

Abstract
Dominant mutations in the valosin‐containing protein (VCP) gene cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia, which is characterized by progressive muscle weakness, dysfunction in bone remodeling, and frontotemporal dementia. More recently, VCP has been linked to 2% of familial amyotrophic lateral sclerosis cases. VCP plays a significant role in a plethora of cellular functions including membrane fusion, transcription activation, nuclear envelope reconstruction, postmitotic organelle reassembly, and cell cycle control. To elucidate the pathological mechanisms underlying the VCP disease progression, we have previously generated a VCPR155H/+ mouse model with the R155H mutation. Histological analyses of mutant muscle showed vacuolization of myofibrils, centrally located nuclei, and disorganized muscle fibers. Global expression profiling of VCPR155H/+ mice using gene annotations by DAVID identified key dysregulated signaling pathways including genes involved in the physiological system development and function, diseases and disorders, and molecular and cellular functions. There were a total of 212 significantly dysregulated genes, several of which are involved in the regulation of proteasomal function and NF‐κB signaling cascade. Findings of the gene expression study were validated by using quantitative reverse transcriptase polymerase chain reaction analyses to test genes involved in various signaling cascades. This investigation reveals the importance of the VCPR155H/+ mouse model in the understanding of cellular and molecular mechanisms causing VCP‐associated neurodegenerative diseases and in the discovery of novel therapeutic advancements and strategies for patients suffering with these debilitating disorders.
Introduction
Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia (IBMPFD, OMIM 167320) is characterized by progressive muscle weakness, bone deformities, and premature dementia.1 Eighty‐two percent of patients manifest weakness and atrophy of the pelvic and shoulder girdle muscles at the mean age of 42 years.1, 2, 3, 4 During disease progression, muscle weakness is also seen in other muscles including limb and respiratory muscles, whereas cardiac failure as well as cardiomyopathy has been observed in later stages. The diagnosis of IBMPFD muscle disease is based on clinical findings including elevated serum creatinine kinase (CK) concentration myopathic electromyograms and skeletal muscle histology findings of rimmed vacuoles and inclusion bodies, which stain positive for ubiquitin and TAR DNA‐binding protein‐43 (TDP‐43) antibodies.1, 4, 5, 6
Paget's disease of bone (PDB) seldom affects individuals under the age of 50 years. However, early‐onset PDB is observed in 49% of IBMPFD patients.1, 3 It typically begins in the 30s to 40s, at a mean age of onset of 42 years. PDB is caused by excessive osteoclastic activity and susceptibility to deformities like bowing and fractures. It involves focal areas of increased bone turnover that typically leads to spine and/or hip pain and deformity of the long bones causing pathological fractures on occasion. The most commonly involved bones are the skull, vertebrae, and pelvis. The diagnosis of PDB is based on elevated concentrations of serum alkaline phosphatase, urine pyridinoline, and deoxypyridinoline, as well as on skeletal radiographs or radionuclide scans.7, 8
Frontotemporal dementia (FTD) comprises about 3% of dementias of the elderly.9, 10, 11 In IBMPFD, premature FTD is observed in 30% of patients with an average age of onset in the mid‐50s. It is characterized by dysnomia, dyscalculia, comprehension deficit, paraphasic errors, and relative preservation of memory. In later stages, patients have auditory comprehension deficits for even one‐step commands, and exhibit alexia and agraphia.1, 3 Affected individuals die from progressive muscle weakness, as well as from cardiac and respiratory failure typically in their 40s to 60s.1, 2, 3, 4
IBMPFD is caused by mutations in the valosin‐containing protein (VCP) gene,5 and more than 31 disease mutations have been identified to date (for review, see Kimonis et al.4). VCP belongs to the family of type II ATPase associated with variety of cellular activities (AAA) having two ATPase domains.12, 13, 14, 15, 16 It has been suggested to be involved in several cellular activities including homotypic membrane fusion, transcription activation, nuclear envelope reconstruction, postmitotic organelle reassembly, cell cycle control, apoptosis, and endoplasmic reticulum‐associated degradation of proteins.17, 18, 19, 20 Disease mutations cluster in the N‐terminal CDC48 domain, which is involved in ubiquitin‐binding and protein‐protein interactions.21, 22 Most of these mutated residues causing IBMPFD are three‐dimensionally located in a close proximity and potentially interact with each other. These findings suggest that these residues may have a similar and specific function within the VCP homohexamer, which binds to several different adapter proteins enabling VCP to target specific substrates for proteasomal degradation.23, 24
In order to investigate the in vivo effects of identified VCP mutations, as well as to understand the pathogenesis of IBMPFD, mouse models have been generated. Both human and mouse VCP proteins consist of 806 amino acids, and the mouse protein differs only by one amino acid residue at position 684 when compared to the corresponding human protein.25 Targeted homozygous deletion of VCP by Cre‐loxP technology was reported to result in early embryonic lethality,26 suggesting an important role for the intact VCP expression in the development of mouse embryos. In contrast, heterozygous mice lacking one VCP allele were indistinguishable from their WT littermates. On the other hand, transgenic mice overexpressing the most common human IBMPFD mutation, R155H, under the regulation of a muscle CK promoter demonstrated progressive muscle weakness in a dose‐dependent manner starting at 6 months of age.27 These mutant mice showed muscle pathology including coarse internal architecture, vacuolization, and disorganized membrane morphology. Also, the level of ubiquitinated protein inclusions was increased even before animals displayed measurable muscle weakness. These results indicate that the phenotype of the generated transgenic mouse model resemble the VCP‐associated disease pathology.
We have previously performed expression profiles using Human Genome Array microarray technology in Vastus lateralis muscles from patients and their first‐degree relatives.28 Genes that were differentially expressed in the patients' muscle were analyzed using gene pathways. The global microarray analyses conducted revealed dysregulation mainly of the genes involved in the actin cytoskeleton cascade, involving several critical growth factor receptors, such as FGFR2 and EGFR; and disruption of the autophagy pathway genes, namely ATG4A, mTOR, and FoxO3.28 The signaling intermediates involved in these pathways are known to be important for the crosstalk between protein breakdown and synthesis in IBMPFD disease. Here, we report the first study to investigate the global microarray expression analysis of the VCPR155H/+ murine model for comparison with human VCP‐associated disease. The mice express the mutant VCP at an endogenous level in the correct location of the mouse genome, and therefore, they are believed to represent an ideal model to study the pathology of the disease. It was hoped that by studying mice we might be able to overcome the variation between individuals with VCP disease. Our analyses revealed that VCPR155H/+ mutant mice demonstrate dysregulated genes and cascades which may aid to study the pathological molecular mechanisms underlying VCP‐associated diseases in patients.
Methods
Generation and validation of VCPR155H/+ mice
Mouse genomic VCP fragment with 7.9 kb of upstream homology sequence and 2.1 kb of downstream homology sequence was subcloned into a targeting vector. Site‐directed mutagenesis using Quick‐Change XL Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA) was used to introduce the R to H mutation at amino acid position 155. The VCPR155H/+ mouse model with the R155H VCP mutation was generated at the inGenious Targeting Laboratory, Inc. (Stony Brook, NY, USA), as described previously.29 All experiments were done with the approval of the Institutional Animal Care and Use Committee (IACUC Protocol #2007–2716–2) of the University of California, Irvine (UCI), and in accordance with the guidelines established by the NIH. All animals were housed in the vivarium of the UCI and maintained under constant temperature (22°C) and humidity with a controlled 12:12‐hour light‐dark cycle. Mice had free access to mouse chow (Harlan Teklad Rodent Diet, Madison, WI, USA) and 0.9% NaCl drinking water ad libitum. Mice were euthanized by CO2 inhalation followed by cervical dislocation for all experiments. For PCR genotyping, tail clips were obtained and sent to Transnetyx, Inc. (Cordova, TN, USA) and analyzed by our laboratory. To follow the development of body mass, the weight of every VCPR155H/+ and WT mouse was measured weekly. Motor coordination and fatigue were assessed by Rotarod testing (Med Associates Inc., St. Albans, VT, USA) accelerating speed analysis and muscle strength of the forelimbs of mice was measured by using a Grip Strength Meter apparatus (TSE Systems Gmbh, Hamburg, Germany).29
Histological analyses
The histopathology of VCPR155H/+ mice was analyzed by hematoxylin and eosin (H&E) staining, immunohistochemistry, and electron microscopy. Ten‐month‐old mice (three WT and three VCPR155H/+) were euthanized by cervical dislocation, dissected quadriceps and brain tissues were mounted in a cryosectioning mounting media (Electron Microscopy Sciences, Hatfield, PA, USA) and stored at ‐80°C before sectioning at 5–10 μm. For H&E staining, cryosections were fixed in 10% formalin for 30 minutes and washed three times with PBS. Thereafter, sections were incubated in hematoxylin/glacial acetic acid (Sigma‐Aldrich, St. Louis, MO, USA) solution for 3 minutes, rinsed with deionized water for 5 minutes, dipped 8–12 times into acid ethanol, rinsed two times for 1 minute with tap water, and once with deionized water for 2 minutes. Thereafter, slides were stained with eosin for 30–45 seconds, incubated in 95% ethanol three times for 5 minutes, three times in 100% ethanol for 5 minutes, and finally three times in xylene for 15 minutes. Slides were mounted with Permount (Fisher Scientific, Pittsburgh, PA, USA) and results were analyzed by a light microscope (Carl Zeiss, Oberkochen, Germany) using an AxioVision image capture system as described previously.30
Gene expression profiling
For comparative gene expression profiling, quadriceps muscle samples from three 10‐month‐old WT and R155H mice were frozen in liquid nitrogen and homogenized. Total RNA was extracted using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) following manufacturer's instructions. RNA extractions were further purified by passage through RNeasy minicolumns (Qiagen, Valencia, CA, USA) according to the manufacturer's protocols for RNA cleanup. Final RNA preparations were resuspended in RNase‐free water and stored at 80°C. The quality of RNA was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) monitoring for the S28/S18 ratio and signs for degradation. Concentrations were measured by spectrophotometry. An aliquot of 2 μg of total high‐quality RNA from each sample was used to generate cDNA containing an initiation site for T7 RNA polymerase. cDNA synthesis and in vitro transcription reactions were performed using Affymetrix GeneChip Expression 3'Amplification One‐Cycle Target Labeling and Control Reagent kit according to manufacturer's instructions (Affymetrix, Santa Clara, CA, USA). Double‐stranded cDNA and Biotinylated cRNA were purified by Gene Chip Sample Cleanup Module (Affymetrix). An aliquot of 17 μg of biotinylated cRNAs from samples were fragmented in 1 x fragmentation buffer (40 mM Tris‐acetate, pH 8.1, 100 mM KOAc, 30 mM MgOAc) at 94°C for 35 minutes, of which 1 μL was subjected to an analysis on 1% agarose gel. Ten μg of biotinylated and fragmented cRNA was hybridized to Mouse Genome 430 2.0 arrays (Affymetrix) for 16 hours. The arrays were washed and stained on the Affymetrix Fluidics Station 450, using instructions and reagents provided by Affymetrix. Signal intensity was amplified by second staining with biotin‐labeled antistreptavidin antibody followed by phycoerythrin‐streptavidin restaining. Fluorescent images were captured using the Affymetrix Gene Chip Scanner 3000 7G.
Microarray data analysis
Expression values were calculated using the Probe Logarithmic Intensity Error Estimation (Affymetrix) algorithm. Hierarchical clustering analysis of that data set was performed using the Hierarchical Clustering Explorer 3,31 which shows how samples are clustered, and allows eliminating suspicious samples from the analyses after checking them on contents of fatty and fibrosis tissue. A gene list of statistically significant up‐ and down‐regulated genes with p‐values <0.01 was generated with Gene Spring GX version 7.3.1. (Agilent Technologies), using ANOVA (Welch, t‐test) with standard operating procedures and quality controls. Normalized signals were calculated using Probe Logarithmic Intensity Error, and SDK Version 2 (Affymetrix). CEL‐processed image files were analyzed for that probe set algorithm.
Q‐real‐time polymerase chain reaction (Q‐RT‐PCR) analysis
Gene expression analyses were performed with the Real‐Time system to simultaneously quantify both rare and abundant genes in the same sample. A two‐step approach was used in which the initial reverse transcription was followed by RT‐PCR amplification. Standard TaqMan two‐step thermocycling conditions with the LightCycler 480 were used: 1 cycle at 95°C for 10 minutes, 45 cycles of 15 seconds at 95°C, 1 minute at 60°C, with a PCR volume of 10 μL as per manufacturer's instructions (Roche Applied Science, Indianapolis, IN, USA). All samples were run in duplicate with both test primer sets and the control genes glyceraldehydes‐3‐phosphate‐dehydrogenase for mRNAs. These genes were used to control for differences in the amount of target material. The Relative Quantification ΔΔCt approach was used for the data analysis where the Threshold Cycle (Ct) reflects the cycle number at which the fluorescence generated within a reaction crosses the threshold. The Ct value assigned to a particular well, thus reflects the point during the reaction at which a sufficient number of amplicons have accumulated, in that well, to be at a statistically significant point above the baseline. Fold changes were calculated by normalizing the test crossing thresholds Ct with the amplified controls Ct as a part of the comparative CT method, which uses arithmetic formula for relative quantitation. The amount of target, normalized to an endogenous control, is given by: 2 –ΔΔCT.
Results
VCPR155H/+ mice share similar histopathological features with human patients
Progressive muscle weakness is one of the leading symptoms in human IBMPFD patients.4 Histological analyses of muscle tissues from IBMPFD patients have revealed that patients' tissues accumulate enlarged vacuoles as well as ubiquitin‐ and TDP‐43‐positive inclusion bodies.6 Similar inclusion pathology has also been reported in patients' brain, including neocortex, as well as in limbic and subcortical nuclei.32, 33 The VCPR155H/+ mice quadriceps depict enlarged vacuoles and centrally located nuclei (Figure 1 ) and have been fully characterized as described previously.29, 30
Figure 1.
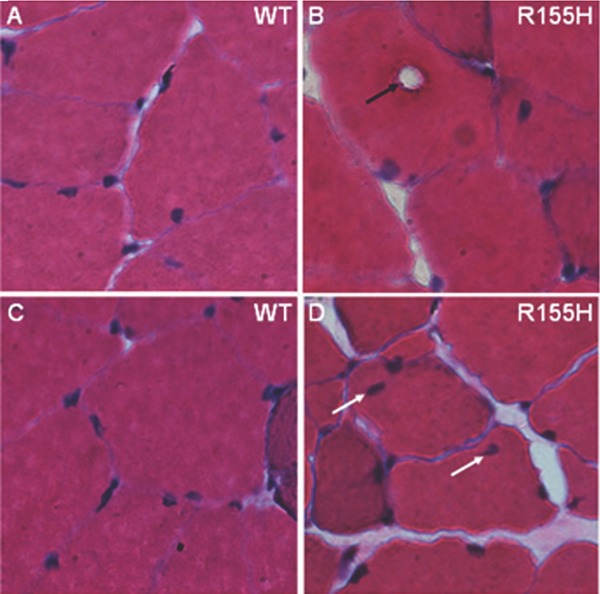
Histological analysis of VCPR155H/+ mice muscle. Quadriceps muscles from 15‐month‐old (A, C) WT and (B, D) VCPR155H/+ mice were analyzed by H&E staining. (Magnification: 630×).
Expression profiling identifies differentially expressed pathways and genes
To obtain some understanding of the consequences of the global gene expression in the mutant skeletal muscle, comparative gene expression profiling was performed in the quadriceps muscle of 10‐month‐old WT and VCPR155H/+ mice. The global expression profiling was performed using Mouse genome 430 2.0 arrays, which have more than 45,000 probe sets. To avoid the influence of estrogen cycle on the expression results, only male mice were selected for the analyses. Significant changes in expression levels were found altogether for 212 genes with p < 0.01, 172 of which were up‐regulated and 40 down‐regulated. Thirty‐nine genes depicted more than a 1.8‐fold change in expressions, 28 of which were up‐regulated and 11 down‐regulated when compared to their WT littermates (Table 1 ). Four genes showed up‐regulation of >3‐fold (killer cell lectin‐like receptor, T‐cell receptor beta, and two unknown transcripts) and six genes showed down‐regulation of >3‐fold (SRY‐box‐containing gene 12, SLAM family member 8, and 4 unknown transcripts).
Table 1.
Dysregulated genes in VCPR155H/+ mouse model
| The most up‐ and down‐regulated genes (>1 .B‐fold change) in the quadriceps muscle of R155H knock‐in mice | ||||
|---|---|---|---|---|
| Fold change | p value | Genbank ID | Gene symbol | Gene description |
| Up‐regulated | ||||
| 5.177 | 0.00477 | BB278809 | Transcribed sequences | |
| 4.773 | 0.00706 | BB375207 | RIKEN cDNA B130035E07 | |
| 4.013 | 0.00961 | AF419250 | Klra6 | Killer cell lectin‐like receptor, subfamily A, member 6 |
| 3.801 | 0.00696 | M31648 | Tcrb‐V13 | T‐cell receptor beta, variable 13 |
| 2.905 | 0.00943 | AV042259 | 493240BB21 | Hypothetical protein 49324Q8B21 |
| 2.400 | 0.00750 | BI736087 | Transcribed hypothetical protein B340B7 | |
| 2.3B5 | 0.00928 | AF087470 | S100a3 | S100 calcium‐binding protein A3 |
| 2.135 | 0.00327 | BE630983 | E43001 9B1 3Rik | RIKEN cDNA E43001 9B1 3 gene |
| 2.1B3 | 0.005B7 | AF397911 | Scml4 | Sex comb on midleg‐like 4 (drosophila) |
| 2.177 | 0.00376 | NM 025741 | 4931412G03Rik | RIKEN CDNA4931412G03 gene |
| 2.124 | 0.00048 | AI647619 | Mus rnusculuscDNA clone IMAGE:1971 621 | |
| 2.066 | 0.00913 | AA415783 | Olfrl 8 | Olfactory receptor 1 8 |
| 2.02B | 0.009B6 | Z31359 | Npn2 | Neoplastic progression 2 |
| 1.993 | 0.00297 | AK020088 | 603049SE09Rik | RIKEN cDNA 603049SE09 gene |
| 1.966 | 0.00073 | BB43B279 | Otx1 | Orthodenticle homolog 1 (drosophila) |
| 1.957 | 0.00104 | BB100151 | RIKEN CDNA9430071J09 | |
| 1.954 | 0.00260 | BG797614 | Transcribed sequences | |
| 1.942 | 0.00366 | AK005952 | 1700013G24Rik | RIKEN cDNA 1 700013G24 gene |
| 1.900 | 0.00243 | BG076799 | PeglO rnRNA for paternally expressed 10 | |
| 1.834 | 0.00203 | AV320422 | L3rnbtl3 | l(3)mbt‐like 3 (drosophila) |
| 1.851 | 0.00146 | W91590 | RIKEN cDNA 5730493B1 9 [mus musculus] | |
| 1.847 | 0.00339 | BI202534 | Nt5c2 | 5'‐nucleotidase, cytosolic II |
| 1.842 | 0.00098 | AVV1 23067 | BC037674 | cDNA sequence BC037674 |
| 1.835 | 0.00469 | AF356B44 | Hrnxl | H6 homeo box 1 |
| 1.832 | 0.00907 | BF3B3739 | Serpina4‐ps1 | Serine (or cysteine) proteinase inhibitor, clade A, member 4, pseudogene 1 |
| 1.820 | 0.00606 | W91754 | Rpl27a | Ribosomal protein L27a |
| 1.817 | 0.00412 | AV2B0058 | 4921517J08Rik | RIKEN CDNA4921517J08 gene |
| 1.813 | 0.005B4 | AV311770 | Hbb‐bh1 | Hemoglobin Z, beta‐like embryonic chain |
| Fold change | p value | Genbank* | Gene symbol | Gene description |
| Down‐regulated | ||||
| –1.876 | 0.00660 | BB787292 | Frk | Fyn‐related kinase |
| –1.908 | 0.00363 | BB750940 | LrpB | Low‐density lipoprotein receptor‐related protein 8, apolipoprotein e receptor |
| –2.068 | 0.00917 | BB241135 | RIKEN CDNAA630085D23 | |
| –2.083 | 0.00482 | BM933756 | Transcribed sequence with strong similarity to protein sp:P00722 | |
| (E. coli) B6AL ECOLI Beta‐galactosidase | ||||
| –2.105 | 0.00935 | BB213608 | RIKEN CDNAA530012F11 | |
| –3.731 | 0.00935 | BC024587 | SlatnfS | SLAM family mem tier 8 |
| –3.861 | 0.00843 | BG070486 | Mus niuscLlus cDNA clone H3087H04 | |
| –7.813 | 0.00768 | AM018140 | C330002l19Rik | RIKEN cDNA 6330407M16 |
| –10.04 | 0.00046 | BQ180104 | Sox12 | SRY‐box‐containing gene 12 |
| –699.3 | 0.00628 | NM 053233 | V1rc3 | Go component: cellular component unknown [gcid 0008372] |
| –1,194.7 | 0.00424 | AW209946 | E130112H22Rik | RIKEN cDNA E1 301 12H22 gene |
To elucidate the biological processes associated with the observed transcriptional profiles, Ingenuity Pathway analysis (Affymetrix) was applied to dysregulated genes with highly significant differences (p < 0.01) between WT and VCPR155H/+ samples. The physiological system development and function (endocrine system development and function, nervous system development and function, tissue development, embryonic development, hematological system development and function) contained 31% of the dysregulated genes, 27.9% of genes were associated with diseases and disorders (genetic disorders, neurological diseases, respiratory diseases, renal and urological diseases, developmental disorders), and 26% of dysregulated genes were associated with different molecular and cellular functions (molecular transport, small molecule biochemistry, lipid metabolism, cell‐to‐cell signaling and interaction, and carbohydrate metabolism; Table 2 ). Further analyses revealed several genes within these biological processes showing differences in expression levels between WT and VCPR155H/+ mice, although not all of these changes reached the >1.8‐fold cutoff level (data not shown).
Table 2.
Ingenuity pathway analysis in VCPR155H/+ mouse model microarray
| Ingenuity pathway analysis of 212 dysregulated genes with p < 0.01 from the quadriceps muscle of R155H knock–in mice | |||
|---|---|---|---|
| Physiological system development and function | |||
| Name | p value | No. of molecules | % of 212 genes |
| Endocrine system development and function | 1.89–04–4.70–02 | 6 | 2.8 |
| Nervous system development and function | 9.33–04–4.70–02 | 22 | 10.4 |
| Tissue development | 9.33–04–4.95–02 | 14 | 6.6 |
| Embryonic development | 1.72–03–4.06–02 | 11 | 5.2 |
| Hematological system development and function | 3.93–03–4.83–02 | 14 | 6.6 |
| Total | 67 | 31.6% | |
| Diseases and disorders | |||
| Name | p value | No. of molecules | % of 212 genes |
| Genetic disorder | 9.33–04–4.70–02 | 21 | 9.9 |
| Neurological disease | 9.33–04–4.83–02 | 15 | 7.1 |
| Respiratory disease | 9.33–04–4.73–02 | 6 | 2.8 |
| Renal and urological disease | 1.30–03–3.16–02 | 5 | 2.4 |
| Developmental disorder | 1.74–03–4.05–02 | 12 | 5.7 |
| Total | 59 | 27.9% | |
| Molecular and cellular functions | |||
| Name | p value | No. of molecules | % of 212 genes |
| Molecular transport | 6.35–05–4.83–02 | 11 | 5.2 |
| Small molecule biochemistry | 6.35–05–4.83–02 | 13 | 6.1 |
| Lipid metabolism | 1.89–04–4.70–02 | 5 | 2.4 |
| Cell–to–cell signaling and interaction | 9.52–04–4.70–02 | 18 | 8.5 |
| Carbohydrate metabolism | 1.30–03–4.83–02 | 8 | 3.8 |
| Total | 55 | 26.0% | |
Finally, we generated Ingenuity networks using dysregulated genes in the quadriceps samples from R155H mice. Network 1 included 24 dysregulated genes with score 49 (amino acid metabolism, molecular transport, small molecule biochemistry). Network 1 includes several differentially expressed genes, which are involved in the regulation of signaling pathways, NF‐κB and ERK, as well as proteasomal function cascades (Figure 2 ). Network 2 included 13 genes with score 21 (cellular development, nervous system, development and function, cellular growth and proliferation), 12 genes belonged to network 3 with score 20 (developmental disorder, genetic disorder, cell cycle), 12 genes belonged to network 4 with score 20 (cellular assembly and organization, cellular function and maintenance, endocrine system development and function), and 12 dysregulated genes belonged to network 5 with score 20 (lipid metabolism, molecular transport, small molecule biochemistry).
Figure 2.
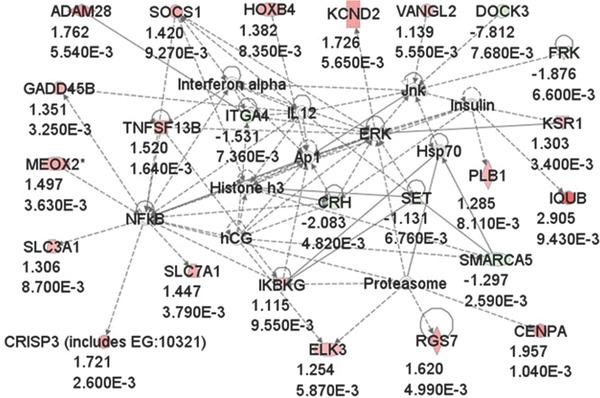
Ingenuity network 1 of the differentially expressed genes. A total of 24 out of 212 dysregulated genes at p < 0.01 belonged to network 1, which includes a number of genes that are involved in the regulation of proteasomal function and NF‐κB signaling. Up‐regulated genes are shown in red and down‐regulated genes in green. Fold changes and p values are indicated below gene names.
Verification of dysregulated genes
Findings of the gene expression study were validated by using quantitative reverse transcriptase polymerase chain reaction analyses to test genes involved in various signaling cascades (Figure 3 ; Table 3 ). These genes S100A3 (S100 calcium‐binding protein A3, up‐regulated 4.19‐fold change), KLRA (killer cell lectin‐like receptor, subfamily A, member 6, up‐regulated 6.574‐fold change), ITGA (integrin alpha 4, down‐regulated ‐7.679‐fold change), LRP8 (low density lipoprotein receptor‐related protein 8, down‐regulated ‐8.463‐fold change), CORO1A (coronin, actin‐binding protein 1A, down‐regulated ‐8.618‐fold change), and SLAMf8 (SLAM family member 8, down‐regulated ‐2.294‐fold change) chosen from our microarray results, represent both increased and decreased expression were selected according to the p values and the FC values for the differentially expressed sequences obtained in the comparisons and statistically significant (p < 0.05; Figure 3 ).
Figure 3.
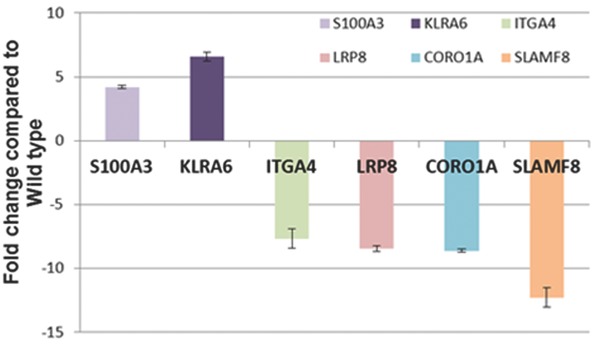
Verification by QPCR of dysregulated genes from VCPR155H/+ mouse model global microarray. Findings of the gene expression study were validated by using quantitative reverse transcriptase polymerase chain reaction analyses to test genes involved in various signaling cascades (S100A3, KLRA, ITGA, LRP8, CORO1A, and SLAMf8).
Table 3.
Q‐PCR analysis of dysregulated genes from mouse model microarray
| Affy ID | Fold change | p value | Common name | Genbank | RefSeq transcript | Gene description |
|---|---|---|---|---|---|---|
| 1452645_x_at | 4.0 | 0.00961 | Klra6 | AF419250 | NM_008464 | Killer cell lectin‐like receptor, subfamily A, member 6 |
| 1427765_a_at | 3.8 | 0.00696 | Tcrb‐V13 | M31648 | — | T‐cell receptor beta, variable 13 |
| 1421856_at | 2.4 | 0.00928 | S100a3 | AF087470 | NM_011310 | S100 calcium‐binding protein A3 |
| 1450155_at | –1.5 | 0.00736 | Itga4 | NM_010576 | NM_010576 | Integrin alpha 4 |
| 1435288_at | –1.7 | 0.00376 | Coro1a | BB740218 | NM_009898 | Coronin, actin‐binding protein 1A |
| 1426570_a_at | –1.9 | 0.0066 | Frk | BB787292 | NM_010237 | Fyn‐related kinase |
| 1440882_at | –1.9 | 0.00363 | Lrp8 | BB750940 | NM_053073 | Low‐density lipoprotein receptor‐related protein 8, apo lipoprotein e receptor |
| 1425294_at | –3.7 | 0.00977 | Slamf8 | BC024587 | — | SLAM family member 8 |
Comparison of mouse and human microarray data
Corelations were performed between the altered genes in the mouse and human muscle studies. In view of the similarity of histopathological features in the VCPR155H/+ mice and VCP patients, we compared the results from the mouse studies with microarray studies in muscle as reported by Nalbandian et al. (2012).28 Pathways that were modified in humans with VCP disease included regulation of actin cytoskeleton, ErbB signaling, cancer, in addition to regulation of autophagy and lysosomal signaling28 and in heterozygous mice proteasomal and NF‐κB signaling as well as genes involved in physiological system development and function. Common genes from both human and mouse studies included: TROAP, F2RL2, GLI2 PHOX2A ANKRD10, and MMD2, and we did not find a consistent pattern across genes that depicted common trends of dysregulation (data not shown).
Discussion
VCP is involved in a number of cellular functions, most of which are related to ubiquitin‐proteasome‐dependent proteolysis of proteins that is mediated by interaction with ubiquitinated polypeptide substrates.21, 34, 35, 36, 37 It is highly conserved in evolution suggesting that it is essential for normal cellular functions by playing a fundamental role within both unicellular (yeast) and multicellular organisms.38, 39, 40 More recently, VCP mutations have been implicated in causing fALS cases. These observations, as well as the finding that inhibition of VCP expression promotes apoptosis,41 suggest that intact VCP is indispensable for normal development and cell survival. Despite intense investigations, the disease molecular mechanisms underlying VCP‐associated diseases remain to be clarified.
To further elucidate tissue and cellular pathology of IBMPFD, we generated and characterized the VCPR155H/+ murine model of VCP‐associated diseases.29 The heterozygous mutation did not affect the fertility or viability of mice since mutant and WT mice were born in Mendelian ratio. These observations also revealed that the more mutant VCP molecules are added to hexamers the more the complex functioning is affected resulting eventually in totally nonfunctional protein complexes. These deficiencies in hexameric functions cannot be compensated by other molecules or protein complexes, which lead to lethality already in embryogenesis.
Although body weight and daily activities of heterozygous VCPR155H/+ mice were not seen to differ from those of WT mice, mutant mice demonstrated several phenotypic and histological changes, all of which are typical of human IBMPFD patients. Muscle strength of forelimbs showed progressive deterioration when measured by grip strength test. Similar progressive weakness has been reported in patients1, 2, 42 and transgenic mice expressing VCPR155H/+.43 Consistently with the transgenic mice and human patients, Badadani et al. (2010)29 demonstrated that the VCPR155H/+ mice did not exhibit weakness in early adulthood (3 months), but only in the later stage of development (from 6 months onward) showing progressive deteriorating of muscle strength.29 Physical performance was also decreased in these VCPR155H/+ mice when tested by Rotarod, further suggesting that the R155H mutation results in accelerated fatigue due to decreased muscle strength. Although no cerebellar dysfunction has been reported in IBMPFD patients, we cannot exclude the possibility of the involvement of defects in the cerebellum of mutant mice because Rotarod also measures the coordination of physical performance, which is controlled by cerebellum, in addition to muscular fatigue. Watts et al. (2007)27 reported severe degeneration of hippocampal region in a postmortem examination of a patient who died at age 52 years,27 and Forman et al. (2006)32 found striking atrophy of the hippocampus in a patient with the R155H mutation. These findings combined with the observation that VCP is abundantly expressed in hippocampus44 propose an important role for VCP in the proper functioning of this brain region, and encourage us to begin more detailed studies of IBMPFD brain focusing on the hippocampal regions. Previous histological analyses, immunohistochemistry, and electron microscopy has shown vacuolization as well as accumulation of ubiquitin‐ and TDP‐43‐positive inclusions in muscle fibers from the quadriceps of VCPR155H/+ mice. Histological analyses of IBMPFD muscle biopsies have shown similar TDP‐43‐ and ubiquitin‐positive sarcoplasmic inclusions6 suggesting that the muscle histopathology of the generated knock‐in mouse model closely resembles that of human IBMPFD patients. These results were consistent with those obtained from the analyses of R155H transgenic mice showing ubiquitin‐positive inclusions bodies.43 VCPR155H/+ mouse muscle also demonstrated increased number of regenerating muscle fibers, which were identified by central nucleation. Although the mutant muscle clearly showed similar defects to human patients, the brain histology of VCPR155H/+ mice seemed to be indistinguishable from the staining patterns of their WT littermates. This may be due to the young age of the mice analyzed (10 months of age). Eighty‐seven percent of patients develop muscle weakness at the mean age of 42 years, whereas only 27% of patients develop FTD at the mean age of 57 years.4 Therefore, it is likely that brain pathology of VCPR155H/+ mice is not histologically detectable before mutant mice reach the age of 18 to 24 months.
Muscle damage was further confirmed by electron microscopic studies of the VCPR155H/+ muscle. Myofilament structures showed disorganization and vacuolization as well as abnormally swollen mitochondria, which is a typical feature of pathophysiological processes45, 46, 47 and suggests that energy metabolism of affected muscle may be disturbed. It can also be assumed that observed increased autophagosome formation in mutant muscle tissue plays a role in muscular dysfunction and cell death. Autophagy is a process that functions as a stress response that is up‐regulated by starvation, oxidative stress, or other harmful conditions. It plays a role in programmed cell death and possesses important housekeeping and quality control functions that contribute to health and longevity (for review, see Bergamini et al.,48 Galluzzi et al.,49 and Scarlatti et al.50). In addition, impaired autophagic degradation contributes to the pathogenesis of several human diseases including muscle diseases and lysosomal storage disorders. Danon disease is an example of a human myopathy that is characterized by accumulation of autophagic vacuoles in the heart and skeletal muscle.51 Mouse model for the disease exhibits similar histological and phenotypic features to human patients showing massive accumulation of autophagic vacuoles in several tissues, including skeletal muscle and heart.52 Mitochondria is a main target for autophagic degradation in muscle diseases resulting in reduced mitochondrial function, which leads to defective energy metabolism and reduced contractility.53 Therefore, increased autophagosome formation in VCPR155H/+ mice may be involved in the increased apoptosis and muscle weakness, which can be due to affected lysosomes‐autophagosome fusion or impaired autophagosome function.
Gene expression profiling of the VCPR155H/+ mice revealed changes on the steady state expression level for several genes that are involved in ubiquitin‐proteasome system. The up‐regulation of these genes (RGS7/regulator of G protein signaling 7, ELK3/member of ETS oncogene family, IKBKG/inhibitor of kappaB kinase gamma, CENPA/unknown transcript) is of great interest because the main function of VCP is believed to be the translocation of defective proteins to proteasomes. These results suggest that proteasome‐related proteins attempt to compensate the dysfunctional ubiquitin‐proteasome system. Defective functioning of VCP has been shown to result in accumulation of ubiquitin‐positive inclusions both in human and mouse.43, 54 These findings were consistent with our results further confirming that the generated VCPR155H/+ mice represent a factual model for human disease. Another interesting finding in expression profiling was the dysregulation of genes which are related to NFκB signaling (SLC7A1/solute carrier family 7, member 1, CRISP3/unknown transcript, SLC3A1/solute carrier family 3, member 1, MEOX2/mesenchyme homeobox 2, GADD45B/unknown transcript, TNFSF13B/tumor necrosis factor superfamily, member 13b, IKBKG/inhibitor of kappaB kinase gamma).
VCP has been shown to regulate NF‐κB signaling pathways by influencing the degradation process of cytoplasmic p‐IκBα, which has an antiapoptotic effect in cells.55 Therefore, the increased apoptosis observed in VCPR155H/+ mouse muscle may, at least partially, be explained by defective NF‐κB signaling. The third interesting finding in expression profiling is the up‐regulation of T‐cell receptor beta (TRB), which is, in general, responsible for recognizing antigens bound to major histocompatibility complex molecules. Microarray analyses of other human myopathies (Inclusion body myositis, polymyositis, and dermatomyositis) demonstrate similar pronounced up‐regulation of TRB in patients' muscle biopsies. More recently, Nalbandian et al. (2012) performed the first global microarray analyses from patients' muscles with VCP mutations versus their first‐degree unaffected and nonmutation carrying relatives.28 This study elucidated several dysregulated signaling transduction pathways in VCP‐associated diseases, including regulation of actin cytoskeleton, regulation of autophagy, ubiquitin/lysosomal, and cancer pathways.28 In our comparative study between human and mouse microarrays, we did not find a consistent pattern across genes that depicted common trends of dysregulation. We studied the literature to find other investigations where microarray was performed in human disease and comparisons made with tissue from animal models of the same disease. Zheng‐Bradley et al. (2010) analyzed data for each normal tissue in humans and mouse separately and found that the global patterns of tissue‐specific expression of orthologous genes were conserved in human and mouse.56 The most variable genes in each tissue were highly enriched with human‐mouse tissue‐specific orthologs and the least variable genes in each tissue were human‐mouse housekeeping orthologs.56 Another study by Chopra et al. (2013) investigated a comparative meta‐analysis between human and mouse cancer microarray revealing critical pathways disrupted in both human and mouse cancers.57 Watchmaker et al. investigated the transcriptional and functional profiling in human and mouse intestinal dendritic cell subsets and identified a few transcription factors whose expression was coordinately regulated, however, several factors represented divergence.58 Similarly, in a study by Turk et al. (2005) in mdx mice and Duchenne muscular dystrophy patient muscle resembled our findings.59 A limited number of overlapping genes were identified, with a number of genes showing opposite expression, suggested that processes active in regenerating mouse muscle are not active in human patients at the time gene expression was profiled, differences in regeneration efficiency and lethal manifestation of the pathology between patients and murine muscles.59
In conclusion, this is the first study to investigate the signaling intermediates and transduction cascades in understanding VCP‐associated disease in an excellent mouse model. Future studies will examine these dysregulated pathways and genes in both patients and VCPR155H/+ animals to elucidate and understand the pathological mechanisms underlying VCP‐associated diseases in hopes of developing novel therapeutic interventions for these currently untreatable neurodegenerative diseases. Further investigations are underway to correlate human and mouse global microarray studies in hopes of better understanding of the pathological cellular and molecular mechanisms underlying VCP‐related neurodegenerative disorders.
Conflict of Interest
The authors have no personal financial or institutional interest or conflicts in any of the drugs, materials, or devices as described in this paper.
Acknowledgments
We are extremely grateful to the CNMC Rehabilitation Core, Children's National Medical Center at Research Center Genetics Medicine in Washington, DC. We thank Ms. Laura Tang for excellent technical assistance. This work was supported by NIH grant AR050236 (V. E. K.) and Muscular Dystrophy Association MDA 175682 (V.E.K.), and MDA training awards (Z.W. and J.V.).
References
- 1. Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, Davis K, Khardori R, Gelber D. Clinical and molecular studies in a unique family with autosomal dominant limb‐girdle muscular dystrophy and Paget disease of bone. Genet Med. 2000; 2: 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, Shanks CA, Gregg G, Al‐Lozi MT, Miller T, et al. Clinical delineation and localization to chromosome 9p13.3‐p12 of a unique dominant disorder in four families: hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. 2001; 74: 458–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Watts GD, Thorne M, Kovach MJ, Pestronk A, Kimonis VE. Clinical and genetic heterogeneity in chromosome 9p associated hereditary inclusion body myopathy: exclusion of GNE and three other candidate genes. Neuromuscul Disord. 2003; 13: 559–567. [DOI] [PubMed] [Google Scholar]
- 4. Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008; 146A: 745–757. doi:10.1002/ajmg.a.31862. [DOI] [PubMed] [Google Scholar]
- 5. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat Genet. 2004; 36: 377–381. [DOI] [PubMed] [Google Scholar]
- 6. Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP‐43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosur Ps. 2008; 79: 1186–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ralston SH. Pathogenesis of Paget's disease of bone. Bone. 2008; 43: 819–825. [DOI] [PubMed] [Google Scholar]
- 8. Ralston SH. Clinical practice. Paget's disease of bone. New Engl J Med. 2013; 368: 644–650. [DOI] [PubMed] [Google Scholar]
- 9. Turner RS, Kenyon LC, Trojanowski JQ, Gonatas N, Grossman M. Clinical, neuroimaging, and pathologic features of progressive nonfluent aphasia. Ann Neurol. 1996; 39: 166–173. [DOI] [PubMed] [Google Scholar]
- 10. Arnold SE, Han LY, Clark CM, Grossman M, Trojanowski JQ. Quantitative neurohistological features of frontotemporal degeneration. Neurobiol Aging. 2000; 21: 913–919. [DOI] [PubMed] [Google Scholar]
- 11. Zhukareva V, Vogelsberg‐Ragaglia V, Van Deerlin VM, Bruce J, Shuck T, Grossman M, Clark CM, Arnold SE, Masliah E, Galasko D, et al. Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol. 2001; 49: 165–175. [DOI] [PubMed] [Google Scholar]
- 12. Confalonieri F, Duguet M. A 200‐amino acid ATPase module in search of a basic function. Bioessays. 1995; 17: 639–650. [DOI] [PubMed] [Google Scholar]
- 13. Patel S, Latterich M. The AAA team: related ATPases with diverse functions. Trends Cell Biol. 1998; 8: 65–71. [PubMed] [Google Scholar]
- 14. Zwickl P, Baumeister W. AAA‐ATPases at the crossroads of protein life and death. Nat Cell Biol. 1999; 1: E97–E98. [DOI] [PubMed] [Google Scholar]
- 15. Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells. 2001; 6: 575–597. [DOI] [PubMed] [Google Scholar]
- 16. Wang Q, Song C, Li CC. Molecular perspectives on p97‐VCP: progress in understanding its structure and diverse biological functions. J Struct Biol. 2004; 146: 44–57. [DOI] [PubMed] [Google Scholar]
- 17. Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Syntaxin 5 is a common component of the NSF‐ and p97‐mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998; 92: 603–610. [DOI] [PubMed] [Google Scholar]
- 18. Hetzer M, Meyer HH, Walther TC, Bilbao‐Cortes D, Warren G, Mattaj IW. Distinct AAA‐ATPase p97 complexes function in discrete steps of nuclear assembly. Nat Cell Biol. 2001; 3: 1086–1091. [DOI] [PubMed] [Google Scholar]
- 19. Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar‐Nun S. AAA‐ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum‐associated protein degradation. Mol Cell Biol. 2002; 22: 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jarosch E, Geiss‐Friedlander R, Meusser B, Walter J, Sommer T. Protein dislocation from the endoplasmic reticulum–pulling out the suspect. Traffic (Copenhagen, Denmark). 2002; 3: 530–536. [DOI] [PubMed] [Google Scholar]
- 21. Dai RM, Li CC. Valosin‐containing protein is a multi‐ubiquitin chain‐targeting factor required in ubiquitin‐proteasome degradation. Nat Cell Biol. 2001; 3: 740–744. [DOI] [PubMed] [Google Scholar]
- 22. Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S. Mobilization of processed, membrane‐tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin‐selective chaperone. Cell. 2001; 107: 667–677. [DOI] [PubMed] [Google Scholar]
- 23. Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. p47 is a cofactor for p97‐mediated membrane fusion. Nature. 1997; 388: 75–78. [DOI] [PubMed] [Google Scholar]
- 24. Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA‐ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000; 19: 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muller JM, Meyer HH, Ruhrberg C, Stamp GW, Warren G, Shima DT. The mouse p97 (CDC48) gene. Genomic structure, definition of transcriptional regulatory sequences, gene expression, and characterization of a pseudogene. J Biol Chem. 1999; 274: 10154–10162. [DOI] [PubMed] [Google Scholar]
- 26. Muller JM, Deinhardt K, Rosewell I, Warren G, Shima DT. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Bioph Res Co. 2007; 354: 459–465. [DOI] [PubMed] [Google Scholar]
- 27. Watts GD, Thomasova D, Ramdeen SK, Fulchiero EC, Mehta SG, Drachman DA, Weihl CC, Jamrozik Z, Kwiecinski H, Kaminska A, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet. 2007; 72: 420–426. [DOI] [PubMed] [Google Scholar]
- 28. Nalbandian A, Ghimbovschi S, Radom‐Aizik S, Dec E, Vesa J, Martin B, Knoblach S, Smith C, Hoffman E, Kimonis VE. Global gene profiling of VCP‐associated inclusion body myopathy. Clin Transl Sci. 2012; 5: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Badadani M, Nalbandian A, Watts GD, Vesa J, Kitazawa M, Su H, Tanaja J, Dec E, Wallace DC, Mukherjee J, et al. VCP associated inclusion body myopathy and Paget disease of bone knock‐in mouse model exhibits tissue pathology typical of human disease. PLoS ONE 2010; 5(10): pii: e13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, Watts G, Mukherjee J, Vesa J, Caiozzo V, et al. A progressive translational mouse model of human valosin‐containing protein disease: the VCP(R155H/+) mouse. Muscle Nerve. 2013; 47: 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seo J, Hoffman EP. Probe set algorithms: is there a rational best bet? BMC Bioinformatics. 2006; 7: 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin‐containing protein gene mutations. J Neuropath Exp Neur. 2006; 65: 571–581. [DOI] [PubMed] [Google Scholar]
- 33. Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP‐43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropath Exp Neur. 2007; 66: 152–157. [DOI] [PubMed] [Google Scholar]
- 34. Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001; 414: 652–656. [DOI] [PubMed] [Google Scholar]
- 35. Bays NW, Hampton RY. Cdc48‐Ufd1‐Npl4: stuck in the middle with Ub. Curr Biol. 2002; 12: R366–R371. [DOI] [PubMed] [Google Scholar]
- 36. Fu X, Ng C, Feng D, Liang C. Cdc48p is required for the cell cycle commitment point at start via degradation of the G1‐CDK inhibitor Far1p. J Cell Biol. 2003; 163: 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005; 120: 73–84. [DOI] [PubMed] [Google Scholar]
- 38. Frohlich KU, Fries HW, Rudiger M, Erdmann R, Botstein D, Mecke D. Yeast cell cycle protein CDC48p shows full‐length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation, and gene expression. J Cell Biol. 1991; 114: 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leon A, McKearin D. Identification of TER94, an AAA ATPase protein, as a Bam‐dependent component of the Drosophila fusome. Mol Biol Cell. 1999; 10: 3825–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamb JR, Fu V, Wirtz E, Bangs JD. Functional analysis of the trypanosomal AAA protein TbVCP with trans‐dominant ATP hydrolysis mutants. J Biol Chem. 2001; 276: 21512–21520. [DOI] [PubMed] [Google Scholar]
- 41. Wojcik C, Yano M, DeMartino GN. RNA interference of valosin‐containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome‐dependent proteolysis. J Cell Sci. 2004; 117: 281–292. [DOI] [PubMed] [Google Scholar]
- 42. Kimonis VE, Watts GD. Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alz Dis Assoc Dis. 2005; 19(Suppl 1): S44–S47. [DOI] [PubMed] [Google Scholar]
- 43. Weihl CC, Miller SE, Hanson PI, Pestronk A. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum Mol Genet. 2007; 16: 919–928. [DOI] [PubMed] [Google Scholar]
- 44. Schröder R, Watts GD, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR. Mutant valosin‐containing protein causes a novel type of frontotemporal dementia. Ann Neurol. 2005; 57: 457–461. [DOI] [PubMed] [Google Scholar]
- 45. Farber JL. Mechanisms of cell injury by activated oxygen species. Environ Health Persp. 1994; 102(Suppl 10): 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bernardi P. Mitochondria in muscle cell death. Ital J Neurol Sci. 1999; 20: 395–400. [DOI] [PubMed] [Google Scholar]
- 47. Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G. Endoplasmic reticulum stress‐induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002; 9: 465–467. [DOI] [PubMed] [Google Scholar]
- 48. Bergamini E, Cavallini G, Donati A, Gori Z. The role of autophagy in aging: its essential part in the anti‐aging mechanism of caloric restriction. Ann NY Acad Sci. 2007; 1114: 69–78. [DOI] [PubMed] [Google Scholar]
- 49. Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. 2008; 8: 78–91. [DOI] [PubMed] [Google Scholar]
- 50. Scarlatti F, Granata R, Meijer AJ, Codogno P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2009; 16: 12–20. [DOI] [PubMed] [Google Scholar]
- 51. Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep. 2003; 3: 64–69. [DOI] [PubMed] [Google Scholar]
- 52. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann‐Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP‐2‐deficient mice. Nature. 2000; 406: 902–906. [DOI] [PubMed] [Google Scholar]
- 53. Stypmann J, Janssen PM, Prestle J, Engelen MA, Kögler H, Lüllmann‐Rauch R, Eckardt L, von Figura K, Landgrebe J, Mleczko A, et al. LAMP‐2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res Cardiol. 2006; 101: 281–291. [DOI] [PubMed] [Google Scholar]
- 54. Greenberg SA. A gene expression approach to study perturbed pathways in myositis. Curr Opin Rheumatol. 2007; 19: 536–541. [DOI] [PubMed] [Google Scholar]
- 55. Asai T, Tomita Y, Nakatsuka S, Hoshida Y, Myoui A, Yoshikawa H, Aozasa K. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J Cancer Res. 2002; 93: 296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng‐Bradley X, Rung J, Parkinson H, Brazma A. Large scale comparison of global gene expression patterns in human and mouse. Genome Biol. 2010; 11: R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chopra P, Kang J, Hong SM. Comparative meta‐analysis between human and mouse cancer microarray data reveals critical pathways. Int J Data Mining Bioinformatics. 2013; 8: 349–365. [DOI] [PubMed] [Google Scholar]
- 58. Watchmaker PB, Lahl K, Lee M, Baumjohann D, Morton J, Kim SJ, Zeng R, Dent A, Ansel KM, Diamond B, et al. Comparative transcriptional and functional profiling defines conserved programs of intestinal DC differentiation in humans and mice. Nat Immunol. 2014; 15: 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Turk R, Sterrenburg E, de Meijer EJ, van Ommen GJ, den Dunnen JT, t Hoen PA. Muscle regeneration in dystrophin‐deficient mdx mice studied by gene expression profiling. BMC Genomics 2005; 6: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]