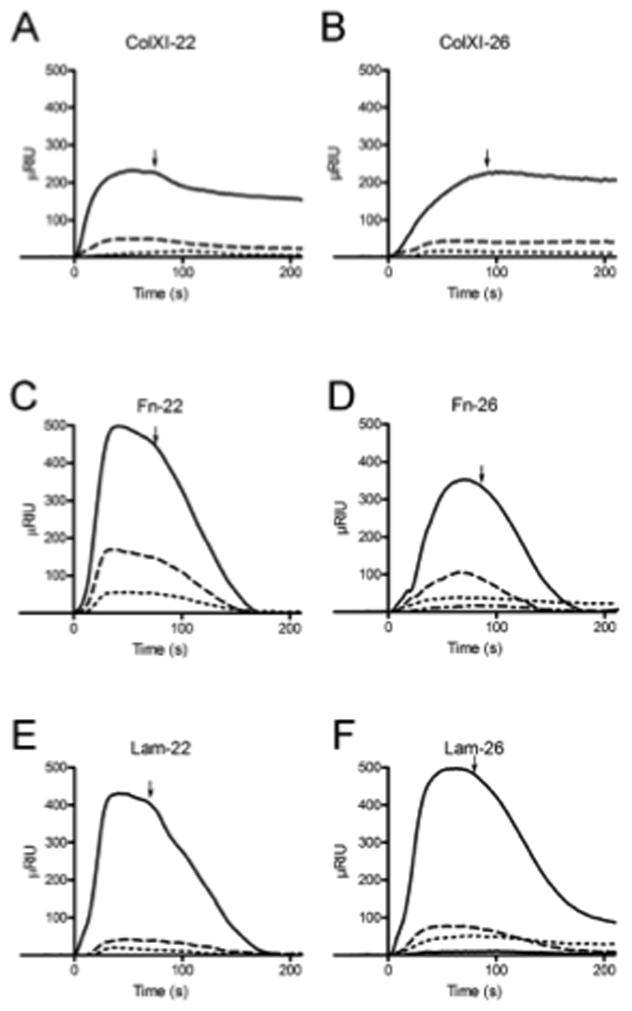
OSM was covalently coupled to the surface of a sensor chip and ECM proteins in solution were able to bind to the OSM. (A, C, E) 22 kDa human OSM covalently coupled to sensor chip, (B, D, F) 26 kDa human OSM covalently coupled to sensor chip. (A, B) Col11a1 fragment sample concentrations ranged from 11 pM to 123 nM. The association and dissociation phases are shown as refractive index units versus time. (C, D) Fibronectin sample concentrations ranged from 104 pM to 1μM. (E, F) Laminin sample concentrations ranged from 39 pM to 720 nM. Arrow indicates time of transition from association to dissociation due to buffer change. The KD, Kon and Koff values calculated from the global fitting of all concentrations are reported in Table 2.
Table 2.
Dissociation constants and on and off rates of interactions
| 26 kDa | |||
|---|---|---|---|
|
| |||
| KD (M) | Kon | Koff | |
| Col XI | 6.73 × 10−9 | 82,300 | 0.000554 |
| Fibronectin | 1.17 × 10−3 | 17 | 0.02 |
| Laminin | 8.44 × 10−7 | 17,100 | 0.0145 |
| 22 kDa | |||
|---|---|---|---|
|
| |||
| KD (M) | Kon | Koff | |
| Col XI | 1.1 × 10−8 | 370,000 | 0.00408 |
| Fibronectin | 2.6 × 10−6 | 10,600 | 0.0275 |
| Laminin | 9.36 × 10−7 | 27,100 | 0.0254 |
3.3 OSM bound to ECM is biologically active
The next set of experiments was designed to determine whether immobilized OSM was bioactive and able to initiate OSM signaling in breast tumor cells (Fig. 4). The first experiment compared the activities of full-length versus 22 kDa OSM in a traditional time-course assay employing soluble OSM (25 ng/ml) to examine the phosphorylation of the OSM signaling protein STAT3. Addition of soluble 26 kDa OSM or 22 kDa OSM to T47D human breast cancer cells resulted in the phosphorylation of STAT3 apparent at 15 minutes; that reached a maximum by 30 minutes and then decreased by 60 minutes, as assessed by immunoblot analysis (Fig. 4A). Both 26 kDa and 22 kDa OSM were able to activate OSM signaling via pSTAT3 to a similar extent, which indicated similar activities for the two OSM forms.
Figure 4. OSM bound to ECM is biologically active.
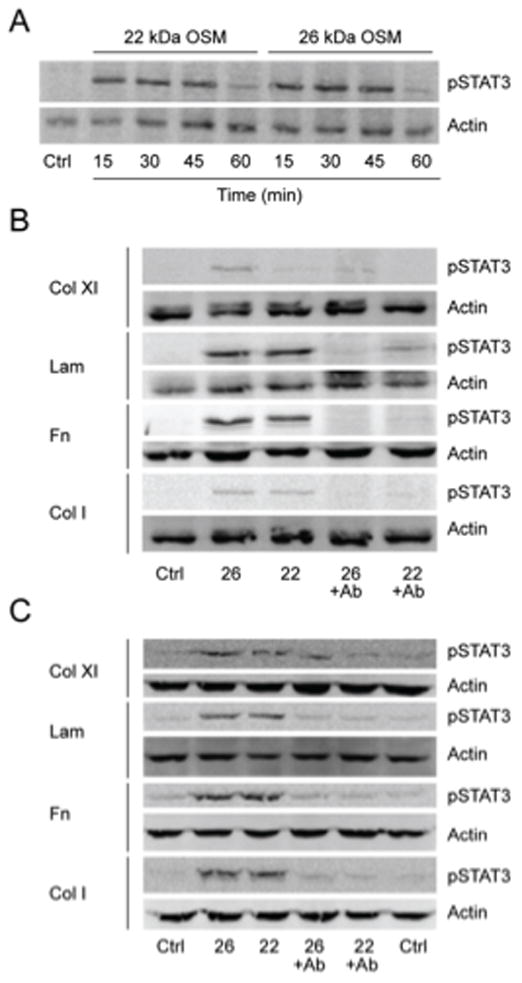
STAT3 phosphorylation by OSM acting as a soluble ligand and as a solid-phase ligand immobilized to ECM was examined. (A) Time course examining the phosphorylation of STAT3 by soluble 26 kDa and 22 kDa OSM. (B) OSM immobilized to ECM proteins (2 h, 37°C, pH 7.5) induced pSTAT3 in T47D cells following a 2h incubation that was inhibited with a neutralizing antibody to OSM. (C) OSM immobilized to ECM (2 h, 37°C, pH 7.5) induced pSTAT3 in ZR-75-1 cells following a 24h incubation that was inhibited by a neutralizing antibody to OSM.
In the next set of experiments, we determined whether OSM immobilized to ECM was biologically active. Full-length 26 kDa and 22 kDa OSM were bound to ECM components for 3 hours in serum-free media (pH 7.4, 37 °C), excess OSM was removed, and T47D or ZR-75-1 human breast cancer cells were added and cultured for 2 or 24 hours, respectively. Two hours was selected as the early time-point, because this was the minimum time required for T47D cells to adhere to substratum and begin to flatten to a control-morphology. Both 26 kDa and 22 kDa OSM immobilized to collagen type XI, laminin, fibronectin, or collagen type I induced pSTAT3 in T47D (Fig. 4B) and ZR-75-1 (Fig. 4C) cells, demonstrating that OSM bound to these ECM proteins in a bioactive conformation. An OSM neutralizing antibody inhibited the induction of pSTAT3 by full-length OSM and 22 kDa OSM in each cell line (Fig. 4B and C). This indicated that induced pSTAT3 was due to OSM receptor (OSMR) signaling and also demonstrated that the OSM-ECM binding site was distinct from the OSMR binding site, which was recognized by the neutralizing antibody.
In Figure 2A, 22 kDa OSM was observed to bind to ECM proteins, particularly collagen-type XI at low levels (pH 6.5–7.5) compared to 26 kDa OSM, as indicated by the low intensity OSM bands assessed by immunoblot. In contrast, in Figure 4, immobilized 22 kDa OSM was observed to generate pSTAT3 bands similar in intensity to 26 kDa OSM. This discrepancy was determined to result from the threshold of detection for the two methods of analysis (Sup. Fig. 1). Two to three nanograms of OSM protein were required to detect immobilized OSM in immunoblots, which was dependent on the sensitivity of the OSM antibody. Faint intensity OSM bands observed in immunoblots from ECM binding experiments, though, were still associated with enough immobilized OSM to generate a strong pSTAT3 band in an OSM activity assay (Sup. Fig. 1). This was particularly true for pSTAT3 assessed at 24 h or longer.
We also demonstrated that 22 kDa and 26 kDa OSM bound to collagens type I and XI to a greater extent than IL-6 or LIF based on pSTAT3 expression (Sup. Fig. 2). IL-6, which contains seven charged acid amino residues but no basic amino acid doublets in its C-terminus (Table 1), did not induce pSTAT3 in T47D cells following incubation with collagens type I and XI. This indicated that IL-6 did not bind to ECM in a bioactive conformation (Sup. Fig. 2). IL-6 does contain one doublet of charged amino acids in its C-terminus that consists of one basic and one acidic residue (Table 1). LIF is associated with eight charged amino acids in its C-terminus, including a cluster of lysines, K-158, K-159 and K-160, as well as lysines at positions K-168 and K-170 (Table 1). LIF was observed to bind to collagens type I and XI as indicated by pSTAT3 bands, but not as well as 22 kDa or 26 kDa OSM (Sup. Fig. 2). This experiment supported a role for pairs or clusters of basic amino acids in mediating IL-6-like cytokine binding to ECM.
3.4 OSM immobilized to ECM induces a prolonged pSTAT3 response and enhances STAT1 and STAT3 protein expression
STAT proteins, particularly STAT3 have been described as key mediators of cancer-associated inflammation (31–36, 61) and are associated with OSM signaling. Therefore, we assessed the effects of immobilized OSM on the expression of STAT3 and STAT1 and phosphorylation of STAT3 (Tyr-705) in T47D breast cancer cells after extended incubation periods. We hypothesized that the prolonged presence of OSM, as well as STAT proteins in a particular microenvironment were necessary to mediate cycles of inflammatory events and tumor progression. OSM immobilized to collagen type XI induced a sustained expression of pSTAT3 in T47D cells that reached a maximum at 1 to 3 days and remained elevated over 5 days (Fig. 5A). This experiment indicated that immobilized OSM could mediate long-term biological effects in tumor cells. The data in Figures 4A and 5A also suggested a bi-phasic pSTAT3 response that increased at 15 to 45 minutes, decreased by 60 minutes to 3 hours, and then increased again from 1 to 3 days. Interestingly, immobilized OSM also enhanced unphosphorylated STAT3 expression that remained elevated after 5 days (Fig. 5A), as well as unphosphorylated STAT1 expression that remained elevated after 4 days (Fig. 5B). These data were supported by protein array analysis of ZR-75-1 breast cancer cells, where cells treated with soluble OSM for 2 days resulted in a greater than 2-fold induction levels of both STAT3 and STAT1 (Fig. 5C).
Figure 5. Immobilized OSM induces and maintains elevated levels of pSTAT3 and enhances the expression of STAT3 and STAT1 in breast cancer cells.
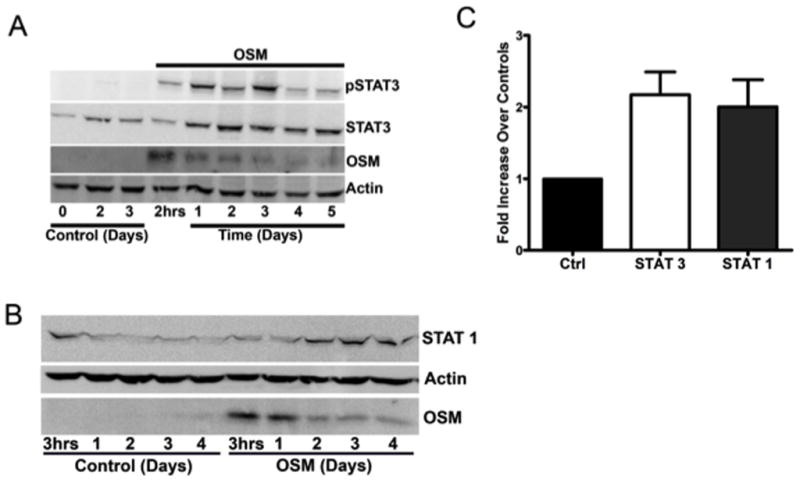
(A) Full-length OSM was immobilized to collagen type XI overnight (37°C, pH 7.5). T47D cells were added and incubated for 5 days. Immobilized OSM induced and sustained elevated levels of pSTAT3 and STAT3. (B) OSM immobilized to collagen type I induced the expression of STAT1 in T47D cells. Following overnight immobilization of OSM to collagen type I (37°C, pH 7.5), T47D cells were added and incubated for 4 days. (C) Protein array analysis indicated that soluble OSM also induced the expression of STAT3 and STAT1 in ZR-75-1 cells. OSM-treated and control ZR-75-1 cells were subjected to a BD Powerblot protein array analysis. ZR-75-1 cells treated with soluble OSM for 2 days were associated with a 2-fold increase in the expression levels of both STAT1 and STAT3 compared to controls.
Immobilized OSM could be recovered and analyzed by immunoblot from the T47D cell lysates for the 5-day culture period (Fig. 5A and B). This indicated that immobilized OSM maintained bioactivity and was able to bind to cellular receptors and that detached, unbound OSM was not necessary to elicit OSM signaling. A decrease in the recovered OSM band was also apparent over time (Fig. 5A and B). OSM bound to ECM via low affinity binding sites may detach at early time points (compare heavy band at 2 hours that decreases at 1 day) versus higher affinity binding sites that maintained prolonged binding of OSM to substratum. In summary, the enhanced and sustained expression of pSTAT3 and STAT3, as well as STAT1 protein levels in breast cancer cells treated with immobilized OSM is a novel observation and suggests an important role for OSM in STAT-mediated inflammation and tumor progression.
3.5 OSM bound to ECM induces morphological changes in breast tumor cells
We also assessed the activity of immobilized OSM by examining morphological changes in T47D breast cancer cells following interaction with immobilized OSM. For this analysis, we developed the OSM ‘circle assay’ (Fig. 6A), which confirmed that immobilized OSM was bioactive and able to interact with cellular receptors and that detached, soluble OSM was not responsible for the biological effects being observed. The differences in light reflection/refraction in T47D breast tumor cells within the ‘OSM circle’ undergoing EMT associated morphological changes (3) were compared to the properties of control cells outside the ‘OSM circle’. In addition, gross morphological changes in tumor cells within the ‘OSM circle’ were compared to the morphology of cells outside the ‘OSM circle’ to assess biological changes in cells induced by immobilized OSM and to determine whether OSM effects remained associated with cells inside the circle over prolonged incubation periods. In these experiments, 26 kDa OSM (50 ng: 100 μg/ml stock) was spotted on an ECM coated dish for 3 hours (pH 7.5, 37 °C), rinsed and incubated for 5 days. After additional washes, T47D cells were added and incubated for 8 days (Fig. 6B–E). OSM immobilized to laminin inside the ‘OSM circle’ induced morphological changes, as well as the detachment of T47D cells (Fig. 6B and D). T47D cells outside the ‘OSM circle’ (Fig 6C and E) remained attached to laminin and maintained a control morphology.
Figure 6. OSM immobilized to ECM induces morphological changes in T47D cells over prolonged incubation periods.
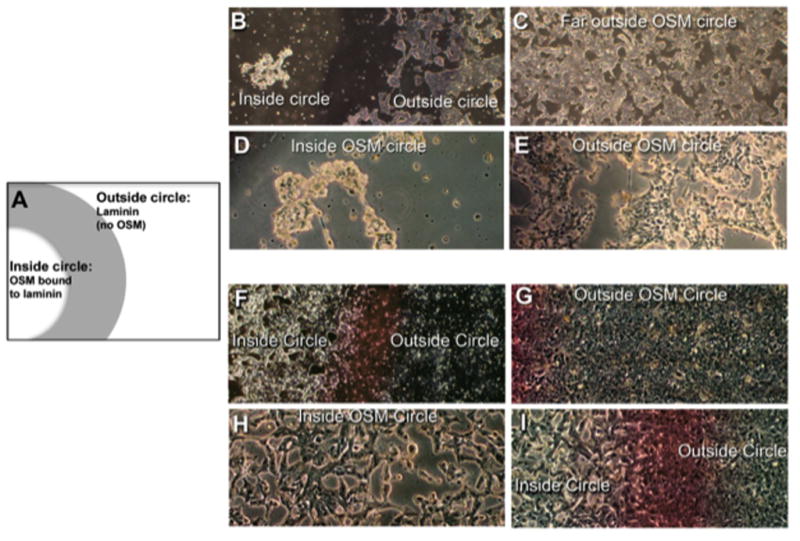
(A) Using the ‘OSM circle’ assay, OSM (50 ng: 100 μg/ml stock) was spotted onto laminin and incubated for 3 h (pH 7.5), followed by incubation of the immobilized OSM for 5 days. After washes, T47D cells were added and incubated for 8 days. Following eight days incubation, cells inside the OSM circle (B; 40× and D; 100×) were rounded-up and almost completely detached compared to cells outside the circle that maintained a control morphology (C; 40× and E; 100×). (F–I) T47D cells were also cultured for 28 days with full-length OSM immobilized to plastic. Phase-contrast microscopy was employed to assess the morphology changes and light reflecting/refracting properties of cells associated inside the ‘OSM circle’ compared to cells outside the circle. After 28 days, differences in morphology and the light scattering properties of tumor cells inside the OSM circle were clearly evident (F; 40× and H; 100×) when compared to cells outside the circle (G; 40× and I; 100×), which also indicated differences in cell number.
Plastic was also employed as a substratum due to concerns that ECM proteins might slowly detach from substratum after prolonged incubation with tumor cells. In this experiment, 26 kDa OSM was immobilized in a circle on plastic tissue culture wells (3-hour binding, 37°C, pH 7.5). T47D cells were then added and cultured for 28 days, with fresh media added every 5 days (Figure 6F–I). After the prolonged incubation, T47D cells associated within the ‘OSM circle’ (Fig. 6F and 6H) were associated with EMT-like morphological differences compared to cells just outside the ‘OSM circle’ (Fig. 6G and 6I), which maintained control morphology. Cells inside the ‘OSM circle’ had a larger cell volume than those outside the circle and were associated with a more stellate morphology with arm-like processes protruding from the cells. Cells inside the ‘OSM circle’ undergoing morphological changes (Fig 6F and 6H) also appeared much brighter under a phase/contrast microscope than cells outside the ‘OSM circle’, which maintained a darker control morphology and refracted/reflected light differently (Fig. 6G and 6I). It should also be noted that the number of cells inside the OSM circle was less than that of cells outside the circle, which were hyper-confluent. The decrease in cell number inside the ‘OSM circle’ resulted from OSM-induced cell detachment from substratum, as well as growth arrest (3). OSM bound to other substratum was also observed to be bioactive in the ‘circle assay’ and further demonstrated that OSM-mediated morphological effects were apparent only in cells within the ‘OSM circle’ (Sup. Fig. 3). In summary, these experiments provided strong evidence for the ability of OSM to bind to substratum in a bioactive conformation that was maintained during a prolonged incubation period and demonstrated that immobilized OSM was able to bind to cellular OSM receptors.
3.6 OSM bound to ECM is protected from proteolysis and maintains stability in an acidic environment
We next examined the effect of extracellular proteases associated with tumor-conditioned media (CM) on the proteolysis of immobilized versus soluble OSM. Overnight incubation of soluble full-length OSM in MCF-7 breast carcinoma cell CM resulted in two major OSM species of MWs 24 kDa and 22 kDa observed by immunoblot analysis (Fig. 7A). These OSM species were not apparent after incubation of OSM in control SFM (Fig. 7A). In contrast to the proteolysis of soluble full-length 26 kDa OSM by MCF-7 CM, OSM bound to collagen type XI was protected from proteolysis (Fig. 7B), and the 26 kDa OSM protein remained intact over time. The protein-processing site in 26 kDa OSM is localized to the C-terminal tail, and binding of full-length OSM to substratum was demonstrated to inhibit the proteolytic processing. This observation provided further evidence that 26 kDa OSM binds to ECM via its C-terminal tail.
Figure 7. Immobilized OSM is protected from proteolysis and maintains activity following prolonged incubation at acidic pH.
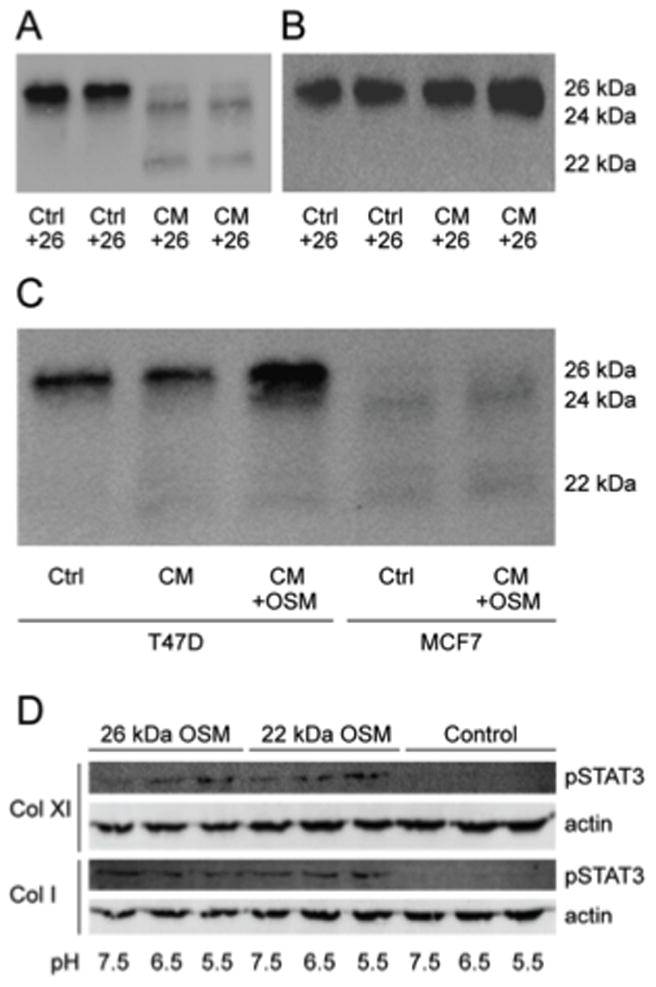
Conditioned medium from MCF-7 breast carcinoma cells (collected after 24h from confluent cells) was incubated with (A) soluble OSM and (B) OSM bound to collagen type XI. Extracellular proteases associated with MCF-7 cells were able to cleave soluble full-length OSM into 24 kDa and 22 kDa OSM species (A). Immobilized OSM was resistant to proteolytic processing form tumor CM associated proteases (B). (C) OSM treatment of T47D cells (left side) induced an extracellular protease activity, constitutively expressed in MCF-7 conditioned medium (right side) that generated 24 kDa OSM. (D) OSM bound to ECM and incubated at pH 5.5–7.5 for 8 days retained the ability to induce pSTAT3 in T47D cells following a 2 h incubation.
MCF-7 breast carcinoma cell CM was utilized because it was found to efficiently process all of the 26 kDa OSM into lower MW species. T47D CM was also observed to process soluble OSM, although the processing of the 26 kDa full-length OSM was less extensive (Fig. 7C). CM from OSM-treated T47D cells, though, was associated with the induction of an extracellular protease activity that was able to generate the 24 kDa OSM species (Fig 7C), which was observed to be constitutively expressed in MCF-7 CM (control in Fig 7A). This experiment demonstrated that the processing of full-length OSM by cell-line specific proteases resulted in different MW OSM species, which were previously observed to be associated with different adhesive properties (Fig. 2).
In an inflammatory environment, OSM may be in an acidic pH for prolonged periods of time. OSM has a basic isoelectric point of 9.97 to 10.71, which is the pH at which it has no net charge. To evaluate its stability at a pH associated with inflammation, OSM was immobilized and incubated at pH 5.5, 6.5 and 7.5 for eight days (37 °C). T47D cells were then added, and pSTAT3 was assessed after 2 hours. Immobilized OSM was observed to induce pSTAT3 in T47D cells following an extended incubation for eight days at pH 5.5, 6.5, and 7.5. This experiment indicated that OSM bound to ECM was able to maintain a bioactive conformation in an acidic environment many pH units from its theoretical pI.
4. DISCUSSION
Several studies have indicated a role for OSM in regulating pathological processes associated with chronic inflammation including cancer (11), arthritis (16, 17, 19, 20), Alzheimer’s disease (21–23), and atherosclerosis (24). Inflammation contributes to almost all aspects of tumor progression and is also a consequence of tumor development (62). Cancer-associated inflammation is a key determinant in tumor progression and has been cited as the seventh hallmark of cancer (27, 32, 62–64). Epidemiological data indicate that over 25% of all cancers are related to chronic infections and other types of chronic inflammation (25). Chronic inflammation may not initiate tumor growth but rather, enhances tumor progression and metastasis by providing a nurturing and pro-tumorigenic microenvironment (65, 66).
OSM is localized to and secreted from tumor cells, lymphocytes (67), macrophages (11, 68), and neutrophils (6, 17, 56). We have previously reported that breast cancer cells induce the secretion of OSM from neutrophils (6). In addition, OSM is reported to be chemotactic for neutrophils (11, 69) and macrophages (70). Each step of tumor progression from initiation through metastasis is promoted via communication between tumor and inflammatory/immune cells through the secretion of cytokines, chemokines, growth factors and proteases that remodel the tumor microenvironment (65–71). It should be noted that ECM proteins play a key role at sites of inflammation where they regulate the inflammatory properties of infiltrating leukocytes (72).
In the current study, evidence is provided employing a co-culture model and immunohistochemical analysis of human tumor tissue suggesting that neutrophils may be an important source of OSM that ultimately ends up immobilized to ECM. This immobilized OSM may function to shape and control a particular microenvironment over an extended period of time into one that is inflammatory and pro-tumorigenic. OSM was also demonstrated to bind in a bioactive conformation to the ECM components collagens I and XI, fibronectin, and laminin, as well as other substratum including plastic. Many proteins and cytokines are sequestered in the ECM, but most such as TGFβ and HGF are bound in a biologically inactive state that requires proteolytic processing to generate a bioactive protein (73–76).
Full-length OSM (227 amino acids), 24 kDa OSM (209 amino acids), and truncated 22 kDa OSM (196 amino acids) were observed to bind to the ECM proteins collagen type I, collagen type XI, laminin, and fibronectin in a pH-dependent fashion, with the greatest overall binding observed at acidic pH. In general, 24 kDa OSM bound to substratum more readily than full-length, or truncated 22 kDa OSM at all pH values examined. Surprisingly, 45 kDa glycosylated OSM bound very weakly to ECM (Sup. Fig. 2C), suggesting a mechanism by which the N-linked glycans sterically hindered the accessibility of ECM binding sites. OSM is glycosylated at two sites, N-106 and N-192, with the latter residue localized to the C-terminal region of OSM that is involved in binding to ECM. The pH-dependent binding of OSM to ECM proteins suggested electrostatic bonds mediated by charged residues in OSM and the ECM proteins.
An OSM derived heparin-binding peptide consisting of the C-terminal amino acids C202--G223) has been reported to bind to fibromodulin and osteoadherin via basic amino acid pairs, or doublets (51). Differences in the distribution of basic amino acids, or doublets in the ECM binding domains of proteins including OSM, as well as the distribution of sulfates in the tyrosine sulfate-binding domains of ECM proteins are reported to influence the binding of these proteins to ECM (51). A higher ‘state of charge’ (charge density) in the sulfates associated with tyrosine sulfate-rich domains of ECM proteins also enhances binding to heparin-binding protein motifs, including the OSM peptide described above (51). In a previous study we established that collagen type XI contains multiple proteoglycan binding sites that include four lysine residues K-147, K-148, K-149 and K-152 (77). Replacement of K-148, K-149 and K-152 decreased binding to heparin sulfate, whereas replacement of K-147 increased binding 5-fold (77). In addition, an essential role for ionic interactions in the binding of fibronectin to collagen has also been reported that involves lysine and arginine residues (78). These studies indicate the similar nature of OSM and ECM electrostatic binding sites and also illustrate the importance of position and composition of particular charged amino acids or amino acid pairs in regulating electrostatic binding interactions.
The pH-dependent binding of OSM to ECM indicated electrostatic interaction and the likely involvement of amino acid side chains that undergo changes in protonation state between pH 5.5 and 7.5 in OSM and/or the ECM molecules. Arginines exist in a protonated state at the pH values used in these studies, and histidine with a pKa of 6.0 will experience changes in its protonation state at pH 5.5 compared to pH 7.5. OSM is a very basic protein with an isoelectric point of 9.97–10.71. With a pI of 9.97–10.71, OSM will be in a highly charged, protonated state between pH 5.5 and 7.5, which are the pH utilized in this study. The high pI of OSM and its ‘state of charge’ at the pH examined (5.5–7.5) could explain why OSM readily binds to many different ECM proteins, as well as plastic. In the current study, IL-6 with a pI of 5.3–7.0 did not bind to collagens type I and XI, whereas LIF with a pI of 8.9–9.6 was observed to bind, although not to the extent that OSM bound. The ‘state of charge’ of these OSM related cytokines at pH 7.5 paralleled their ability to bind to collagens type I and XI.
The qualitative differences observed between the binding of 22 kDa, 24 kDa, and 26 kDa OSM to ECM may result from the particular complement of charged amino acids associated with the different OSM C-terminal tails. IL-6 contains seven charged amino acids but no doublets of basic amino acids in its C-terminus, and it did not bind to collagens type I and XI. LIF contains a cluster of three consecutive lysines in its C-terminus and it did bind to collagens type I and XI, but not as well as OSM. The thirty-one amino acid C-terminal extension of 26 kDa OSM contains thirteen charged amino acids including five pairs of basic amino acids, versus six charged residues associated with the C-terminus of 22 kDa OSM, which contains only one pair of arginine residues. This suggests that the greater number of charged amino acids, or pairs of basic amino acids associated with the C-terminal extension of full-length OSM compared to 22 kDa OSM enhanced its ability to bind to ECM at pH 6.5 and pH7.5. At pH 5.5, though, similar levels of 22 kDa and 26 kDa OSM bound to ECM indicating binding sites not associated with the C-terminal extension, such as the cluster of three arginines associated with the 22 kDa OSM C-terminus, or alternatively, differences in the ‘state of charge’ and reactivity of sulfates associated with tyrosine sulfate rich binding domains of ECM proteins.
Compared to 22 kDa OSM, the 24 kDa OSM C-terminal tail contains an additional thirteen amino acids that includes six basic residues. These include two histidines, in addition to two basic amino acid doublets in the last six amino acids of the 24 kDa C-terminus (197-HSPHQALRKGVRR-209). The greater overall binding of 24 kDa OSM to ECM proteins indicated the importance of this particular complement of basic amino acids in OSM immobilization. The observation that 24 kDa OSM bound to ECM more readily than 26 kDa OSM suggested that the composition or position of basic amino acids in the 24 kDa C-terminus may be associated with a more flexible conformation and have easier access to ECM binding sites, or bind to ECM sites with a higher affinity. If particular basic amino acids, or doublets associated with the C-terminal extension of 26 kDa OSM are the first to come into contact with, and bind to the primary ECM binding site then this would prevent the association of basic amino acids of the 24 kDa and 22 kDa C-terminal binding domains within 26 kDa OSM with the same ECM binding site. The 26 kDa OSM C-terminal extension contains a lysine-arginine, as well as an arginine-lysine pair of amino acids that may readily bind to ECM but with a lower affinity than the arginine-arginine doublet, or histidines associated with the cluster of basic amino acids in the 24 kDa OSM C-terminal extension. Low affinity binding sites may result in the loss of OSM during the extensive washes that follow the binding incubation.
Peptides derived from various heparin-binding protein motifs, including the C202--G223 amino acid binding domain of OSM, which contain different patterns of basic amino acids are associated with different heparin binding affinities (51). A peptide with two arginine-arginine doublets has a different binding affinity than a peptide with a lysine-lysine doublet and a lysine-arginine doublet. The peptide derived from OSM that is associated with an arginine-arginine doublet, as well as two arginine-lysine doublets and a lysine-arginine doublet had the highest heparin binding affinity (51). The different positions of basic amino acids in the peptides, as well as the positions and ‘state of charge’ of sulfates in the tyrosine sulfate binding domains of ECM proteins are also associated with differences in binding affinities (51). It is important to note that the binding experiments in the current study were carried out in SFM, which contains physiological salt concentrations. These ions will also interact with charged amino acid residues in OSM and the ECM and affect their conformation and accessibility to potential binding sites, as well as binding affinities.
It was uncertain whether electrostatic bonds that immobilize OSM to ECM remain stable over the course of a prolonged period of incubation, thus we examined the potential role of disulfide bonds in OSM immobilization. OSM contains five cysteine residues, with three localized to its surface (60). The intramolecular disulfide bridge at C49-C167 is crucial for OSM activity (60), which would prevent the immobilization of bioactive OSM. However, the free cysteine residue C80 and the C6-C127 intramolecular disulfide bridge of human OSM are not required for OSM activity, are localized to the molecular surface of OSM, and are suggested to mediate the covalent binding of OSM to gold nanoclusters (52).
In the current study, the disulfide reducing agent DTT was observed to inhibit or reduce the levels of 24 and 26 kDa OSM binding to ECM with a greater inhibitory effect on full-length 26 kDa OSM binding. It is interesting to note that the presence of a disulfide bond in a protein is reported to regulate its ‘z number’, which is the number of charged amino acid residues that are able to interact with a particular surface (79). This observation suggests a mechanistic link between the presence of a disulfide bond in a protein such as OSM and its ability to electrostatically bind to ECM via charged amino acid residues. OSM, as well as the ECM proteins examined in this study contain disulfide bonds, which may include intra-, as well as inter-molecular bonds that regulate and maintain the conformations of these proteins (80). In our study, DTT was observed to reduce the binding of OSM to four different ECM proteins, which supported an effect on OSM conformation rather than on the ECM proteins. It is difficult to distinguish the effects of DTT from the electrostatic binding of OSM to ECM. Future studies will investigate the contribution of specific sulfhydryl groups (cysteines 6, 80, and 127) on OSM binding to ECM by site-directed mutagenesis to generate OSM cysteine mutants, however these studies are beyond the scope of this paper.
STAT3 signaling in an inflammatory microenvironment is a major factor in determining whether an elicited immune response is tumor promoting or inhibitory (66). In addition, the STAT3 signaling pathway is an important factor in the regulation of cancer-associated inflammation (31–36, 81, 82). STAT3 signaling shapes and controls a particular microenvironment into a pro-tumorigenic inflammatory microenvironment (31, 34, 82) and is constitutively activated at sites of chronic inflammation (36, 83–85). Furthermore, sustained STAT3 signaling in tumor cells has been shown to enhance proliferation, survival, angiogenesis, invasion, and tumor-promoting inflammation (32, 61). OSM contributes to the chronic inflammation associated with the progression of atherosclerosis via STAT1 and STAT3 signaling (24), and OSM-expressing macrophages are observed in lesions from all the patients examined (24). Unphosphorylated STAT3 has also been reported to mediate cell signaling (38, 86, 87) and to regulate and activate cellular programs associated with inflammation (37, 38). In this study, we established that OSM immobilized to ECM mediated a sustained and enhanced expression of pSTAT3, as well as STAT3 and STAT1 protein levels in breast cancer cells. The induction of pSTAT3 in breast cancer cells by immobilized OSM was inhibited by an OSM neutralizing antibody, which indicated that the OSM-ECM binding site was distinct from the OSM receptor binding site. The enhanced expression of STAT1 and STAT3 suggests an important role for OSM in cancer-associated inflammation and tumor progression. Future experiments will establish whether the increases in STAT1 and STAT3 protein levels result from an induction of expression, or an enhanced stability and increase in half-life.
The activity of OSM immobilized to ECM was also demonstrated by OSM-induced morphology changes in T47D cells. These changes were observed using the ‘OSM circle’ assay, where OSM was immobilized in a small circle of ECM and then incubated with T47D tumor cells for up to 28 days. Under these conditions, OSM induced EMT-like morphological changes in tumor cells that included rounding-up and detachment from substratum. These changes were observed almost exclusively in cells inside the OSM circle, while cells outside the circle maintained control morphology. Cells inside the ‘OSM circle’ undergoing OSM induced morphological changes could also be distinguished from control cells outside the ‘OSM circle’ by their light refraction/reflection properties under a phase contrast microscope. Cells inside the ‘OSM circle’ appeared much brighter than cells outside the circle, which maintained control morphology and were darker in intensity. Together, these experiments indicated that OSM immobilized to ECM was bioactive and able to bind to cellular OSM receptors and that detachment from ECM was not necessary to elicit an OSM response. The ability of OSM to remain localized in a particular microenvironment for prolonged periods of time indicated that immobilized OSM may mediate the long-term behavior of cells that come into contact with this cytokine, including an EMT in tumor cells. The binding of OSM to ECM in a bioactive conformation is a novel observation and suggests that immobilized OSM may help shape a microenvironment into one that is conducive to inflammation and metastasis.
An epithelial-mesenchymal transition is considered a hallmark of a metastatic tumor and involves changes in gene expression that result in coordinated changes in cell morphology, which are associated with enhanced motility, protease secretion, and the disruption of intra- and inter-cellular junctions (88–92). OSM is reported to mediate an EMT in tumor cells via STAT3 signaling (2, 61, 93, 94). We previously described EMT-like morphological changes induced in T47D cells by OSM that involved a disruption of intra- and inter-cellular junctions, enhanced invasion, and secretion of extracellular proteases that culminated in cell detachment from substratum (3). These morphological changes were reversible upon elimination of OSM, although single cells (but not multi-cell islands) induced to detach by OSM generally became apoptotic unless OSM was removed. Recently, a number of EMT markers induced by OSM in T47D and MCF-7 cells have been reported including snail and slug, as well as the CD44+/CD24− phenotype (2, 7). IL-6 is also reported to induce morphological changes in T47D and ZR-75-1 breast cancer cells, as well as detachment from substratum and enhanced motility that is associated with an EMT (95–98).
In the current study, we demonstrated that OSM immobilized to ECM was protected from proteolysis due to tumor-associated proteases and remained active after prolonged incubation at an acidic pH associated with an inflammatory environment. This suggested that the stability of OSM was maintained by immobilization, which also likely increased its half-life, although since OSM will bind to substratum during prolonged incubation it is difficult to establish a soluble OSM control for comparison. Since the proteolytic processing site in full-length OSM is associated with the C-terminal tail, the inhibition of OSM processing by immobilization to substratum provided further evidence that the C-terminal OSM tail was involved in binding to ECM and was not accessible to proteases. In addition, we observed that OSM induced the expression of a protein processing activity in T47D CM, constitutively expressed in MCF-7 CM that mediated the conversion of soluble full-length OSM into 24 kDa OSM. It is well established that proteases are key mediators of metastasis as well as inflammation, and we have previously observed that OSM induces the secretion of cathepsins D and L from breast cancer cells (3). The protection from proteolysis and the stability at acidic pH for prolonged periods displayed by immobilized OSM represents another mode of OSM regulation by the ECM and cell microenvironment. By binding to ECM, the biological effects of OSM may be prolonged by preventing its proteolysis from proteases associated with tumors and an inflammatory environment, as well as by maintaining its stability in different physicochemical environments. Surprisingly, very little in known regarding the MWs and properties of OSM species associated with different pathological processes, including cancer, arthritis, and Alzheimer’s disease. The processing of glycosylated or full-length OSM in a specific microenvironment that is associated with a particular complement of extracellular proteases may generate different MW OSM species with different biological properties such as adhesiveness.
The binding of OSM to ECM in a bioactive conformation may represent an early inflammatory event, which over time may exert an influence over a particular microenvironment culminating in chronic inflammation or metastasis. Chronic inflammation, in general, involves a temporal increase in the concentration of pro-inflammatory cytokines in an environment that is also associated with a constant diffusion of cytokines out of the area. Immobilization of a pro-inflammatory cytokine in a discrete microenvironment will increase the local cytokine concentration and reduce the amount of cytokine necessary to maintain inflammation. In addition, the immobilization of OSM to substratum may serve to concentrate and restrict the activity of OSM to discrete microenvironments where it can maintain long-term biological effects. The immobilization of OSM to ECM may also sequester OSM away from certain populations of cells in a microenvironment. The pH dependency of OSM binding to ECM suggests a plausible role for pH in regulating the state of OSM attachment to ECM occurring during the development and cessation of inflammation. As inflammation develops in a microenvironment, the pH decreases and OSM will bind more readily to ECM; as inflammation subsides, the pH will rise and OSM will detach and be cleared from the microenvironment.
Our model describing the role of immobilized OSM in the acquisition of chronic inflammation and metastasis is depicted in Figure 8, where infiltrating inflammatory cells (or tumor cell subpopulations) secrete OSM. The inflammatory or tumor cells expressing OSM receptors may then interact with the immobilized OSM to initiate signaling (Fig. 8, upper panel). OSM is reported to be chemotactic for neutrophils and macrophages (11, 69, 70), and the slow release of OSM from ECM may enhance the infiltration of additional inflammatory cells (Fig. 8, lower panel). Infiltrating inflammatory cells, migrating tumor cells, or cancer cells at the invasive edge of a tumor may come into contact with the immobilized OSM and initiate another round of inflammatory events resulting in chronic inflammation or an EMT and metastasis in tumor cells.
Figure 8. Model for OSM role in chronic inflammation and tumor metastasis.
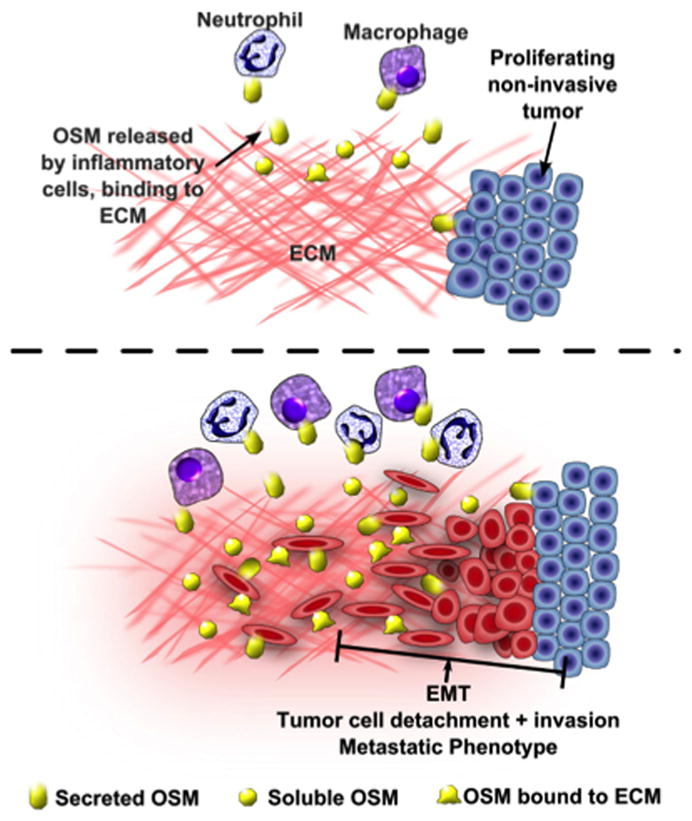
(A) Inflammatory cells infiltrate into a particular microenvironment and secrete OSM, which binds to ECM and helps to nurture an inflammatory or pro-metastatic microenvironment. (B) Immobilized bioactive OSM maintains a prolonged OSM response ultimately resulting in chronic inflammation or tumor metastasis. A non-OSM secreting, proliferating tumor may also come into contact with immobilized OSM secreted from inflammatory cells. The interaction of immobilized OSM with proliferating tumor cells may induce an EMT in the tumor cells and enhance tumor metastasis.
In conclusion, the binding of OSM to ECM represents the first of several inflammatory events in a particular microenvironment that may result in chronic inflammation, if inflammatory cells are involved, or tumor metastasis and tumor associated-inflammation, if tumor cells interact with the immobilized OSM. OSM bound to ECM proteins via electrostatic bonds associated with basic amino acids in the OSM C-terminus. Immobilized OSM was bioactive and induced a sustained expression of pSTAT3, as well as STAT3 and STAT1 protein in T47D and ZR-75-1 breast tumor cells. The OSM ‘circle assay’ demonstrated that immobilized OSM remained localized in a discrete microenvironment where it mediated prolonged biological effects in tumor cells. Immobilized OSM was also observed to be resistant to proteolysis from tumor-associated proteases, and OSM was observed to induce an extracellular protease activity in T47D cells capable of processing soluble full-length OSM. Lastly, the data in this study provided further evidence for the role of inflammatory processes in cancer.
Supplementary Material
HIGHLIGHTS.
OSM binding to ECM is pH-dependent and involves electrostatic bonds.
Immobilized OSM is bioactive and induces prolonged phosphorylation of STAT3.
The novel ‘OSM circle’ assay validates immobilized OSM bioactivity.
Immobilized OSM is protected from proteolysis.
A model for the role of immobilized OSM in inflammation and metastasis is developed
Acknowledgments
Authors wish to thank Raquel Brown, Chris Chandler, and Barbara Jibben for technical and editorial support, and the Department of Veterans Affairs Medical Center, Boise ID, for archival tissues. This work was supported in part by the National Institutes of Health (R15CA137510), Idaho INBRE Program (P20GM103408), Mountain States Medical Research Institute Small Project Grant, the Susan G. Komen Foundation grant (KG100513), the American Cancer Society Grant (RSG-09-276-01-CSM, and Research Corporation Cottrell College Scholars grant.
ABBREVIATIONS
- OSM
oncostatin M
- IL-6
interleukin-6
- LIF
leukemia inhibitory factor
- PBS
phosphate-buffered saline
- TBS
Tris-buffered saline
- TTBS
Tween-20/Tris-buffered saline
- DTT
dithiothreitol
- TX
Triton X-100
- NC4
Col-I, collagen type I
- Col-XI
collagen type XI
- Lam
laminin
- Fn
fibronectin
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- SPR
surface plasmon resonance
- MWs
molecular weights
- TGF-β
transforming growth factor-β
- HGF
hepatocyte growth factor
- SFM
serum free medium
- CM
tumor conditioned media
- kDa
kilodaltons
- FBS
fetal bovine serum
Footnotes
AUTHORS’ CONTRIBUTION
All authors read and approved the final manuscript. RR, BM, and JTO carried out data collection and analysis. RR, KT, OMM, JTO, and CLJ carried out data interpretation and modeling. RR, JTO, and CLJ participated in the conception of the study and research question.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bolin C, Tawara K, Sutherland C, Redshaw J, Aranda P, Moselhy J, Anderson R, Jorcyk CL. Oncostatin m promotes mammary tumor metastasis to bone and osteolytic bone degradation. Genes & cancer. 2012;3(2):117–30. doi: 10.1177/1947601912458284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo L, Chen C, Shi M, Wang F, Chen X, Diao D, Hu M, Yu M, Qian L, Guo N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013;32(45):5272–82. doi: 10.1038/onc.2012.573. [DOI] [PubMed] [Google Scholar]
- 3.Jorcyk CL, Holzer RG, Ryan RE. Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine. 2006;33(6):323–36. doi: 10.1016/j.cyto.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Li QH, Zhu JH, Sun F, Liu L, Liu XX, Yue Y. Oncostatin M promotes proliferation of ovarian cancer cells through signal transducer and activator of transcription 3. Int J Mol Med. 2011;28(1):101–8. doi: 10.3892/jimm.2011.647. [DOI] [PubMed] [Google Scholar]
- 5.Ng DCH, Lim CP, Lin BH, Zhang T, Cao XM. SCG10-like protein (SCLIP) is a STAT3-interacting protein involved in maintaining epithelial morphology in MCF-7 breast cancer cells. Biochem J. 2010;425:95–105. doi: 10.1042/Bj20091213. [DOI] [PubMed] [Google Scholar]
- 6.Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer research. 2005;65(19):8896–904. doi: 10.1158/0008-5472.CAN-05-1734. [DOI] [PubMed] [Google Scholar]
- 7.West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene. 2014;33(12):1485–94. doi: 10.1038/onc.2013.105. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F, Li C, Halfter H, Liu JW. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene. 2003;22(6):894–905. doi: 10.1038/sj.onc.1206158. [DOI] [PubMed] [Google Scholar]
- 9.Linsley PS, Bolton-Hanson M, Horn D, Malik N, Kallestad JC, Ochs V, Zarling JM, Shoyab M. Identification and characterization of cellular receptors for the growth regulator, oncostatin M. The Journal of biological chemistry. 1989;264(8):4282–9. [PubMed] [Google Scholar]
- 10.deMiguel MP, deBoerBrouwer M, deRooij DG, Paniagua R, vanDisselEmiliani FMF. Ontogeny and localization of an oncostatin M-like protein in the rat testis: Its possible role at the start of spermatogenesis. Cell Growth Differ. 1997;8(5):611–8. [PubMed] [Google Scholar]
- 11.Modur V, Feldhaus MJ, Weyrich AS, Jicha DL, Prescott SM, Zimmerman GA, McIntyre TM. Oncostatin M is a proinflammatory mediator - In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J Clin Invest. 1997;100(1):158–68. doi: 10.1172/Jci119508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malik N, Evans B, Greenfield BW, Shapiro RA, Hanson M, Shoyab M. Autocrine/paracrine induction of tissue inhibitor of metalloproteinase-1 in Chinese hamster ovary cells by oncostatin M. Matrix biology : journal of the International Society for Matrix Biology. 1995;14(8):677–80. doi: 10.1016/s0945-053x(05)80031-9. [DOI] [PubMed] [Google Scholar]
- 13.Richards CD, Shoyab M, Brown TJ, Gauldie J. Selective regulation of metalloproteinase inhibitor (TIMP-1) by oncostatin M in fibroblasts in culture. Journal of immunology. 1993;150(12):5596–603. [PubMed] [Google Scholar]
- 14.Cichy J, Potempa J, Chawla RK, Travis J. Stimulatory effect of inflammatory cytokines on alpha 1-antichymotrypsin expression in human lung-derived epithelial cells. J Clin Invest. 1995;95(6):2729–33. doi: 10.1172/JCI117975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko HS, Kang HK, Kim HS, Choi SK, Park IY, Shin JC. The effects of oncostatin M on trophoblast cells: influence on matrix metalloproteinases-2 and -9, and invasion activity. Placenta. 2012;33(11):908–13. doi: 10.1016/j.placenta.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Carroll G, Bell M, Hui W. Role of oncostatin M in the regulation of cartilage macromolecule metabolism: comment on the article by Nemoto et al. Arthritis and rheumatism. 1997;40(3):589–90. doi: 10.1002/art.1780400332. [DOI] [PubMed] [Google Scholar]
- 17.Cross A, Edwards SW, Bucknall RC, Moots RJ. Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis and rheumatism. 2004;50(5):1430–6. doi: 10.1002/art.20166. [DOI] [PubMed] [Google Scholar]
- 18.Hui W, Bell M, Carroll G. Oncostatin M (OSM) stimulates resorption and inhibits synthesis of proteoglycan in porcine articular cartilage explants. Cytokine. 1996;8(6):495–500. doi: 10.1006/cyto.1996.0067. [DOI] [PubMed] [Google Scholar]
- 19.Hui W, Bell M, Carroll G. Detection of oncostatin M in synovial fluid from patients with rheumatoid arthritis. Annals of the rheumatic diseases. 1997;56(3):184–7. doi: 10.1136/ard.56.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okamoto H, Yamamura M, Morita Y, Harada S, Makino H, Ota Z. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis and rheumatism. 1997;40(6):1096–105. doi: 10.1002/1529-0131(199706)40:6<1096::AID-ART13>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 21.Kordula T, Rydel RE, Brigham EF, Horn F, Heinrich PC, Travis J. Oncostatin M and the interleukin-6 and soluble interleukin-6 receptor complex regulate alpha1-antichymotrypsin expression in human cortical astrocytes. The Journal of biological chemistry. 1998;273(7):4112–8. doi: 10.1074/jbc.273.7.4112. [DOI] [PubMed] [Google Scholar]
- 22.Reale M, Iarlori C, Gambi F, Lucci I, Salvatore M, Gambi D. Acetylcholinesterase inhibitors effects on oncostatin-M, interleukin-1 beta and interleukin-6 release from lymphocytes of Alzheimer’s disease patients. Experimental gerontology. 2005;40(3):165–71. doi: 10.1016/j.exger.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Schulz I, Engel C, Niestroj AJ, Zeitschel U, Menge K, Kehlen A, Meyer A, Rossner S, Demuth HU. Heteroarylketones inhibit astroglial interleukin-6 expression via a STAT3/NF-kappaB signaling pathway. Journal of neuroinflammation. 2011;8:86. doi: 10.1186/1742-2094-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albasanz-Puig A, Murray J, Preusch M, Coan D, Namekata M, Patel Y, Dong ZM, Rosenfeld ME, Wijelath ES. Oncostatin M is expressed in atherosclerotic lesions: A role for Oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis. 2011;216(2):292–8. doi: 10.1016/j.atherosclerosis.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Amedei A, Prisco D, D’Elios MM. The Use of Cytokines and Chemokines in the Cancer Immunotherapy. Recent Pat Anti-Canc. 2013;8(2):126–42. [PubMed] [Google Scholar]
- 26.de Ferranti S, Mozaffarian D. The perfect storm: obesity, adipocyte dysfunction, and metabolic consequences. Clinical chemistry. 2008;54(6):945–55. doi: 10.1373/clinchem.2007.100156. [DOI] [PubMed] [Google Scholar]
- 27.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 28.Waldner MJ, Neurath MF. Colitis-associated cancer: the role of T cells in tumor development. Seminars in immunopathology. 2009;31(2):249–56. doi: 10.1007/s00281-009-0161-8. [DOI] [PubMed] [Google Scholar]
- 29.Das Roy L, Curry JM, Sahraei M, Besmer DM, Kidiyoor A, Gruber HE, Mukherjee P. Arthritis augments breast cancer metastasis: role of mast cells and SCF/c-Kit signaling. Breast Cancer Res. 2013;15(2):R32. doi: 10.1186/bcr3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das Roy L, Pathangey LB, Tinder TL, Schettini JL, Gruber HE, Mukherjee P. Breast cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res. 2009;11(4) doi: 10.1186/Bcr2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer cell. 2009;15(2):79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guthrie GJ, Roxburgh CS, Horgan PG, McMillan DC. Does interleukin-6 link explain the link between tumour necrosis, local and systemic inflammatory responses and outcome in patients with colorectal cancer? Cancer treatment reviews. 2013;39(1):89–96. doi: 10.1016/j.ctrv.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sansone P, Bromberg J. Environment, inflammation, and cancer. Current opinion in genetics & development. 2011;21(1):80–5. doi: 10.1016/j.gde.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 36.Demaria M, Poli V. Pro-malignant properties of STAT3 during chronic inflammation. Oncotarget. 2012;3(4):359–60. doi: 10.18632/oncotarget.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes & development. 2007;21(11):1396–408. doi: 10.1101/gad.1553707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell research. 2008;18(4):443–51. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 39.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–9. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hynes RO. The evolution of metazoan extracellular matrix. The Journal of cell biology. 2012;196(6):671–9. doi: 10.1083/jcb.201109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka Y, Kimata K, Adams DH, Eto S. Modulation of cytokine function by heparan sulfate proteoglycans: sophisticated models for the regulation of cellular responses to cytokines. Proceedings of the Association of American Physicians. 1998;110(2):118–25. [PubMed] [Google Scholar]
- 42.Bamber B, Reife RA, Haugen HS, Clegg CH. Oncostatin M stimulates excessive extracellular matrix accumulation in a transgenic mouse model of connective tissue disease. Journal of molecular medicine. 1998;76(1):61–9. doi: 10.1007/s001090050191. [DOI] [PubMed] [Google Scholar]
- 43.Duncan MR, Hasan A, Berman B. Oncostatin M stimulates collagen and glycosaminoglycan production by cultured normal dermal fibroblasts: insensitivity of sclerodermal and keloidal fibroblasts. The Journal of investigative dermatology. 1995;104(1):128–33. doi: 10.1111/1523-1747.ep12613623. [DOI] [PubMed] [Google Scholar]
- 44.Ihn H, LeRoy EC, Trojanowska M. Oncostatin M stimulates transcription of the human alpha2(I) collagen gene via the Sp1/Sp3-binding site. The Journal of biological chemistry. 1997;272(39):24666–72. doi: 10.1074/jbc.272.39.24666. [DOI] [PubMed] [Google Scholar]
- 45.Levy MT, Trojanowska M, Reuben A. Oncostatin M: a cytokine upregulated in human cirrhosis, increases collagen production by human hepatic stellate cells. Journal of hepatology. 2000;32(2):218–26. doi: 10.1016/s0168-8278(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 46.Scaffidi AK, Mutsaers SE, Moodley YP, McAnulty RJ, Laurent GJ, Thompson PJ, Knight DA. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. British journal of pharmacology. 2002;136(5):793–801. doi: 10.1038/sj.bjp.0704769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freise C, Erben U, Muche M, Farndale R, Zeitz M, Somasundaram R, Ruehl M. The alpha 2 chain of collagen type VI sequesters latent proforms of matrix-metalloproteinases and modulates their activation and activity. Matrix Biology. 2009;28(8):480–9. doi: 10.1016/j.matbio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Tatti O, Vehvilainen P, Lehti K, Keski-Oja J. MT1-MMP releases latent TGF-beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Experimental cell research. 2008;314(13):2501–14. doi: 10.1016/j.yexcr.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 49.Wu YH, Chang TH, Huang YF, Huang HD, Chou CY. COL11A1 promotes tumor progression and predicts poor clinical outcome in ovarian cancer. Oncogene. 2014;33(26):3432–40. doi: 10.1038/onc.2013.307. [DOI] [PubMed] [Google Scholar]
- 50.Somasundaram R, Ruehl M, Schaefer B, Schmid M, Ackermann R, Riecken EO, Zeitz M, Schuppan D. Interstitial collagens I, III, and VI sequester and modulate the multifunctional cytokine oncostatin M. The Journal of biological chemistry. 2002;277(5):3242–6. doi: 10.1074/jbc.M110011200. [DOI] [PubMed] [Google Scholar]
- 51.Tillgren V, Onnerfjord P, Haglund L, Heinegard D. The tyrosine sulfate-rich domains of the LRR proteins fibromodulin and osteoadherin bind motifs of basic clusters in a variety of heparin-binding proteins, including bioactive factors. The Journal of biological chemistry. 2009;284(42):28543–53. doi: 10.1074/jbc.M109.047076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prisco U, Leung C, Xirouchaki C, Jones CH, Heath JK, Palmer RE. Residue-specific immobilization of protein molecules by size-selected clusters. Journal of the Royal Society, Interface / the Royal Society. 2005;2(3):169–75. doi: 10.1098/rsif.2005.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Warner LR, Blasick CM, Brown RJ, Oxford JT. Expression, purification, and refolding of recombinant collagen alpha1(XI) amino terminal domain splice variants. Protein expression and purification. 2007;52(2):403–9. doi: 10.1016/j.pep.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radka SF, Kallestad JC, Linsley PS, Shoyab M. The Binding Pattern of a Neutralizing Monoclonal-Antibody to Mutant Oncostatin-M Molecules Is Correlated with Functional-Activity. Cytokine. 1994;6(1):55–60. doi: 10.1016/1043-4666(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 55.Radka SF, Naemura JR, Shoyab M. Abrogation of the antiproliferative activity of oncostatin M by a monoclonal antibody. Cytokine. 1992;4(3):221–6. doi: 10.1016/1043-4666(92)90059-z. [DOI] [PubMed] [Google Scholar]
- 56.Grenier A, Dehoux M, Boutten A, Arce-Vicioso M, Durand G, Gougerot-Pocidalo MA, Chollet-Martin S. Oncostatin M production and regulation by human polymorphonuclear neutrophils. Blood. 1999;93(4):1413–21. [PubMed] [Google Scholar]
- 57.Opdenakker G, Vandamme J. Cytokines and Proteases in Invasive Processes - Molecular Similarities between Inflammation and Cancer. Cytokine. 1992;4(4):251–8. doi: 10.1016/1043-4666(92)90064-X. [DOI] [PubMed] [Google Scholar]
- 58.Fukamachi T, Chiba Y, Wang X, Saito H, Tagawa M, Kobayashi H. Tumor specific low pH environments enhance the cytotoxicity of lovastatin and cantharidin. Cancer letters. 2010;297(2):182–9. doi: 10.1016/j.canlet.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Fukamachi T, HS, Tagawa M, Kobayashi H. In: The Impact of Extracellular Low pH on the Anti-Tumor Efficacy Against Mesothelioma, Mesotheliomas - Synonyms and Definition, Epidemiology, Etiology, Pathogenesis, Cyto-Histopathological Features, Clinic, Diagnosis, Treatment, Prognosis. Zubritsky Alexander., Dr, editor. InTech; 2012. [DOI] [Google Scholar]
- 60.Kallestad JC, Shoyab M, Linsley PS. Disulfide bond assignment and identification of regions required for functional activity of oncostatin M. The Journal of biological chemistry. 1991;266(14):8940–5. [PubMed] [Google Scholar]
- 61.Junk DJ, Bryson BL, Jackson MW. HiJAK’d Signaling; the STAT3 Paradox in Senescence and Cancer Progression. Cancers. 2014;6(2):741–55. doi: 10.3390/cancers6020741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Seminars in cancer biology. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 64.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 65.DeNardo DG, Coussens LM. Inflammation and breast cancer - Balancing immune response: crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007;9(4):Artn 212. doi: 10.1186/Bcr1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. Journal of molecular medicine. 2013;91(4):411–29. doi: 10.1007/s00109-013-1021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clegg CH, Rulffes JT, Wallace PM, Haugen HS. Regulation of an extrathymic T-cell development pathway by oncostatin M. Nature. 1996;384(6606):261–3. doi: 10.1038/384261a0. [DOI] [PubMed] [Google Scholar]
- 68.Vlaicu P, Mertins P, Mayr T, Widschwendter P, Ataseven B, Hogel B, Eiermann W, Knyazev P, Ullrich A. Monocytes/macrophages support mammary tumor invasivity by co-secreting lineage-specific EGFR ligands and a STAT3 activator. BMC cancer. 2013;13:197. doi: 10.1186/1471-2407-13-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kerfoot SM, Raharjo E, Ho M, Kaur J, Serirom S, McCafferty DM, Burns AR, Patel KD, Kubes P. Exclusive neutrophil recruitment with oncostatin M in a human system. The American journal of pathology. 2001;159(4):1531–9. doi: 10.1016/S0002-9440(10)62538-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lam D, Harris D, Qin Z. Inflammatory mediator profiling reveals immune properties of chemotactic gradients and macrophage mediator production inhibition during thioglycollate elicited peritoneal inflammation. Mediators of inflammation. 2013;2013:931562. doi: 10.1155/2013/931562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008;43(3):374–9. doi: 10.1016/j.cyto.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 72.Kremlev SG, Chapoval AI, Evans R. Cytokine release by macrophages after interacting with CSF-1 and extracellular matrix proteins: characteristics of a mouse model of inflammatory responses in vitro. Cellular immunology. 1998;185(1):59–64. doi: 10.1006/cimm.1998.1276. [DOI] [PubMed] [Google Scholar]
- 73.Lamszus K, Joseph A, Jin L, Yao Y, Chowdhury S, Fuchs A, Polverini PJ, Goldberg ID, Rosen EM. Scatter factor binds to thrombospondin and other extracellular matrix components. The American journal of pathology. 1996;149(3):805–19. [PMC free article] [PubMed] [Google Scholar]
- 74.Munger JS, Sheppard D. Cross Talk among TGF-beta Signaling Pathways, Integrins, and the Extracellular Matrix. Csh Perspect Biol. 2011;3(11):ARTN a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Todorovic V, Rifkin DB. LTBPs, more than just an escort service. J Cell Biochem. 2012;113(2):410–8. doi: 10.1002/Jcb.23385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tritschler I, Gramatzki D, Capper D, Mittelbronn M, Meyermann R, Saharinen J, Wick W, Keski-Oja J, Weller M. Modulation of TGF-beta activity by latent TGF-beta-binding protein 1 in human malignant glioma cells. International journal of cancer Journal international du cancer. 2009;125(3):530–40. doi: 10.1002/ijc.24443. [DOI] [PubMed] [Google Scholar]
- 77.Warner LR, Brown RJ, Yingst SM, Oxford JT. Isoform-specific heparan sulfate binding within the amino-terminal noncollagenous domain of collagen alpha1(XI) The Journal of biological chemistry. 2006;281(51):39507–16. doi: 10.1074/jbc.M608551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vuento M, Salonen E, Osterlund K, Stenman UH. Essential charged amino acids in the binding of fibronectin to gelatin. Biochem J. 1982;201(1):1–8. doi: 10.1042/bj2010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kunitani M, Johnson D, Snyder LR. Model of protein conformation in the reversed-phase separation of interleukin-2 muteins. Journal of chromatography. 1986;371:313–33. doi: 10.1016/s0021-9673(01)94716-8. [DOI] [PubMed] [Google Scholar]
- 80.Markovichousley Z, Schulthess T, Engel J, Richter H, Hormann H. Different Susceptibility of Inter-Chain and Intra-Chain Disulfide Bonds to Reductive Cleavage in Native Fibronectin and Effect of Their Cleavage on Conformation. Biol Chem H-S. 1985;366(10):985–91. doi: 10.1515/bchm3.1985.366.2.985. [DOI] [PubMed] [Google Scholar]
- 81.Lowe DB, Storkus WJ. Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy. 2011;3(10):1265–74. doi: 10.2217/imt.11.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pini M, Rhodes DH, Castellanos KJ, Hall AR, Cabay RJ, Chennuri R, Grady EF, Fantuzzi G. Role of IL-6 in the resolution of pancreatitis in obese mice. Journal of leukocyte biology. 2012;91(6):957–66. doi: 10.1189/jlb.1211627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert opinion on therapeutic targets. 2012;16(9):889–901. doi: 10.1517/14728222.2012.709238. [DOI] [PubMed] [Google Scholar]
- 84.Narayan C, Kumar A. Constitutive over expression of IL-1beta, IL-6, NF-kappaB, and Stat3 is a potential cause of lung tumorgenesis in urethane (ethyl carbamate) induced Balb/c mice. Journal of carcinogenesis. 2012;11:9. doi: 10.4103/1477-3163.98965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. P Natl Acad Sci USA. 2007;104(18):7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brown S, Zeidler MP. Unphosphorylated STATs go nuclear. Current opinion in genetics & development. 2008;18(5):455–60. doi: 10.1016/j.gde.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Cheon H, Yang JB, Stark GR. The Functions of Signal Transducers and Activators of Transcriptions 1 and 3 as Cytokine-Inducible Proteins. J Interf Cytok Res. 2011;31(1):33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boyer B, Valles AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochemical pharmacology. 2000;60(8):1091–9. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 89.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 90.Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO molecular medicine. 2009;1(6–7):303–14. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nature reviews Molecular cell biology. 2006;7(2):131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 92.Yang J, Weinberg RA. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev Cell. 2008;14(6):818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 93.Argast GM, Mercado P, Mulford IJ, O’Connor M, Keane DM, Shaaban S, Epstein DM, Pachter JA, Kan JLC. Cooperative Signaling between Oncostatin M, Hepatocyte Growth Factor and Transforming Growth Factor-beta Enhances Epithelial to Mesenchymal Transition in Lung and Pancreatic Tumor Models. Cells Tissues Organs. 2011;193(1–2):114–32. doi: 10.1159/000320179. [DOI] [PubMed] [Google Scholar]
- 94.Wang ML, Pan CM, Chiou SH, Chen WH, Chang HY, Lee OK, Hsu HS, Wu CW. Oncostatin m modulates the mesenchymal-epithelial transition of lung adenocarcinoma cells by a mesenchymal stem cell-mediated paracrine effect. Cancer research. 2012;72(22):6051–64. doi: 10.1158/0008-5472.CAN-12-1568. [DOI] [PubMed] [Google Scholar]
- 95.Sehgal PB, Tamm I. Interleukin-6 enhances motility of breast cancer cells. Cancer investigation. 1990;8(6):661–3. doi: 10.3109/07357909009018940. [DOI] [PubMed] [Google Scholar]
- 96.Tamm I, Cardinale I, Krueger J, Murphy JS, May LT, Sehgal PB. Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. The Journal of experimental medicine. 1989;170(5):1649–69. doi: 10.1084/jem.170.5.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tamm I, Cardinale I, Murphy JS. Decreased Adherence of Interleukin-6-Treated Breast-Carcinoma Cells Can Lead to Separation from Neighbors after Mitosis. P Natl Acad Sci USA. 1991;88(10):4414–8. doi: 10.1073/pnas.88.10.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tamm I, Kikuchi T, Cardinale I, Krueger JG. Cell-adhesion-disrupting action of interleukin 6 in human ductal breast carcinoma cells. Proc Natl Acad Sci U S A. 1994;91(8):3329–33. doi: 10.1073/pnas.91.8.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.