

Abstract
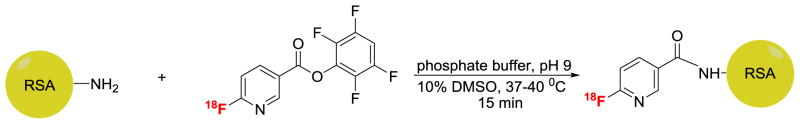
We sought to develop a practical, reproducible and clinically translatable method of radiolabeling serum albumins with fluorine-18 for use as a PET blood pool imaging agent in animals and man. Fluorine-18 radiolabeled fluoronicotinic acid-2,3,5,6-tetrafluorophenyl ester, [18F]F-Py-TFP was prepared first by the reaction of its quaternary ammonium triflate precursor with [18F]tetrabutylammonium fluoride ([18F]TBAF) according to a previously published method for peptides, with minor modifications. The incubation of [18F]F-Py-TFP with rat serum albumin (RSA) in phosphate buffer (pH 9) for 15 min at 37–40 °C produced fluorine-18-radiolabeled RSA and the product was purified using a mini-PD MiniTrap G-25 column. The overall radiochemical yield of the reaction was 18–35% (n = 30, uncorrected) in a 90-min synthesis. This procedure, repeated with human serum albumin (HSA), yielded similar results.
Fluorine-18-radiolabeled RSA demonstrated prolonged blood retention (biological half-life of 4.8 hours) in healthy awake rats. The distribution of major organ radioactivity remained relatively unchanged during the 4 hour observation periods either by direct tissue counting or by dynamic PET whole-body imaging except for a gradual accumulation of labeled metabolic products in the bladder.
This manual method for synthesizing radiolabeled serum albumins uses fluorine-18, a widely available PET radionuclide, and natural protein available in both pure and recombinant forms which could be scaled up for widespread clinical applications. These preclinical biodistribution and PET imaging results indicate that [18F]RSA is an effective blood pool imaging agent in rats and might, as [18F]HSA, prove similarly useful as a clinical imaging agent.
1. Introduction
The regional distribution of blood in the vascular space of both humans and animals is a critical physiological variable and, as a result, a variety of non-invasive imaging methods have been devised for assessing regional blood volume [1–5]. However, while a variety of PET blood pool labels have been developed [6–10], no PET blood volume imaging agent is today in general widespread use for routine measurement and visualization of regional blood volume in either clinical or animal studies. Accordingly, the goal of the present work was to identify and synthesize a blood pool-imaging agent with potential for commercialization and wide-spread use in both clinical and animal PET imaging applications. In order that a radiolabeled agent achieves these goals, it must exhibit prolonged in vivo stability and imaging and dosimetry properties appropriate to both animals and man. At the same time, synthesis of this agent must be highly reproducible and easily adapted to automated manufacturing methods. Driven directly by these requirements we chose fluorine-18 as the isotopic label to meet the imaging and dosimetry conditions and native albumin as the labeled compound in order to meet the in vivo stability, compatibility, reproducibility and manufacturing requirements. We report here the synthesis of fluorine-18 albumins (human serum albumin, HSA, and rat serum albumin, RSA) and the biological evaluation of [18F]RSA in healthy rats.
Labeling with fluorine-18 can be accomplished directly or indirectly. In direct labeling, a suitable precursor is reacted with activated [18F]fluoride, usually at elevated temperature in the presence of base. Direct fluorine-18 labeling of protein is generally not feasible because of the harsh conditions needed for radiofluorination reactions. Therefore, fluorine-18 labeling of proteins has been performed usually as a multi-step process by first labeling a prosthetic group and then conjugating that to the protein, a process that can take several hours. Serum albumin has been labeled with various prosthetic groups such as 18F-2,3,5,6-tetrafluorophenyl pentafluorobenzoate [11], 4-nitrophenyl-2-18F-fluoropropionate [12], methyl 3-18F-fluoro-5-nitrobenzimidate [13], 4-18F-fluorophenacylbromide [13], 18F-APF [12], 18F-pentafluorobenzaldehyde, 18F-FBA [14], and N-succinimidyl 4-[18F]fluorobenzoate (18F-SFB) [12]. However, while each of these synthesis methods has certain advantages, other complications have limited clinical application and there continues to be a need for rapid, clean and mild synthesis techniques for the radiolabeling of proteins.
In recent years click chemistry has been widely applied in many fields of chemical sciences, including radiopharmaceutical chemistry [15–20]. The copper(I)-catalyzed cycloaddition of azides with terminal alkynes to form 1,2,3-triazole has proved to be particularly valuable for efficient 18F labeling of biomolecules. Ramenda et al. has reported 18F-labelled sulfonamide-based click chemistry building blocks to radiolabel HAS [21], A non-conventional Si-18F bonded prosthetic group has also been used to radiolabel serum albumin [22], [23]. In this approach a prosthetic group for non-site-specific protein labeling was designed and synthesized in one step by simple 19F–18F isotopic exchange. However, all these methods suffer from drawbacks, including difficult radiosyntheses and purifications, the generation of non-natural HSA-adducts, handling and stability issues and product contamination questions. In choosing our approach we sought after a method that could ultimately be performed simply within a radiopharmacy setting and with minimal regulatory concerns regarding injectable composition. Recently, Olberg et al. reported an one-step straightforward synthesis of [18F]fluoronicotinate ([18F]F-Py-TFP) for radiolabeling peptides [24]. Herein, we have adopted this approach and report the radiosyntheisis of fluorine-18 labeled rat ([18F]RSA) and human ([18F]HSA) serum albumins via the conjugation of [18F]F-py-TFP with amine groups of the protein. Further, we evaluated in vivo the biodistribution of [18F]RSA in healthy rats by direct tissue counting and PET imaging.
2. Material and Methods
Tetrabutylammonium hydrogen carbonate (0.075 M) used for radiochemical work was purchased from ABX (Radeberg, Germany). Whole human serum was obtained from MP Biomedicals, LLC (Solon, OH, USA). All other chemicals and solvents were received from Sigma Aldrich (St. Louis, MO, USA) and used without further purification. The precursor N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate was prepared according to a previously described method [24]. The precursor was further purified by crystallization from a solution of acetonitrile and diethylether at 4 °C. Fluorine-18 was purchased from National Institutes of Health cyclotron facility (Bethesda, MD, USA). PBS 1X buffer (Gibco) was obtained from Life Technologies (Carlsbad, CA, USA). PD10 MiniTrap™ columns were obtained from GE Healthcare Bioscience (Pittsburg, PA, USA). Other columns and all the Sep-Pak cartridges used in this synthesis were obtained from Agilent Technologies (Santa Clara, CA, USA) and Waters (Milford, MA, USA), respectively. Mass spectrometry (MS) was performed on a 6130 Quadrupole LC/MS Agilent Technologies instrument equipped with a diode array detector. 1H, 13C and 19F NMR spectra were recorded on a Varian spectrometer (400 MHz). Chemical shifts (ppm) were reported relative to the solvent residual peaks of chloroform (δ 1H, 7.26 ppm; 13C 77.36). 19F NMR spectra were reported with reference to the trifluoroacetic acid (δ 19F, −76.55 ppm). High-resolution mass spectra (HRMS) were collected on an Agilent Time-Of-Flight Mass Spectrometer (TOF, Agilent Technologies) following HPLC purification. A 3-min gradient from 4 to 100% Acetonitrile (0.1% formic acid) in water (0.1% formic acid) was used with a 4-min run time at a flow rate of 1 mL/min. A Zorbax SB-C18 column (2.1 × 30 mm, 3.5 μm) was used at a temperature of 50°C. Molecular mass was confirmed using APCI in the positive mode with the Agilent Masshunter software (version B.02). Radiosyntheses were performed manually. Purification of the radiolabeled product was done by HPLC on a Beckman Coulter System Gold instrument equipped with a multi-wavelength detector using an Agilent Eclipse C18 column (9.4 × 250 mm, 5 μm). Analytical HPLC analyses for radiochemical work were performed on an Agilent 1200 Series instrument equipped with multi-wavelength detectors using an Agilent Eclipse XDB C18 column (4.6 × 150 mm, 5 μm) with a flow rate of 1.0 mL/min. Oasis MCX Plus cartridge was conditioned by passing 5 mL ethanol, 10 mL air and 10 mL water.
NCI-Frederick is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council; 1996; National Academy Press; Washington, D.C.).
2.1. Chemistry
2.1.1. Synthesis of N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate
The compound N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate was prepared according to a previously published method [24] and further purified by recrystallization.
2.1.2. Synthesis of 2,3,5,6-tetrafluorophenyl 6-fluoronicotinate
To the mixture of 6-fluoronicotinic acid (250 mg, 1.77 mmol) and tetrafluorophenol (294.5 mg, 177 mmol) in dioxane (10 mL) was added N, N –dicyclohexylcarbodiimide (350 mg, 1.70 mmol) and the reaction was stirred for 18 h. The mixture was filtered and dioxane from the filtrate was removed under reduced pressure. The crude product was purified by crystallization from hexane (258 mg, 0.90 mmol, 51%).
1H NMR (CDCl3, 400 MHz): δ 9.08 (d, J = 2.7 Hz, 1H), 8.56 (ddd, J = 2.3, 7.4, 8.6 Hz, 1H), 7.14-7.06 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ 167.8, 165.4, 160.3, 151.6, 147.3, 144.9, 143.4, 121.6, 110.1, 103.7. 19F NMR (CDCl3, 100 MHz): δ −58.04 (dd, J = 2.73, 6.65 Hz), −138.47 (m), 152.59 (m). MS (APCI, m/z) calculated for C13H5F5O2, 288.02, found 289.1 (M+H)+.
2.1.3. Synthesis of [18F]albumin
2.1.3.1. Preparation of [18F]2,3,5,6-tetrafluorophenyl 6-fluoronicotinate
Fluorine-18 labeled 2,3,5,6-tetrafluorophenyl 6-fluoronicotinate was prepared according to a previously published method [24] with a minor modifications. Briefly, to the azeotropically dried [18F]TBAF (10–100 mCi) was added N,N,N-trimethyl-5-((2,3,5,6-tetrafluorophenoxy)carbonyl)pyridin-2-aminium trifluoromethanesulfonate (5–10 mg) in 1 mL 2:8 acetonitrile, tert-butanol and stirred for 10 min at 40 °C. To the solution was added 5 mL of water and passed through a preconditioned Oasis MCX Plus cartridge and the cartridge washed with 5 mL of water. The trapped [18F]2,3,5,6-tetrafluorophenyl 6-fluoronicotinate was eluted from the Sep-Pak cartridge into a V-shaped microwave vial with 5 mL diethyl ether and the solvent was evaporated under nitrogen. Purity of the compound was checked by semi-preparative HPLC {Agilent Eclipse C18 5 μm, 9.4 x 250 mm column, eluent: 60% acetonitrile, 40% water (with 0.1% TFA), flow rate = 4 mL/min} with the product fraction eluted at 10.6 min (>97% radiochemically pure).
2.1.3.2. Conjugation with [18F]albumin
Rat serum albumin (RSA, 40 mg) was dissolved in 350 μL of 0.2 M phosphate buffer (pH 10). The pH of the solution was adjusted to 9 adding 50 μL of 0.5 M phosphate buffer (pH 10). For each conjugation 200 μL of the RSA solution was used. To the solution of [18F]2,3,5,6-tetrafluorophenyl 6-fluoronicotinate (~250 μL water + 50 μL dimethylsulfoxide) was added a solution of RSA (20 mg in 200 μL of phosphate buffer of pH 9) and incubated at 40 °C. The product was purified by PD10 Minitrap size exclusion column using phosphate buffer (pH 7.4) as an eluent. In PD10 Minitrap PD10 size exclusion column, sample loading volume is 0.5 mL and collection volume is 1 mL. Therefore, the final concentration of albumin supplied for the animal study was 20 mg/mL.
2.1.4. Stability test
To test the serum stability, 1.4 mCi of [18F]albumin in 300 μL of PBS (1X, pH 7.4) was added to human serum (2 mL), kept at room temperature or 37 °C and monitored by size exclusion HPLC up to 4 h.
2.2. Rat Biodistribution Studies
Awake or anesthetized (isoflurane/O2, 1.5% to 3% v/v) rats (Fisher 344/NCr) were injected IV (50 to 100 μCi; 1.85 to 3.7 MBq per rat; 0.05 to 0.11 mg; volume: 0.1 mL) and euthanized at 0.5, 1, 2 or 4 h post injection; blood and tissues were excised, weighed and the radioactivity content determined by gamma counting (Perkin Elmer 2480 Wizard 3). Blood and tissue radioactivity content was expressed as differential uptake ratio (DUR): [(%Injected Dose/tissue (g)) x (body weight (g))]/100. These DUR values were taken to represent the “true” quantitative biodistribution of [18F]RSA in these animals over time Statistical analysis was performed using a Welch’s t test (GraphPad InStat version 3.00 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com).
2.3. PET Imaging Studies
Rats were anesthetized using isoflurane/O2 (1.5% to 3% v/v) and imaged (BioPET/CT, Bioscan, Washington, D.C.) either by continuous, repetitive whole-body scanning or at various discrete times after IV injection (tail vein) of 18F- RSA (0.5 to 1.6 mCi; 18.5 to 59 MBq per rat; 0.55 to 1.76 mg; volume = 0.1 to 0.4 mL). Whole body images were obtained in four bed positions at 10 minutes per position for a total imaging time of 40 min per whole-body scan and reconstructed (4 iterations, 40 subsets) with a three-dimensional ordered-subset expectation maximum (3D-OSEM) algorithm. PET imaging in these animals was carried out primarily for qualitative purposes: to ascertain the degree of detail in the rat vasculature that could be visualized with 18F- RSA using a commercially available small animal PET scanner; the extent to which the cardiac chambers could be seen as separate structures; to assess the amount of activity in structures not easily sampled by direct tissue counting methods, e.g. the bladder; and to compare qualitatively with the biodistribution measurements.
3. Results
3.1. Chemistry and Radiochemistry
The radiolabeling precursor was prepared according to the method described in the literature but unlike the literature [24] method, a non-radioactive cold standard (F-Py-TFP) was prepared from commercially available 2,3,5,6-tetrafluorophenol. For the cold standard, commercially available 6-fluoronicotinic acid and 2,3,5,6-tetrafluorophenol and DCC in dioxane were stirred at room temperature for 18 h. After removal of the solvent the product was crystallized from hexanes. Fluorine-18 labeling was performed according to the above-referenced literature with minor modifications. Conjugation of [18F]F-Py-TFP with peptide has been reported in phosphate buffer/DMSO/acetonitrile (pH 9) by directly eluting it from the Oasis MCX Plus cartridge. However, in our current manual synthesis arrangement we were limited by the total amount of activity due to radiation exposure concerns. To reach high radioactive concentrations of the labeled albumins we needed to reduce the total volume of the conjugation reaction mixture, therefore the [18F]F-Py-TFP was eluted from the Sep-Pak cartridge with diethyl ether and the solvent was evaporated under nitrogen. The total radiochemical yield of [18F]F-Py-TFP was 50 ± 10% (uncorrected, n = 18) starting from the fluorination of the precursor with very high radiochemical (> 98%) purity (supporting information, Figure S2A). The identity of the [18F]F-Py-TFP was confirmed by comparing the HPLC retention time with co-injected, authentic nonradioactive standard (supporting information, Figure S2B).
Since one of the goals of this study was to develop a facile methodology for preparing [18F]HSA for clinical blood pool imaging, test conjugations were performed with a view to optimizing the conjugation yields by examining the effects of pH. In these studies reaction mixtures containing HSA (200 μL of 100 mg/mL)) in phosphate buffer/10% DMSO (total reaction volume was 500 μL) at 40 °C for 30 min with pHs of 8.0, 8.6 and 9.0 had conjugation yields of 89, 94 and 97% respectively, determined by size exclusion HPLC (Figure 1).
Figure 1.
Fluorine-18 labeling of albumin.
The highest conjugation yield [at pH 9.0] with 200 μL of 100 mg/mL solution (total reaction volume 500 μL) was then checked at different time intervals (10, 15, 30 min), which showed that the conjugation yield reached its highest level (97%) within 15 min. Conjugation reactions were then tested at different concentrations of both HSA and RSA with the finding that radiochemical yields were highly dependent on protein concentration, similar to the observations of others [13], [24]. Further, HSA yields appeared slightly higher than RSA yields at the higher albumin concentrations. However, since the biological system under investigation is non-saturable [25], the specific activity of fluorine-18 labeled albumin is not expected to impact in vivo targeting and studies were continued with fluorine-18 radiolabeled RSA prepared from 200 μL of 100 mg/mL RSA solution.
Fluorine-18 labeled RSA was purified by PD10 Minitrap size exclusion column using phosphate buffer of pH 7.4 as an eluent. The overall radiochemical yield was 18–35% (n = 30, uncorrected) in a 90-min synthesis time with a radiochemical purity ≥99% (Figure 2). The compound was stable up to 4 h of post-synthesis in human serum at 37 °C (Figure 3), and no other peaks were generated over time.
Figure 2.

HPLC profile of pure [18F]albumin. HPLC conditions: Agilent Eclipse GF 250, 5 μm, 4.6 x 250 mm column, PBS buffer (pH 7.4), flow rate: 1 ml/min; solid line, in-line radiodetector; dotted line, UV detector at 254 nm.
Figure 3.

Stability study of [18F]RSA at 37 °C; a) 0 min; b) 1h; c) 2h; d) 4h. HPLC conditions: Agilent Zorbax GF-250 4.6 x 250 mm, PBS (1X, pH 7.4), flow: 0.3 ml/min; red line, in-line radiodetector; blue line, UV detector at 254 nm.
3.2. Rat Biodistribution Studies
The highest retention of [18F]RSA (Figure 4A) occurred in the blood at all times with DURs of 11.5, 10.0, 8.5, and 6.8 at 0.5 h, 1h, 2 h and 4 h, respectively from which the blood biological half life (Tb) was calculated to be 4.8 h (Figure 4B). The next highest [18F]RSA retentions were observed in the lungs (DURs: 5.1 to 3.0) and heart (DURs: 3.4 to 2.4), which were significantly (P < 0.001) decreased 2 to 3 fold compared to blood DURs at the same times (Figure 4A). Similarly significant decreases (P < 0.001) in radioactive content ranging from 3.5 to 6.5 fold were observed in the DURs of the kidney (2.5 to 2.0), liver (2.4 to 1.5) and spleen (1.9 to 1.1) compared to the blood DURs at the same times (Figure 4A–B). The rank order of these DURs for lung, heart, kidney, liver and spleen corresponded to the rank order of published findings for blood volumes determined in these rat tissues [26]. Femur DURs (1.5 to 1.1) over the time course exhibited little change indicating minimal de-fluorination (Figure 4A–B).
Figure 4.


A) Biodistribution of [18F]RSA in rats from 0.5 to 4 h. Each bar represents the mean DUR ± SD of [18F]RSA (n =5); B) Blood and tissue radioactive content (DUR) at 0.5, 1, 2, 3 and 4 h (each time point represents the mean DUR ± SD; n =5); the blood data points were fitted with a one component exponential equation: y = 11.81e-0.00237x; R2 = 0.967, blood t1/2= 4.87 h; C) Tissue (DUR) to blood ratios (T:B) of [18F]RSA from 0.5 to 4 h. Bars represent mean T:B ± SD (n= 5).
Tissue (DUR) to blood (DUR) (T:B) ratios did not exceed 1 and were significantly decreased (P < 0.001) 2 to 9 fold at all times compared to the blood with the lung and heart T:B ratios the highest (0.30 to 0.44) and the muscle the lowest (0.02 to 0.05) (Figure 4C.). The T:B ratios of the lungs, heart, liver and spleen remained relatively constant over the entire time course with no significant differences (P> 0.05) observed whereas kidney, femur and muscle T:B ratios were significantly increased 37%, 42% and 180%, respectively from 0.5 h to 4 h (Table 1). These findings suggest that [18F]RSA has suitable in vivo blood retention and stability for tissue blood volume determinations in the lungs, heart, liver and spleen but may be problematic for the kidney, femur and muscle where radioactivity content increases slowly with time. Early imaging (prior to 1 h post-injection), however, might permit blood volume determinations in these tissues (Figure 4C).
Table 1.
Comparison of changes in tissue (DURs): Blood (DURs) Ratios to determine the effects of time on retention of [18F]RSA in rat organs and tissues
| % Change in Tissue: Blood Ratios over time ((+) =% increase in ratio; (−) =% decrease in ratio) | |||
|---|---|---|---|
| Awake Rats after 0.5h uptake vs 1h, 2 h or 4h [% Change from 0.5 h to 1, 2 or 4 h)] | |||
| 0.5 to 1 h | 0.5 to 2 h | 0.5 to 4 h | |
| Heart | 0.35 | 9.8 | 21 |
| Lungs | −5.2 | −9.9 | −4.3 |
| Liver | 5.6 | 1.4 | 2.7 |
| Spleen | −0.12 | −3.8 | −3.2 |
| Kidney | 16 | 27 | 37* |
| Muscle | 86 | 74 | 180* |
| Femur | 8.5 | 40* | 42* |
P< 0.05 (n=5 per group, Welch’s T test), represents a significant increase in T:B ratio from 0.5 h.
3.3. PET Imaging Studies
A representative rat whole-body dynamic PET imaging study is shown in Figure 5. The peripheral vasculature and the ventricles of the heart are well visualized and are prominent throughout the imaging period.
Figure 5.

Consecutive whole-body posterior view maximum intensity projection images of a rat (150 g) following injection of 1.6 mCi of [18F]RSA. Data were acquired for 10 minutes at each of 4 bed positions in each whole body scan. The start time of each 40-min whole body scan relative to the time of injection is shown below each scan. LV: left ventricle; RV: right ventricle; LU: lungs; L: liver; K: kidneys; S: spleen; B: bladder.
The lungs, spleen and liver can also be discerned in these images but at lower radioactivity concentrations compared to the central vasculature. Tissue concentrations appeared relatively stable over all imaging periods and were consistent with the biodistribution results at comparable times. Note in Figure 5 that apparent bone activity does not increase significantly with time suggesting that kidney activity and small variations in bladder activity are due to labeled metabolic products, rather than free fluoride.
4. Discussion
Fluorine-18 labeled prosthetic groups are generally incorporated into proteins via conjugation with amine, carboxyl or thiol groups, with most such prosthetic groups designed for coupling with amino groups (N terminus R-NH2 or, more commonly, side-chain lysine -NH2 residues). Among the amine reactive prosthetic groups [12, 27–36], N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) [12, 31–36] is the most extensively studied and several modifications have been made to improve the synthesis procedure since it was first reported [31] by Vaidyanathan et al. in 1992. However, the preparation of [18F]SFB is a time consuming multi-step synthesis procedure that includes an HPLC purification step. In this work, we used the alternative 18F synthon method first reported by Olberg et al. [24] to prepare fluorine-18 labeled serum albumins to avoid HPLC purification, because of the better stability of the tetrafluorophenyl ester at higher pH’s [37, 38] and its better solubility characteristics compared to [18F]SFB. Also advantageously, the synthesis of synthon [18F]F-Py-TFP is a one step process. [18F]F-Py-TFP was successfully conjugated with serum albumins in 18–35% yields with protein concentration an important determinant of final yield. The method’s simplicity and reproducibility (>50 repetitions) are particularly advantageous in the context of routine production of [18F]HSA in a clinical setting.
The choices of albumin, isotopic label and, ultimately, the synthesis method needed to create the labeled product, were guided by other requirements we believe should also be met by an “ideal” PET blood pool imaging agent intended for widespread clinical and research use: first, the agent should label only the blood pool and no organs should contain the compound in excess of the amount of blood in that organ: second, the “effective” half-life of the agent (Te), a combination of the physical half life (Tp) of the isotopic label and the biological half life of the compound (Tb), should be of the order of hours where Te = (Tb) (Tp)/(Tb + Tb); third, the positron emitting isotope should possess favorable PET imaging and dosimetry properties; and fourth, the compound, isotope and manufacturing technology needed for reproducible synthesis of the agent should be widely available and able to fulfill clinical criteria. This synthesis method of radiolabeling albumins with fluorine-18 and the resulting PET radiopharmaceutical largely achieves these goals. The first and second requirements are met since the tissue counting and PET imaging studies demonstrate prolonged blood retention (effective half-life of 80 min, biological half-life of 4.8 h) with no abnormal distribution of the agent outside the blood pool except for slowly accumulating activity in the bladder. This result implies that individual organ blood volumes can be determined by imaging, since the agent is present only in the blood of that organ. Moreover, blood retention persists long enough for clinical and research imaging studies to be completed without excessive increases in imaging time. The third requirement is also met in the choice of fluorine-18 as the labeling isotope. With a half life of 109 minutes, a maximum positron energy of 0.633 MeV, positron branching ratio of 0.97 and mean positron range in tissue of 0.6 mm, fluorine-18 is near ideal for PET imaging in both humans and rodent-size animals where structures are small and in close proximity to one another. In addition, the choice of fluorine-18 reduces radiation exposure to the subject by virtue of its modest positron kinetic energy and relatively short half-life. The fourth requirement also appears to be met. As noted above, serum albumin is the major natural protein present in plasma, is readily available in pure and recombinant form for clinical (and experimental) use, possesses no known toxicological properties and does not require high specific activity radiolabeling. Fluorine-18 itself is also available commercially throughout the U.S. and is manufactured on-site at many centers. A clinical synthesis of this compound would, ideally, be carried out using a synthesis “box” adapted to the various steps described above and this methodology is well-suited to this approach. The cost of implementing this box-based synthesis method is also likely to be acceptable.
Finally, many different methods for labeling serum albumins and other compounds with fluorine-18 for PET blood pool imaging have been reported [6–10]. While any of these methods might offer some advantages in certain circumstances, these methods do not appear to meet simultaneously the criteria for pre-clinical and clinical blood pool imaging we have set forth. 68Ga blood pool agents [6, 8], for example, do not require a cyclotron (a significant advantage) but the positron range of this isotope is excessive (2.2 mm vs. 0.6 mm for fluorine-18) for small animal studies and quantitative accuracy will be compromised. Some fluorine-18 albumin preparations, while sharing the imaging advantages of fluorine-18, can have shorter synthesis times but may also exhibit tissue uptake in excess of the amount in blood [22]. Such agents violate the primary requirement of a blood pool agent and limit applications of this measurement/imaging technology. In short, while other literature methods may offer certain advantages in certain circumstances, we believe the method described here offers the prospect of general and widespread use.
5. Conclusions
Fluorine-18 labeled RSA and HSA were prepared in 18–35% yields via a conjugation reaction with [18F]F-Py-TFP. This [18F]albumin exhibits favorable in vivo stability, dosimetry, imaging and manufacturing properties that make it suitable for both clinical and research PET blood pool imaging applications.
Supplementary Material
Acknowledgments
The authors thank Christine Enders for her excellent technical assistance. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sourbron S, Ingrisch M, Siefert A, Reiser M, Herrmann K. Quantification of Cerebral Blood Flow, Cerebral Blood Volume, and Blood-Brain-Barrier Leakage with DCE-MRI. Magn Reson Med. 2009;62:205–17. doi: 10.1002/mrm.22005. [DOI] [PubMed] [Google Scholar]
- 2.Chen CG, Guerrero JL, Deprada JAV, Padial LR, Schwammenthal E, Chen MH, et al. Intracardiac Ultrasound Measurement of Volumes and Ejection Fraction in Normal, Infarcted, and Aneurysmal Left-Ventricles Using a 10-Mhz Ultrasound Catheter. Circulation. 1994;90:1481–91. doi: 10.1161/01.cir.90.3.1481. [DOI] [PubMed] [Google Scholar]
- 3.Inoue Y, Momose T, Machida K, Honda N, Mamiya T, Takahashi T, et al. Quantitation of cerebral blood volume by 99mTc-DTPA-HSA SPECT. Radiat Med. 1992;10:184–8. [PubMed] [Google Scholar]
- 4.Croteau E, Benard F, Cadorette J, Gauthier ME, Aliaga A, Bentourkia M, et al. Quantitative gated PET for the assessment of left ventricular function in small animals. J Nucl Med. 2003;44:1655–61. [PubMed] [Google Scholar]
- 5.Vanhove C, Franken PR, Defrise M, Momen A, Everaert H, Bossuyt A. Automatic determination of left ventricular ejection fraction from gated blood-pool tomography. J Nucl Med. 2001;42:401–7. [PubMed] [Google Scholar]
- 6.Todica A, Brunner S, Boning G, Lehner S, Nekolla SG, Wildgruber M, et al. [Ga-68]-Albumin-PET in the Monitoring of Left Ventricular Function in Murine Models of Ischemic and Dilated Cardiomyopathy: Comparison with Cardiac MRI. Mol Imaging Biol. 2013;15:441–9. doi: 10.1007/s11307-013-0618-y. [DOI] [PubMed] [Google Scholar]
- 7.Wangler C, Niedermoser S, Chin J, Orchowski K, Schirrmacher E, Jurkschat K, et al. Onestep (18)F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat Protoc. 2012;7:1946–55. doi: 10.1038/nprot.2012.109. [DOI] [PubMed] [Google Scholar]
- 8.Hoffend J, Mier W, Schuhmacher J, Schmidt K, Dimitrakopoulou-Strauss A, Strauss LG, et al. Gallium-68-DOTA-albumin as a PET blood-pool marker: experimental evaluation in vivo. Nucl Med Biol. 2005;32:287–92. doi: 10.1016/j.nucmedbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Saatchi K, Gelder N, Gershkovich P, Sivak O, Wasan KM, Kainthan RK, et al. Long-circulating non-toxic blood pool imaging agent based on hyperbranched polyglycerols. Int J Pharm. 2012;422:418–27. doi: 10.1016/j.ijpharm.2011.10.036. [DOI] [PubMed] [Google Scholar]
- 10.Todica A, Boning G, Lehner S, Weidl E, Cumming P, Wangler C, et al. Positron emission tomography in the assessment of left ventricular function in healthy rats: A comparison of four imaging methods. J Nucl Cardiol. 2013;20:262–74. doi: 10.1007/s12350-012-9663-1. [DOI] [PubMed] [Google Scholar]
- 11.Herman LW, Fischman AJ, Tompkins RG, Hanson RN, Byon C, Strauss HW, et al. The Use of Pentafluorophenyl Derivatives for the F-18 Labeling of Proteins. Nucl Med Biol. 1994;21:1005–10. doi: 10.1016/0969-8051(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 12.Wester HJ, Hamacher K, Stocklin G. A comparative study of NCA fluorine-18 labeling of proteins via acylation and photochemical conjugation. Nucl Med Biol. 1996;23:365–72. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]
- 13.Kilbourn MR, Dence CS, Welch MJ, Mathias CJ. F-18 Labeling of Proteins. J Nucl Med. 1987;28:462–70. [PubMed] [Google Scholar]
- 14.Chang YS, Jeong JM, Lee YS, Kim HW, Rai GB, Lee SJ, et al. Preparation of 18F-human serum albumin: a simple and efficient protein labeling method with 18F using a hydrazone-formation method. Bioconjug Chem. 2005;16:1329–33. doi: 10.1021/bc050086r. [DOI] [PubMed] [Google Scholar]
- 15.Mamat C, Ramenda T, Wuest FR. Recent Applications of Click Chemistry for the Synthesis of Radiotracers for Molecular Imaging. Mini-Rev Org Chem. 2009;6:21–34. [Google Scholar]
- 16.Glaser M, Robins EG. ‘Click labelling’ in PET radiochemistry. J Labelled Compd Rad. 2009;52:407–14. [Google Scholar]
- 17.Marik J, Sutcliffe JL. Click for PET: rapid preparation of [F-18]fluoropeptides using Cu-I catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6681–4. [Google Scholar]
- 18.Campbell-Verduyn LS, Mirfeizi L, Dierckx RA, Elsinga PH, Feringa BL. Phosphoramidite accelerated copper(I)-catalyzed [3+2] cycloadditions of azides and alkynes. Chem Commun. 2009:2139–41. doi: 10.1039/b822994e. [DOI] [PubMed] [Google Scholar]
- 19.Li ZB, Wu Z, Chen K, Chin FT, Chen X. Click chemistry for F-18-labeling of RGD peptides and microPET Imaging of tumor integrin alpha(v)beta(3) expression. Bioconjug Chem. 2007;18:1987–94. doi: 10.1021/bc700226v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramenda T, Bergmann R, Wuest F. Synthesis of F-18-labeled neurotensin(8–13) via copper-mediated 1,3-dipolar [3+2] cycloaddition reaction. Lett Drug Des Discov. 2007;4:279–85. [Google Scholar]
- 21.Ramenda T, Kniess T, Bergmann R, Steinbach J, Wuest F. Radiolabelling of proteins with fluorine-18 via click chemistry. Chem Commun. 2009:7521–3. doi: 10.1039/b916075b. [DOI] [PubMed] [Google Scholar]
- 22.Wangler B, Quandt G, Iovkova L, Schirrmacher E, Wangler C, Boening G, et al. Kit-Like F-18-Labeling of Proteins: Synthesis of 4-(Di-tert-butyl[F-18]fluorosilyl)benzenethiol (Si[F-18]FA-SH) Labeled Rat Serum Albumin for Blood Pool Imaging with PET. Bioconjug Chem. 2009;20:317–21. doi: 10.1021/bc800413g. [DOI] [PubMed] [Google Scholar]
- 23.Kostikov AP, Chin J, Orchowski K, Niedermoser S, Kovacevic MM, Aliaga A, et al. Oxalic Acid Supported Si-F-18-Radiofluorination: One-Step Radiosynthesis of N-Succinimidyl 3-(Di-tert-butyl[F-18]fluorosilyl) benzoate ([F-18]SiFB) for Protein Labeling. Bioconjug Chem. 2012;23:106–14. doi: 10.1021/bc200525x. [DOI] [PubMed] [Google Scholar]
- 24.Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM, et al. One Step Radiosynthesis of 6-[F-18]Fluoronicotinic Acid 2,3,5,6-Tetrafluorophenyl Ester ([F-18]F-Py-TFP): A New Prosthetic Group for Efficient Labeling of Biomolecules with Fluorine-18. J Med Chem. 2010;53:1732–40. doi: 10.1021/jm9015813. [DOI] [PubMed] [Google Scholar]
- 25.Jagoda EM, Vaquero JJ, Seidel J, Green MV, Eckelman WC. Experiment assessment of mass effects in the rat: implications for small animal PET imaging. Nucl Med Biol. 2004;31:771–9. doi: 10.1016/j.nucmedbio.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Altman PL, Dorothy DS. Blood and other body fluids: analysis and compilation. Federation of American Societies for Experimental Biology. 1961:9–10. [Google Scholar]
- 27.Block D, Coenen HH, Stocklin G. Nca F-18-Fluoroacylation Via Fluorocarboxylic Acid-Esters. J Labelled Compd Rad. 1988;25:185–200. [Google Scholar]
- 28.Guhlke S, Coenen HH, Stocklin G. Fluoroacylation Agents Based on Small Nca [F-18] Fluorocarboxylic Acids. Appl Radiat Isotopes. 1994;45:715–27. [Google Scholar]
- 29.Aloj L, Lang LX, Jagoda E, Neumann RD, Eckelman WC. Evaluation of human transferrin radiolabeled with N-succinimidyl 4-[fluorine-18](fluoromethyl) benzoate. J Nucl Med. 1996;37:1408–12. [PubMed] [Google Scholar]
- 30.Garg PK, Garg S, Zalutsky MR. F-18 Labeling of Monoclonal-Antibodies and Fragments with Preservation of Immunoreactivity. Bioconjug Chem. 1991;2:44–9. doi: 10.1021/bc00007a008. [DOI] [PubMed] [Google Scholar]
- 31.Vaidyanathan G, Zalutsky MR. Labeling Proteins with F-18 Using N-Succinimidyl 4-[F-18]Fluorobenzoate. Nucl Med Biol. 1992;19:275–81. doi: 10.1016/0883-2897(92)90111-b. [DOI] [PubMed] [Google Scholar]
- 32.Wust F, Hultsch C, Bergmann R, Johannsen B, Henle T. Radiolabelling of isopeptide N-epsilon-(gamma-glutamyl)-L-lysine by conjugation with N-succinimidyl-4-[F-18]fluorobenzoate. Appl Radiat Isotopes. 2003;59:43–8. doi: 10.1016/s0969-8043(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 33.Li ZB, Wu ZH, Chen K, Ryu EK, Chen XY. F-18-labeled BBN-RGD heterodimer for prostate cancer imaging. J Nucl Med. 2008;49:453–61. doi: 10.2967/jnumed.107.048009. [DOI] [PubMed] [Google Scholar]
- 34.Vaidyanathan G, Zalutsky MR. Synthesis of N-succinimidyl 4-[F-18] fluorobenzoate, an agent for labeling proteins and peptides with F-18. Nat Protoc. 2006;1:1655–61. doi: 10.1038/nprot.2006.264. [DOI] [PubMed] [Google Scholar]
- 35.Tang G, Zeng WB, Yu MX, Kabalka G. Facile synthesis of N-succinimidyl 4-[(18)F]fluorobenzoate ([(18)F]SFB) for protein labeling. J Labelled Compd Rad. 2008;51:68–71. [Google Scholar]
- 36.Tang GH, Tang XL, Wang XL. A facile automated synthesis of N-succinimidyl 4-[F-18]fluorobenzoate ([F-18]SFB) for F-18-labeled cell-penetrating peptide as PET tracer. J Labelled Compd Rad. 2010;53:543–7. [Google Scholar]
- 37.Wilbur DS, Hamlin DK, Srivastava RR, Burns HD. Synthesis and Radioiodination of N-Boc-P-(Tri-N-Butylstannyl)-L-Phenylalanine Tetrafluorophenyl Ester - Preparation of a Radiolabeled Phenylalanine Derivative for Peptide-Synthesis. Bioconjug Chem. 1993;4:574–80. doi: 10.1021/bc00024a024. [DOI] [PubMed] [Google Scholar]
- 38.Lockett MR, Phillips MF, Jarecki JL, Peelen D, Smith LM. A tetrafluorophenyl activated ester self-assembled monolayer for the immobilization of amine-modified oligonucleotides. Langmuir. 2008;24:69–75. doi: 10.1021/la702493u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.