

Abstract
Pneumocystis jirovecii is a common opportunistic infection in the HIV-positive population and is re-emerging as a growing clinical concern in the HIV-negative immunosuppressed population. Newer targeted immunosuppressive therapies and the discovery of rare genetic mutations have furthered our understanding of the immunity required to clear Pneumocystis infection. The immune system can also mount a pathologic response against Pneumocystis following removal of immunosuppression and result in severe damage to the host lung. The current review will examine the most recent epidemiologic studies about the incidence of Pneumocystis in the HIV-positive and HIV-negative populations in the developing and developed world and will detail methods of diagnosis for Pneumocystis pneumonia. Finally, this review aims to summarize the known mediators of immunity to Pneumocystis and detail the pathologic immune response leading to Pneumocystis-related immune reconstitution inflammatory syndrome.
Keywords: Pneumocystis, Diagnosis, Immunity, Immune Reconstitution Syndrome, Therapy
Introduction
Pneumocystis was first described in the lungs histologically by Dr. Carlos Chagas in the early 1900’s. Approximately forty years later during World War II, the first cases of a diffuse interstitial pneumonia caused by Pneumocystis were documented in malnourished infants in orphanages (1). Pneumocystis at that time was considered a rare infection observed in patients with genetic immunodeficiencies. Fast forwarding another forty years, the CDC released the first case report of Pneumocystis pneumonia in homosexual men in Los Angeles in 1981 (2). Pneumocystis was and remains one of the most common and most devastating opportunistic infections in the HIV/AIDS population. Currently, thirty years after the connection between Pneumocystis and HIV was elucidated, Pneumocystis is re-emerging onto the clinical scene in the HIV-negative population. The use of newer immunosuppressive agents and chemotherapeutics has left patients with autoimmune conditions, transplantation, and hematologic malignancies at-risk to developing Pneumocystis pneumonia. Given the clinical problem that Pneumocystis presents, we will discuss the epidemiology, clinical features, and diagnostic methods for Pneumocystis in the current review. We will also review the protective immunity responsible for eliminating Pneumocystis infection, as well as the pathologic immune response following reconstitution of the immune system.
Epidemiology
The epidemiology of Pneumocystis can be categorized into the HIV and non-HIV populations. As described above, Pneumocystis first emerged as a common opportunistic infection in the HIV/AIDS population. As a result, anti-Pneumocystis prophylaxis was recommended for any individual with low CD4+ T cell counts (<200 cells/μL), which led to a reduction in the incidence of infection (3). This reduction was furthered by the implementation of combined anti-retroviral therapy (cART) regimens (3). Within three years of the use of cART, Pneumocystis incidence (as measured by infection rates per 1,000 person-years) decreased by approximately half (3). Although the incidence of Pneumocystis has been reduced, a study by Walzer et al. demonstrated that the mortality of Pneumocystis pneumonia has largely been unchanged by the implementation of antiretroviral therapy (4). Prior to cART, mortality rates of Pneumocystis pneumonia in HIV patients were 10.1%; following cART, mortality rates were modestly reduced to 9.7% (4).
Despite the use of cART and anti-Pneumocystis prophylaxis, Pneumocystis pneumonia remains the most common serious opportunistic infection in HIV patients in the United States (3, 5, 6). One study reported 322 cases of Pneumocystis pneumonia in 2,622 patients with AIDS-defining events (5). Unsurprisingly, most cases of Pneumocystis in the developed world are in patients unaware of their HIV-positive status and/or patients not receiving prophylaxis or antiretroviral therapy (6).
In the developing world, Pneumocystis pneumonia is a common complicating factor in the HIV-positive population. Pneumocystis was detected in the bronchoalveolar lavage (BAL) fluid of 33% of HIV-infected patients presenting with a diffuse pneumonia in southern Africa (7). Furthering those findings, additional studies in Africa have shown that HIV-positive patients with symptoms (e.g. cough/dyspnea) of pneumonia are likely to have Pneumocystis infection; the incidence of Pneumocystis in such populations were found to be between 37.2% and 48.6% in South Africa and Kenya, respectively (8, 9). Asian countries, such as Thailand, India, and Malaysia, also have high incidences of Pneumocystis infection in the HIV-positive population with diagnosis rates between 12.2–25% (10–12). Developing countries in South America, such as Chile and Venezuela, also report high incidences of Pneumocystis in HIV-positive patients with respiratory symptoms (~37%) (13, 14). More alarming than any individual percentage, the above studies all further the point that Pneumocystis remains a global clinical concern for patients with HIV/AIDS where prophylaxis and/or cART use is limited for a variety of reasons.
Pneumocystis is also re-emerging in developed countries in the HIV-negative population. A study conducted in Sweden demonstrated that 75% of patients presenting to the hospital with Pneumocystis pneumonia were HIV-negative (15). Another study conducted in the United Kingdom between 2000–2010 found that the number of hospital episodes of Pneumocystis pneumonia more than doubled during the study period, with transplant and hematologic malignancy patients representing the highest risk groups (16). In addition to malignancy and transplantation, several other conditions including autoimmune conditions and inherited immunodeficiencies have been implicated as emerging risk factors for Pneumocystis infection (Table 1) (5, 17–29).
Table 1.
Summary of conditions and procedures, along with the therapeutic agents used to treat those conditions, which have been implicated in increasing the patient’s risk for developing Pneumocystis pneumonia (5, 17–29).
| Conditions/procedures associated with Pneumocystis infection | Therapeutic agents associated with Pneumocystis infection |
|---|---|
|
| |
| HIV Hematologic malignancy Solid tumors Hematopoietic stem cell transplantation Solid organ transplantation Rheumatoid arthritis Severe combined immunodeficiency Hyper-IgM syndrome Wegener’s granulomatosis Inflammatory Bowel Disease Collagen vascular disorders |
Corticosteroids Alkylating agents (e.g. cyclophosphamide) Antimetabolite chemotherapeutics (e.g. methotrexate) TNF inhibitors (e.g. Etanercept) Azathioprine Alemtuzumab Rituximab Sirolimus/Tacrolimus Cyclosporine |
Although there are several immunologic changes associated with each of the above conditions, the immunosuppressive therapy for each disease undoubtedly contributes to the risk of developing Pneumocystis pneumonia (Table 1). One such example is the use of Rituximab, a monoclonal antibody against the B-cell marker CD20, for the treatment of hematological malignancies such as diffuse large B-cell lymphoma. Martin-Garrido et al. found that approximately 30% of patients receiving Rituximab went on to develop Pneumocystis pneumonia over the course of the study period (30). Perhaps more troubling, acute respiratory failure was seen in 40% of patients with Rituximab-associated Pneumocystis, while mortality in these patients was as high as 30% (30). Although several other agents can increase a patient’s risk for Pneumocystis (Table 1), this example of Rituximab highlights two important points. First, the use of these targeted immunosuppressive agents has lead to a greater understanding of the immune response required to protect against Pneumocystis (see Immunity against Pneumocystis section). Rituximab selectively targets B-cells and leaves the often-implicated CD4+ T cells intact; however, these patients are exquisitely susceptible to Pneumocystis to the point where universal prophylaxis is being discussed. Second, much like the above study found with Rituximab, non-HIV cases of Pneumocystis tend to have increased morbidity (e.g. higher mechanical ventilation rates) and mortality than HIV-positive cases (31, 32). At this time, it remains unclear if the direct cause of the increased mortality is due to changes in the disease or differences in clinical management.
Clinical Features
Pneumocystis pneumonia in the HIV-positive population is generally characterized by a sub-acute onset of low-grade fever, nonproductive cough, and progressive dyspnea (33). Further nonspecific findings, such as tachypnea and tachycardia, can be found on physical exam, while the lung exam may range from normal to diffuse crackles upon auscultation (33). While the above characteristic presentation of Pneumocystis pneumonia is common in HIV-positive patients, HIV-negative patients can present much differently. Typically, the HIV-negative patients will have a more acute or fulminant presentation with substantial dyspnea, fever, and chills (17). Furthermore, HIV-negative patients have a wider alveolar-arterial oxygen gradient and are more likely to require mechanical ventilation (17).
Diagnosis
Radiologic methods are a useful first step in making a diagnosis of Pneumocystis, as most patients presenting with fever and dyspnea will receive a chest x-ray (CXR). The classic CXR of patients with Pneumocystis shows diffuse, bilateral interstitial and alveolar infiltrates (Figure 1A). Less common findings, such as pneumatoceles, lobar infiltrates, and pneumothoraxes have been reported (17). Some patients presenting with Pneumocystis will have near normal or unimpressive radiographic findings, in which case higher resolution radiologic approaches are recommended, such as high-resolution chest CT (6, 17). Chest CT most commonly demonstrates ground glass opacity with relative peripheral sparing, although mosaic and diffuse patterns can be observed (34). Notably, the findings described above are not specific for Pneumocystis and a broad differential for opportunistic pneumonias (e.g. Aspergillus, Mycobacterium avium-complex) should be maintained (6).
Figure 1.
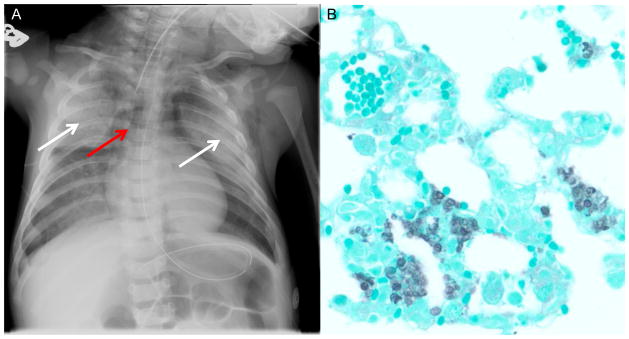
Radiographic and microbiologic diagnosis of Pneumocystis. A. Chest radiograph of child with X-linked severe combined immunodeficiency showing bilateral ground glass infiltrates (white arrows) and air bronchograms consistent with Pneumocystis pneumonia. Note that this patient also has a pneumomediastinum (red arrow) with air dissecting into the soft tissue of the neck and an absent thymic shadow in the mediastinum consistent with athymia. B. Gomori-methenamine silver stain (GMS) on Pneumocystis infected mouse lung, showing lung architecture (green) with Pneumocystis organisms (black) filling the alveolar spaces.
It is also important to interpret radiologic findings in the context of the underlying immunosuppression. HIV-negative patients with Pneumocystis tend to have a greater extent of ground-glass opacity on chest CT (34). Moreover, HIV-negative Pneumocystis patients are more likely to have lung consolidations, perhaps reflecting a more robust host immune response (34).
Although radiologic methods, coupled with the appropriate clinical context, can highly suggest a diagnosis of Pneumocystis, the gold standard of diagnosis remains harvesting organism from bronchoalveolar lavage (BAL) fluid. Several stains can be utilized to identify Pneumocystis microscopically: Gomori-methenamine silver stain (GMS) (Figure 1B), Wright-Giemsa, toluidine blue O, or Calcofluor white (33). Monoclonal antibodies conjugated to a fluorescent marker are also available to stain Pneumocystis. In fact, these conjugated monoclonal antibodies have greater sensitivity and specificity for detecting Pneumocystis than most non-immunofluorescent stains (33). In addition to BAL sampling, induced sputum samples can also be analyzed for Pneumocystis by the above stains, with monoclonal antibodies again having the highest sensitivity and specificity (18). Importantly, HIV-negative patients with suspected Pneumocystis infection may have a negative induced sputum sample, as HIV-negative patients tend to have a lower organism burden than their HIV-positive counterparts (18). In HIV-negative patients, BAL is recommended (18).
Several molecular techniques have been tested as means to diagnose Pneumocystis from BAL or sputum samples. One such method developed in the 1990’s was the use of a single-round polymerase chain reaction (PCR) to amplify the mitochondrial small subunit rRNA of Pneumocystis (35, 36). Since that time, the use of nested-PCR (two round PCR) has been used on several gene targets, such as dihydropteroate synthase (DHPS), dihydrofolate reductase (DHFR), major surface glycoprotein (MSG), and loci within the region coding for rRNAs (36). While using the nested approach increases sensitivity to nearly 100%, this often comes at the cost of decreasing specificity due to the ability to detect colonized individuals who may not have active Pneumocystis pneumonia (36).
Newer techniques include the use of quantitative real-time PCR (qPCR) on many of the same targets described above. In addition to reduced turnaround times, qPCRs provide semi-quantitative data to discriminate the cases of colonized individuals from the truly infected individuals. As such, qPCRs for Pneumocystis tend to have high sensitivities, along with increased specificities when compared to nested techniques (36). One study that illustrates this point was conducted by Flori et al. and examined the diagnostic value of a qPCR test on MSG (37). In this study, which examined both HIV-positive and HIV-negative patients, the MSG qPCR test had a sensitivity of 100% and specificity of 98.6% (37). While this indicates such diagnostic tests can be optimized to have value in the clinical setting, implementing a qPCR test requires validating the gradations of Pneumocystis PCR product to distinguish colonization from infection in the heterogeneous population of the immunocompromised (36). At this time, this challenge still persists in the field and limits the clinical use of such tests.
Two serum markers, β-1,3-glucan and KL-6, have been evaluated as diagnostics for Pneumocystis. β-1,3-glucan is a component of the fungal wall, particularly of the ascus (see Microbiology of Pneumocystis below) and can enter the serum upon active infection. One study demonstrated that serum β-1,3-glucan above 100 pg/mL had a sensitivity of 100% and a specificity of 96.4% for diagnosing Pneumocystis pneumonia using a retrospective analysis (38). However, specificity for serum β-1,3-glucan testing is difficult to establish, as this test does not discriminate between fungal species. As such, serum β-1,3-glucan is often used as an adjunct to clinical suspicion and other diagnostic tests to confirm a Pneumocystis infection rather than a stand alone diagnostic. Similarly, KL-6, a glycoprotein expressed on pneumocytes, can enter the serum in the setting of infectious lung disease. One study has shown that KL-6 levels are elevated in HIV-positive cases of Pneumocystis, but the generalizability of serum KL-6 to the HIV-negative population has yet to be demonstrated (39).
Microbiology of Pneumocystis
One of the unique features shared by all the Pneumocystis species is the multiphasic life cycle that occurs within the alveolar space of the host. The ascus (cyst) form of Pneumocystis is circular or ovoid in shape and is approximately 4–7 μm in diameter (1, 40). The ascus form has a distinctive thick outer wall made of β-1,3-D-glucan, while within the ascus, eight ascospores mature (1, 40, 41). Following maturation, the ascospores will leave the ascus through a small pore and become the troph life form (40, 42). The troph life form appears to be the more metabolically active and replicative form of Pneumocystis. Trophs range in size from 2–8 μm and are more irregular in shape. Trophs are thought to replicate in both an asexual and sexual manner. Although most fungal species replicate asexually through a process known as “budding,” trophs are thought to propagate via binary fission (43). Two trophs can also conjugate via the use of pheromone receptors and replicate sexually by fusing. Following fusion, the two previous trophs are now a single diploid early sporocyte, which divides using meiosis. This meiotic process is then followed mitosis, generating the eight ascospores (44). During the division processes, the wall of the sporocyte thickens and hardens and returns the life cycle to the ascus stage.
One study by Cushion et al. examined the life cycle in vivo; in particular, they examined the effects of β-1,3-D-glucan synthase inhibitors on the ascus and troph population within the lung (41). Mice treated with anidulafungin had a decrease burden of asci in the lung, while the level of trophs remained the same. More importantly, mice depleted of asci were no longer able to aerially transmit infection to immunodeficient mice, implicating the ascus as the infectious form. A second study examined the Pneumocystis life cycle in vitro, despite the fact that a continuous axenic culture method for Pneumocystis has yet to be discovered (45). In this study, Martinez et al. showed that asci were capable of producing new trophs while the reverse (trophs becoming asci) did not occur. These results demonstrated that information regarding the Pneumocystis life cycle could be gleamed from in vitro studies, although the viability in culture is undoubtedly a confounding variable. Recently, a novel mechanism to grow Pneumocystis jirovecii has been reported using differentiated pseudostratified CuFi-8 cells, although the utility of such a culture system for propagating infection and/or directed therapy selection has yet to be determined (46). Further studies on the Pneumocystis life cycle would be greatly enhanced by any sustainable Pneumocystis culture method.
In addition to elucidating the life cycle of Pneumocystis, the genetic makeup of Pneumocystis has also been heavily studied. In fact, even though Pneumocystis was first described in the early 1900’s as a protozoan, it would not be until the end of the century that Pneumocystis was correctly classified as a fungus (1, 47, 48). Early studies demonstrated homology between Pneumocystis and fungi, such as Saccharomyces cerevesiae, using alignments of mitochondrial and ribosomal gene sequences. Furthermore, several studies demonstrated that Pneumocystis is a unique genus that encompasses several host-specific species, including Pneumocystis jirovecii, the human pathogen (49–51). Despite these early genetic studies, it was only recently that the genome of Pneumocystis had been sequenced (52).
Immunity of Pneumocystis Infection
Much of what we have learned of the immune response to Pneumocystis has been gleaned from acquired and congenital immunodeficiencies leading to susceptibility and many of these human conditions have also been successfully replicated in animal models. High dose corticosteroid treatment remains a risk factor and steroid induce immunosuppression has been a widely used tool to induce infection in rodents (53, 54). Prior to the epidemic of the acquired immunodeficiency syndrome (AIDS), Pneumocystis infection in humans was associated with significant malnutrition or myelosuppressive chemotherapy for acute leukemia (55). When the epidemic of Pneumocystis infection was observed in AIDS, it was realized that the prevalence of Pneumocystis inversely correlated with the peripheral blood CD4+ T-cell lymphocyte count (56). This was recapitulated in a murine model where CD4+ T-cell depletion resulted in Pneumocystis pneumonia whereas CD4+ T-cell replete mice cleared the infection (57). Although CD4+ T-cells are essential the specific T-cell subsets required remain unclear. Experimental Pneumocystis infection induces Th1, Th2, and Th17 responses in mice. Mice deficient in Th17 immunity have delayed clearance of the pathogen but ultimately clear in the infection (58). Analogous to these findings, patients with STAT3 mutations that have reduced Th17 cells to Candida albicans (59) rarely develop clinical Pneumocystis infection (60). IL-21 is a cytokine produced by T-follicular helper cells in the germinal center of secondary lymphoid tissues (61) as well as Th17 cells. IL-21 appears to potentially play a key role in susceptibility to Pneumocystis as evidenced by a recent patient with an IL-21 receptor mutation who subsequently developed clinical Pneumocystis pneumonia (62).
B-cells also play a key role in susceptibility to infection. B-cell deficient mice are susceptible to infection (63), as well as patients with hyper IgM syndrome due to either mutations in CD40 or CD40 ligand (64, 65). Consistent with these findings, both CD40−/− or CD40L−/− mice are also susceptible to Pneumocystis. In addition to obvious effects on antibody production, the increased susceptibility of these patients may also be due to the fact that B-cells can function as critical antigen presenting cells during the infection (63). Antigen presentation by B-cells may also explain the fact that patients with mutations that affect antibody production such as common-variable or X-linked agammaglobulinemia can, but rarely, develop clinical Pneumocystis pneumonia (66). One caveat to the low incidence of Pneumocystis pneumonia in these patients, however, is that intravenous immunoglobulin is typically given prophylactically, which may mask some of the susceptibility to Pneumocystis. Despite the ambiguity associated with genetic immunodeficiencies, the use of anti-CD20 monoclonal antibodies in humans has emerged as a strong risk factor for Pneumocystis pneumonia, which further implicates the B-cell as an important cell type for the normal host defense against Pneumocystis (30). Thus, the B-cell appears to be critical and the dual functions of antigen presentation and antibody production are likely important.
Further evidence suggests antibodies can provide protection against Pneumocystis. It has been demonstrated that antibodies can provide protective immunity by passive transfer of serum elicited by immunization (67) or of monoclonal antibodies that recognize surface epitopes on the organism (68) to immunodeficient mice. Thus, although the role of humoral immunity in conferring susceptibility in humans remains unclear, antibodies could still be exploited for prevention or therapy.
It is thought that ultimately macrophages are the key effector cells that actually clear the infection. Indeed, macrophage depletion increases organism burden in the lung (69). Pneumocystis has also been shown to induce apoptosis of lung macrophages and this could be a major host evasion strategy of the organisms (70). Non-opsonic phagocytosis and killing of the organism requires the c-type lectin receptor Clec7a (71). This pathway can be bypassed if the organism is opsonized with IgG (71). Complement may also play a role in control of Pneumocystis (72). GM-CSF treatment of macrophages also increases their fungicidal activity (73). Recently, it has been demonstrated that macrophages that have an alternative activation program have greater fungicidal activity. Thus, given that CD4+ T-cell depletion is sufficient to confer susceptibility, CD4+ T-cells must be required for T-cell dependent antibody production as well as the recruitment of fungicidal macrophages.
Immune Reconstitution Inflammatory Syndrome
Immune Reconstitution Inflammatory Syndrome (IRIS) is a clinical phenomenon that occurs within the context of an opportunistic infection acquired during an immunosuppressive state. Upon treatment to reconstitute the immune system or address the underlying immunosuppressive condition, the immune response to an opportunistic infection can actually become pathologic. Typically, IRIS is observed in the context of HIV/AIDS following treatment with combination antiretroviral therapy (cART) and can present as worsening dyspnea, fever, and cough. Depending on the treatment status of the opportunistic infection, there are two different classifications of IRIS (74, 75). First, unmasking IRIS is characterized by a smoldering, undetected opportunistic infection acquired during the immunosuppressive state that is untreated upon the start of cART. After initiating treatment, an exaggerated immune response to the active infection can cause damage to host tissues while failing to clear the infection. The second form of IRIS, paradoxical IRIS, occurs despite the fact that adequate treatment for the opportunistic infection has already been received. The immune system, however, can still target residual non-self antigens and an overzealous response can again cause damage to the host.
IRIS is defined clinically by the temporal relationship between cART initiation and a subsequent inflammatory condition associated with a positive response to cART (75). IRIS remains a fairly common condition, as approximately 16% of HIV-positive patients will develop IRIS following initiation of cART (76). A number of different opportunistic infections (and even some non-infectious conditions) can result in IRIS, including Mycobacterium tuberculosis, Cryptococcus, herpes infections, and Pneumocystis (74). Despite remaining one of the most common opportunistic infections in HIV, retrospective studies demonstrate that Pneumocystis only accounts for approximately 2.7–4% of IRIS cases (77, 78). One prospective study examined the incidence and causative agent of IRIS in 282 patients (79). Interestingly, 63% of the patients enrolled in the study were diagnosed with Pneumocystis pneumonia prior to the initiation of cART; out of the 177 patients with Pneumocystis, 13 (7%) developed IRIS (79). However, the true incidence rate of Pneumocystis-related IRIS and IRIS in general are difficult to calculate for a number of reasons. First, there are several different functional definitions of IRIS used in research, making the diagnosis of IRIS variable across studies. Second, most patients with Pneumocystis-related IRIS present two months after initiation of cART (with some patients presenting closer to a year after therapy), thereby requiring that the patients are extensively followed (77, 79).
Clinical studies, both retrospective and prospective, have elucidated certain risk factors for the development of IRIS. Several studies have demonstrated that individuals with lower CD4+ T cell counts upon cART implementation are more likely to develop IRIS (74, 75, 77–79). Similarly, patients with increased HIV viral loads at the start of therapy are more susceptible to IRIS development (78, 79). Following cART, individuals who experience IRIS tend to have a more rapid decline in HIV viral load, followed by a sharp increase in CD4+ T cell counts (74, 75). The above risk factors are highlighted in a case series following three patients, each of whom presented with low CD4+ counts and high HIV viral loads, and subsequently developed a life-threatening Pneumocystis-related IRIS requiring mechanical ventilation (80). Interestingly, this case series suggested that early use of cART may increase the likelihood of severe Pneumocystis-related IRIS, calling into question the timing of cART implementation. However, one of the largest studies to date found an increase in overall and Pneumocystis-related mortality associated with delayed cART (81).
The pathophysiology of Pneumocystis-related IRIS has been further studied in mouse models of the disease. One of the first studies of Pneumocystis-related IRIS demonstrated that reconstitution of a SCID mouse infected with Pneumocystis resulted in increased expression of pro-inflammatory cytokines such as IL-1, IL-3, IL-6, TNF-α, TNF-β, and IFN-γ (82). Pneumocystis-related IRIS appears to be a T cell mediated phenomenon, as Pneumocystis-infected SCID mice receiving CD4+ T cells alone develop severe lung pathology (83). Further studies using an anti-CD3 antibody targeting T cells abrogated the inflammation associated with IRIS (84). CD8+ T cells have also been implicated in IRIS, as CD8+ cells have been shown to modulate the CD4+ response, while sensitized CD8+ cells may result in pathology independent of CD4+ cells (85, 86). Another subset of T cells, T regulatory cells, has been shown to dampen the lung inflammation secondary to a robust anti-Pneumocystis CD4+ T cell response (87). Local expression of IL-10, a hallmark cytokine of T regulatory cells, ameliorated the inflammation of Pneumocystis-related IRIS, while selective depletion of T regulatory cells worsened the course of the disease (88, 89). Reconstitution also appears to modulate lung mechanics, particularly surfactant regulation. Wright et al. demonstrated that reconstitution increased the protein:phospholipid ratios and minimum surface tension of bronchoalveolar lavage fluid when compared to wild-type mice (90). A more recent study further characterized the changes in the lung, as reconstituted animals had impaired surfactant biophysical function and decreased amounts of surfactant protein B and surfactant phospholipid (91). In addition, the S-nitrosylated form of surfactant protein D (SP-D) was increased; S-nitrosylated SP-D exists mostly as a monomer and has pro-inflammatory functions, such as increased cellular recruitment (91). While the above studies focus on the host’s immune response in IRIS, the properties of Pneumocystis leading to the development of IRIS have also been explored. Mice infected with asci (cysts) and trophs of Pneumocystis have greater immunopathology than mice infected with trophs alone, as measured by increased cellularity and pro-inflammatory cytokine profile (92). These results were corroborated by the finding that treatment of Pneumocystis-related IRIS with an ascus-targeting dectin:Fc fusion protein limited hypoxemia in reconstituted mice (93).
While the definitive mechanism of IRIS remains elusive, a model of Pneumocystis-related IRIS can be proposed based off of the above murine models and clinical findings (Figure 2). Prior to cART, a high Pneumocystis burden is observed in the lung due to the lack of functional CD4+ T cells capable of controlling the infection. Following cART (+/− treatment with antifungals), Pneumocystis antigen, likely derived from asci, remains prevalent in the lung while the high-affinity memory T cell compartment shows increased proliferation early in reconstitution (as reviewed in (74, 75)). Furthermore, the early expansion of memory T cells is facilitated by the lack of T regulatory cells, as these cells show a slower response to cART (as reviewed in (74, 75)). Due to the abundance of antigen and the lack of cellular regulators, T cells in the lung undergo antigen induced cellular death, again limiting the number of functional T cells in the lung (as reviewed in (74, 75)). Reconstitution in the context of Pneumocystis also alters the surfactant properties of the lung and leads to an increase in cellular recruitment and pro-inflammatory cytokines such as IL-1, IL-6, TNF-α, and IFN-γ. The recruited macrophages and neutrophils, coupled with T cell dysregulation, ultimately result in a pro-inflammatory state and damage to the host tissues. Importantly, the above model needs further validation in Pneumocystis-specific IRIS, as it is evident that the nature of the underlying opportunistic infection can alter the host response.
Figure 2.

Model of Immune Reconstitution Inflammatory Syndrome. In a patient with less than 200 cells/uL of CD4+ T cells, Pneumocystis (PC) can propagate in the lungs and produce high PC burdens. Following reconstitution, high levels of PC asci or PC antigen can persist in the lungs and lead to activation of T cells through IL-2, which can induce apoptosis in the absence of regulatory T cell (Treg) inhibition. The lack of Tregs also allows for increased inflammatory cell recruitment due to PC and resultant surfactant changes, leading to systemic release of pro-inflammatory cytokines.
Treatment
The first line therapy for active Pneumocystis infection and for Pneumocystis prophylaxis is trimethoprim-sulfamethoxazole (TMP-SMX). However, TMP-SMX can be associated with several side effects (e.g. rash, cytopenia) and is not recommended for patients with sulfa allergy (94–96). Interestingly, HIV-infected patients appear to be more likely to develop adverse side effects to sulfa drugs, further limiting the efficacy of TMP-SMX in the population at-risk for Pneumocystis infection (97). Several other treatments are indicated as second line therapies (e.g. pentamidine and dapsone) but such regimens tend to have much higher treatment failure rates (94, 95).
Treatment for Pneumocystis-related IRIS typically consists of eradicating the underlying infection with an anti-Pneumocystis agent described above, such as TMP-SMX (98–100). Identifying the cause of immunosuppression and providing adequate care and therapy for the underlying condition is also crucial for treating IRIS. As described above, early initiation of cART in the setting of an HIV-positive patient is thought to reduce overall mortality (81). Because some cases can become life threatening, glucocorticoid therapy can be used to reduce the inflammatory environment in the lung and supportive care and airway maintenance should be provided when needed (99, 100).
References
- 1.Chabe M, et al. Pneumocystis: from a doubtful unique entity to a group of highly diversified fungal species. FEMS Yeast Res. 2011;11:2–17. doi: 10.1111/j.1567-1364.2010.00698.x. [DOI] [PubMed] [Google Scholar]
- 2.Centers for Disease C. Pneumocystis pneumonia--Los Angeles. MMWR Morb Mortal Wkly Rep. 1981;30:250–2. [PubMed] [Google Scholar]
- 3.Morris A, et al. Current epidemiology of Pneumocystis pneumonia. Emerg Infect Dis. 2004;10:1713–20. doi: 10.3201/eid1010.030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walzer PD, et al. Early predictors of mortality from Pneumocystis jirovecii pneumonia in HIV-infected patients: 1985–2006. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2008;46:625–33. doi: 10.1086/526778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antiretroviral Therapy Cohort C et al. Variable impact on mortality of AIDS-defining events diagnosed during combination antiretroviral therapy: not all AIDS-defining conditions are created equal. Clin Infect Dis. 2009;48:1138–51. doi: 10.1086/597468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang L, et al. HIV-associated Pneumocystis pneumonia. Proc Am Thorac Soc. 2011;8:294–300. doi: 10.1513/pats.201009-062WR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malin AS, et al. Pneumocystis carinii pneumonia in Zimbabwe. Lancet. 1995;346:1258–61. doi: 10.1016/s0140-6736(95)91862-0. [DOI] [PubMed] [Google Scholar]
- 8.Ruffini DD, Madhi SA. The high burden of Pneumocystis carinii pneumonia in African HIV-1-infected children hospitalized for severe pneumonia. AIDS. 2002;16:105–12. doi: 10.1097/00002030-200201040-00013. [DOI] [PubMed] [Google Scholar]
- 9.Chakaya JM, et al. Pneumocystis carinii pneumonia in HIV/AIDS patients at an urban district hospital in Kenya. East Afr Med J. 2003;80:30–5. doi: 10.4314/eamj.v80i1.8663. [DOI] [PubMed] [Google Scholar]
- 10.Tansuphasawadikul S, et al. Clinical features, etiology and short term outcomes of interstitial pneumonitis in HIV/AIDS patients. Southeast Asian J Trop Med Public Health. 2005;36:1469–78. [PubMed] [Google Scholar]
- 11.Nissapatorn V, et al. Spectrum of opportunistic infections among HIV-infected patients in Malaysia. Southeast Asian J Trop Med Public Health. 2004;35(Suppl 2):26–32. [PubMed] [Google Scholar]
- 12.Udwadia ZF, et al. Pneumocystis carinii pneumonia in HIV infected patients from Mumbai. J Assoc Physicians India. 2005;53:437–40. [PubMed] [Google Scholar]
- 13.Panizo MM, et al. Pneumocystosis in Venezuelan patients: epidemiology and diagnosis (2001–2006) Rev Iberoam Micol. 2008;25:226–31. doi: 10.1016/s1130-1406(08)70054-8. [DOI] [PubMed] [Google Scholar]
- 14.Chernilo S, et al. Lung diseases among HIV infected patients admitted to the “Instituto Nacional del Torax” in Santiago, Chile. Rev Med Chil. 2005;133:517–24. doi: 10.4067/s0034-98872005000500002. [DOI] [PubMed] [Google Scholar]
- 15.Mikaelsson L, et al. Pneumocystis pneumonia--a retrospective study 1991–2001 in Gothenburg, Sweden. The Journal of infection. 2006;53:260–5. doi: 10.1016/j.jinf.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Maini R, et al. Increasing Pneumocystis pneumonia, England, UK, 2000–2010. Emerg Infect Dis. 2013;19:386–92. doi: 10.3201/eid1903.121151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmona EM, Limper AH. Update on the diagnosis and treatment of Pneumocystis pneumonia. Ther Adv Respir Dis. 2011;5:41–59. doi: 10.1177/1753465810380102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Catherinot E, et al. Pneumocystis jirovecii Pneumonia. Infect Dis Clin North Am. 2010;24:107–38. doi: 10.1016/j.idc.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 19.Goto N, Oka S. Pneumocystis jirovecii pneumonia in kidney transplantation. Transpl Infect Dis. 2011;13:551–8. doi: 10.1111/j.1399-3062.2011.00691.x. [DOI] [PubMed] [Google Scholar]
- 20.Mussini C, et al. Patients presenting with AIDS in the HAART era: a collaborative cohort analysis. AIDS. 2008;22:2461–9. doi: 10.1097/QAD.0b013e328314b5f1. [DOI] [PubMed] [Google Scholar]
- 21.Neff RT, et al. Analysis of USRDS: incidence and risk factors for Pneumocystis jiroveci pneumonia. Transplantation. 2009;88:135–41. doi: 10.1097/TP.0b013e3181aad256. [DOI] [PubMed] [Google Scholar]
- 22.Sepkowitz KA. Opportunistic infections in patients with and patients without Acquired Immunodeficiency Syndrome. Clin Infect Dis. 2002;34:1098–107. doi: 10.1086/339548. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe RA, et al. Trends in organ donation and transplantation in the United States, 1999–2008. Am J Transplant. 2010;10:961–72. doi: 10.1111/j.1600-6143.2010.03021.x. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez M, Fishman JA. Prevention of infection due to Pneumocystis spp. in human immunodeficiency virus-negative immunocompromised patients. Clin Microbiol Rev. 2004;17:770–82. doi: 10.1128/CMR.17.4.770-782.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Louie GH, et al. Trends in hospitalizations for Pneumocystis jiroveci pneumonia among patients with rheumatoid arthritis in the US: 1996–2007. Arthritis Rheum. 2010;62:3826–7. doi: 10.1002/art.27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol. 2010;37:686–8. doi: 10.3899/jrheum.091426. [DOI] [PubMed] [Google Scholar]
- 27.Falagas ME, et al. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol. 2007;26:663–70. doi: 10.1007/s10067-006-0441-9. [DOI] [PubMed] [Google Scholar]
- 28.Poppers DM, Scherl EJ. Prophylaxis against Pneumocystis pneumonia in patients with inflammatory bowel disease: toward a standard of care. Inflamm Bowel Dis. 2008;14:106–13. doi: 10.1002/ibd.20261. [DOI] [PubMed] [Google Scholar]
- 29.De Castro N, et al. Occurrence of Pneumocystis jiroveci pneumonia after allogeneic stem cell transplantation: a 6-year retrospective study. Bone Marrow Transplant. 2005;36:879–83. doi: 10.1038/sj.bmt.1705149. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Garrido I, et al. Pneumocystis pneumonia in patients treated with rituximab. Chest. 2013;144:258–65. doi: 10.1378/chest.12-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monnet X, et al. Critical care management and outcome of severe Pneumocystis pneumonia in patients with and without HIV infection. Crit Care. 2008;12:R28. doi: 10.1186/cc6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mori S, Sugimoto M. Pneumocystis jirovecii infection: an emerging threat to patients with rheumatoid arthritis. Rheumatology (Oxford) 2012;51:2120–30. doi: 10.1093/rheumatology/kes244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas CF, Jr, Limper AH. Pneumocystis pneumonia. The New England journal of medicine. 2004;350:2487–98. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- 34.Kanne JP, et al. Pneumocystis jiroveci pneumonia: high-resolution CT findings in patients with and without HIV infection. AJR Am J Roentgenol. 2012;198:W555–61. doi: 10.2214/AJR.11.7329. [DOI] [PubMed] [Google Scholar]
- 35.Wakefield AE, et al. Detection of Pneumocystis carinii with DNA amplification. Lancet. 1990;336:451–3. doi: 10.1016/0140-6736(90)92008-6. [DOI] [PubMed] [Google Scholar]
- 36.Reid AB, et al. Pneumocystis jirovecii pneumonia in non-HIV-infected patients: new risks and diagnostic tools. Curr Opin Infect Dis. 2011;24:534–44. doi: 10.1097/QCO.0b013e32834cac17. [DOI] [PubMed] [Google Scholar]
- 37.Flori P, et al. Comparison between real-time PCR, conventional PCR and different staining techniques for diagnosing Pneumocystis jiroveci pneumonia from bronchoalveolar lavage specimens. J Med Microbiol. 2004;53:603–7. doi: 10.1099/jmm.0.45528-0. [DOI] [PubMed] [Google Scholar]
- 38.Desmet S, et al. Serum (1–3)-beta-D-glucan as a tool for diagnosis of Pneumocystis jirovecii pneumonia in patients with human immunodeficiency virus infection or hematological malignancy. J Clin Microbiol. 2009;47:3871–4. doi: 10.1128/JCM.01756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura H, et al. Clinical utility of serum beta-D-glucan and KL-6 levels in Pneumocystis jirovecii pneumonia. Intern Med. 2009;48:195–202. doi: 10.2169/internalmedicine.48.1680. [DOI] [PubMed] [Google Scholar]
- 40.Aliouat-Denis CM, et al. The Pneumocystis life cycle. Mem Inst Oswaldo Cruz. 2009;104:419–26. doi: 10.1590/s0074-02762009000300004. [DOI] [PubMed] [Google Scholar]
- 41.Cushion MT, et al. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. PLoS One. 2010;5:e8524. doi: 10.1371/journal.pone.0008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itatani CA. Ultrastructural demonstration of a pore in the cyst wall of Pneumocystis carinii. J Parasitol. 1994;80:644–8. [PubMed] [Google Scholar]
- 43.Cushion MT. Pneumocystis: unraveling the cloak of obscurity. Trends Microbiol. 2004;12:243–9. doi: 10.1016/j.tim.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Martinez A, et al. Ploidy of cell-sorted trophic and cystic forms of Pneumocystis carinii. PLoS One. 2011;6:e20935. doi: 10.1371/journal.pone.0020935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez A, et al. Growth and airborne transmission of cell-sorted life cycle stages of Pneumocystis carinii. PLoS One. 2013;8:e79958. doi: 10.1371/journal.pone.0079958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schildgen V, et al. Pneumocystis jirovecii Can Be Productively Cultured in Differentiated CuFi-8 Airway Cells. MBio. 2014:5. doi: 10.1128/mBio.01186-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edman JC, et al. Ribosomal RNA sequence shows Pneumocystis carinii to be a member of the fungi. Nature. 1988;334:519–22. doi: 10.1038/334519a0. [DOI] [PubMed] [Google Scholar]
- 48.Stringer SL, et al. Sequence from ribosomal RNA of Pneumocystis carinii compared to those of four fungi suggests an ascomycetous affinity. J Protozool. 1989;36:14S–6S. doi: 10.1111/j.1550-7408.1989.tb02670.x. [DOI] [PubMed] [Google Scholar]
- 49.Aliouat-Denis CM, et al. Pneumocystis species, co-evolution and pathogenic power. Infect Genet Evol. 2008;8:708–26. doi: 10.1016/j.meegid.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 50.Frenkel JK. Pneumocystis jiroveci n. sp. from man: morphology, physiology, and immunology in relation to pathology. Natl Cancer Inst Monogr. 1976;43:13–30. [PubMed] [Google Scholar]
- 51.Keely SP, et al. Evolution and speciation of Pneumocystis. J Eukaryot Microbiol. 2003;50(Suppl):624–6. doi: 10.1111/j.1550-7408.2003.tb00655.x. [DOI] [PubMed] [Google Scholar]
- 52.Cisse OH, et al. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. MBio. 2012;4:e00428–12. doi: 10.1128/mBio.00428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kluge RM, et al. Combination of pentamidine and trimethoprim-sulfamethoxazole in therapy of Pneumocystis carinii pneumonia in rats. Antimicrob Agents Chemother. 1978;13:975–8. doi: 10.1128/aac.13.6.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandler FW, et al. Pneumocystis pneumonia. Animal model: pneumocystis cartinii pneumonia in the immunosuppressed rat. Am J Pathol. 1979;95:571–4. [PMC free article] [PubMed] [Google Scholar]
- 55.Hughes WT, et al. Pneumocystis carinii pneumonitis in children with malignancies. J Pediatr. 1973;82:404–15. doi: 10.1016/s0022-3476(73)80113-1. [DOI] [PubMed] [Google Scholar]
- 56.Phair J, et al. The risk of Pneumocystis carinii pneumonia among men infected with human immunodeficiency virus type 1. Multicenter AIDS Cohort Study Group. N Engl J Med. 1990;322:161–5. doi: 10.1056/NEJM199001183220304. [DOI] [PubMed] [Google Scholar]
- 57.Shellito J, et al. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J Clin Invest. 1990;85:1686–93. doi: 10.1172/JCI114621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rudner XL, et al. Interleukin-23 (IL-23)-IL-17 cytokine axis in murine Pneumocystis carinii infection. Infect Immun. 2007;75:3055–61. doi: 10.1128/IAI.01329-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mogensen TH. STAT3 and the Hyper-IgE syndrome: Clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAKSTAT. 2013;2:e23435. doi: 10.4161/jkst.23435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 62.Kotlarz D, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. 2013;210:433–43. doi: 10.1084/jem.20111229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lund FE, et al. B cells are required for generation of protective effector and memory CD4 cells in response to Pneumocystis lung infection. J Immunol. 2006;176:6147–54. doi: 10.4049/jimmunol.176.10.6147. [DOI] [PubMed] [Google Scholar]
- 64.Al-Saud BK, et al. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J Clin Immunol. 2013;33:1325–35. doi: 10.1007/s10875-013-9951-9. [DOI] [PubMed] [Google Scholar]
- 65.Tsai HY, et al. X-linked hyper-IgM syndrome with CD40LG mutation: Two case reports and literature review in Taiwanese patients. J Microbiol Immunol Infect. 2012 doi: 10.1016/j.jmii.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 66.Costa-Carvalho BT, et al. Pulmonary complications in patients with antibody deficiency. Allergol Immunopathol (Madr) 2011;39:128–32. doi: 10.1016/j.aller.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Zheng M, et al. CD4+ T cell-independent vaccination against Pneumocystis carinii in mice. J Clin Invest. 2001;108:1469–74. doi: 10.1172/JCI13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gigliotti F, et al. Passive intranasal monoclonal antibody prophylaxis against murine Pneumocystis carinii pneumonia. Infect Immun. 2002;70:1069–74. doi: 10.1128/IAI.70.3.1069-1074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Limper AH, et al. The role of alveolar macrophages in Pneumocystis carinii degradation and clearance from the lung. J Clin Invest. 1997;99:2110–7. doi: 10.1172/JCI119384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lasbury ME, et al. Polyamine-mediated apoptosis of alveolar macrophages during Pneumocystis pneumonia. J Biol Chem. 2007;282:11009–20. doi: 10.1074/jbc.M611686200. [DOI] [PubMed] [Google Scholar]
- 71.Steele C, et al. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J Exp Med. 2003;198:1677–88. doi: 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wells J, et al. Complement and Fc function are required for optimal antibody prophylaxis against Pneumocystis carinii pneumonia. Infect Immun. 2006;74:390–3. doi: 10.1128/IAI.74.1.390-393.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McAllister F, et al. T cytotoxic-1 CD8+ T cells are effector cells against pneumocystis in mice. J Immunol. 2004;172:1132–8. doi: 10.4049/jimmunol.172.2.1132. [DOI] [PubMed] [Google Scholar]
- 74.Mori S, Levin P. A brief review of potential mechanisms of immune reconstitution inflammatory syndrome in HIV following antiretroviral therapy. Int J STD AIDS. 2009;20:447–52. doi: 10.1258/ijsa.2009.008521. [DOI] [PubMed] [Google Scholar]
- 75.Martin-Blondel G, et al. Pathogenesis of the immune reconstitution inflammatory syndrome in HIV-infected patients. Curr Opin Infect Dis. 2012;25:312–20. doi: 10.1097/QCO.0b013e328352b664. [DOI] [PubMed] [Google Scholar]
- 76.Muller M, et al. Immune reconstitution inflammatory syndrome in patients starting antiretroviral therapy for HIV infection: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:251–61. doi: 10.1016/S1473-3099(10)70026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Achenbach CJ, et al. Paradoxical immune reconstitution inflammatory syndrome in HIV-infected patients treated with combination antiretroviral therapy after AIDS-defining opportunistic infection. Clin Infect Dis. 2012;54:424–33. doi: 10.1093/cid/cir802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Novak RM, et al. Immune reconstitution inflammatory syndrome: incidence and implications for mortality. AIDS. 2012;26:721–30. doi: 10.1097/QAD.0b013e3283511e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grant PM, et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416. doi: 10.1371/journal.pone.0011416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jagannathan P, et al. Life-threatening immune reconstitution inflammatory syndrome after Pneumocystis pneumonia: a cautionary case series. AIDS. 2009;23:1794–6. doi: 10.1097/QAD.0b013e32832d9b20. [DOI] [PubMed] [Google Scholar]
- 81.Zolopa A, et al. Early antiretroviral therapy reduces AIDS progression/death in individuals with acute opportunistic infections: a multicenter randomized strategy trial. PLoS One. 2009;4:e5575. doi: 10.1371/journal.pone.0005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wright TW, et al. Analysis of cytokine mRNA profiles in the lungs of Pneumocystis carinii-infected mice. Am J Respir Cell Mol Biol. 1997;17:491–500. doi: 10.1165/ajrcmb.17.4.2851. [DOI] [PubMed] [Google Scholar]
- 83.Wright TW, et al. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J Clin Invest. 1999;104:1307–17. doi: 10.1172/JCI6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bhagwat SP, et al. Anti-CD3 antibody decreases inflammation and improves outcome in a murine model of Pneumocystis pneumonia. J Immunol. 2010;184:497–502. doi: 10.4049/jimmunol.0901864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gigliotti F, et al. Sensitized CD8+ T cells fail to control organism burden but accelerate the onset of lung injury during Pneumocystis carinii pneumonia. Infect Immun. 2006;74:6310–6. doi: 10.1128/IAI.00668-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swain SD, et al. CD8 T cells modulate CD4 T-cell and eosinophil-mediated pulmonary pathology in pneumocystis pneumonia in B-cell-deficient mice. Am J Pathol. 2006;168:466–75. doi: 10.2353/ajpath.2006.050724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hori S, et al. CD25+CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur J Immunol. 2002;32:1282–91. doi: 10.1002/1521-4141(200205)32:5<1282::AID-IMMU1282>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 88.Ruan S, et al. Local delivery of the viral interleukin-10 gene suppresses tissue inflammation in murine Pneumocystis carinii infection. Infect Immun. 2002;70:6107–13. doi: 10.1128/IAI.70.11.6107-6113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKinley L, et al. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol. 2006;177:6215–26. doi: 10.4049/jimmunol.177.9.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wright TW, et al. Pulmonary inflammation disrupts surfactant function during Pneumocystis carinii pneumonia. Infect Immun. 2001;69:758–64. doi: 10.1128/IAI.69.2.758-764.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Atochina-Vasserman EN, et al. Immune reconstitution during Pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J Immunol. 2009;182:2277–87. doi: 10.4049/jimmunol.0802775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Linke MJ, et al. Characterization of a distinct host response profile to Pneumocystis murina asci during clearance of pneumocystis pneumonia. Infect Immun. 2013;81:984–95. doi: 10.1128/IAI.01181-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ricks D, et al. Dectin immunoadhesins and Pneumocystis pneumonia. Infect Immun. 2013 doi: 10.1128/IAI.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bellamy RJ. HIV: treating Pneumocystis pneumonia (PCP) Clin Evid (Online) 2008;2008 [PMC free article] [PubMed] [Google Scholar]
- 95.Helweg-Larsen J, et al. Clinical efficacy of first- and second-line treatments for HIV-associated Pneumocystis jirovecii pneumonia: a tri-centre cohort study. J Antimicrob Chemother. 2009;64:1282–90. doi: 10.1093/jac/dkp372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jick H. Adverse reactions to trimethoprim-sulfamethoxazole in hospitalized patients. Rev Infect Dis. 1982;4:426–8. doi: 10.1093/clinids/4.2.426. [DOI] [PubMed] [Google Scholar]
- 97.Phillips E, Mallal S. Drug hypersensitivity in HIV. Curr Opin Allergy Clin Immunol. 2007;7:324–30. doi: 10.1097/ACI.0b013e32825ea68a. [DOI] [PubMed] [Google Scholar]
- 98.Hirsch HH, et al. Immune reconstitution in HIV-infected patients. Clin Infect Dis. 2004;38:1159–66. doi: 10.1086/383034. [DOI] [PubMed] [Google Scholar]
- 99.Barry SM, et al. Immune reconstitution pneumonitis following Pneumocystis carinii pneumonia in HIV-infected subjects. HIV Med. 2002;3:207–11. doi: 10.1046/j.1468-1293.2002.00115.x. [DOI] [PubMed] [Google Scholar]
- 100.Koval CE, et al. Immune reconstitution syndrome after successful treatment of Pneumocystis carinii pneumonia in a man with human immunodeficiency virus type 1 infection. Clin Infect Dis. 2002;35:491–3. doi: 10.1086/341974. [DOI] [PubMed] [Google Scholar]