

Abstract
Current hypothesis suggests that genetic, immunological and bacterial factors contribute essentially to the pathogenesis of inflammatory bowel disease (IBD). Variations within the gene loci encoding protein tyrosine phosphatases (PTPs) have been associated with the onset of IBD. PTPs modulate the activity of their substrates by dephosphorylation of tyrosine residues and are critical for the regulation of fundamental cellular signalling processes.Evidence emerges that expression levels of PTPN2, PTPN11 and PTPN22 are altered in actively inflamed intestinal tissue. PTPN2 seems to be critical for protecting intestinal epithelial barrier function, regulating innate and adaptive immune responses and finally for maintaining intestinal homeostasis. These observations have been confirmed in PTPN2 knock-out mice in vivo. Those animals are clearly more susceptible to intestinal and systemic inflammation and feature alterations in innate and adaptive immune responses. PTPN22 controls inflammatory signalling in lymphocytes and mononuclear cells resulting in aberrant cytokine secretion pattern and autophagosome formation. PTPN22-deficiency in vivo results in more severe colitis demonstrating the relevance of PTPN22 for intestinal homeostasis in vivo. Of note, loss of PTPN22 promotes MAPK-induced cytokine secretion, but limits secretion of NFκB-associated cytokines and autophagy in mononuclear cells. Loss of PTPN11 is also associated with increased colitis severity in vivo. In summary, dysfunction of those PTPs results in aberrant and uncontrolled immune responses that result in chronic inflammatory conditions. This way, it becomes more and more evident that dysfunction of PTPs displays an important factor in the pathogenesis of chronic intestinal inflammation, in particular IBD.
INTRODUCTION
Current hypothesis suggests that genetic, immunological and bacterial factors contribute essentially to the pathogenesis of inflammatory bowel disease (IBD). An epithelial barrier defect, coupled with a dysfunctional immune response of the innate as well as the acquired immune system to commensal flora results in either excessive up- or impaired downregulation of inflammatory events, what finally drives the development of chronic intestinal inflammation (1). The predispositions are genetically determined and genome wide association studies (GWAS) identified variations in about 160 gene loci being associated with IBD (2). Many of the identified risk genes are critically involved in bacterial recognition, induction of antimicrobial factors, activation and modulation of innate as well as adaptive immune responses and in the maintenance of intestinal epithelial barrier function.
Phosphorylation and dephosphorylation of specific amino acid residues, in particular tyrosine residues, represents a fundamental mechanism for the activation and inactivation of intracellular signalling molecules. Dephosphorylation is carried out by a large number of different protein phosphatases. One important family of such proteins are protein tyrosine phosphatases (PTPs). Members of this family play an essential role in the regulation of critical cell signalling events, i.e. proliferation, differentiation and cell survival. Recent studies demonstrated a pivotal role for PTPs in the regulation of inflammatory and antibacterial responses (3).
In this review we will summarize the most recent knowledge about the role for PTPNs in the pathogenesis of chronic intestinal inflammation, in particular IBD.
PROTEIN TYROSINE PHOSPHATASE NON-RECEPTOR TYPE 22
In the past decade, GWAS revealed a strong association between two different single nucleotide polymorphisms (SNP) in the gene locus encoding protein tyrosine phosphatase non-receptor type 22 (PTPN22) and several inflammatory disorders, including the IBD subforms, Crohn's disease (CD) (2, 4) and ulcerative colitis (UC) (2, 4), systemic lupus erythematodes (SLE) (5), rheumatoid arthritis (RA) (6, 7), type 1 diabetes (T1D) (8) and others (9). One disease associated SNP (C1858>T; SNP ID: rs2476601) results in the substitution of Arginine 620 with a tryptophan residue in the protein product (referred to as 620W variant); while in the other SNP (G788>C; SNP ID: rs33996649), arginine 263 is substituted with a glutamine residue (referred to as 263Q variant).
Arginine 263 lies within the catalytic domain of the protein and substitution with a glutamine residue results in decreased phosphatase activity (10). Of note, presence of the 263Q variant reduces the risk to develop SLE and RA (10, 11), and protects from UC onset (4). PTPN22 620W in contrast is associated with increased risk for developing RA (6, 7), SLE (5) and T1D(8), but reduces the risk to develop CD (4). Initial studies by Vang et al. demonstrated that presence of this genetic variation results in a gain-of-function PTPN22 variant causing inhibition of the T cell receptor signalling cascade (12). Further, this variant features increased in vitro de-phosphorylation capacity (12), possibly resulting from reduced PTPN22 phosphorylation at an inhibitory residue (13, 14). In contrast, one study using mice that express the disease associated PTPN22 variant, suggested that the PTPN22 619W variant (the murine orthologue of human PTPN22 620W) results in reduced protein stability and therefore in a loss-of-function effect of this variant (15). However, this finding could not be confirmed in other studies using an independently generated PTPN22 619W knock-in strain (16).
In vivo silencing of PTPN22 by siRNA interference caused phenotypically opposite effects on B cell differentiation when compared with the effects of the presence of the PTPN22 620W allele in humans (17). This further supports the gain-of-function hypothesis. Wang et al recently proposed that the 620W variant might be an altered-function, rather than a loss- or gain-of function variant (18), proposing altered substrate specificity of the variant as possible mechanism for the observed effects (18). Nevertheless, in general, the PTPN22 620W variant is nowadays regarded as a gain-of-function variant.
PTPN22 in lymphocytes
PTPN22 is expressed in both, myeloid and lymphoid immune cells, but barely present in non-hematopoietic cell types (19). Given the strong association with inflammatory diseases, several studies addressed the role of PTPN22 in immune cell signalling. In T cells, PTPN22 activity attenuates T cell antigen receptor (TCR) signalling by interacting with and de-phosphorylating the TCR-associated kinases lymphocyte-specific protein tyrosine kinase (Lck) and zeta-chain associated protein (ZAP)-70 (20, 21). Presence of the gain-of-function variant results in reduced T cell receptor signalling in T1D patients and healthy individuals (14), while PTPN22 deficient T cells are hyper-responsive to TCR ligation (22). PTPN22 deficient mice, in contrast, display increased levels of effector and memory T cells, but interestingly do not show signs of autoimmunity (22). This observation was explained in additional studies, which revealed that PTPN22 deficient regulatory T cells show enhanced suppressor functions and thereby are able to counteract the increased T cell activation found in PTPN22 deficient effector T cells (23).
Presence of the PTPN22 620W variant leads to an increased frequency of auto-reactive B cell clones in healthy individuals (24, 25) and T1D patients (25). Several studies argue that this is the result of B cell intrinsic defects (16), such as enhanced survival of auto-reactive B cells at the transition from immature to naïve B cells (24, 25) due to changed BCR signalling (24, 25). A more recent study however claims that this effect might result from changes in follicular Th cell activation, which drives altered activation of B cells in germinal centres, rather than from a B cell intrinsic effect (26).
PTPN22 in myeloid immune cells
Most studies addressing PTPN22 function have been carried out in lymphocytes, but PTPN22 expression is very high in other immune cell types as well, including neutrophils, macrophages, and dendritic cells (15, 19). We showed that loss of PTPN22 in monocytes results in profound changes in IFN-γ-induced signalling with reduced signal transducer and activator of transcription (STAT) 1, but strongly enhanced p38 mitogen-activated protein kinase (MAPK) activation, ultimately resulting in drastically enhanced interleukin (IL)-6 secretion together with reduced IL-12 levels (27). Further, loss of PTPN22 also resulted in an altered response to the bacterial wall product, muramyl-dipeptide (MDP), with enhanced autophagy induction but also increased secretion of pro-inflammatory IL-6 and IL-8 (28), hence loss of PTPN22 in myeloid immune cells results in an increase in the secretion of pro-inflammatory cytokines (Figure 1).
Figure 1. PTPN22 controls IFN-γ and MDP induced signaling cascades.
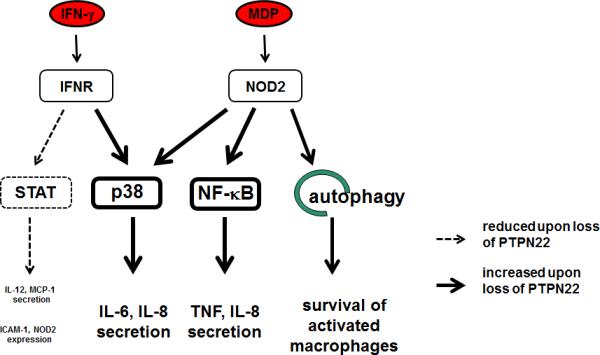
Loss of PTPN22 promotes p38 MAPK and canonical NF-κB activation, and increases MDP induced autophagy. On the other hand, loss of PTPN22 results in decreased STAT activity. Taken together, this results in a highly imbalanced cytokine secretion pattern. ICAM-1: intercellular adhesion molecule 1; IFN: interferon; IFNR: interferon receptor; MCP-1: monocyte chemoattractant protein 1; MDP: muramyl-dipeptide; NF-κB: nuclear factor κB; NOD2: nucleotide oligomerization containing protein 2; p38: mitogen activated protein kinase p38; STAT; signal transducer and activator of transcription; TNF: tumor necorsis factor.
Wang et al. demonstrated that presence of the PTPN22 620W variant results in impaired type-1 IFN production upon TLR4 ligation (18), and loss of PTPN22, or the presence of the 620W variant, promotes differentiation of monocytes into pro-inflammatory M1 macrophages (29). Dendritic cells (DC) from PTPN22 deficient mice expressed higher levels of the co-stimulatory molecule CD40 and induced higher proliferation of OT-II T cells in in vitro co-culture experiments (15).
PTPN22 in IBD patients
The PTPN22 gain-of-function variant is regarded as a general autoimmune risk allele, as it is associated with increased risk to develop several autoimmune disorders (9). Interestingly, the gain-of-function variant at the same time results in a reduced risk to develop CD (4). This opposite effect on intestinal inflammation evokes the question about the underlying mechanism(s). We found that PTPN22 expression is reduced in both UC and CD patients, with a dominant down-regulation in CD68+ mononuclear cells. However, only a slight difference in PTPN22 expression in T or B cells was observed (28), indicating an important role of PTPN22 in the myeloid cell compartment for maintaining intestinal homeostasis (Figure 2).
Figure 2. Reduced PTPN22 expression in IBD patients.
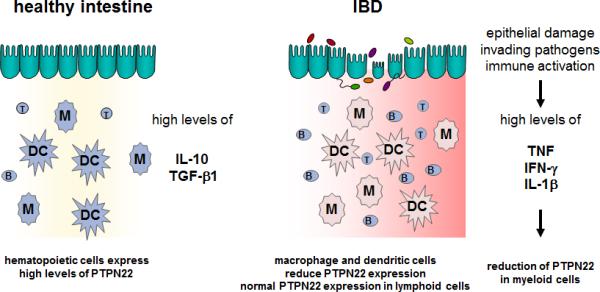
In the healthy intestine, high levels of IL-10 and TGF-β result in high expression of PTPN22 in all immune cells. Upon epithelial damage, the pro-inflammatory cytokine milieu results in reduced PTPN22 expression specifically in cells of the myeloid linage, while lymphoid cells still express high amounts of PTPN22. B: B cell, DC: dendritic cell; IFN: interferon; M/MF: Macrophage; M1: inflammatory macrophage; T: T cell; TNF: Tumor necrosis factor.
PTPN22 in experimental colitis
Although several association studies revealed a relation between PTPN22 and IBD susceptibility (4, 30), only a few studies addressed its functional role during intestinal inflammation. One study showed that in a T cell mediated colitis model, where naive T cells were transferred into immune-deficient mice, naive PTPN22 deficient T cells aggravated intestinal inflammation and tissue damage (23). Interestingly, the transfer of PTPN22 deficient regulatory T cells – but not the transfer of wild-type regulatory T cells – rescued this phenotype (23). This effect is attributed to increased suppressive competence of PTPN22 deficient regulatory T cells (23). A recent study further revealed that PTPN22 deficient naive T cells proliferate faster, acquire full effector functions and loos self-tolerance in immune deficient hosts (31). This might explain how PTPN22 deficient T cells are able to mediate increased inflammation when injected into lymphopenic mice.
The important protective role of PTPN22 during intestinal inflammation has further been confirmed in two independent studies using a dextran-sodium sulphate (DSS) induced model of acute colitis. In both studies, loss of PTPN22 resulted in increased reaction to DSS with aggravated weight loss and enhanced colitis severity (18, 29). Enhanced colitis susceptibility has been attributed to altered TLR4 signalling and type I interferon production in one study (18), or altered macrophage polarization in the other report (29), although the cell-type specific contribution to intestinal inflammation has not been addressed directly. We further observed that PTPN22 deficiency results in reduced myelin peroxidase activation and decreased granulocyte infiltration into the inflamed colon during DSS colitis (own unpublished data), suggesting a role for PTPN22 in mediating granulocyte function / recruitment.
As PTPN22 expression is reduced mainly in myeloid cells within the intestine of IBD patients (28), transfer of PTPN22 deficient T cells, or colitis induction in a setting where PTPN22 is missing in all cells, might however be an incomplete approach to address PTPN22 function during intestinal inflammation. Additional in vivo studies including PTPN22 deficiency specifically in non-lymphoid immune cells would greatly improve our general understanding on the role of PTPN22 in IBD.
In summary, IBD patients feature decreased levels of PTPN22 in myeloid cells in the intestine. Several studies demonstrated, that loss of PTPN22 in myeloid cells changes the response towards pro-inflammatory cytokines and bacterial products. Hence, reduction of PTPN22, as observed in IBD patients importantly affects the cytokine balance within the inflamed intestine, ultimately promoting disease persistence and progression (Figure 3).
Figure 3. Proposed mechanism, how PTPN22 is involved in IBD pathogenesis.
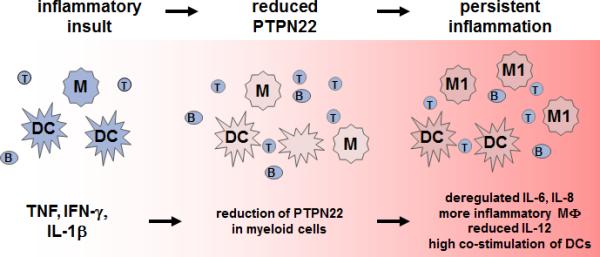
Inflammatory insults result in high levels of TNF, IFN-γ and IL-1β. TNF and IL-1β reduce PTPN22 expression in myeloid immune cells. Upon loss of PTPN22, monocytes/macrophages secrete increased levels of IL-6 and IL-8 and differentiate preferentially into pro-inflammatory M1 macrophages. The increased expression of co-stimulatory molecules on DC results in an enhanced activation of the adaptive immune system. B: B cell, DC: dendritic cell; IFN: interferon; M/MF: Macrophage; M1: inflammatory macrophage; T: T cell; TNF: Tumor necrosis factor.
PROTEIN TYROSINE PHOSPHATASE NON-RECEPTOR TYPE 2
The protein tyrosine phosphatase non-receptor type 2 (PTPN2) gene encodes the tyrosine phosphatase PTPN2, also known as T-cell protein tyrosine phosphatase (TCPTP). PTPN2 was named due to being originally cloned from a T-cell cDNA library. However, it is ubiquitously expressed (32, 33). In humans there are two functional variants of the PTPN2 enzyme that occur through alternative PTPN2 splicing. The larger 48 kD form is restricted to the endoplasmic reticulum by a hydrophobic C-terminus that masks a nuclear localization sequence (NLS). The more enzymatically active 45 kD variant lacks the hydrophobic C-terminus and can therefore transit to the nucleus via the NLS and is thus regarded as the mobile form of PTPN2 (34, 35). Indeed, in response to an appropriate stimulus the 45 kD form can also exit the nucleus and dephosphorylate target substrates in the cytoplasm and at the plasma membrane (36). PTPN2 is capable of dephosphorylating many protein tyrosine kinases such as the insulin receptor, epidermal growth factor receptor (EGFR), Src family kinases as well as several janus kinase (JAK) and STAT family members (36-42). The PTPN2 gene has emerged as a site of distinct clinical significance due to the association of a number of SNPs in the PTPN2 locus (18p11) with chronic inflammatory conditions such as CD, UC, T1D and celiac disease (43-45). The rs2542151 SNP has been the most widely identified PTPN2 SNP associated with IBD. It was first reported in the defining Welcome Trust Case Control Consortium (WTCCC) paper published in 2007 demonstrating a link with CD (46). Follow-up studies confirmed this association and also identified links between the rs2542151 SNP with CD and UC (44, 47-51). Additional SNPs in the PTPN2 gene locus have also been associated with IBD and disease outcomes. This includes the rs7234029 SNP that has a potential association with a stricturing disease phenotype in CDsubjects, and may be linked to early onset CD and UC (52). The rs1893217 SNP was originally associated with T1D, however it has emerged as a candidate SNP in both adult and early-onset pediatric CD as well as UC although the effect of this SNP on the PTPN2 gene and function remains to be determined (43, 44, 53). Nevertheless, a first study points towards the presence of a loss-of-function PTPN2 protein in variant carrying cells (54).
PTPN2 and Barrier Function Regulation
With respect to PTPN2 expression in IBD, colonic PTPN2 mRNA and protein expression is increased in active CD with expression being most prominent in the epithelium (55, 56). These findings were supported by another study that also showed increased PTPN2 protein expression in intestinal tissues (colon and ileum) from patients with active CD (57). Consistent with this, we have shown that in intestinal epithelial cell lines (IEC) the IBD-associated inflammatory cytokines, IFN-γ and tumor necrosis factor (TNF) are capable of increasing expression of PTPN2 (55, 58). This suggests that these inflammatory cytokines induce expression of a negative regulator of their own signalling in an apparent negative feedback loop. While expression of PTPN2 is increased in CD, the impact of non-coding SNPs appears to manifest in a loss of enzymatic activity or efficacy, although the full impact of individual SNPs on PTPN2 expression and function has yet to be fully determined. A loss of PTPN2 expression has been shown to have dramatic consequences for intestinal epithelial cells (IEC) and their ability to form an effective barrier. siRNA knockdown of PTPN2 resulted in accentuated barrier defects induced by IFN-γ-treatment of IEC (55). This included a greater decrease in transepithelial electrical resistance (TER) in response to IFN-γ coupled with a pronounced increase in expression of the cation-selective pore-forming molecule, claudin-2 (55). Claudin-2 expression is elevated in colonic tissues in IBD patients, especially UC, and functionally this could contribute to symptoms of disease by permitting increased paracellular passage of sodium ions into the intestinal lumen thus tangibly contribute to intestinal fluid loss associated with IBD (59-61). In addition to a further decrease in TER, PTPN2-deficient cells also displayed increased macromolecule permeability following IFN-γ treatment as determined by increased apical-basolateral passage of FITC-dextran across polarized IEC monolayers (55). Due to the width of the pore size generated by claudin-2 being insufficient to permit passage of FITC-dextran (10 kD), this strongly suggests that additional mechanisms capable of modifying tight junction components responsible for regulation of macromolecule permeability are recruited by IFN-γ in cells lacking PTPN2 (62). Conclusive evidence of this has not yet been determined as PTPN2 knockdown did not cause further decreases in expression of the tight junction proteins occludin or zonula occludens (ZO)-1 by IFN-γ, while a possible influence of PTPN2 on relocalization of tight junction proteins has not been investigated (55). These data suggest that PTPN2 plays an important role in protecting intestinal epithelial barrier function. A protective role for PTPN2 in intestinal barrier function has also been indicated in vivo. Ptpn2 knockout mice exhibit a pronounced phenotype at 3-5 weeks of age that is characterized by systemic inflammation, hematopoietic defects, increased levels of pro-inflammatory cytokines, splenomegaly and diarrhea. The severity and kinetics of the inflammation in mice are strain-dependent with an accelerated morbidity for mice on a Balb/c background compared with mice on a C57BL/6 background (63, 64). Bone marrow chimeric studies indicated that the inflammation and mortality in mice on the Balb/c background were governed by loss of Ptpn2 in the non-hematopoietic compartment (65). Studies with heterozygous Ptpn2-deficient mice showed that these mice have no overt inflammatory phenotype and exhibit normal survival rates. However, they did exhibit increased sensitivity to DSS-induced colitis suggesting that Ptpn2/PTPN2 deficiency increases the susceptibility of the intestine to agents that disrupt the epithelial barrier (66).
One additional area of epithelial function that can contribute to the broader concept of “barrier function” is appropriate regulation of electrolyte transport. This is essential for absorption and secretion of electrolytes and fluids as well as being responsible for the absorption of nutrients, maintenance of luminal pH and preserving the sterility of intestinal crypts (67). Epithelial electrolyte transport is suppressed in IBD and this is believed to contribute to overall fluid loss through decreased absorptive capacity, as well as creating an environment conducive to increased bacterial interactions with the intestinal epithelium (68-70). PTPN2 has been shown to play a role in regulating fluid secretion. Specifically, PTPN2 knockdown in T84 colonic epithelial cells accentuated epidermal growth factor (EGF) inhibition of Ca2+-stimulated chloride secretion thus promoting EGFR suppression of electrolyte secretion (71). Thus, it is possible that PTPN2 mutations resulting in a loss of enzymatic activity could encourage elevated or prolonged EGFR phosphorylation and exacerbate overall dysregulation of intestinal fluid homeostasis.
As discussed later in this manuscript, loss of PTPN2 expression also has consequences for other aspects of innate and adaptive immunity including autophagy and T-cell function (54, 72). Given that PTPN2 represents a point of convergence for multiple aspects of intestinal homeostasis it may therefore play a key role in multiple IBD-associated physiological events and thus, like other IBD candidate genes, make a greater cumulative contribution to IBD pathogenesis than suggested by the genetic prevalence of PTPN2 SNPs in sampled populations. Therefore, clinically relevant loss-of-function mutations in the PTPN2 gene may contribute, at least in part, to the development of IBD and other chronic inflammatory intestinal diseases via a compromised epithelial barrier. Indeed, given that increased intestinal permeability is a feature of IBD, T1D and celiac disease, this may be one avenue to explore commonality in the etiology of these conditions arising from PTPN2 mutations (Figure 4).
Figure 4. PTPN2 functions in IEC.
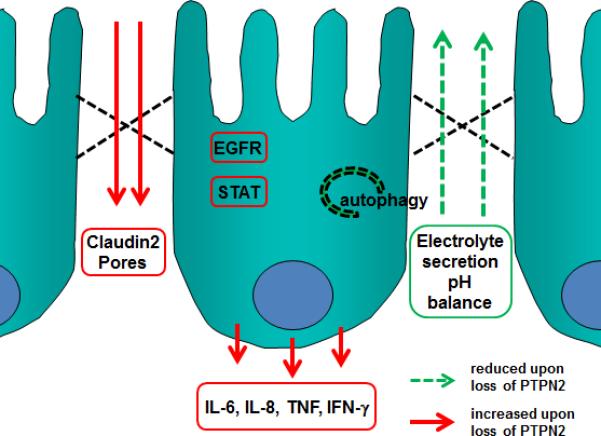
Loss of PTPN2 or presence of PTPN2 variants results in reduced barrier function, and suppression of electrolyte secretion resulting in a disturbed liquid/electrolyte balance. Further, PTPN2 deficiency results in enhanced autophagy and increased secretion of pro-inflammatory cytokines. EGFR: epidermal growth factor receptor; IFN-γ: interferon gamma; IL: interleukin; STAT: signal transducer and activator of transcription; TNF: tumor necrosis factor.
PTPN2 and innate immune system
Studies with Ptpn2−/− mice confirmed that PTPN2 is a key negative regulator of cytokine signalling. These animals develop progressive systemic inflammatory disease as demonstrated by chronic myocarditis, gastritis, nephritis, and sialadenitis as well as elevated serum levels of IFN-γ, IL-12, TNF and nitric oxide (NO). These cytokines seem to be mostly produced by mononuclear cells. Interestingly, PTPN2 deficient mice also present increased sensitivity to the bacterial wall component, lipopolysaccharide (LPS), in vivo and in vitro cultured macrophages from Ptpn2−/− mice are hypersensitive to LPS (65).
Recent data have demonstrated that PTPN2 regulates IFNγ-induced signalling and effects in cell models of chronic intestinal inflammation. Treatment of human IEC with IFN-γ increases PTPN2 mRNA and protein levels, elevates enzymatic PTPN2 activity and causes cytoplasmic accumulation of PTPN2. Similar as in fibroblasts, these effects are mediated via the cellular energy sensor, adenosine-monophosphate activated protein kinase (AMPK). Knock-down studies using PTPN2-specific siRNA constructs revealed that dephosphorylation of STAT1+3 is dependent on PTPN2 in these cells (55).
In PTPN2-deficient human THP-1 monocytic cells IFNγ-induced activity of the MAPK-subunit p38. Additionally, secretion of monocyte chemo-attractant protein 1 (MCP-1) and IL-6 were enhanced (73). PTPN2 also regulates signalling responses to the bacterial wall component MDP in human monocytes finally resulting in enhanced secretion of IFN-γ, but reduced secretion of IL-8 and TNF from these cells. This might be due to the fact that dysfunction of PTPN2 in human monocytes causes enhanced MAPK signalling in response to MDP. Of note, PTPN2 dysfunction also resulted in enhanced cleavage of caspase-1 and increased IL-1β secretion, indicative of increased inflammasome activation, in response to MDP (54). Further studies demonstrated that TNF induces PTPN2 protein and mRNA levels in human IEC via a NFκB-dependent mechanism. PTPN2 in turn regulates TNF-induced ERK- and p38-MAPK activity as well as IL-6 and IL-8 secretion (56). A further study also demonstrated that PTPN2 controls TNF-induced IL-6 secretion in mouse embryonic fibroblasts (40). These findings strongly suggest that PTPN2 is crucial for maintaining intestinal homeostasis and controlling cytokine secretion form intestinal epithelial cells, fibroblasts and mononuclear cells.
PTPN2 regulates autophagosome formation
Autophagy is a fundamental process for bulk degradation of cytoplasmic compartments, damaged organelles and/or misfolded proteins. In autophagosomes, these structures are sequestered into double-membrane-enclosed vesicles and delivered to lysosomes for final degradation (74-76). Autophagy is activated by stress conditions, such as starvation or hypoxia, and dysfunction of autophagy has been implicated in numerous pathologies, such as cancer or neurodegeneration (77). Autophagy is also critically involved in host defence against intracellular pathogens, such as Listeria monocytogenes (LM) or Salmonella typhimurium (78-80). This observation further supports the hypothesis that abnormal immune responses to luminal bacteria or bacterial antigens play an essential role for the manifestation of CD. Previous data have well demonstrated that presence of genetic variations within autophagy genes results in a defective bacterial handling, prolonged intracellular survival of pathogenic bacteria and an uncontrolled inflammatory situation.
Recent data demonstrated that PTPN2 not only regulates the cytokine-induced activation and expression of autophagy-related molecules, but is also involved in the regulation of autophagosome formation in IEC (81). siRNA-induced knock-out of PTPN2 in IEC inhibits the expression of several autophagy-associated molecules, including beclin-1, ATG5, ATG7, ATG12, ATG16L1 and IRGM in response to IFN-γ and TNF co-treatment. Of note, reduced protein levels of all of these autophagy markers have also been observed in intestinal tissue samples derived from patients with active CD when compared to tissue samples from non-IBD control patients (81).
On a functional level, loss of PTPN2 in human IEC completely abrogated the increase in LC3B-II protein levels as well as the formation of LC3B-positive (LC3B+) subcellular structures in the cells, indicating reduced autophagosome formation, in response to TNF and IFN-γ co-treatment. Further, PTPN2-deficient cells feature only a small number of LC3B+ vesicles overall and TNF and IFN-γ co-treatment caused the formation of fewer, but larger LC3B+ vacuoles that were localized close to cell borders (81). The appearance of such abnormal, large autophagic vacuoles has been regarded as a marker of an ineffective formation of dysfunctional autophagosomes due to a defective autophagy process in these cells (82). Similar likely dysfunctional autophagosomes were observed in PTPN2-deficient MDP-treated human monocytes (54). Interestingly, the effects of PTPN2 on autophagosome formation seemed to be mediated by controlling the phosphorylation status of the EGFR and subsequently of phosphatidyl-inositol 3-kinase (PI3K), Akt and molecular target of rapamycine (mTOR) activity (81).
Using primary colonic lamina propria fibroblasts (CLPF) isolated from CD patients featuring either the PTPN2-WT or the CD-associated PTPN2 variant, rs2542151, it was demonstrated that presence of the disease-associated PTPN2 variant exerts similar effects than siRNA-induced loss of PTPN2 expression (81). In particular, CLPFs featuring the CD-associated PTPN2 variant revealed reduced basal levels of PTPN2 protein when compared to PTPN2-WT fibroblasts and the increase in PTPN2 protein in response to TNF and IFN-γ treatment was absent in the variant cells. As in IEC, PTPN2 dysfunction also prevented the cytokine induced rise in the expression of autophagy markers, such as IRGM and also resulted in diminished formation of autophagosomes in PTPN2-variant carrying CLPF (81).
Impaired autophagy has been described to result in defective handling of invading bacteria and a defective handling of luminal and/or invading bacteria might critically contribute to the onset of IBD. Therefore, the question arises whether PTPN2 dysfunction might result in ineffective clearance of invading bacteria due to the altered autophagosome formation. Interestingly, using GFP-labelled Listeria monocytogenes it has been demonstrated that defective PTPN2 function results in enhanced levels of intracellular Listeria monocytogenes and reduced numbers of autophagosome as well as of LC3B-II protein levels in IEC. These findings were fully confirmed using CLPF from patients being either PTPN2 wild-type or variant (81). Presence of the CD-associated PTPN2 variant in CLPF caused similar effects with respect to autophagosome formation and the number of invading bacteria as PTPN2 deficiency in IEC. In PTPN2 variant CLPF, LC3B-staining was clearly less detectable while a strong GFP+ staining, suggestive for a large number of intracellular Listeria monocytogenes, was observed (81). These data demonstrate that dysfunction of PTPN2 results in impaired autophagosome formation and defective handling of invading bacteria and suggest how presence of the CD-associated PTPN2 variant within intestinal cells could critically contribute to the onset of IBD by causing a defective function of the innate immune system.
PTPN2 and adaptive immune system
PTPN2 had initially been termed T-cell protein tyrosine phosphatase (TC-PTP) which indicates that it was first detected and characterized in the T-cell subset of the adaptive immune system (83, 84). Subsequently a number of functions for PTPN2 in the adaptive immune system have been described in the following years. T-cells express PTPN2 at especially high levels indicating a specific role in this cell type (85). But also B-cell development is crucially regulated by PTPN2: The knock down of PTPN2 leads - besides other phenotypic changes in the respective animals - to an early bone marrow B-cell deficiency which is caused by a block of the transition from pre-B cells to immature B-cells (85). This is associated with the secretion of high amounts of IFN-γ, which in turn is followed by phosphorylation of STAT molecules in the pre-B cell compartment (86). Normally an important function of PTPN2 is to dephosphorylate STAT1 leading to its inactivation (41). Upon ligand binding, cytokine-receptor associated kinases phosphorylate STAT molecules which subsequently dimerize and translocate to the nucleus, where they act as transcription factors. PTPN2 counteracts the activity of receptor-associated kinases by de-phosphorylation of STAT molecules, ultimately repressing cytokine signalling (42). Similar to the innate immune system PTPN2 has an anti-inflammatory role in the adaptive immune system, both in the T- and B-cell compartment. Not surprisingly mice homozygous deficient of PTPN2 suffer from uncontrolled inflammation and die 3-5 weeks after birth (63). They show severe alterations in organs of the adaptive immune system including splenomegaly and lymphadenopathy including impaired T and B cell functions (63). These data indicate an important role of PTPN2 in the adaptive immune system and in the maturation of function of B- and T-cells and subsequently in autoimmunity are supported by data from human autoimmune diseases. In GWAS in patient cohorts with different autoimmune diseases SNPs of PTPN2 have been linked with the development of the disease such as T1D and CD (72). Besides B- and T-cell maturation as well as STAT1 de-phosphorylation this may be caused by the important role PTPN2 has as a key negative regulator of TCR signalling (72). PTPN2 dephosphorylates and inactivates Src family kinases to regulate T cell responses (72). By interfering with TCR signalling PTPN2 attenuates T cell activation and proliferation and limits antigen-induced responses. Controlled TCR activation and signalling is important for the priming of cytotoxic CD8+ T cells to pathogens and pathologic antigens on one hand, as well as mediating peripheral tolerance to self-antigens or commensal microbes in the gut on the other. PTPN2-deficient antigen stimulated CD8+ T cells are no longer self-tolerant and are able to destroy cells such as pancreatic β cells in an auto-reactive manner finally resulting in the development of diabetes even in the absence of CD4+ T helper cells (87). Thus, PTPN2 variants can re-direct a normally tolerogenic CD8+ T cell response into an auto-reactive and destructive response.
In addition a deficiency of PTPN2 was reported to enhance naive T cell responses to low-affinity ligands (72). This may partially be associated with the fact that STAT3 and STAT5 are further substrates for de-phosphorylation by PTPN2. Wiede et al reported that in the periphery PTPN2 deficiency resulted in a memory phenotype of CD4+ T-cells (72). In their mouse model where PTPN2 lacks specifically in the T cell compartment, the number of T cells with an effector/memory phenotype increased progressively from 4 to 12 weeks of age, which was paralleled by a decrease in naive T cell numbers. This may lead to a selection of high-affinity, potentially self-reactive T cells which would represent another pathway of auto-immune disease induction by a lack of PTPN2 function. In contrast, a lack of PTPN2 function does not seems to influence the number of regulatory T cells and does not interfere with regulatory T cell function.
The increase in secretion of pro-inflammatory cytokines found in other cell types certainly can also be found in T-cell. Misbalanced JAK and STAT signalling may play an important role here. An upregulation of IFN-γ secretion, IL-12 secretion and other cytokines may play an important role for T-cell activation (85). Additionally, it has been demonstrated that PTPN2 is elevated in naive T cells that leave the thymus. PTPN2-deficient CD8+ T cells undergo rapid lymphopenia-induced proliferation after transfer into lymphopenic hosts acquiring characteristics of antigen-experienced effector T-cells. This increase in lymphopenia-induced proliferation is associated with elevated TCR-dependent, but not IL-7-dependent responses. This finally results in aberrant TCR function and the development of autoimmunity (88) (Figure 6).
Figure 6. The effect of loss of (functional) PTPN2 during intestinal inflammation.
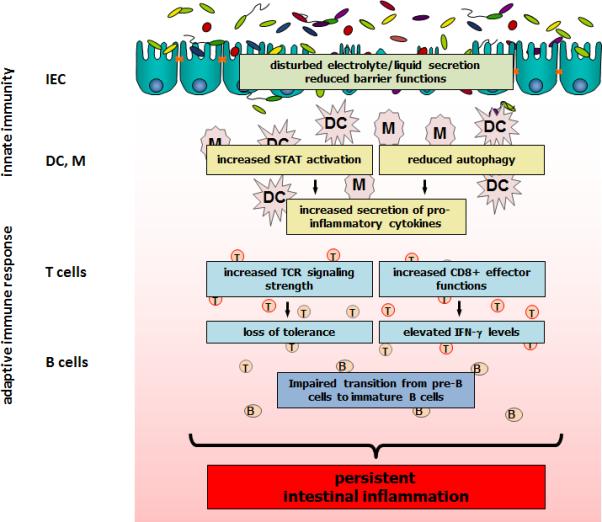
Loss of PTPN2 in experimental models or the presence of PTPN2 variants in humans results in several disease driving mechanisms ultimately maintaining intestinal disease. B: B cell; DC: dendritic cell; IEC: intestinal epithelial cell; M: macrophage; STAT: signal transducer and activator of transcription; T: T cell.
Despite the important role of PTPN2 and the finding of relevant variants in auto-immune diseases such as CD and UC a detailed analysis of modifications of T-cell functions by those variants or a complete lack of PTPN2 has not been performed yet. Animal models may not be optimally suited as there are complex interactions between the immune cells and also between adaptive and innate immune mechanisms.
A ROLE FOR OTHER PTPNs IN INTESTINAL INFLAMMATION
In addition to PTPN2 and PTPN22, some other PTPNs have also been associated with intestinal inflammation. In particular PTPN1 (PTP1B), PTPN6 (SHP-1) and PTPN11 (SHP-2) have already been demonstrated to play a role for the development of intestinal inflammation.
PTP1B is a key molecule in modulating low-degree inflammatory conditions such as diabetes. A recent study demonstrated that PTP1B deficiency ameliorates DSS-induced murine experimental colitis via expanding CD11b+Gr-1+ myeloid-derived suppressor cells (MDSCs). PTP1B-null mice demonstrate greater resistance to DSS-induced colitis, as reflected by slower weight-loss, greater survival rates and decreased macrophage infiltration into the colon. This resistance of PTP1B deficient mice to DSS-induced colitis is based on the expansion of MDSCs regulating cytokine secretion and activation of signalling molecules (89).
A further study demonstrated a significant association between PTPN6 and UC in a collective of Tunisian IBD patients (90). On a functional level, PTPN6 has been identified as a critical regulator of Th17 development. Genetically or pharmacologically caused dysfunction of PTPN6 activity in T cells makes these cells hyper-responsive to stimulation via IL-6 and IL-21. Additionally, PTPN6 controls cytokine-induced phosphorylation of STAT3 in primary CD4+ T helper cells. These data suggest a role for PTPN6 in the pathogenesis of IBD by modulating Th17 cell development (91).
PTPN11 is a ubiquitously expressed cytoplasmic protein tyrosine phosphatase. Genetic associations exist between UC, but not CD, and PTPN11. The Hap 1 haplotype and its homozygous Hap 1/Hap 1 diplotype of PTPN11 is significantly increased in UC patients compared to control subjects (92). The expression of PTPN11 protein in the intestinal tissue of UC patients is reduced (93). On a functional level, it has been demonstrated that pharmacologically induced interaction between PTPN11 and cytosolic STAT1, what finally prevents the recruitment of STAT1 to the IFN-γ receptor and inhibits STAT1 signalling, results in reduced Th1 cytokine production and an improvement in 2, 4, 6-trinitrobenzene sulfonic acid (TNBS)-induced colitis in mice. Inhibition of PTPN11, vice versa, worsens colitis symptoms in vivo (94).
Mice featuring an IEC-specific PTPN11 deletion feature growth retardation and rapidly develop clinical and histological signs of severe colitis. These animals also feature decreased expression of claudins accompanied by enhanced intestinal permeability and an upregulation of STAT3 and NF-κB activity as well as an induction of several epithelial chemokines and cytokines. Interestingly, the development of colitis in PTPN11 IEC specific knock-out mice was remarkably impaired by antibiotic treatment. A further study demonstrated that the number of goblet cells in both the small intestine and colon of theses mice is clearly reduced compared to control mice. Furthermore, the PTPN11 deficient mice showed marked impairment of both, IEC migration along the crypt-villus axis in the small intestine and development of intestinal organoids from isolated crypts (93, 95). These findings suggest that intestinal epithelial PTPN11 might play a critical role for protecting the intestinal epithelium and thereby for the prevention of gut inflammation.
SUMMARY
Protein tyrosine phosphatases play a crucial role for regulating intracellular signalling events. Recent GWAS have associated a number of PTPs with the onset of IBD. Evolving evidence emerges that expression levels of PTPs, mainly PTPN2, PTPN22 and PTPN11 are altered in actively inflamed intestinal tissue. Further, the PTPs seem to be critical for protecting intestinal epithelial barrier function, regulating innate and adaptive immune responses and finally for maintaining intestinal homeostasis. Dysfunction of those PTPs results in aberrant and uncontrolled immune responses that result in chronic inflammatory conditions. This way, it becomes more and more evident that dysfunction of PTPs displays an important factor in the pathogenesis of chronic intestinal inflammation, in particular IBD.
Figure 5. Signalling pathways regulated by PTPN2.
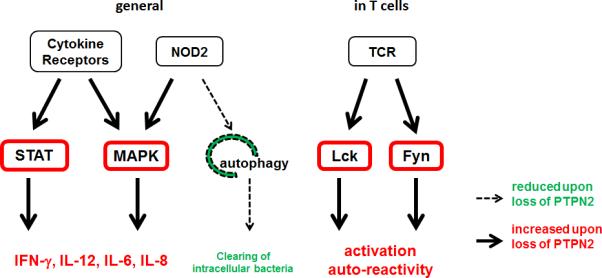
PTPN2 dephosphorylates STAT1, STAT3 and STAT5 to reduce cytokine signalling. In addition, loss of PTPN2 results in enhanced MAPK activation and decreased autophagy induction. In T cells, PTPN2 counteracts TCR associated kinases and thereby controls the reaction towards antigens maintaining tolerance towards self-antigens. IL: interleukin; Lck: lymphocyte tyrosine kinase; MAPK: mitogen activated protein kinase; NOD2: nucleotide oligomerization domain containing protein 2; STAT: signal transducer and activator of transcription; TCR: T cell antigen receptor.
Acknowledgments
Funding: This research was supported by a grant from Fonds zur Förderung des akademischen Nachwuchses (FAN) of the Zürcher Universitätsverein (ZUNIV) to MS, the Abbvie IBD Award 2013 to MS, a research grant from the Swiss Philanthropy Foundation to MS and GR, research grants from the Swiss National Science Foundation (SNF) to MS (Grant No. 314730-146204 and Grant No. CRSII3_154488/1), GR (Grant No. 310030-120312) and the Swiss IBD Cohort (Grant No. 3347CO-108792) and by the Zurich Center for Integrative Human Physiology (ZIHP) of the University of Zurich.
Footnotes
COMPETING INTERESTS
The authors have no competing financial interests to disclose.
REFERENCES
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Lees CW, Barrett JC, Parkes M, et al. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
- 3.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 4.Diaz-Gallo LM, Espino-Paisan L, Fransen K, et al. Differential association of two PTPN22 coding variants with Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2287–2294. doi: 10.1002/ibd.21630. [DOI] [PubMed] [Google Scholar]
- 5.Kyogoku C, Langefeld CD, Ortmann WA, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet. 2004;75:504–507. doi: 10.1086/423790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michou Lt, Lasbleiz S, Rat A-C, et al. Linkage proof for PTPN22, a rheumatoid arthritis susceptibility gene and a human autoimmunity gene. Proceedings of the National Academy of Sciences. 2007;104:1649–1654. doi: 10.1073/pnas.0610250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Begovich AB, Carlton VE, Honigberg LA, et al. A missense singlenucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337–338. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 9.Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. 2014 doi: 10.1038/nrrheum.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orru V, Tsai SJ, Rueda B, et al. A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet. 2009;18:569–579. doi: 10.1093/hmg/ddn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodríguez-Rodríguez L, Taib WRW, Topless R, et al. The PTPN22 R263Q polymorphism is a risk factor for rheumatoid arthritis in Caucasian case–control samples. Arthritis & Rheumatism. 2011;63:365–372. doi: 10.1002/art.30145. [DOI] [PubMed] [Google Scholar]
- 12.Vang T, Congia M, Macis MD, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 13.Yu X, Sun JP, He Y, et al. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci U S A. 2007;104:19767–19772. doi: 10.1073/pnas.0706233104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiorillo E, Orru V, Stanford SM, et al. Autoimmune-associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem. 2010;285:26506–26518. doi: 10.1074/jbc.M110.111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Zahir N, Jiang Q, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43:902–907. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 16.Dai X, James RG, Habib T, et al. A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest. 2013;123:2024–2036. doi: 10.1172/JCI66963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng P, Kissler S. PTPN22 silencing in the NOD model indicates the type 1 diabetes-associated allele is not a loss-of-function variant. Diabetes. 2013;62:896–904. doi: 10.2337/db12-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Shaked I, Stanford SM, et al. The Autoimmunity-Associated Gene PTPN22 Potentiates Toll-like Receptor-Driven, Type 1 Interferon-Dependent Immunity. Immunity. 2013;39:111–122. doi: 10.1016/j.immuni.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Science signaling. 2010;3:rs1. doi: 10.1126/scisignal.2000966. [DOI] [PubMed] [Google Scholar]
- 20.Gjorloff-Wingren A, Saxena M, Williams S, et al. Characterization of TCRinduced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. 1999;29:3845–3854. doi: 10.1002/(SICI)1521-4141(199912)29:12<3845::AID-IMMU3845>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 21.Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med. 1999;189:111–121. doi: 10.1084/jem.189.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasegawa K, Martin F, Huang G, et al. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303:685–689. doi: 10.1126/science.1092138. [DOI] [PubMed] [Google Scholar]
- 23.Brownlie RJ, Miosge LA, Vassilakos D, et al. Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Science signaling. 2012;5:ra87. doi: 10.1126/scisignal.2003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menard L, Saadoun D, Isnardi I, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. J Clin Invest. 2011;121:3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Habib T, Funk A, Rieck M, et al. Altered B cell homeostasis is associated with type I diabetes and carriers of the PTPN22 allelic variant. J Immunol. 2012;188:487–496. doi: 10.4049/jimmunol.1102176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maine CJ, Marquardt K, Cheung J, et al. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol. 2014;192:1415–1424. doi: 10.4049/jimmunol.1302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spalinger MR, Lang S, Weber A, et al. Loss of protein tyrosine phosphatase nonreceptor type 22 regulates interferon-gamma-induced signaling in human monocytes. Gastroenterology. 2013;144:978–988. e910. doi: 10.1053/j.gastro.2013.01.048. [DOI] [PubMed] [Google Scholar]
- 28.Spalinger MR, Lang S, Weber A, et al. Loss of Protein Tyrosine Phosphatase Non-Receptor Type 22 Regulates Interferon-gamma-Induced Signaling in Human Monocytes. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.01.048. [DOI] [PubMed] [Google Scholar]
- 29.Chang HH, Miaw SC, Tseng W, et al. PTPN22 Modulates Macrophage Polarization and Susceptibility to Dextran Sulfate Sodium-Induced Colitis. J Immunol. 2013;191:2134–2143. doi: 10.4049/jimmunol.1203363. [DOI] [PubMed] [Google Scholar]
- 30.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmond RJ, Brownlie RJ, Morrison VL, et al. The tyrosine phosphatase PTPN22 discriminates weak self peptides from strong agonist TCR signals. Nat Immunol. 2014;15:875–883. doi: 10.1038/ni.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cool DE, Tonks NK, Charbonneau H, et al. cDNA isolated from a human Tcell library encodes a member of the protein-tyrosine-phosphatase family. Proc Natl Acad Sci U S A. 1989;86:5257–5261. doi: 10.1073/pnas.86.14.5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorenzen JA, Dadabay CY, Fischer EH. COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus. J Cell Biol. 1995;131:631–643. doi: 10.1083/jcb.131.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibarra-Sanchez MJ, Simoncic PD, Nestel FR, et al. The T-cell protein tyrosine phosphatase. Semin Immunol. 2000;12:379–386. doi: 10.1006/smim.2000.0220. [DOI] [PubMed] [Google Scholar]
- 36.Tiganis T. PTP1B and TCPTP--nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013;280:445–458. doi: 10.1111/j.1742-4658.2012.08563.x. [DOI] [PubMed] [Google Scholar]
- 37.Galic S, Hauser C, Kahn BB, et al. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol Cell Biol. 2005;25:819–829. doi: 10.1128/MCB.25.2.819-829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiganis T, Bennett AM, Ravichandran KS, et al. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol Cell Biol. 1998;18:1622–1634. doi: 10.1128/mcb.18.3.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattila E, Pellinen T, Nevo J, et al. Negative regulation of EGFR signalling through integrin-alpha1beta1-mediated activation of protein tyrosine phosphatase TCPTP. Nat Cell Biol. 2005;7:78–85. doi: 10.1038/ncb1209. [DOI] [PubMed] [Google Scholar]
- 40.van Vliet C, Bukczynska PE, Puryer MA, et al. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat Immunol. 2005;6:253–260. doi: 10.1038/ni1169. [DOI] [PubMed] [Google Scholar]
- 41.ten Hoeve J, de Jesus Ibarra-Sanchez M, Fu Y, et al. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol Cell Biol. 2002;22:5662–5668. doi: 10.1128/MCB.22.16.5662-5668.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simoncic PD, Lee-Loy A, Barber DL, et al. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and. 3. Curr Biol. 2002;12:446–453. doi: 10.1016/s0960-9822(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 43.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to. 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smyth DJ, Plagnol V, Walker NM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkes M, Barrett JC, Prescott NJ, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Franke A, Balschun T, Karlsen TH, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 49.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weersma RK, Stokkers PC, Cleynen I, et al. Confirmation of multiple Crohn's disease susceptibility loci in a large Dutch-Belgian cohort. Am J Gastroenterol. 2009;104:630–638. doi: 10.1038/ajg.2008.112. [DOI] [PubMed] [Google Scholar]
- 51.Waterman M, Xu W, Stempak JM, et al. Distinct and overlapping genetic loci in Crohn's disease and ulcerative colitis: correlations with pathogenesis. Inflamm Bowel Dis. 2011;17:1936–1942. doi: 10.1002/ibd.21579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glas J, Wagner J, Seiderer J, et al. PTPN2 gene variants are associated with susceptibility to both Crohn's disease and ulcerative colitis supporting a common genetic disease background. PLoS One. 2012;7:e33682. doi: 10.1371/journal.pone.0033682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amre DK, Mack DR, Morgan K, et al. Susceptibility loci reported in genome-wide association studies are associated with Crohn's disease in Canadian children. Aliment Pharmacol Ther. 2010;31:1186–1191. doi: 10.1111/j.1365-2036.2010.04294.x. [DOI] [PubMed] [Google Scholar]
- 54.Scharl M, Mwinyi J, Fischbeck A, et al. Crohn's disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm Bowel Dis. 2012;18:900–912. doi: 10.1002/ibd.21913. [DOI] [PubMed] [Google Scholar]
- 55.Scharl M, Paul G, Weber A, et al. Protection of epithelial barrier function by the Crohn's disease associated gene protein tyrosine phosphatase n2. Gastroenterology. 2009;137:2030–2040. e2035. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scharl M, McCole DF, Weber A, et al. Protein tyrosine phosphatase N2 regulates TNF{alpha}-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut. 2011;60:189–197. doi: 10.1136/gut.2010.216606. [DOI] [PubMed] [Google Scholar]
- 57.Marcil V, Mack DR, Kumar V, et al. Association between the PTPN2 gene and Crohn's disease: dissection of potential causal variants. Inflamm Bowel Dis. 2013;19:1149–1155. doi: 10.1097/MIB.0b013e318280b181. [DOI] [PubMed] [Google Scholar]
- 58.Scharl M, McCole DF, Weber A, et al. Protein tyrosine phosphatase N2 regulates TNFalpha-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut. 2011;60:189–197. doi: 10.1136/gut.2010.216606. [DOI] [PubMed] [Google Scholar]
- 59.Yu W, Hegarty JP, Berg A, et al. PTPN2 is associated with Crohn's disease and its expression is regulated by NKX2-3. Disease markers. 2012;32:83–91. doi: 10.3233/DMA-2011-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weber CR, Raleigh DR, Su L, et al. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–12046. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCole DF. Regulation of epithelial barrier function by the inflammatory bowel disease candidate gene, PTPN2. Ann N Y Acad Sci. 2012;1257:108–114. doi: 10.1111/j.1749-6632.2012.06522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.You-Ten KE, Muise ES, Itie A, et al. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatasedeficient mice. J Exp Med. 1997;186:683–693. doi: 10.1084/jem.186.5.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiede F, Chew SH, van Vliet C, et al. Strain-dependent differences in bone development, myeloid hyperplasia, morbidity and mortality in ptpn2-deficient mice. PLoS One. 2012;7:e36703. doi: 10.1371/journal.pone.0036703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinonen KM, Nestel FP, Newell EW, et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood. 2004;103:3457–3464. doi: 10.1182/blood-2003-09-3153. [DOI] [PubMed] [Google Scholar]
- 66.Hassan SW, Doody KM, Hardy S, et al. Increased susceptibility to dextran sulfate sodium induced colitis in the T cell protein tyrosine phosphatase heterozygous mouse. PLoS One. 2010;5:e8868. doi: 10.1371/journal.pone.0008868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Current opinion in gastroenterology. 2007;23:647–654. doi: 10.1097/MOG.0b013e3282f0153b. [DOI] [PubMed] [Google Scholar]
- 68.Greig ER, Boot-Handford RP, Mani V, et al. Decreased expression of apical Na+ channels and basolateral Na+, K+-ATPase in ulcerative colitis. J Pathol. 2004;204:84–92. doi: 10.1002/path.1613. [DOI] [PubMed] [Google Scholar]
- 69.Sugi K, Musch MW, Field M, et al. Inhibition of Na+,K+-ATPase by interferon gamma down-regulates intestinal epithelial transport and barrier function. Gastroenterology. 2001;120:1393–1403. doi: 10.1053/gast.2001.24045. [DOI] [PubMed] [Google Scholar]
- 70.Xiao F, Juric M, Li J, et al. Loss of downregulated in adenoma (DRA) impairs mucosal HCO3(-) secretion in murine ileocolonic inflammation. Inflamm Bowel Dis. 2012;18:101–111. doi: 10.1002/ibd.21744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scharl M, Rudenko I, McCole DF. Loss of protein tyrosine phosphatase N2 potentiates epidermal growth factor suppression of intestinal epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol. 2010;299:G935–945. doi: 10.1152/ajpgi.00106.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wiede F, Shields BJ, Chew SH, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scharl M, Hruz P, McCole DF. Protein tyrosine phosphatase non-receptor Type 2 regulates IFN-gamma-induced cytokine signaling in THP-1 monocytes. Inflamm Bowel Dis. 2010;16:2055–2064. doi: 10.1002/ibd.21325. [DOI] [PubMed] [Google Scholar]
- 74.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 76.Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Birmingham CL, Canadien V, Gouin E, et al. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy. 2007;3:442–451. doi: 10.4161/auto.4450. [DOI] [PubMed] [Google Scholar]
- 79.Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 80.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 81.Scharl M, Wojtal KA, Becker HM, et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm Bowel Dis. 2011 doi: 10.1002/ibd.21891. [DOI] [PubMed] [Google Scholar]
- 82.Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 83.Miyasaka H, Li SS. Molecular cloning, nucleotide sequence and expression of a cDNA encoding an intracellular protein tyrosine phosphatase, PTPase-2, from mouse testis and T-cells. Molecular and cellular biochemistry. 1992;118:91–98. doi: 10.1007/BF00249698. [DOI] [PubMed] [Google Scholar]
- 84.Johnson CV, Cool DE, Glaccum MB, et al. Isolation and mapping of human T-cell protein tyrosine phosphatase sequences: localization of genes and pseudogenes discriminated using fluorescence hybridization with genomic versus cDNA probes. Genomics. 1993;16:619–629. doi: 10.1006/geno.1993.1239. [DOI] [PubMed] [Google Scholar]
- 85.Doody KM, Bourdeau A, Tremblay ML. T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: lessons from the knockout mouse model and implications in human disease. Immunol Rev. 2009;228:325–341. doi: 10.1111/j.1600-065X.2008.00743.x. [DOI] [PubMed] [Google Scholar]
- 86.Bourdeau A, Dube N, Heinonen KM, et al. TC-PTP-deficient bone marrow stromal cells fail to support normal B lymphopoiesis due to abnormal secretion of interferon-{gamma}. Blood. 2007;109:4220–4228. doi: 10.1182/blood-2006-08-044370. [DOI] [PubMed] [Google Scholar]
- 87.Wiede F, Ziegler A, Zehn D, et al. PTPN2 restrains CD8(+) T cell responses after antigen cross-presentation for the maintenance of peripheral tolerance in mice. Journal of autoimmunity. 2014;53:105–114. doi: 10.1016/j.jaut.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 88.Wiede F, La Gruta NL, Tiganis T. PTPN2 attenuates T-cell lymphopeniainduced proliferation. Nature communications. 2014;5:3073. doi: 10.1038/ncomms4073. [DOI] [PubMed] [Google Scholar]
- 89.Zhang J, Wang B, Zhang W, et al. Protein tyrosine phosphatase 1B deficiency ameliorates murine experimental colitis via the expansion of myeloidderived suppressor cells. PLoS One. 2013;8:e70828. doi: 10.1371/journal.pone.0070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bouzid D, Fourati H, Amouri A, et al. Association of ZAP70 and PTPN6, but Not BANK1 or CLEC2D, with inflammatory bowel disease in the Tunisian population. Genetic testing and molecular biomarkers. 2013;17:321–326. doi: 10.1089/gtmb.2012.0372. [DOI] [PubMed] [Google Scholar]
- 91.Mauldin IS, Tung KS, Lorenz UM. The tyrosine phosphatase SHP-1 dampens murine Th17 development. Blood. 2012;119:4419–4429. doi: 10.1182/blood-2011-09-377069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Narumi Y, Isomoto H, Shiota M, et al. Polymorphisms of PTPN11 coding SHP-2 as biomarkers for ulcerative colitis susceptibility in the Japanese population. J Clin Immunol. 2009;29:303–310. doi: 10.1007/s10875-008-9272-6. [DOI] [PubMed] [Google Scholar]
- 93.Coulombe G, Leblanc C, Cagnol S, et al. Epithelial tyrosine phosphatase SHP-2 protects against intestinal inflammation in mice. Mol Cell Biol. 2013;33:2275–2284. doi: 10.1128/MCB.00043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu X, Guo W, Wu L, et al. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis. J Immunol. 2012;189:3497–3507. doi: 10.4049/jimmunol.1201006. [DOI] [PubMed] [Google Scholar]
- 95.Yamashita H, Kotani T, Park JH, et al. Role of the protein tyrosine phosphatase Shp2 in homeostasis of the intestinal epithelium. PLoS One. 2014;9:e92904. doi: 10.1371/journal.pone.0092904. [DOI] [PMC free article] [PubMed] [Google Scholar]