

Abstract
In the majority of breast cancers, overexpression and hyperactivation of Ras in the tumor microenvironment play significant role in promoting cancer cell growth, angiogenesis, and metastasis. We have previously shown that vascular endothelial growth factor (VEGF) expression in triple negative breast cancer cells is regulated, at least in part, by SAF-1 (serum amyloid A activating factor 1) transcription factor. In this study we show that transformation of normal MCF-10A breast epithelial cells by constitutively active, oncogenic Ras, induces the DNA-binding activity and transcription function of SAF-1. Furthermore, we show that inhibition of MEK/MAPK-signaling pathway prevents Ras-mediated activation of SAF-1. Interestingly, silencing of SAF-1 expression in breast cancer cells by SAF-1-specific short hairpin RNAs (shRNAs) significantly reduced H-Ras and K-Ras mRNA level. We show that SAF-1 is a direct transcriptional regulator of H-Ras and K-Ras and overexpression of SAF-1 increases H-Ras and K-Ras gene expression. Chromatin immunoprecipitation (ChIP) analyses demonstrated in vivo interaction of SAF-1 at highly purine-rich sequences present at the proximal promoter region, upstream of the transcription start site, in H-Ras and K-Ras genes. Previous studies have shown that these sequences are nuclease hypersensitive and capable of forming G4 quadruplex structure. Together, our results show the presence of a novel transactivating loop, in which, Ras and SAF-1 are interconnected. These findings will help defining molecular mechanisms of abnormal overexpression of Ras in breast tumors, which seldom show genetic Ras mutations.
Keywords: Angiogenesis, breast cancer, Ras, SAF-1, VEGF expression
Introduction
Tumor microenvironment (TME) is a dynamic entity which determines tumor growth, invasion, and metastasis. A number of cellular processes regulate different elements of the TME and determine the status of the tumor ranging from dormancy to aggressive growth. Angiogenesis is one of these distinct elements that trigger tumor growth by promoting vascular endothelial growth factor (VEGF) expression and subsequent vascular growth 1,2. In the TME, activated Ras proteins have been shown to play an important role in VEGF synthesis by the tumor cells and this finding was further validated by the ectopic expression of a constitutively active mutant Ras plasmid DNA which has been shown to directly promote VEGF expression and in vitro angiogenesis 3–5. In correlation, farnesyltransferase inhibitors that prevent posttranslational modification and activation of Ras proteins have been shown to inhibit VEGF expression 6 and its secretion from tumor cells 7. Ras-mediated VEGF synthesis has implicated Raf-1/MEK/MAPK and PI3K/Akt pathways in the activation of several transcription factors including Sp1, AP2, and HIF-1/2α in the induction of VEGF expression 8–10. Sp1 has been shown to induce VEGF in an HIF-1-independent mechanism 11 and a strong association between Sp1 and gastric cancer has been reported 12. Interestingly, the Sp1-binding sites in VEGF promoter is not restricted for Sp1 alone as other transcription factors with similar DNA-binding motif, namely, Cys2-His2 type zinc finger, often bind to this promoter region 13.
Recently, we reported a novel mechanism of VEGF induction in aggressive triple negative breast cancer cells by transcription factor serum amyloid A activating factor 1 (SAF-1) 14 which belongs to the family of Cys2-His2 type zinc finger and binds to the VEGF proximal promoter region that overlaps the Sp1-binding site. SAF-1 and its human homolog, myc-associated zinc finger protein (MAZ) 15 have been shown to be overexpressed in many human cancers 14,16–19. Marked increase of MAZ mRNA is observed at the terminal phase of human chronic myelogenous leukemia (CML) and an increased transcription from the MAZ gene has been closely linked to the malignant phenotype of this disease 20. SAF-1/MAZ is reported to be activated via phosphorylation by various protein kinases. Previous reports have indicated that in response to various inflammatory stimuli, DNA binding and transcriptional function of SAF-1/MAZ are further increased via its phosphorylation by various protein kinases, including MAPK, PKC, PKA, and CK2 21–24. These studies, however, did not address whether oncogenic signaling could activate SAF-1/MAZ in the TME. Since SAF-1/MAZ is highly induced and activated in breast cancer cells and breast tumors 14 and Ras-mediated signaling pathway is highly prevalent in these tumor cells, raises the possibility of this pathway in SAF-1/MAZ activation. In the present study, we demonstrate that in breast cancer cells, constitutively activated, oncogenic ras increases DNA-binding and transcriptional activities of SAF-1/MAZ via phosphorylation through the MAP-kinase pathway. In this context, SAF-1/MAZ could be a target for Ras/MAPK-directed therapeutics.
Human Ras family contains three genes (H-, N-, and K-Ras), which encode four highly homologous Ras proteins of ∽21 kDa size. H-Ras and K-Ras genes were first identified as cellular homologs of Harvey and Kirsten rat sarcoma virus oncogenes while N-Ras was identified in human Neuroblastoma cells 25. The Ras proteins are guanine-binding proteins and involved in relaying various extracellular signals to cytoplasm via GTP/GDP on-off switching 26. In normal cells, Ras proteins are only transiently activated, which then act on multiple downstream effector pathways to regulate cell proliferation, survival, and differentiation. Mutations of the Ras family of proto-oncogenes are very common; being found in 20–30% of many human cancers, including bladder, pancreas, colon, thyroid, and lung 27. However, breast cancer has very low, <5%, incidence of Ras gene mutations 27. Oncogenic mutations in ras genes confer gain-of-function by providing constitutive activation that drives uncontrolled cell proliferation and malignant transformation. Besides mutation, overexpression of Ras that facilitates Ras-mediated signaling has been reported in human cancer 28–30. Surprisingly, the mechanisms regulating how overexpression of Ras occurs are less understood. During our studies on Ras-mediated SAF-1 induction, we have observed that once activated, SAF-1 acts as a transcriptional inducer of H-Ras and K-Ras and we reveal the presence of a transactivation loop in which oncogenic Ras increases DNA-binding and transcriptional activities of SAF-1 which in turn, increases transcription of Ras forming a feed-forward regulatory pathway.
Material and Methods
Cell lines
MCF-10A and MDA-MB-468 cells were obtained from American Type Culture Collection (ATCC), cultured and stored following ATCC protocol of authentication by short terminal repeat analysis. The cells were maintained in Dulbecco's modified Eagle's medium containing high glucose (4.5 g/L) and supplemented with 7% fetal bovine serum.
Establishment of cell lines and transfection assay
To generate MCF-10A-ras cells, MCF-10A cells were transfected with pCMVrasV12 plasmid, expressing constitutively active ras oncogene (Clontech Laboratories, Mountain View, CA, USA). As a control, MCF-10A cells were transfected with the same vector containing only the neo gene. The transformed cells were selected by culturing in medium containing 400 μg/mL geneticin (G418). In some transfection assays, SAF-1/MAZ short hairpin RNAs (shRNAs), and control shRNAs (Santa Cruz Biotechnologies, Dallas, TX, USA) were used. For overexpression of SAF-1, cells were transfected with either pcD-SAF-1 expression plasmid or a mutant SAF-1 plasmid construct. For transfection analysis, the cells were transfected with different reporter plasmid DNA (0.5 μg) as described before 14 and cell extracts were used chroramphenicol acetyl transferase (CAT) assay as described before 14.
Reporter gene and expression plasmids
Wild-type and mutant SAF-CAT reporter plasmids, designated as wt SAF-CAT and mt SAF-CAT, respectively, were constructed by ligating into the pBLCAT2 vector, three tandem copies of the wild-type SAF-binding element of SAA gene promoter from nucleotide position −254 to −226 containing the following sequences: wild-type: 5′-CCCTTCCTCTCCACCCACAGCCCCCATGG-3′ and mutant: 5′-CAATGAGTCGAGACCGTCGACATCCATGG-3′ as described earlier 31. 1.2 VEGF-CAT reporter plasmid, that contains human VEGF promoter sequences from nucleotide position −1179 to +21, was prepared by ligating this DNA into pBLCAT3 vector. A tandem copy of two SAF-1-binding sites is located within nucleotide position −130 and −30 of this VEGF promoter DNA 14. 1.2 Δ(−130/−30) VEGF-CAT reporter plasmid, containing a deletion of DNA sequences from −130 to −30 of VEGF promoter, was prepared as described earlier 14 by ligating polymerase chain reaction (PCR)-generated fragments of VEGF promoter DNA into pBLCAT3 vector. The 0.6H-Ras-CAT reporter was prepared by ligating, into pBLCAT3 vector, a promoter region of human Ha-ras 32, from nucleotide position −552 to +28 that corresponds to the junction of the noncoding first exon called exon 0 and intron 1. Similarly, the 0.68K-Ras-CAT reporter plasmid was prepared by ligating, in pBLCAT3 vector, a promoter region of human Ki-ras 33, from nucleotide position −465 to +215 that corresponds to the junction between noncoding exon 0 and intron 1. Both, H-Ras and K-Ras promoter DNA fragments were prepared by PCR amplification and verified by DNA sequencing. SAF-1 expression plasmid, pcD-SAF-1, was prepared by inserting full-length SAF-1 cDNA with a FLAG tag into pcDNA3 vector (Invitrogen Corporation, Grand Island, NY, USA). A mutant SAF-1, mut pcD-SAF-1(V71), containing a mutation at the MAP kinase phosphorylation site by replacement of threonine at amino acid position 71 of SAF-1 with valine was used. Both the wild-type pcD-SAF-1 and mutant pcD-SAF-1(V71) were described earlier 21.
Preparation of nuclear extract and DNA-binding assay
Nuclear extract preparation and DNA-binding assays were performed as described before 14. As 32P-labeled probes, sequences from −135 to +29 of VEGF promoter that contains SAF-1-binding elements 14 and a SAF-1-binding oligonucleotide of SAA promoter as described before 14 were used. In some binding reactions, antibody (Ab) against SAF-1 and Sp1 or normal IgG (Santa Cruz Biotechnology) were added to the reaction mixtures during a preincubation period of 30 min on ice. As a competitor of SAF-1 binding, some binding reactions contained 50 pmol of a canonical SAF-binding double-stranded oligonucleotide having an upper strand sequence of 5′-CCCTTCCTCTCCACCC-3′. As a nonspecific oligonucleotide competitor, a random sequence of oligonucleotide with an upper strand sequence of 5′-TCGAACTGAACTTGAG-3′ was used.
RNA isolation and qRT-PCR
Total RNA was isolated using a RNA isolation kit (Qiagen, Germantown, MD, USA). Relative expression levels of SAF-1, H-Ras, and K-Ras was determined by real-time RT-PCR using gene-specific primers. The cDNAs were prepared by reverse transcription from 0.5 μg of total RNA using TaqMan reverse transcription reagents and analyzed for SAF-1, Ha-Ras, K-Ras, and glyceraldehyde-3-phosphate (GAPDH) according to the manufacturer's protocol (Applied Biosystems, Life Technologies, Grand Island, NY, USA). Transcript levels were normalized against GAPDH. All experiments were done using biological triplicates and experimental duplicates. The primers were: forward, 5′-GCTCAGGACTTAGCAAGAAG-3′ and reverse, 5′-GTATTTACATAATTACACACTTTG-3′ for human K-Ras; forward, 5′-GGGGCAGTCGCGCCTGTGAA-3′ and reverse, 5′-CCGGCGCCCACCACCACCAG-3′ for human H-Ras; forward, 5′-CTCCAGTCCCGCTTCT-3′ and reverse, 5′-GGGAGCAAGTCCACCT-3′ for human SAF-1/MAZ; forward, 5′-CCCTTCATTGACCTCAACTACATG-3′ and reverse, 5′-TGGGATTTCCATTGATGACAAGC-3′ for human GAPDH.
ChIP assay
Chromatin immunoprecipitation (ChIP) assays were performed following a method as described 14 with minor modifications. Cells grown in culture were cross-linked with 1% formaldehyde for 10 min followed by addition of 0.125 mol/L glycine for 5 min and washed in PBS buffer. Following lysis of the cells and sonication, DNA-protein complexes in the lysates were subjected to immunoprecipitation using anti-SAF-1 or normal IgG. After precipitation of the immunocomplex with protein G-agarose, followed by washing and extraction with elution buffer, immunoprecipitated DNA was used as template in PCR with specific primers spanning the target region of human K-ras and human H-ras gene promoters. Primers used for amplification of human K-Ras promoter were 5′-TCCGGGTCAGAATTGGCG-3′ and 5′-GTTCCGCGCTCGATTCTTCT-3′, which yields an amplicon of 313-bp. Primers used for amplification of human H-Ras promoter were 5′-GTCTCCAGCCAAGCCCAAC-3′ and 5′-ACTCACCGTTCACAGGCG-3′, which yields an amplicon of 363-bp.
Statistics
To compare multiple sets of data, a one-way analysis of variance (ANOVA) with post hoc Fisher's least significant difference test was used. For paired data sets, a two-tailed t test was used. Values of P < 0.05 were considered to represent a significant difference.
Results
SAF-1/MAZ DNA-binding and transcriptional activities are induced in oncogenic rasV12 transformed MCF-10A cells
To understand whether in the TME, SAF-1/MAZ activity is increased by oncogenic signaling, we stably transduced MCF-10A cells, which has a low basal SAF-1/MAZ activity 34, with a constitutively active oncogenic pCMVrasV12 expression vector or a control empty vector DNA. MCF-10A is a spontaneously immortalized cell line originated from nonmalignant breast epithelial cells and these cells have many characteristics of normal breast epithelium, including inability to form colonies in soft agar and tumors in nude mice 35. Previous studies have shown that upon the introduction of Ras oncogene, MCF-10A cells undergo transformation and these transformed cells exhibit hall marks of transformation and epithelial-mesenchymal transition (EMT), including anchorage-independent growth, loss of requirement of hormones, invasiveness, and tumorigenicity in nude mice 36,37. As shown in Figure1, DNA-binding activity of SAF-1 was low in untransfected or empty vector transfected MCF-10A cells, but in oncogenic pCMVrasV12 transfected MCF-10A cells, the DNA-binding activity became markedly higher (Fig.1, lanes 2–4). Presence of SAF-1 in this DNA-protein complex was verified by using molar excess of competitor SAF-1-binding oligonucleotide and an anti-SAF-1 antibody, both of which inhibited the DNA-protein complex formation (Fig.1, lanes 7 and 9). Involvement of Sp1 transcription factor in this DNA-protein complex was evident by the appearance of a super-shifted complex (ss) when anti-Sp1 antibody was used (Fig.1, lanes 10 and 11). It is likely that SAF-1 and Sp1 together bind to the VEGF promoter. Indeed, such interaction of SAF-1 and Sp1 may have a synergistic effect on the VEGF expression as these two transcription factors have earlier been shown to synergize in the transcriptional induction process 38. A minor DNA-protein complex, complex A, was not affected by either SAF-1 or Sp1-specific antibodies and is composed of KLF-4 as described earlier 34. Ras expression seems to have little effect in this complex formation.
Figure 1.
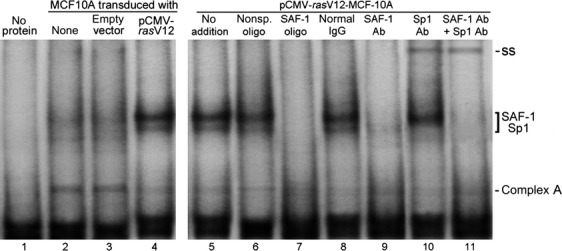
Increase of SAF-1 DNA-binding activity in oncogenic ras-transformed MCF-10A cells. Nuclear extracts (10 μg of protein) prepared from untreated (lane 2), empty vector transformed (lane 3) or pCMV-rasV12 transformed (lanes 4–11) MCF-10A cells, as indicated, were incubated with 32P-labeled VEGF DNA (−135 to +29) containing SAF-1-binding element. Lane 1 contains no nuclear extract. Resulting DNA-protein complexes were fractionated in a 6% nondenaturing polyacrylamide gel. In some assays, 50-fold molar excess of either nonspecific oligonucleotide (lane 6) or SAF-1-binding competitor oligonucleotide (lane 7) or normal IgG (lane 8) or antibody to SAF-1 (lanes 9 and 11) or antibody to Sp1 (lanes 10 and 11) were included during a preincubation reaction. Migration positions of SAF-1-specific complex are indicated. Super-shift (ss) of Sp1-specific DNA-protein complex is indicated. Complex A represents a DNA-protein complex present in normal MCF-10A cell nuclear extract is not affected by either SAF-1 competitor oligonucleotide, or the antibodies. SAF-1, serum amyloid A activating factor 1; VEGF, vascular endothelial growth factor.
To assess whether increased DNA-binding activity of SAF-1 in Ras-transformed MCF-10A-ras cells leads to an alteration in its transcriptional function, we evaluated SAF-1-driven promoter activity using a CAT reporter gene that contains three tandem copies of a bona fide SAF-1-binding element 31. Transcription from this promoter was evaluated by transfection of this reporter plasmid in MCF-10A, MCF-10A-ras, or empty vector transformed MCF-10A cells (Fig.2A). The CAT expression was significantly higher in Ras-transformed MCF-10A cells as compared to untransformed or empty vector transformed cells (Fig.2A). The reporter gene containing mutated SAF-binding element, mt SAF-CAT, showed no response to oncogenic ras, indicating that Ras-activated SAF-1 that binds to a bona fide SAF-1-binding element can drive CAT expression. To assess whether Ras-activated SAF-1 can promote angiogenesis-associated gene promoter activity, we used VEGF promoter-driven CAT reporter system (Fig.2B). This assay revealed that CAT activity is highly increased in ras-transformed MCF-10A-ras cells that are transfected with the 1.2 VEGF-CAT plasmid. This region of VEGF promoter contains a SAF-1-binding site with two tandem SAF-binding elements located between nucleotide position −130 and −30 14. Involvement of Ras-activated SAF-1 in the promoter activation was further verified when the VEGF promoter with a deletion in the SAF-1-binding site, failed to show a significant induction in the ras-transformed cells (Fig.2B). Together, these results suggested that Ras increases both the DNA-binding activity and transcription function of SAF-1.
Figure 2.
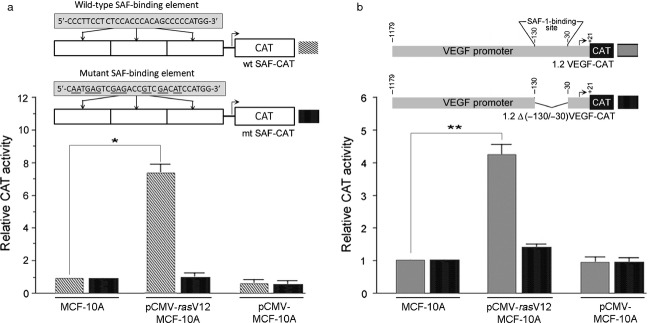
Induction of SAF-1 promoter function in oncogenic ras-transformed MCF-10A cells. (A) MCF-10A or pCMV-rasV12 or empty vector, pCMV, transformed MCF-10A cells were transfected with 1.0 μg each of wild type (wt) or mutant (mt) SAF-CAT reporter construct. The wild-type SAF-CAT reporter contains three tandem repeats of SAF-1-binding elements ligated in pBLCAT2 vector. The mutant SAF-CAT contains three tandem copies of mutant SAF-1-binding elements ligated to pBLCAT2. Relative CAT activity in ras- and empty vector-transformed cells was determined by comparing it to that in normal MCF-10A cells. Results represent an average of three separate experiments. *P < 0.05. (B) The same three sets of cells, as those in (A), were transfected with 1.0 μg each of 1.2 VEGF-CAT containing a 1200 base pair VEGF promoter with a SAF-1-binding site located within nucleotide position −130 and −30 or a deletion mutant of VEGF promoter lacking the SAF-1-binding site and ligated in pBLCAT3 vector. Relative CAT activity in ras- and empty vector-transformed cells was determined by comparing it to that in normal MCF-10A cells. Results represent an average of three separate experiments. **P < 0.02. SAF-1, serum amyloid A activating factor 1; CAT, chroramphenicol acetyl transferase; VEGF, vascular endothelial growth factor.
Ras activates SAF-1/MAZ activity through ERK/MAPK-signaling
The Ras oncogenes utilize various signaling pathways to modify downstream effectors that drive tumorigenesis. Among these pathways, the Raf-MEK-ERK-MAPK cascade is a major one. Raf serine/threonine kinases (c-Raf-1, A-Raf, and B-Raf) phosphorylate and activate MEK1/2 dual specificity kinases, which then phosphorylate and activate the ERK1 and ERK2 MAPKs. Activated ERKs translocate to the nucleus where they phosphorylate and regulate various transcription factors. To determine if Ras-mediated activation of SAF-1 proceeds through Raf-MEK-ERK-MAPK cascade, MCF-10A-ras cells were grown with or without PD98059, which is a specific inhibitor of mammalian MEK-1/2 and ERK (P42/44 MAP kinase). PD98059 markedly inhibited DNA-binding activity of SAF-1/MAZ in MCF-10A-ras cells (Fig.3A). U0126, a specific inhibitor of MEK1/2 and SB203580 which inhibits both p42/44 and p38 MAP kinases also inhibited the binding activity. Identity of the protein in the DNA-protein complex was verified as SAF-1 by the ablation of the complex by anti SAF-1 antibody (Fig.3A, lane 8).
Figure 3.
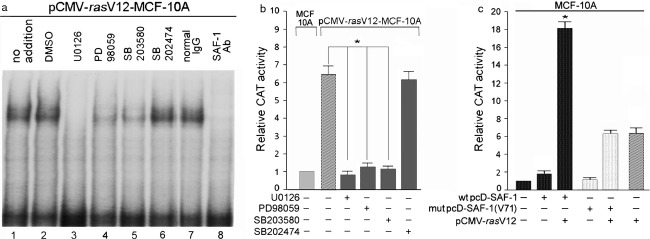
Oncogenic ras-mediated activation of SAF-1 is regulated via MEK/MAPK pathway. (A) Oncogenic pCMV-rasV12 transformed MCF-10A cells were either left untreated or incubated with DMSO (vehicle), MEK 1/2 inhibitor U0126 (20 μmol/L), MAP kinase inhibitors PD98059 (20 μmol/L), SB203580 (20 μmol/L), or SB202474 (20 μmol/L) for 24 h. Nuclear extracts (10 μg of protein) prepared from these cells, as indicated, were incubated with 32P-labeled DNA containing SAF-1-binding element of the SAA promoter. Resulting DNA-protein complexes were fractionated in a 6% nondenaturing polyacrylamide gel. (B) MCF-10A or pCMV-rasV12-transformed MCF-10A cells were transfected with equal amount (1.0 μg) of wt SAF-CAT reporter plasmid. Cells were grown for an additional 24 h in absence or presence of 20 μmol/L each of U0126, PD98059, SB203580, or SB202474, as indicated. Relative CAT activity in ras-transformed cells was determined by comparing it to that in normal MCF-10A cells. Results represent an average of three separate experiments. *P < 0.05. (C) MCF-10A cells were transfected with equal amount (1.0 μg) of wt SAF-CAT reporter plasmid. In addition, some cells were cotransfected with either a wild-type SAF-1 expression plasmid, wt pcD-SAF-1 (0.5 μg) or a mutant SAF-1 expression plasmid, mut pcD-SAF-1(V71) (0.5 μg). Also, some cells were transfected with Ras expression plasmid, pCMVrasV12 (0.5 μg), as indicated. Relative CAT activity was determined by measuring CAT activity of cotransfected MCF-10A cells compared to that of SAF-CAT transfected MCF-10A cells. Results represent an average of three separate experiments. *P < 0.05. SAF-1, serum amyloid A activating factor 1; CAT, chroramphenicol acetyl transferase.
To test whether changes in the DNA-binding activity of SAF-1 in response to the MEK and MAPK inhibitors also affects its transcriptional function, we used CAT reporter assay using wt SAF-CAT, where CAT expression is driven by the SAF-binding promoter element (Fig.3B). Treatment of the CAT vector-transfected cells with U0126, PD98059, and SB203580 but not by SB202474, which is an inactive analog of SB203580, significantly reduced the ras-mediated induction of SAF-1/MAZ function, suggesting involvement of MEK-ERK-MAPK cascade in the activation of SAF-1/MAZ by Ras.
To examine whether SAF-1/MAZ is directly targeted by Ras, we used a mutant SAF-1 construct, SAF-1(V71), that contains a defective MAP-kinase phosphorylation site 21. The DNA-binding activity and transactivation potential of this mutated SAF-1 protein has earlier been shown to be markedly less than that of wt SAF-1 21. We cotrasnfected MCF-10A cells with the reporter plasmid wt SAF-CAT along with oncogenic pCMVrasV12 and wild-type pcD-SAF-1 or mutant pcD-SAF-1(V71) plasmid DNAs (Fig.3C). There was marked increase in the reporter gene expression in the cells cotransfected with wild-type pcD-SAF-1 and oncogenic pCMVrasV12 as compared to the cells cotransfected with mutant pcD-SAF-1(V71) and oncogenic pCMVrasV12 plasmid DNAs (Fig.3C). The level of induction in cells transfected with mutant pcD-SAF-1(V71) + pCMVrasV12 was same as that with pCMVrasV12 alone. This level of induction is the result of Ras-mediated activation of endogenous SAF-1 in the MCF-10A cells. These data suggested that MAPK site of SAF-1 is necessary for Ras-mediated activation of SAF-1 and confirmed the involvement of MEK-ERK-MAPK-signaling in Ras-mediated activation of SAF-1/MAZ.
Knockdown of SAF-1/MAZ in breast cancer cells represses Ras gene expression
To further validate the role of Ras in SAF-1 activation, we knocked-down SAF-1 in the breast cancer cells. The evidences that overexpression of SAF-1/MAZ occurs in many human cancers and SAF-1/MAZ transcription factor is a target of activated Ras proteins, raised the questions, what downstream pathways are affected during aberrant activation of SAF-1 in cancer. MDA-MB-468 breast cancer cells are triple negative and contain no mutation in Ras genes 39. Also, presence of high level of SAF-1/MAZ in MDA-MB-468 cells 14 renders it suitable to assess the effect of silencing SAF-1/MAZ. To examine, SAF-1/MAZ expression was silenced in MDA-MB-468 cells by transfection of SAF-1/MAZ shRNA and global gene expression profile was examined. These data revealed down-regulation of several prominent cancer-linked genes that included MMP-1, MMP-9, MMP-14, p21WAF1/CIP1, hTERT, PPARg1, Prox-1 and VEGF (data not shown), and surprisingly, it showed downregulation of Ras mRNA expression. Quantitative RT-PCR analysis revealed downregulation of both H-Ras and K-Ras mRNAs by about 50% in SAF-1 knockdown MDA-MB-468 cells (Fig.4A). Reciprocally, overexpression of SAF-1 in MCF-10A cells that normally contain low level of SAF-1 34 and therefore suitable to examine the effect of overexpression of SAF-1, showed increased H-Ras and K-Ras mRNA levels by more than threefold compared to empty vector transfected cells (Fig.4B).
Figure 4.
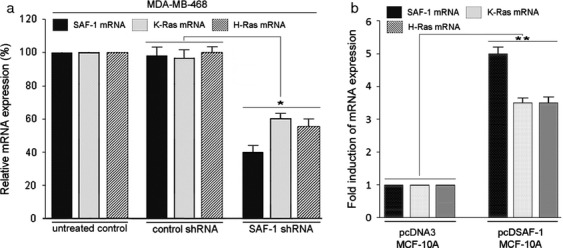
Silencing of SAF-1/MAZ reduces mRNA level of K-Ras and H-Ras. (A) MDA-MB-468 breast cancer cells were transfected with MAZ/SAF-1 short hairpin RNA (shRNA) lentiviral particles, control shRNA lentiviral particles (Santa Cruz Biotechnology), or left untreated. Total RNA isolated from these cells was subjected to qRT-PCR analysis with primers specific for SAF-1/MAZ, K-Ras and H-Ras. The results were normalized to the level of GAPDH in each sample and represent an average of three separate experiments. *P < 0.05. (B) MCF-10A cell were transfected with pcDNA3 empty vector or pcD-SAF-1 expression plasmid DNAs, as indicated. Total RNA isolated from these cells was subjected to qRT-PCR analysis with primers specific for SAF-1/MAZ, K-Ras, and H-Ras, as described in (A). The results were normalized to the level of GAPDH in each sample and represent an average of three separate experiments. **P < 0.03. SAF-1, serum amyloid A activating factor 1; qRT-PCR, quantitative reverse transcription-polymerase chain reaction.
SAF-1 acts as a transcriptional regulator of Ras
A strong correlation between SAF-1 and Ras, in vivo, suggested that SAF-1 could be involved in regulating Ras gene expression. It has long been known that human breast tumors have low percentage of activating Ras mutations 27,28 but that does not necessarily mean that Ras plays no role in breast cancer. In fact there is extensive experimental evidence demonstrating overexpression of wild-type Ras proteins and high level of activated Ras proteins in human breast tumor tissues 29,30, suggesting activation of upstream mechanisms regulating Ras in breast cancer. Overexpression of Ras proteins is also very common in other human cancers that harbor genetic Ras mutations. Thus, upregulation of Ras may be one mechanism leading to carcinogenic transformation in many types of human cancer. To determine, if SAF-1 is a direct transcriptional regulator of Ras, we examined the promoter function of human H-Ras and K-Ras genes in SAF-1 overexpressing cells. It is worthy of mention that like the protein coding regions, the proximal promoter region of human Ras genes are highly similar. The expression 0.6HRas-CAT and 0.68KRas-CAT reporter genes was consistently increased, in a dose-dependent manner, in pcD-SAF-1 expression plasmid transfected cells (Fig.5A and B). These data suggested that SAF-1 is a transcriptional regulator of human H-Ras and K-Ras genes.
Figure 5.
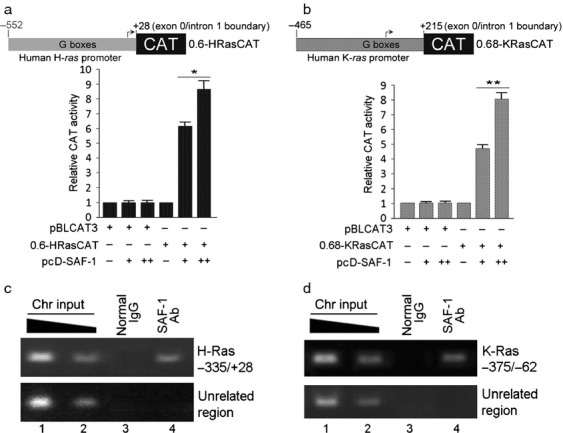
SAF-1 increases H-Ras and K-Ras promoter function by binding to the G-box promoter elements. MDA-MB-468 cells were cotransfected with equal amount (1.0 μg) of pBLCAT3 or 0.6HRas-CAT reporter plasmid DNA (A) or 0.68KRas-CAT reporter plasmid DNA (B), as indicated, and increasing concentration (0.5 and 1.0 μg) of pcD-SAF-1 plasmid DNA. Relative CAT activity was determined by comparing the CAT activities of transfected plasmids with that of pBLCAT3 alone. (C and D). MDA-MB-468 cells were cross-linked with formaldehyde and chromatin isolated from these cells was subjected to ChIP analysis by immunoprecipitating with anti-SAF-1 antibody or control IgG, as indicated. The precipitated chromatin DNA or input DNA was used for PCR amplification using H-Ras (C) and K-Ras (D) gene-specific primers. An unrelated upstream region was amplified to serve as negative control. SAF-1, serum amyloid A activating factor 1; CAT, chroramphenicol acetyl transferase; CHIP, chromatin immunoprecipitation; PCR, polymerase chain reaction.
SAF-1 interacts at a purine-rich region in the proximal promoters of H-Ras and K-Ras
The proximal promoter regions of the three functional Ras-genes are highly similar and contain many copies of the GGGC/A/TGGG element (G-boxes) or its inverted complement, which have been shown to be essential for the promoter transcriptional activity 40. SAF-1/MAZ also binds to highly purine-rich sequences and the G-boxes in Ras promoters appeared to bear considerable similarity with the consensus SAF-1/MAZ -binding element, RGGGRAGGRR, in which R is a purine 31. We employed ChIP analysis, which readily detects in vivo interaction of a transcription factor with the DNA in the chromosomal context. Formalin-fixed and SAF-1 antibody immunoprecipitated chromatin from MDA-MB-468 breast cancer cells showed specific enrichment of the purine-rich promoter regions of H-ras and K-ras (Fig.5C and D). There was no enrichment of these sequences when a nonspecific antibody was used, indicating specificity of SAF-1 interaction. Together these results demonstrated that SAF-1 interacts with Rasgenes in vivo and is capable of inducing expression of H-Ras and K-Ras genes.
Discussion
Angiogenesis is crucial for tumor cell growth and proliferation for which cancer cells employ multiple pathways to increase VEGF transcription. Identification of novel mechanisms of VEGF-mediated angiogenesis in the context of cancer biology is therefore very important for improving cancer therapy. We provide evidence and a model (Fig.6) illustrating how oncogenic, activated Ras can increase the DNA-binding and transcription function of SAF-1/MAZ transcription factor, a transcriptional regulator of VEGF. In turn, high level of SAF-1/MAZ, that is present in the breast cancer cells, increases Ras gene expression allowing more SAF-1/MAZ activation by activating MAP kinase pathway. These findings provide an explanation for persistently high level of expression of Ras proteins in many types of human cancer. In this context, SAF-1/MAZ could be a target for Ras/Raf/MAPK-directed therapeutics.
Figure 6.
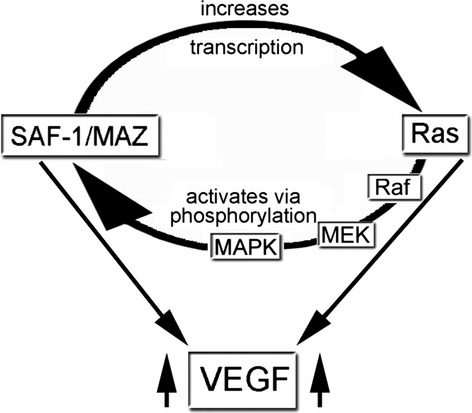
Model illustrating the Ras→SAF-1/MAZ→VEGF signaling network axis. Ras activates SAF-1/MAZ by Raf-MEK-ERK-MAPK pathway of phosphorylation. Activated SAF-1/MAZ promotes Ras expression by transcriptional induction of ras gene. The cyclical feed-forward regulation of SAF-1/MAZ and Ras promotes angiogenesis by promoting VEGF expression 14,27. SAF-1, serum amyloid A activating factor 1; VEGF, vascular endothelial growth factor.
SAF-1 transcription factor originally was isolated as an inflammation-responsive, Cys2-His2-type zinc finger protein 31. Its human homolog MAZ was identified as a regulator of c-myc gene 15. Regulation of SAF-1 activity is mediated via phosphorylation of this protein by various protein kinases, which markedly increase its DNA-binding activity 21–24. In this paper, we show for the first time, that Ras is involved in the activation of SAF-1 and utilizes MAPK-signaling pathway. Based on the results obtained from the use of a series of inhibitors, namely U0126, PD98059, and SB22025 that inhibit the MEK-MAPK-signaling, it was evident that Ras-mediated activation of SAF-1 proceeds through modulation of the Raf-MEK-ERK-MAPK signaling cascade. In the Ras-Raf-MEK-ERK-MAPK cascade Ras binds to and promote activation of Raf and activated Raf, in turn, phosphorylates and activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase MEK1 and MEK2 dual specificity kinases. MEKs then phosphorylate and activate the ERK1 and ERK2 mitogen activated protein kinases. It is noteworthy that the role of PI3K-Akt kinase signaling in mediating Ras-mediated activation of SAF-1 has not been examined and awaits further investigation.
We showed that silencing of SAF-1 reduces K-Ras and H-Ras mRNA level (Fig.4) and SAF-1 is a direct transcriptional regulator of these two Ras genes (Fig.5A and B). These results were confirmed by in vivo ChIP assays, which demonstrated that SAF-1 interacts at the G-rich sequences that are present upstream of the transcription start site in these two Ras promoters (Fig.5C and D). Like the protein coding sequences, the proximal promoters of three human Ras genes are highly similar and conserved and contain highly G-rich sequences, including multiple repeats of GGGCGGG and its inverted complements. Guanine-rich nucleic acid sequences are known to self-associate and form unusual structures known as G-quartets, G-4 DNA, or G-quadruplexes. G-quadruplexes have been shown to be present in regions of biological significance, such as human telomeres and in the promoter regions of several cancer-linked genes, including c-myc, bcl-2, c-kit, VEGF, Ras, HIF-1α, and Rb 41,42. The G-rich sequences that are capable of forming G-quadruplexes are not rigid and can somewhat vary. For example, in human telomere, tandem repeats of the hexanucleotide d(TTAGGG)n form G-quadruplexes 43, while the G-quadruplexes in gene promoters are composed of G-tracts with unequal numbers of guanines and various numbers of intervening bases 44. Among these, the G3NG3 motif appears to be widespread and evolutionarily selected to serve as a stable core in promoter intramolecular sequences of G-quadruplex structure. In correlation with our observation, Pur-1, the mouse homolog of SAF-1, is found to interact with G-quartets in the insulin-linked polymorphic region (ILPR, also known as IDDM2), located in the promoter region of the human insulin gene 45, while MAZ, the human homolog of SAF-1 is shown to interact with quadruplex forming GA-element in K-Ras promoter 46. The SAF-1-binding element in the VEGF promoter 14 also forms a G-quadruplex structure 47. Our finding of the G-rich sequences and a possible G-quadruplex structure in Ras gene promoter as a site of interaction of SAF-1/MAZ and involvement of such interaction in ras expression could have a significant impact in understanding the role of SAF-1/MAZ in breast cancer and perhaps in other cancer as well due to ubiquitous presence of Ras in many cancers and its direct link to SAF-1/MAZ which is revealed in the current study.
A common feature of transcription networks is the presence of feedback regulatory loops used for amplification of an initial signal through positive reinforcement or through a negative regulation. Our investigation elucidated an important aspect of cancer cell signaling that cancer thrives on multiple intermolecular conjunctions to sustain oncogenicity by depicting (Ras→SAF-1/MAZ→VEGF) signaling network axis (Fig.6). Downregulation of Ras mRNAs in SAF-1 silenced cells further showed the range of downstream networks that can be affected during aberrant silencing or overexpression of a transcription factor. In this context, a number of clinical trials have been aimed at disrupting Ras signal transduction and inhibiting VEGF function, independently. Inhibitors of farnesylation that disrupt activation of Ras were not very efficient because of specificity of the transfer of farnesyl/geranyl moiety 48,49. Similarly, VEGF inhibitors although prolonged survival rate in cancer patients, also caused increase in the red blood cells. As inhibition of SAF-1 can suppress Ras and VEGF expression, these findings offer a new possibility for cancer therapy by targeting SAF-1/MAZ function that may provide an improved treatment option for cancer. Targeting SAF-1/MAZ can also benefit other diseases that are linked with aberrant expression of VEGF, including arthritis, defective wound repair, endometriosis, and blindness.
Acknowledgments
This work was supported by United States Army Medical Research and Material Command grant W81XWH-09-1-0084 and University of Missouri Research Council grant.
Conflict of Interest
None declared.
References
- Brown LF, Berse B, Jackman RW, Tognazzi K, Guidi AJ, Dvorak HF, et al. Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in breast cancer. Hum. Pathol. 1995;26:86–91. doi: 10.1016/0046-8177(95)90119-1. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin. Oncol. 2002;29:10–14. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. USA. 1997;94:861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, et al. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- Serban D, Leng J. Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ. Res. 2008;102:1350–1358. doi: 10.1161/CIRCRESAHA.107.169664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Prendergast GC. Fenton RG. Farnesyltransferase inhibitors reverse Ras-mediated inhibition of Fas gene expression. Cancer Res. 2002;62:450–458. [PubMed] [Google Scholar]
- Kim CK, Choi YK, Lee H, Ha KS, Won MH, Kwon YG, et al. The farnesyltransferase inhibitor LB42708 suppresses vascular endothelial growth factor-induced angiogenesis by inhibiting ras-dependent mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signal pathways. Mol. Pharmacol. 2010;78:142–150. doi: 10.1124/mol.110.063586. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D. Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin. Cell Biol. 2004;14:123–130. doi: 10.1016/j.semcancer.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Berra E, Pages G. Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- Milanini-Mongiat J, Pouyssegur J. Pages G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: their implication in vascular endothelial growth factor gene transcription. J. Biol. Chem. 2002;277:20631–20639. doi: 10.1074/jbc.M201753200. [DOI] [PubMed] [Google Scholar]
- Pore N, Liu S, Shu H-K, Li B, Haas-Kogan D, Stokoe D, et al. Sp1 is involved in Akt-mediated induction of VEGF expression through an HIF-1-independent mechanism. Mol. Biol. Cell. 2004;15:4841–4853. doi: 10.1091/mbc.E04-05-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JC, Wang L, Wei D, Gong W, Hassan M, Wu T-T, et al. Association between expression of transcription factor Sp1 and increased vascular endothelial growth factor expression, advanced stage, and poor survival in patients with resected gastric cancer. Clin. Cancer Res. 2004;10:4109–4117. doi: 10.1158/1078-0432.CCR-03-0628. [DOI] [PubMed] [Google Scholar]
- Pages G. Pouyssegur J. Transcriptional regulation of the vascular endothelial growth factor gene–a concert of activating factors. Cardiovasc. Res. 2005;65:564–573. doi: 10.1016/j.cardiores.2004.09.032. [DOI] [PubMed] [Google Scholar]
- Ray A, Dhar S. Ray BK. Control of VEGF expression in triple-negative breast carcinoma cells by suppression of SAF-1 transcription factor activity. Mol. Cancer Res. 2011;9:1030–1041. doi: 10.1158/1541-7786.MCR-10-0598. [DOI] [PubMed] [Google Scholar]
- Bossone SA, Asselin C, Patel AJ. Marcu KB. MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc. Natl. Acad. Sci. USA. 1992;89:7452–7456. doi: 10.1073/pnas.89.16.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Southard RC, Allred CD, Talbert DR, Wilson ME. Kilgore MW. MAZ drives tumor-specific expression of PPAR gamma 1 in breast cancer cells. Breast Cancer Res. Treat. 2008;111:103–111. doi: 10.1007/s10549-007-9765-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas J, Mansuroglu T, Moriconi F, Haller F, Wilting J, Lorf T, et al. Altered regulation of Prox1-gene-expression in liver tumors. BMC Cancer. 2008;8:92. doi: 10.1186/1471-2407-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits M, Wurdinger T, van het Hof B, Drexhage JA, Geerts D, Wesseling P, et al. Myc-associated zinc finger protein (MAZ) is regulated by miR-125b and mediates VEGF-induced angiogenesis in glioblastoma. FASEB J. 2012;26:2639–2647. doi: 10.1096/fj.11-202820. [DOI] [PubMed] [Google Scholar]
- Jiao L, Li Y, Shen D, Xu C, Wang L, Huang G, et al. The prostate cancer-up-regulated Myc-associated zinc-finger protein (MAZ) modulates proliferation and metastasis through reciprocal regulation of androgen receptor. Med. Oncol. 2013;30:570. doi: 10.1007/s12032-013-0570-3. [DOI] [PubMed] [Google Scholar]
- Daheron L, Salmeron S, Patri S, Brizard A, Guilhot F, Chomel JC, et al. Identification of several genes differentially expressed during progression of chronic myelogenous leukemia. Leukemia. 1998;12:326–332. doi: 10.1038/sj.leu.2400923. [DOI] [PubMed] [Google Scholar]
- Ray A, Yu GY. Ray BK. Cytokine-responsive induction of SAF-1 activity is mediated by a mitogen-activated protein kinase signaling pathway. Mol. Cell. Biol. 2002;22:1027–1035. doi: 10.1128/MCB.22.4.1027-1035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Ray P, Guthrie N, Shakya A, Kumar D. Ray BK. Protein kinase A signaling pathway regulates transcriptional activity of SAF-1 by unmasking its DNA-binding domains. J. Biol. Chem. 2003;278:22586–22595. doi: 10.1074/jbc.M300705200. [DOI] [PubMed] [Google Scholar]
- Ray A, Fields AP. Ray BK. Activation of transcription factor SAF involves its phosphorylation by protein kinase C. J. Biol. Chem. 2000;275:39727–39733. doi: 10.1074/jbc.M007907200. [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Geltinger C, Murata T, Itakura K, Wada T, Handa H, et al. The DNA-binding and transcriptional activities of MAZ, a myc-associated zinc finger protein, are regulated by casein kinase II. Biochem. Biophys. Res. Commun. 1999;262:198–205. doi: 10.1006/bbrc.1999.1130. [DOI] [PubMed] [Google Scholar]
- Ellis RW, Defeo D, Shih TY, Gonda MA, Young HA, Tsuchida N, et al. The p21 src genes of Harvey and Kirsten sarcoma viruses originate from divergent members of a family of normal vertebrate genes. Nature. 1981;292:506–511. doi: 10.1038/292506a0. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D. Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–238. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Barbacid M. Ras genes. Annu. Rev. Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE. Boss GR. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000;62:51–62. doi: 10.1023/a:1006491619920. [DOI] [PubMed] [Google Scholar]
- Watson DM, Elton RA, Jack WJ, Dixon JM, Chetty U. Miller WR. The H-ras oncogene product p21 and prognosis in human breast cancer. Breast Cancer Res. Treat. 1991;17:161–169. doi: 10.1007/BF01806365. [DOI] [PubMed] [Google Scholar]
- Ray A. Ray BK. Isolation and functional characterization of cDNA of serum amyloid A-activating factor that binds to the serum amyloid A promoter. Mol. Cell. Biol. 1998;18:7327–7335. doi: 10.1128/mcb.18.12.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Merlino GT. Pastan I. Promoter region of the human Harvey ras proto-oncogene: similarity to the EGF receptor proto-oncogene promoter. Science. 1985;230:1378–1381. doi: 10.1126/science.2999983. [DOI] [PubMed] [Google Scholar]
- McGrath JP, Capon DJ, Smith DH, Chen EY, Seeburg PH, Goeddel DV, et al. Structure and organization of the human Ki-ras proto-oncogene and a related processed pseudogene. Nature. 1983;304:501–506. doi: 10.1038/304501a0. [DOI] [PubMed] [Google Scholar]
- Ray A, Alalem M. Ray BK. Loss of epigenetic Kruppel-like factor 4 histone deacetylase (KLF-4-HDAC)-mediated transcriptional suppression is crucial in increasing vascular endothelial growth factor (VEGF) expression in breast cancer. J. Biol. Chem. 2013;288:27232–27242. doi: 10.1074/jbc.M113.481184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol. Carcinog. 1991;4:25–35. doi: 10.1002/mc.2940040106. [DOI] [PubMed] [Google Scholar]
- Dawson PJ, Wolman SR, Tait L, Heppner GH. Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am. J. Pathol. 1996;148:313–319. [PMC free article] [PubMed] [Google Scholar]
- Ray A, Schatten H. Ray BK. Activation of Sp1 and its functional co-operation with serum amyloid A-activating sequence binding factor in synoviocyte cells trigger synergistic action of interleukin-1 and interleukin-6 in serum amyloid A gene expression. J. Biol. Chem. 1999;274:4300–4308. doi: 10.1074/jbc.274.7.4300. [DOI] [PubMed] [Google Scholar]
- Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW. Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol. Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- Ishii S, Kadonaga JT, Tjian R, Brady JN, Merlino GT. Pastan I. Binding of the Sp1 transcription factor by the human Harvey ras1 proto-oncogene promoter. Science. 1986;232:1410–1413. doi: 10.1126/science.3012774. [DOI] [PubMed] [Google Scholar]
- Burge S, Parkinson GN, Hazel P, Todd AK. Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam EY, Beraldi D, Tannahill D. Balasubramanian S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013;4:1796. doi: 10.1038/ncomms2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert JL. Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [Erratum appears in. 2007; (6):2105]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew A, Rutter WJ. Kennedy GC. Unusual DNA structure of the diabetes susceptibility locus IDDM2 and its effect on transcription by the insulin promoter factor Pur-1/MAZ. Proc. Natl. Acad. Sci. USA. 2000;97:12508–12512. doi: 10.1073/pnas.97.23.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogoi S, Paramasivam M, Membrino A, Yokoyama KK. Xodo LE. The KRAS promoter responds to Myc-associated zinc finger and poly(ADP-ribose) polymerase 1 proteins, which recognize a critical quadruplex-forming GA-element. J. Biol. Chem. 2010;285:22003–22016. doi: 10.1074/jbc.M110.101923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Liu WJ, Guo K, Rusche JJ, Ebbinghaus S, Gokhale V, et al. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol. Cancer Ther. 2008;7:880–889. doi: 10.1158/1535-7163.MCT-07-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD. Sebti SM. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 1997;15:1283–1288. doi: 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- James G, Goldstein JL. Brown MS. Resistance of K-RasBV12 proteins to farnesyltransferase inhibitors in Rat1 cells. Proc. Natl. Acad. Sci. USA. 1996;93:4454–4458. doi: 10.1073/pnas.93.9.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]