

Abstract
Aims
Alcohol problems (AP) contribute substantially to the global disease burden. Twin and family studies suggest that AP are genetically influenced, though few studies have identified variants or genes that are robustly associated with risk. This study identifies genetic and genomic influences on AP during young adulthood, which is often when drinking habits are established.
Design
We conducted a genome-wide association study of AP. We further conducted gene-based tests, gene ontology analyses, and functional genomic enrichment analyses to assess genomic factors beyond single variants that are relevant to AP.
Setting
The Avon Longitudinal Study of Parents and Children, a large population-based study of a UK birth cohort.
Participants
Genetic and phenotypic data were available for 4304 participants.
Measurements
The AP phenotype was a factor score derived from items from the Alcohol Use Disorders Identification Test, symptoms of DSM-IV alcohol dependence, and three additional problem-related items.
Findings
One variant met genome-wide significance criteria. Four out of 22,880 genes subjected to gene-based analyses survived a stringent significance threshold (q< .05); none of these have been previously implicated in alcohol-related phenotypes. Several biologically plausible gene ontologies were statistically over-represented among implicated SNPs. SNPs on the Illumina 550K SNP chip accounted for ~5% of the phenotypic variance in AP.
Conclusions
Genetic and genomic factors appear to play a role in alcohol problems in young adults. Genes involved in nervous system-related processes, such as signal transduction and neurogenesis, potentially contribute to liability to alcohol problems, as do genes expressed in non-brain tissues.
Keywords: alcohol problems, ALSPAC, GWAS, polygenic
Introduction
Alcohol problems (AP) represent a significant public health concern with immense personal, social, and economic costs [1]. Twin and family studies have consistently suggested that alcohol-related phenotypes are genetically influenced, with heritability estimates for alcohol abuse and dependence estimated at ~50–60% [2–4]; estimates for other alcohol-related phenotypes are sometimes lower [5–7]. The heritability of alcohol use typically increases beyond adolescence [8, 9]. Alcohol-related phenotypes are complex genetic traits [10, 11], influenced by hundreds to thousands of genetic variants, each of small effect; these genetic factors are likely to interact with each other and the environment to impact risk, and to be heterogeneous [2], with phenotypically similar individuals harboring genetic risk variants with potentially little overlap.
Though linkage and candidate gene studies have identified genomic regions and genetic variants associated with liability to AP, these findings have been inconsistently replicated, with the exception of variants in ADH and ALDH genes [12, 13]. More recently, numerous genome-wide association studies (GWAS) have been conducted on alcohol-related phenotypes (e.g., [14–25]). Single nucleotide polymorphisms (SNPs) that meet stringent genome-wide significance criteria are seldom identified in these studies. Although small sample sizes almost certainly contribute to the paucity of “significant” results, the complex genomic nature of alcohol-related phenotypes likely also plays a role.
Thus, studies that adopt a more comprehensive approach, considering not just significant SNP-level findings but also suggestive evidence for a role of individual genes or enrichment for gene ontologies, could be critical in furthering our understanding of the etiology of AP. Rather than focusing on individual SNPs, SNP-level data can be used to conduct gene-based tests [26], which combine evidence across multiple SNPs; test for enrichment of gene ontologies; and investigate whether genes expressed in different tissue types are of particular relevance to AP. These approaches can be complemented by analyses of the aggregate effects of risk variants.
Here, we apply a comprehensive approach to the investigation of genomic influences on AP in a population-based sample of emerging adults in the United Kingdom. This is a critical time frame for the establishment of drinking behaviors [27] and the development of alcohol use disorders [28]. We use GWAS results to identify genes and gene ontologies likely to play a role in the etiology of AP. We further test whether the implicated SNPs map to hypothesized regions of regulatory significance across a variety of tissues. Finally, we examine the aggregate effects of common variants.
Materials and Methods
Sample
The Avon Longitudinal Study of Parents and Children (ALSPAC) total sample included 15,247 pregnancies from women residing in Avon, UK with expected due dates between April 1991 and December 1992, resulting in 15,458 fetuses. Of this total sample, 14,775 were live births and 14,701 were alive at 1 year of age. Additional details are available from [29]. Please note that the study website contains details of all the data that is available through a fully searchable data dictionary (http://www.bris.ac.uk/alspac/researchers/data-access/data-dictionary/). Ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee, Bristol University, and Virginia Commonwealth University.
Phenotype
Using data collected at the age 17y9mo clinic assessment (referred to as age 18 hereafter), we derived an alcohol problems factor score, as previously described [30]. Briefly, participants were administered (via computer) 10 items from the Alcohol Use Disorders Identification Test [31] along with 7 items aimed at assessing DSM-IV [32] symptoms of alcohol dependence. Three additional measures (getting into fights, police involvement, and drinking to alleviate withdrawal symptoms) were also included. To improve sample size, we used IVEware [33] to impute age 18 alcohol problems data for participants who completed the AUDIT at age 16y6mo, but not the age 18 assessment (N=1993). See Supplementary Material for additional information. Frequency and correlation checks after imputation showed that all imputations kept similar frequency distributions and that imputed and original variables were closely correlated. Factor scores were created for 5952 participants after imputation, using Mplus 6.11 [34]. Of these, genetic data were available for 4304 individuals after quality control screens were applied (see below).
Genotyping
Samples were genotyped using the Illumina HumanHap550 quad genome-wide SNP genotyping platform as previously described [35]. Individuals were excluded from analyses on the basis of excessive or minimal heterozygosity, gender mismatch, individual missingness (3%), cryptic relatedness as measured by identity by descent (genome-wide IBD 10%) and sample duplication. Individuals were assessed for population stratification using multi-dimensional scaling modeling seeded with HapMap Phase II release 22 reference populations, and those of non-European ancestry were excluded from further analysis [35]. SNPs with a final call rate of <95%, minor allele frequency <1% and evidence of departure from Hardy–Weinberg equilibrium (p<5 × 10−7) were also excluded from analyses. Individuals were imputed to 1000 Genomes Phase 1 Version 3, using MACH for phasing and Minimac for imputation. Imputed polymorphisms with an r-square imputation quality metric of <.5 were excluded from further analyses. The genome-wide association analysis was conducted using MACH2QTL [36], including sex as a covariate. After quality controls were applied, genetic data were available for 4304 of the individuals with phenotypic data (42.9% male).
Gene-based analyses
We used KGG 2.5 [37, 38] to conduct gene-based tests of GWAS results. We used publicly available 1000 Genomes Phase 1 Version 3 (European subsample) linkage disequilibrium (LD) files to build the “analysis genome” by position, for autosomes only. We included extended gene lengths of 5kb at the 5’ and 3’ ends. SNPs in high LD (r2>.9) were considered connected; those in low LD (r2<.02) were considered independent. We used the HYST test option. A Benjamini and Hochberg [39] false discovery rate (FDR) of 0.05 was used, and q-values are reported where appropriate.
Gene set enrichment analyses
We used i-GSEA4GWAS [40] to assess enrichment across canonical pathways (defined in [40]) and gene ontologies, using only measured SNPs; of those submitted, 235,574 variants mapped to within 5kb of 16,296 genes and were included in the analysis. We set the minimum number of genes per category to 3, and the maximum to 200.
Epigenetic enrichment analyses
We utilized publicly available data generated by the ENCODE Project [41] to identify SNPs overlapping regulatory DNA as denoted by the presence of a DNase I hypersensitivity site (DHS) or H3K4me4 histone modification (see Supplemental Material). We examined whether SNPs with lower p-values were more likely to localize within these regulatory regions by performing an enrichment analysis using an increasingly selective inclusion criterion based on association p-value.
Additional genomic analyses
We ran Genome-wide Complex Trait Analysis (GCTA, [42]) to estimate the amount of variance tagged by SNPs on the Illumina 550K platform in the total sample. In addition, we assessed the predictive utility of polygenic risk scores by splitting the sample in half, and using one half as the discovery sample and the other half as the replication sample. These analyses utilized the polygenic risk score method available in Plink [43, 44], employing p-value cutoffs of 0.1- 0.5 in increments of 0.1.
Results
Alcohol problems factor score
The Eigenvalues for the first three factors from the alcohol problems factor analysis exceeded 1 (6.78, 1.69, and 1.29), and the scree plot and fit statistics indicated that a one-factor model provided an adequate fit to the data. We then ran a confirmatory factor analysis to calculate factor scores for use in subsequent analyses. Resulting alcohol problems factor scores (n = 5952) ranged from −0.45 to 4.18 (Figure S1) and were positively skewed (skewness=2.82).
Genome-wide association analysis
After data cleaning, results were obtained for 8,458,542 polymorphisms (Figure 1). The marker rs193135937 on chromosome 4 had a p-value of 1.28 × 10−8, surpassing the genome-wide significance threshold (3.06 × 10−8 for imputed data [45]). The q-value for rs193135937 was 0.11. This intergenic SNP is >500kb downstream from the pseudogene LOC100288337. No surrounding markers approached this significance level. No other markers achieved genome-wide significance. The QQ-plot is depicted in Figure 2. The genomic inflation value was λ=1.015 (SE=3.031 × 10−6).
Figure 1.
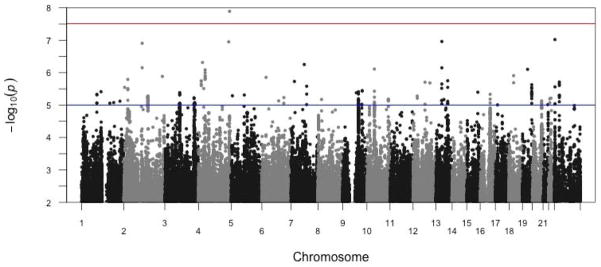
Manhattan plot of primary GWAS results. Only SNPs with p ≤ 0.01 are depicted. The top horizontal line represents the genome-wide significance cut-off (3.06 × 10−8 for imputed data [45]). The lower horizontal line represents the threshold for suggestive SNPs (1 × 10−5).
Figure 2.

QQ-plot of primary GWAS results. Diagonal line represents the null expectation.
Shaded areas represent 95% confidence intervals.
Gene-based analyses
Results were available for 22,880 genes. Four genes survived the Benjamini and Hochberg FDR (q<0.05). The gene with the most significant p-value (p=6.24 × 10−6) encodes the NLRP12 protein, which is involved in the regulation of various immune responses and signal transduction. The signal is localized to the 3’ region of the gene (Figure 3). It is expressed across many tissues, with highest levels of expression in the blood (www.genecards.org). The next most significant gene (p=6.44 × 10−6) encodes the glucose transporter SLC2A2, which is involved in carbohydrate metabolism, and has been implicated in noninsulin dependent diabetes mellitus. It is preferentially expressed in the liver and the blood. Multiple SNPs with modest p-values are distributed across SLC2A2, and in the downstream gene EIF5A2 (gene-based p<0.001). The gene EFCAB4B (p=7.14 × 10−6) encodes a calcium ion binding protein and is broadly expressed. The strongest signal (from rs182211455, p=1.89 × 10−5) is intronic. SNPs 5’ to that marker have similar p-values; they are in relatively low LD (r2<0.2) with rs182211455 and therefore may represent independent signals. Finally, TBCE (p=8.52 × 10−6) is also widely expressed; it is involved in the folding of beta-tubulin. The signals in TBCE are distributed widely across the gene, but are of modest significance (largely p~0.01). To our knowledge, none of these genes have been previously associated with alcohol-related outcomes; of these four genes, only TBCE exhibited a trend toward significance in an independent sample (p=0.09; see Supplementary Material). A complete list of gene-based results is available in Table S1; the genes with q<0.10 are listed in Table 1.
Figure 3.

Map of SNPs falling within 10 kilobases of NLRP12. Figure was constructed using LocusZoom (Pruim et al., 2010). rs10403709 represents the SNP with the lowest p-value. Other SNPs are color-coded according to their pairwise r2 with rs10403709 (indicated by the legend in the upper right corner). The solid blue line represents the regional recombination rate (see right-side y-axis) based on the 1000 Genomes hg19 CEU reference panel.
Table 1.
Genes with q<0.10 in gene-based analyses.
| Gene | p-value | q-value |
|---|---|---|
| NLRP12 | 6.24e-6 | 4.87e-2 |
| SLC2A2 | 6.44e-6 | 4.87e-2 |
| EFCAB4B | 7.14e-6 | 4.87e-2 |
| TBCE | 8.52e-6 | 4.87e-2 |
| ZP2 | 1.27e-5 | 5.79e-2 |
| LRP1B | 1.89e-5 | 5.88e-2 |
| SP8 | 1.95e-5 | 5.88e-2 |
| MCTP1 | 2.05e-5 | 5.88e-2 |
| GMNN | 3.37e-5 | 8.50e-2 |
| RANBP1 | 4.90e-5 | 8.50e-2 |
| MIR371A | 5.20e-51 | 8.50e-2 |
| MIR371B | 5.20e-51 | 8.50e-2 |
| MIR373 | 5.20e-51 | 8.50e-2 |
| MIR372 | 5.20e-51 | 8.50e-2 |
| C20orf26 | 6.17e-5 | 9.27e-2 |
| CPM | 6.48e-5 | 9.27e-2 |
| NKAPP1 | 7.43e-5 | 9.70e-2 |
| GLYATL2 | 7.95e-5 | 9.70e-2 |
| GRID1 | 8.05e-5 | 9.70e-2 |
| DEFA3 | 8.54e-5 | 9.77e-2 |
| B3GALNT2 | 9.10e-5 | 9.92e-2 |
These microRNA-encoding genes are located 3’ to NLRP12; given the structure of microRNA transcripts, the SNPs to which this signal is attributable are more likely to be relevant to NLRP12 than to the microRNA genes (see Figure 3).
Gene set enrichment analyses
The 21 categories with q<0.25 in the i-GSEA4GWAS analysis are presented in Table 2; complete results are available in Table S3. The top gene set, alpha-linolenic acid metabolism, included 8 genes (of 15 in the gene set) that span variants nominally associated with the alcohol problems factor score. Ontologies related to immune function/inflammation response (e.g., negative regulation of cytokine biosynthetic process, interferon gamma pathway) were also implicated in these analyses. Multiple ontologies related to nervous system development or function were over-represented, with modest overlap in the gene members across these categories.
Table 2.
Gene set categories with q<0.25 prior to rounding.
| Gene Set Name | Gene Set q-value |
|---|---|
| Alpha linolenic acid metabolism | 0.05 |
| Integrator complex | 0.05 |
| Negative regulation of cytokine biosynthetic process | 0.06 |
| Sensory organ development | 0.07 |
| Secretin like receptor activity | 0.07 |
| Caffeine metabolism | 0.07 |
| Ribonuclease activity | 0.08 |
| Synapse part | 0.10 |
| Structural constituent of ribosome | 0.10 |
| Detection of abiotic stimulus | 0.11 |
| Homophilic cell adhesion | 0.12 |
| Hydrogen ion transmembrane transporter activity | 0.12 |
| Regulation of neurogenesis | 0.13 |
| Interferon gamma pathway | 0.14 |
| Phagocytosis | 0.14 |
| Heterophilic cell adhesion | 0.16 |
| Negative regulation of signal transduction | 0.19 |
| Regulation of neuron apoptosis | 0.20 |
| Regulation of axonogenesis | 0.22 |
| Ribosome | 0.25 |
| Endoribonuclease activity | 0.25 |
Epigenetic enrichment analyses
We examined whether SNPs with lower p-values were more likely to localize to regions of potential regulatory significance in the genome (DHS or the H3K4me3 histone mark). When collapsing all cell types together, there was a clear trend toward general enrichment (Figure 4): as the p-value threshold becomes more stringent, variants were more likely to be located in DHS or H3K4me3 sites. When tissue types were examined separately, the most pronounced evidence of enrichment for DHS was in monocytes; for H3K4me3 histone marks, the strongest enrichment was in spinal cord cell lines derived from astrocytes. The second-highest degree of enrichment among nervous system-related tissue types was for DHS in cerebellar tissue. Complete results are available in Table S4.
Figure 4.
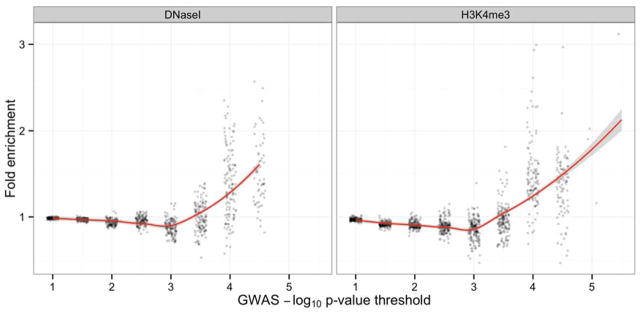
Enrichment scores (y-axis) for SNPs meeting increasingly stringent significance thresholds (x-axis) across all tissue types. Results are presented separately for DHS (left panel) and H3K4me3 histone marks (right panel). For ease of presentation, SNPs at each –log10 p-value cut-off are distributed horizontally slightly beyond the defined cut-off. The solid line represents the mean enrichment; shaded areas represent 95% confidence intervals.
Additional genomic analyses
GCTA indicated that the measured variants explained a low proportion of variance of the alcohol factor score (h2=0.052, SE=0.093, p=0.3). We next tested whether genetic risk variants identified in one half of the sample predicted alcohol problem factor scores in the other half of the sample. We applied a series of p-value cutoffs, as described by Purcell and colleagues [44]. Polygenic risk scores derived from Plink accounted for a very small proportion of the variance – up to r2=0.6% (p’s>0.05) – with markers meeting a p<0.10 threshold providing the highest estimate. This estimate did not differ significantly from 0, and varied widely depending on which subsample was used for discovery versus replication.
Discussion
These analyses evaluated genomic influences on alcohol problems in ALSPAC, a population-based sample of emerging adults. Results from GCTA suggest that such influences are modest in this sample, accounting for ~5% of the variance in an alcohol problems factor score. Although only one individual polymorphism met genome-wide significance criteria, several genes surpassed a stringent multiple testing threshold, two of which (NLRP12 and SLC2A2) have putative roles in immune function. Furthermore, gene ontologies related to immune and nervous system processes were statistically over-represented, along with categories that warrant further consideration. Bioinformatic analyses suggest that SNPs meeting increasingly stringent p-value thresholds are generally more likely than is expected by chance to map to regulatory regions; tissue derived from monocytes and spinal cord exhibited the most striking evidence of such enrichment. Our findings provide potential insight into the biological context of genomic influences on this complex behavior, and suggest that environmental factors remain strongly influential on liability to alcohol problems in this sample.
As is true of the current results, few previous studies of alcohol related phenotypes have identified replicable individual polymorphisms that meet genome-wide significance criteria. This is consistent with our understanding of the highly polygenic nature of alcohol problems: it is unlikely that individual common variants (MAF≥0.01) would have a large effect on the phenotype. Rather, hundreds if not thousands of variants influence risk. Risk variants are hypothesized to act in concert with one another in an additive or multiplicative fashion, and to be differentially influential in the context of environmental risk or protective factors.
Multiple gene ontology categories with q<0.25 are biologically plausible candidates for influencing alcohol-related phenotypes. The top gene ontology category, alpha-linolenic acid metabolism, is of interest given the role of polyunsaturated fatty acids in brain composition and function [46]. Alpha-linolenic acid is the precursor to omega-3 fatty acids, which have been associated with beneficial outcomes on several immune disorders [47]. Furthermore, studies in animal models have demonstrated a relationship between ethanol consumption and levels of polyunsaturated fatty acids in the liver [48–51].
Additional evidence for a role of the immune system in alcohol problems comes from the implication of the gene ontologies negative regulation of cytokine biosynthetic process, detection of abiotic stimulus, and interferon gamma pathway, all with q ≤ 0.14. Despite some degree of functional overlap among the categories related to immune function – for example, interferons are a type of cytokine – there is no overlap in the genes driving “significance” across these top categories. This suggests that each represents a relatively independent implication of immune-related processes. Cytokines may be biomarkers of alcohol consumption/problems [52]: elevated levels of some cytokines have been observed in individuals with alcoholic liver disease [53], and patients with cirrhosis who are actively consuming alcohol have low levels of cytokines compared to abstinent patients with cirrhosis [54].
These links raise the possibility that variation in genes with roles in fatty acid metabolism or immune function could contribute to liability to alcohol problems. This possibility is bolstered by the preliminary evidence for enrichment of localization to DHS sites in monocytes: those results indicate that genes relevant to alcohol problems are actively transcribed in certain blood cells, which again suggests a role of immune response. The mechanism(s) through which these genes/variants might exert their effects is beyond the scope of the current study, though given the nature of the identified systems, one might speculate that the physiological response to ethanol exposure would be involved. Though previous research provides evidence that immune function and fatty acids are related to alcohol-related medical problems, which are typically a consequence of problem drinking, the current study raises the possibility that these systems are associated with problem drinking itself. Alcohol consumption impacts the immune system in myriad ways, with distinct effects in different tissues [55, 56]. Additional work is needed to determine whether variation in immune-related loci is causally related to alcohol consumption or impacts downstream processes (e.g., physiological responses that might increase liability to misuse).
The gene ontology results are suggestive of multiple processes impacting genetic liability to alcohol problems. As described above, immune functioning and fatty acid metabolism are implicated, suggesting that the physiological response to/processing of ethanol is related to alcohol problems. Those findings are complemented by enrichment of ontologies directly related to nervous system functioning, which are more likely to be related to behavioral or cognitive characteristics and processes related to the misuse of alcohol. Additional studies could derive physiology-specific and behavior/cognitive-specific alcohol-related phenotypes for genome-wide studies, the results of which could be compared to determine the extent to which genetic liability overlaps across these processes. Here, the highest factor loadings for the alcohol problems factor score were related to frequency and quantity of alcohol use, but factor loadings onto items related to loss of control and experiencing negative consequences of drinking were also substantial. Thus, our factor score phenotype is capturing risk that is unlikely to be limited to the physiological response to ethanol exposure.
The genetic variance accounted for by non-imputed variants did not differ significantly from zero; however, we do not interpret this result as indicating that genetic factors are not relevant for alcohol problems. First, the current results are consistent with twin studies demonstrating that environmental factors remain strongly influential on alcohol-related phenotypes into early adulthood [9]. Genetic influences remain relevant but account for less of the variance. Second, previous studies that have derived heritabilities based on measured genetic factors have also reported lower estimates than those derived from biometric structural equation modeling [57–59], inspiring the term “missing heritability” [10]. Our prediction of 0.5–0.6% of the variance in the split-sample analyses is comparable to the r2 estimate reported by Kos et al. [60], who used common alleles to predict risk for alcohol dependence in a European-American sample. Unmeasured common and rare variants likely contribute to missing heritability in the current and previous studies.
Adding further evidence to the relevance of genetic factors identified in the current study are the results of our replication efforts. First, we examined genes with q<0.05 in an independent sample consisting of Finnish twins assessed for alcohol problems at age 14, and found a trend toward significance for TBCE. Furthermore, as reported by Salvatore and colleagues [30], polygenic risk scores derived using SNP weights from the ALSPAC sample were significantly correlated with alcohol problems in the Finnish sample. Thus, we are confident that despite the small magnitude of effects, we are capturing some degree of “true” genetic risk.
Limitations
The current results are not without limitations. First, individuals in the ALSPAC sample were, on average, only around 18 years old when they reported on alcohol-related problems. This is a relatively early stage of one’s drinking “career” and participants are not yet through the risk period for the development of alcohol problems, which stretches well into adulthood. In addition, the age of participants could be related to the rather low estimates of heritability, as previous studies have demonstrated that the heritability of alcohol-related phenotypes typically increases from adolescence into adulthood [9]. These changes are typically thought to be due to greater autonomy in adulthood, enabling individuals’ genetic liability to develop to a greater degree than is possible in the context of the social controls often present during adolescence. It is possible that genetic effects could play a more substantial role in risk for alcohol problems later in participants’ lives, and that alcohol problems may be more pronounced among the unobserved sample.
Although our sample size is substantial relative to others that have been the subject of alcohol-related GWAS (e.g., [14, 15, 21, 22]), a subset of the data was imputed which could have introduced error into the alcohol problems phenotype. It is evident that quite large sample sizes are necessary to detect the small effect sizes expected for variants influencing complex traits. Thus, it is critical that the current findings be replicated in additional, and larger, samples. In addition, the findings presented here reflect a particular socio-cultural context of a predominantly white sample in southwest England, and these results might not generalize to other ethnicities or cultures. Although our use of a continuous phenotype is more statistically powerful than a dichotomous phenotype, the factor score is comprised of heterogeneous components (e.g., social consequences of drinking, development of tolerance, frequency of drinking, etc.), making it potentially less powerful than a homogeneous phenotype. We note that the number of SNPs for which data were available for some cell lines/tissue types limited our analyses examining the localization of implicated SNPs to genomic regions of regulatory relevance. In addition, previous work suggests that there is substantial overlap in regulatory regions across tissues [41], so “tissue-specific” findings should be interpreted with caution. Finally, the exploration of epistatic interactions, gene-environment interactions, or main effects of environmental risk factors on alcohol problems were beyond the scope of this study.
Summary
In conclusion, we present here preliminary but converging evidence across genes, gene ontologies, and other bioinformatic approaches, that liability to alcohol problems is influenced by genomic variation related to immune function. We observed evidence of enrichment in gene ontologies related to nervous system processes, which was complemented by findings that SNPs with lower p-values were enriched for localization to regulatory regions in nervous system tissues. Common genetic variants accounted for a small proportion of the phenotypic variance in alcohol problems in this population-based sample of emerging UK adults. The sample could be underpowered to detect variants of small effect; however, genes and gene ontologies meeting both suggestive and more stringent p-value thresholds provide insight to the etiology of alcohol use problems in this sample.
Supplementary Material
Acknowledgments
We are grateful to all the families who took part in this study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. ALSPAC GWAS data was generated by Sample Logistics and Genotyping Facilities at the Wellcome Trust Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. The UK Medical Research Council and the Wellcome Trust (Grant refs: 092731 and 074268/Z/07/Z) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and the corresponding author will serve as guarantor for the contents of this paper. This research was specifically funded by the National Institutes of Health (K01AA021399, T32MH020030, UL1RR031990, K02AA018755, F32AA22269, and R01AA018333).
Footnotes
Conflicts of Interest: None to declare.
References
- 1.Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGue M. The behavioral genetics of alcoholism. Current Directions in Psychological Science. 1999;8:109–15. [Google Scholar]
- 3.Heath AC, Bucholz KK, Madden PA, Dinwiddie SH, Slutske WS, Bierut LJ, et al. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol Med. 1997;27:1381–96. doi: 10.1017/s0033291797005643. [DOI] [PubMed] [Google Scholar]
- 4.Prescott CA, Kendler KS. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. Am J Psychiatry. 1999;156:34–40. doi: 10.1176/ajp.156.1.34. [DOI] [PubMed] [Google Scholar]
- 5.Agrawal A, Lynskey MT, Heath AC, Chassin L. Developing a genetically informative measure of alcohol consumption using past-12-month indices. J Stud Alcohol Drugs. 2011;72:444–52. doi: 10.15288/jsad.2011.72.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fowler T, Lifford K, Shelton K, Rice F, Thapar A, Neale MC, et al. Exploring the relationship between genetic and environmental influences on initiation and progression of substance use. Addiction. 2007;102:413–22. doi: 10.1111/j.1360-0443.2006.01694.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poelen EA, Engels RC, Scholte RH, Boomsma DI, Willemsen G. Similarities in drinking behavior of twin’s friends: moderation of heritability of alcohol use. Behav Genet. 2009;39:145–53. doi: 10.1007/s10519-008-9250-z. [DOI] [PubMed] [Google Scholar]
- 8.van Beek JH, Kendler KS, de Moor MH, Geels LM, Bartels M, Vink JM, et al. Stable genetic effects on symptoms of alcohol abuse and dependence from adolescence into early adulthood. Behav Genet. 2012;42:40–56. doi: 10.1007/s10519-011-9488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergen SE, Gardner CO, Kendler KS. Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood: a meta-analysis. Twin Research and Human Genetics. 2007;10:423–33. doi: 10.1375/twin.10.3.423. [DOI] [PubMed] [Google Scholar]
- 10.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kendler KS, Aggen SH, Prescott CA, Crabbe J, Neale MC. Evidence for multiple genetic factors underlying the DSM-IV criteria for alcohol dependence. Mol Psychiatry. 2012;17:1306–15. doi: 10.1038/mp.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luczak SE, Glatt SJ, Wall TL. Meta-analyses of ALDH2 and ADH1B with alcohol dependence in Asians. Psychol Bull. 2006;132:607–21. doi: 10.1037/0033-2909.132.4.607. [DOI] [PubMed] [Google Scholar]
- 13.Thomasson HR, Edenberg HJ, Crabb DW, Mai XL, Jerome RE, Li TK, et al. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am J Hum Genet. 1991;48:677–81. [PMC free article] [PubMed] [Google Scholar]
- 14.Bierut LJ, Agrawal A, Bucholz KK, Doheny KF, Laurie C, Pugh E, et al. A genome-wide association study of alcohol dependence. Proc Natl Acad Sci U S A. 2010;107:5082–7. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edenberg HJ, Koller DL, Xuei X, Wetherill L, McClintick JN, Almasy L, et al. Genome-Wide Association Study of Alcohol Dependence Implicates a Region on Chromosome 11. Alcohol Clin Exp Res. 2010;34:840–52. doi: 10.1111/j.1530-0277.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank J, Cichon S, Treutlein J, Ridinger M, Mattheisen M, Hoffmann P, et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict Biol. 2012;17:171–80. doi: 10.1111/j.1369-1600.2011.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heath AC, Whitfield JB, Martin NG, Pergadia ML, Goate AM, Lind PA, et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biol Psychiatry. 2011;70:513–8. doi: 10.1016/j.biopsych.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kendler KS, Kalsi G, Holmans PA, Sanders AR, Aggen SH, Dick DM, et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res. 2011;35:963–75. doi: 10.1111/j.1530-0277.2010.01427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lind PA, Macgregor S, Vink JM, Pergadia ML, Hansell NK, de Moor MH, et al. A genomewide association study of nicotine and alcohol dependence in Australian and Dutch populations. Twin Res Hum Genet. 2010;13:10–29. doi: 10.1375/twin.13.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schumann G, Coin LJ, Lourdusamy A, Charoen P, Berger KH, Stacey D, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc Natl Acad Sci U S A. 2011;108:7119–24. doi: 10.1073/pnas.1017288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P, et al. Genome-wide Association Study of Alcohol Dependence. Arch Gen Psychiatry. 2009;66:773–84. doi: 10.1001/archgenpsychiatry.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo L, Gelernter J, Zhang CK, Zhao H, Lu L, Kranzler HR, et al. Genome-wide association study of alcohol dependence implicates KIAA0040 on chromosome 1q. Neuropsychopharmacology. 2012;37:557–66. doi: 10.1038/npp.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han S, Yang BZ, Kranzler HR, Liu X, Zhao H, Farrer LA, et al. Integrating GWASs and human protein interaction networks identifies a gene subnetwork underlying alcohol dependence. Am J Hum Genet. 2013;93:1027–34. doi: 10.1016/j.ajhg.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry. 2014;19:41–9. doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wetherill L, Kapoor M, Agrawal A, Bucholz K, Koller D, Bertelsen SE, et al. Family-based association analysis of alcohol dependence criteria and severity. Alcohol Clin Exp Res. 2014;38:354–66. doi: 10.1111/acer.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neale BM, Sham PC. The future of association studies: gene-based analysis and replication. Am J Hum Genet. 2004;75:353–62. doi: 10.1086/423901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Englund MM, Egeland B, Oliva EM, Collins WA. Childhood and adolescent predictors of heavy drinking and alcohol use disorders in early adulthood: a longitudinal developmental analysis. Addiction. 2008;103 (Suppl 1):23–35. doi: 10.1111/j.1360-0443.2008.02174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nixon K, McClain JA. Adolescence as a critical window for developing an alcohol use disorder: current findings in neuroscience. Curr Opin Psychiatry. 2010;23:227–32. doi: 10.1097/YCO.0b013e32833864fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, et al. Cohort Profile: the ‘children of the 90s’--the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2013;42:111–27. doi: 10.1093/ije/dys064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salvatore J, Aliev F, Edwards A, Evans D, Macleod J, Hickman M, et al. Polygenic Scores Predict Alcohol Problems in an Independent Sample and Show Moderation by the Environment. Genes. 2014;5:330–46. doi: 10.3390/genes5020330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babor T, Higgins-Biddle J, Saunders J, Monteiro M. Guidelines for Use in Primary Health Care. Geneva, Switzerland: World Health Organization; 2001. The Alcohol Use Disorders Identification Test. [Google Scholar]
- 32.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders DSM-IV Fourth Edition. 4. American Psychiatric Association; 1994. [Google Scholar]
- 33.Raghunathan TE, Solenberger PW, Van Hoewyk J. IVEware: Imputation and Variance Estimation Software. Ann Arbor, MI: Survey Methodology Program, Survey Research Center, Institute for Social Research, University of Michigan; 2007. Available from: http://www.isr.umich.edu/src/smp/ive/ [Google Scholar]
- 34.Muthén LK, Muthén BO. Mplus User’s Guide. 6. Los Angeles, CA: Muthén & Muthén; 1998–2011. [Google Scholar]
- 35.Fatemifar G, Hoggart CJ, Paternoster L, Kemp JP, Prokopenko I, Horikoshi M, et al. Genome-wide association study of primary tooth eruption identifies pleiotropic loci associated with height and craniofacial distances. Hum Mol Genet. 2013;22:3807–17. doi: 10.1093/hmg/ddt231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li MX, Gui HS, Kwan JS, Sham PC. GATES: a rapid and powerful gene-based association test using extended Simes procedure. Am J Hum Genet. 2011;88:283–93. doi: 10.1016/j.ajhg.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MX, Kwan JS, Sham PC. HYST: a hybrid set-based test for genome-wide association studies, with application to protein-protein interaction-based association analysis. Am J Hum Genet. 2012;91:478–88. doi: 10.1016/j.ajhg.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc Ser B (Stat Method) 1995;57:289–300. [Google Scholar]
- 40.Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res. 2010;38:W90–5. doi: 10.1093/nar/gkq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.International Schizophrenia C. Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li MX, Yeung JM, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131:747–56. doi: 10.1007/s00439-011-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yehuda S, Rabinovitz S, Mostofsky DI. Essential fatty acids are mediators of brain biochemistry and cognitive functions. J Neurosci Res. 1999;56:565–70. doi: 10.1002/(SICI)1097-4547(19990615)56:6<565::AID-JNR2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 47.Fogaca MN, Santos-Galduroz RF, Eserian JK, Galduroz JC. The effects of polyunsaturated fatty acids in alcohol dependence treatment--a double-blind, placebo-controlled pilot study. BMC Clin Pharmacol. 2011;11:10. doi: 10.1186/1472-6904-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham CC, Bottenus RE, Spach PI, Rudel LL. Ethanol-related changes in liver microsomes and mitochondria from the monkey, Macaca fascicularis. Alcohol Clin Exp Res. 1983;7:424–30. doi: 10.1111/j.1530-0277.1983.tb05500.x. [DOI] [PubMed] [Google Scholar]
- 49.Pawlosky RJ, Salem N., Jr Ethanol exposure causes a decrease in docosahexaenoic acid and an increase in docosapentaenoic acid in feline brains and retinas. Am J Clin Nutr. 1995;61:1284–9. doi: 10.1093/ajcn/61.6.1284. [DOI] [PubMed] [Google Scholar]
- 50.Pawlosky RJ, Salem N., Jr Alcohol consumption in rhesus monkeys depletes tissues of polyunsaturated fatty acids and alters essential fatty acid metabolism. Alcohol Clin Exp Res. 1999;23:311–7. [PubMed] [Google Scholar]
- 51.Villanueva J, Chandler CJ, Shimasaki N, Tang AB, Nakamura M, Phinney SD, et al. Effects of ethanol feeding on liver, kidney and jejunal membranes of micropigs. Hepatology. 1994;19:1229–40. [PubMed] [Google Scholar]
- 52.Torrente MP, Freeman WM, Vrana KE. Protein biomarkers of alcohol abuse. Expert Rev Proteomics. 2012;9:425–36. doi: 10.1586/epr.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol. 2010;5:83–91. doi: 10.1007/s11481-009-9185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laso FJ, Vaquero JM, Almeida J, Marcos M, Orfao A. Production of inflammatory cytokines by peripheral blood monocytes in chronic alcoholism: relationship with ethanol intake and liver disease. Cytometry B Clin Cytom. 2007;72:408–15. doi: 10.1002/cyto.b.20169. [DOI] [PubMed] [Google Scholar]
- 55.Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S, et al. Cytokines and alcohol. Alcohol Clin Exp Res. 2006;30:720–30. doi: 10.1111/j.1530-0277.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- 56.Szabo G. Consequences of alcohol consumption on host defence. Alcohol Alcohol. 1999;34:830–41. doi: 10.1093/alcalc/34.6.830. [DOI] [PubMed] [Google Scholar]
- 57.McGue M, Zhang Y, Miller MB, Basu S, Vrieze S, Hicks B, et al. A genome-wide association study of behavioral disinhibition. Behav Genet. 2013;43:363–73. doi: 10.1007/s10519-013-9606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Llewellyn CH, Trzaskowski M, Plomin R, Wardle J. Finding the missing heritability in pediatric obesity: the contribution of genome-wide complex trait analysis. Int J Obes (Lond) 2013;37:1506–9. doi: 10.1038/ijo.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trzaskowski M, Eley TC, Davis OS, Doherty SJ, Hanscombe KB, Meaburn EL, et al. First genome-wide association study on anxiety-related behaviours in childhood. PLoS One. 2013;8:e58676. doi: 10.1371/journal.pone.0058676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kos MZ, Yan J, Dick DM, Agrawal A, Bucholz KK, Rice JP, et al. Common biological networks underlie genetic risk for alcoholism in African- and European-American populations. Genes Brain Behav. 2013;12:532–42. doi: 10.1111/gbb.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.