

Abstract
Repeated administration of peroxisome proliferator-activated receptor gamma (PPARγ) agonists reduces neuropathic pain-like behavior and associated changes in glial activation in the spinal cord dorsal horn. As PPARγ is a nuclear receptor, sustained changes in gene expression are widely believed to be the mechanism of pain reduction. However, we recently reported that a single intrathecal injection of pioglitazone, a PPARγ agonist, reduced hyperalgesia within 30 minutes, a time frame that is typically less than that required for genomic mechanisms. To determine the very rapid anti-hyperalgesic actions of PPARγ activation we administered pioglitazone to rats with spared nerve injury (SNI) and evaluated hyperalgesia. Pioglitazone inhibited hyperalgesia within 5 min of injection, consistent with a non-genomic mechanism. Systemic or intrathecal administration of GW9662, a PPARγ antagonist, inhibited the anti-hyperalgesic actions of intraperitoneal or intrathecal pioglitazone, suggesting a spinal PPARγ-dependent mechanism. To further address the contribution of non-genomic mechanisms, we blocked new protein synthesis in the spinal cord with anisomycin. When co-administered intrathecally, anisomycin did not change pioglitazone anti-hyperalgesia at an early 7.5 min timepoint, further supporting a rapid non-genomic mechanism. At later timepoints anisomycin reduced pioglitazone anti-hyperalgesia, suggesting a delayed recruitment of genomic mechanisms. Pioglitazone reduction of SNI-induced increases in GFAP expression occurred more rapidly than expected, within 60 min. We are the first to show that activation of spinal PPARγ rapidly reduces neuropathic pain independent from canonical genomic activity. We conclude that acute pioglitazone inhibits neuropathic pain in part by reducing astrocyte activation, and via both genomic and non-genomic PPARγ mechanisms.
Keywords: neuropathic, pain, PPAR gamma, astrocyte, pioglitazone, non-genomic
INTRODUCTION
Peroxisome proliferator-activated receptor gamma (PPARγ) is activated by thiazolidinedione (TZD) drugs such as pioglitazone [74] that cross the blood brain barrier [55]. TZDs reduce molecular and behavioral sequelae of many neurological diseases [see [23;42;54;93]]. It is widely assumed that genomic PPARγ activity mediates reduction of inflammatory [59;64] and neuropathic [53;60] pain after repeated TZD dosing. However, agonists for PPARγ and other nuclear receptors (e.g. estrogen, glucocorticoid, PPARα) can modulate neuronal excitability [24;41;46] and pain [13;22;48;95] within minutes. Thus, the anti-hyperalgesic effects of nuclear receptor agonists may occur through multiple mechanisms including: 1) genomic, receptor-dependent transcription/translation, 2) non-genomic, receptor-dependent activation of membrane-bound receptors [41;50;73], 3) receptor-independent “off-target” effects at GPCRs [56;61;62].
Traumatic nerve injury produces changes in the spinal cord such as astrocyte activation [88;94], increased post-synaptic responses to glutamate [91], and long-term potentiation [30] resulting in central sensitization [77]. During chronic pain states, activated spinal astrocytes release pro-nociceptive mediators [63;90] such as TNFα that facilitate pain sensitization [for review see [27]]; therefore, pharmacotherapeutic approaches that decrease astrocyte activation may reduce chronic pain. TZDs agonize PPARγ expressed in spinal cord [58] and brain [17] astrocytes to reduce activation [6] and GFAP upregulation [34]. Dosing over several weeks produced anti-hyperalgesia that was associated with reductions in spinal GFAP [39;53;60], injury-induced pro-inflammatory cytokine expression [40], and in vitro astrocyte TNFα release [31;82]. A single injection of PPARγ agonist reduces neuropathic pain [13]; however, an important gap is whether there are acute effects on spinal astrocytes.
Here, we characterized the effects of PPARγ activation in neuropathic (spared nerve injury; SNI) and acute nociceptive (capsaicin) conditions after acute drug administration. We hypothesized that rapid inhibition of pain by PPARγ agonists is mediated by receptor-dependent, spinal mechanisms and is independent of translation (i.e. non-genomic). We used systemic and spinal agonist/antagonist administration to determine the site of PPARγ anti-hyperalgesia. We investigated pain-like hypersensitivity as early as 5 min after intrathecal injection of pioglitazone in the presence or absence of anisomycin, an inhibitor of protein synthesis [25;29;76]. Finally, we tested whether a single pioglitazone injection, as opposed to repeated administration [60], would reduce astrocyte activation in the form of GFAP overexpression after SNI.
MATERIALS & METHODS
Animals
Male Sprague-Dawley rats (CD-IGS, Charles River Laboratories, Inc., Wilmington, MA) weighing 300–450g at the time of behavioral procedures were housed 2 per cage on a 12-hour light/dark cycle (7am lights on / 7pm lights off) in a temperature (68–72 °F) and humidity controlled room with food and water provided ad libitum. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize alternatives to in vivo techniques, in accordance with the International Association for the Study of Pain and the National Institutes of Health Office of Laboratory Animal Welfare Guide for the Care and Use of Laboratory Animals. All behavioral procedures were performed between 8am–6pm (lights on) and approved by an Institutional Animal Care and Use Committee (IACUC) protocol. Behavioral measurements and immunohistochemistry quantification were performed by an observer blinded to experimental treatments (e.g. injury and/or drug).
Spared Nerve Injury (SNI) surgery
Surgical anesthesia was achieved with isoflurane (5% induction and 1.5% maintenance diluted in oxygen). As previously described [18;85], the skin was incised on the left hindlimb over the sciatic nerve trifurcation. The overlying muscles were retracted to expose the common peroneal, tibial, and sural nerves. The common peroneal and tibial nerves were ligated with 6-0 silk (Ethicon, Somerville, NJ) and transected 1mm proximal and 1mm distal to the ligation. The ligation knot and adjacent nerve were removed. Sural nerve perturbations were avoided. The muscle and skin were closed with loosely tied 5-0 absorbable sutures (Ethicon) and 9mm stainless steel wound clips, respectively. During sham surgeries, all steps were performed except ligation and transection of the common peroneal and tibial nerves. SNI or sham surgery day is referred to as day 0.
Pain-like behavior
Animals were acclimated in individual Plexiglas boxes (4″ × 8″ × 4″) on top of a raised stainless steel mesh grid (mechanical and cold) or Plexiglas floor (Hargreaves) for 1 h. Fluctuations in noise, vibrations, temperature, and other distractors in the behavioral testing room were avoided to optimize reliable measurements between cohorts of animals tested on different days.
Mechanical hypersensitivity was assessed using von Frey filaments (Stoelting, Inc., Wooddale, IL). The lateral aspect of the hindpaw plantar surface (sural receptive field) was stimulated with an incremental series of 8 monofilaments of logarithmic stiffness using a modified up-down method [10;20]. Each filament was applied to the sural receptive field three times at proximal, intermediate, and distal locations with respect to the heel. Testing began by applying an intermediate von Frey monofilament (number 4.31, exerts 2.0 g of force) perpendicular to the glabrous skin, causing a slight bending. In the case of a positive response (withdraw of the paw) a filament exerting less force was applied. In the case of a negative response, a filament exerting greater force was applied. The calculated 50% withdraw threshold is reported.
Cold hypersensitivity was assessed after application of a drop of acetone to the sural receptive field. We used a 3 mL syringe attached to an 8 cm length of PE-10 tubing flared to a diameter of 3.5 mm at the distal end. Surface tension maintained the volume of the drop to 10–12 μl. The amount of time the animal lifted, shook, or licked the affected hindpaw was recorded with a cutoff of 30 s after each acetone application. The average of three trials per subject at each timepoint is reported.
Heat hypersensitivity was assessed by recording paw withdraw latencies using the Hargreaves method and apparatus [32]. An adjustable infrared heat source (8 V, 50 W lamp, Ugo Basile, Italy) was positioned under the Plexiglas floor directly beneath the uninjured hindpaw. Prior to each behavioral testing session, intensity was adjusted so that the average latency to paw withdraw was 9 ± 2 s. If an animal did not respond within 20 s, the radiant beam was shut off to avoid tissue damage. The average of three trials per subject at each timepoint is reported.
Motor coordination
Motor coordination was assessed by placing the animals on an accelerating rotarod (Stoelting, Wood Dale, IL). Beginning at 2 revolutions per minute (rpm), the rotarod machine was programed to accelerate 0.5 rpm every 5 s until reaching 60 rpm. Animals were acclimated to the rotarod and subjected to one training session per d for 2 consecutive d prior to drug administration. During training and testing, the rats were placed on the rotating bar at 2 rpm, the acceleration program was started, and the time spent on the rotarod prior to fall was recorded. During the first training session, the animals were repeatedly placed on the rotarod until they performed 3 consecutive trials ≥ 150 s or reached a predetermined cutoff of 20 trials, whichever occurred first. By the second day of training, all rats successfully completed 3 consecutive trials ≥ 150 s prior to reaching 20 trials. Baseline measurement of time spent on the rotarod was recorded 7 d after SNI followed by reassessment at 1, 2, and 3 h after i.p. or i.t. drug administration. Three trials per timepoint were averaged.
Intracerebroventricular cannulation
Surgery was performed one week before injury to allow for 5 μl of drug or vehicle injection directly into the ventricle. Surgical anesthesia was achieved with isoflurane (5% for induction, 1.5% for maintenance in oxygen). Rats were placed in a stereotaxic apparatus fitted with blunt ear bars (Stoelting, Kiel, WI). After an incision to expose the cranium, the dorsal surface of the skull was leveled by zeroing the dorsoventral coordinate at lambda and bregma. A 26Ga stainless steel guide cannula (Plastics One, Roanoke, VA) was lowered to the right lateral brain ventricle using the following stereotaxic coordinates: 0.7 mm posterior to bregma, 1.5 mm lateral from midline and 3.3–4.0 mm below the skull surface [70]. Correct placement was indicated by the observation of the movement of 1μl sterile saline from a piece of PE-10 tubing attached to the cannula into the ventricle [86]. The cannula was fixed to the skull with 3 small screws and dental cement and after suturing the incision a 30 Ga stylet (Plastics One) was secured within the guide cannula.
Drugs
Pioglitazone potassium salt (10028, Cayman Chemical, Ann Arbor, MI) was dissolved in a 0.9 % saline slurry. GW9662 (70785, Cayman Chemical) was dissolved in a mixture of ethanol, ethoxylated castor oil, and saline (2:2:6) for i.t. administration and 50% DMSO in saline for i.p. administration. Neither vehicle solution altered pain-like behavior (see Fig 2). Anisomycin (A9789, Sigma-Aldrich, St. Louis, MO) was first dissolved in a small volume of 1 M HCl and then an equal volume of 0.9% saline was added. Next, the solution was titrated with NaOH to obtain a physiological pH of 7.0–7.4. Finally, the anisomycin solution was further diluted in saline to a final concentration of 20 mg/ml. Capsaicin (M2028, Sigma-Aldrich) was first dissolved in 100% ethanol (5% final v/v) followed by addition of a Tween 80 (P1754, Sigma-Aldrich) in saline (8% v/v) solution to a final capsaicin concentration of 50 mg/ml (w/v). Capsaicin (50 μl) was injected into the subdermal space between the 2nd and 3rd digits on the ventral plantar surface using a 30 Ga ½” needle.
Figure 2. Anti-hyperalgesic actions of systemic pioglitazone are mediated by spinal PPARγ.

Systemic GW9662 (GW), a PPARγ antagonist, blocked pioglitazone (Pio) reduction of (A–B) mechanical and (C–D) cold hypersensitivity 14d after SNI. (E–F) Spinal GW completely abolished pioglitazone reduction of mechanical hyperalgesia and (G–H) partially blocked pioglitazone alleviation of cold hypersensitivity. GW was administered i.p. or i.t. 15 min prior to i.p. pioglitazone. ★ significantly different from “i.p. GW 10 mg/kg” or “i.p. Pio 100 mg/kg + i.t. GW 300μg”. † significantly different from “i.p. GW 2 mg/kg” and “i.p. GW 10 mg/kg”. # significantly different from “i.p. Saline + i.t. Vehicle”. ‘n’ are shown in parentheses.
Drug Injections
Rats were anesthetized with isoflurane. Vehicle or drug solution (10–20 μl) was injected into the subarachnoid space using a 27 Ga 1″ needle inserted into a stretch of PE-20 tubing and attached to a Hamilton micro syringe [57]. We confirmed needle placement by both visualization of cerebrospinal fluid aspiration within the tubing and/or a reflexive tail/hindpaw flick. For systemic pioglitazone studies, i.p. (2 or 10 mg/kg) or i.t. GW9662 (300 μg) was administered 15 min prior to pioglitazone. For intrathecal pioglitazone studies, GW9662 (300 μg) or anisomycin (0–200 μg) was co-administered with pioglitazone to avoid multiple i.t. injections.
Capsaicin-induced nociception
Intrathecal vehicle (saline), pioglitazone (0–300 μg), or anisomycin (0–200 μg) were injected 20 min before intraplantar (i.pl.) capsaicin (50 μg in 50 μl). Behavior was recorded for 0–2 min following i.pl. capsaicin. The number of flinches and the time spent licking or lifting (s) the affected hindpaw were combined and reported as the number of nociceptive responses, where 1 s of licking/lifting is equal to one flinch response [1;49;78]. Sham injections consisted of capsaicin vehicle as a control. Rats were perfused 60 min following i.pl. capsaicin for Fos immunohistochemistry.
Immunohistochemical quantification of Fos and GFAP in the dorsal horn
Rats were anesthetized with pentobarbital (Fatal Plus, Med-Vet International, Mettawa, IL) and perfused through the left ventricle with 250 ml of room temperature 0.1 M phosphate buffered saline (PBS) with heparin (10,000 USP units/L) followed by 250 ml of ice-cold fixative (10% phosphate buffered formalin). The lumbar spinal cord was removed and post-fixed overnight in 10% phosphate buffered formalin and then cryoprotected in 30% sucrose in 0.1 M PBS for several days. Transverse sections (30 μm) from L4-L5 were cut on a freezing microtome and collected in 0.1 M PBS. The sections were washed three times in 0.1 M PBS and then pretreated with blocking solution (3% normal goat serum and 0.3% Triton X-100 in 0.1 M PBS) for 1 h. Sections were incubated overnight at room temperature in blocking solution containing either rabbit anti-Fos (1:500, SC-52, Santa Cruz Biotechnology, Santa Cruz, CA) for capsaicin studies or anti-GFAP (1:1000, ab7779, Abcam, Cambridge, MA) for SNI studies. The slices were washed three times in 0.1 M PBS, incubated in goat anti-rabbit (1:800, Alexa 488 or 568, Molecular Probes, Grand Island, NY) for 90 min, washed in 0.1 M PBS then 0.01 M PB, mounted onto Superfrost Plus slides, air dried, and cover-slipped using Prolong Gold with DAPI mounting medium (Molecular Probes, Grand Island, NY).
All images were captured and analyzed on a Nikon Eclipse TE2000-E microscope using 4× or 10× objectives using NIS-Elements Advanced Research software. In capsaicin studies we quantified the number of Fos+ cell profiles in lamina I-V of the dorsal horn. In SNI studies the dorsal horn was separated into lamina I-II, lamina III-IV, and lamina V as well as medial, central, and lateral regions of interest (ROI). Medial-lateral ROI correspond to afferent input from the tibial (medial), common peroneal (central), and sural (lateral) receptive fields as previously described [15]. Each of the six ROI were quantified and analyzed separately. For GFAP, pixel intensity values were summed, normalized to ROI area, and background subtracted. Background intensity was determined by selecting a ROI in the devoid of GFAP+ cells. This method accounts for changes in both the number and the fluorescence intensity of GFAP+ cells. Slices were analyzed by an observer blinded to treatment. Quantification of 4–6 slices per subject was averaged for each ROI and n=3–4 rats per group are reported.
Western blotting of lumbar spinal GFAP
Western blot analyses were performed on lumbar spinal cord quadrants that were sonicated on ice in 50 mM Tris buffer containing 100 mM 6-amino-n-caproic acid, 1 mM EDTA, 5 mM benzamidine, 0.2 mM phenylmethyl sulfonyl fluoride (in 100% ethanol), and protease inhibitors. After extraction, proteins were subjected to NuPAGE Bis–Tris (4–12%) gel electrophoresis under reducing conditions (Invitrogen, Carlsbad, CA) and then transferred to nitrocellulose membranes electrophoretically (Invitrogen, Carlsbad, CA). Nonspecific binding sites on the membrane were blocked with Odyssey Blocking Buffer (50%; LI-COR Biosciences, Lincoln, NE) in TBS containing 0.1% Tween-20, 0.05% Tris-Chloride, and 0.03% 5 M NaCl for 1 h at 22–24°C. Membranes were subsequently incubated with primary antibodies in Odyssey Blocking buffer containing 0.1% Tween-20 overnight at 4°C. The membranes were then washed with PBS containing 0.1% Tween-20, and probed with appropriate IRDye secondary antibodies (LI-COR Biosciences) in Odyssey Blocking buffer containing 0.1% Tween-20 for 1 h at 22–24°C, protected from light. Following washing with PBS containing 0.1% Tween-20, membranes were scanned on an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Primary antibodies and dilution ratios used were rabbit GFAP (1:1000, z0334, Dako, Carpinteria, CA), and mouse β actin (1:100,000, A5316, Sigma-Aldrich, St. Loius, MO). Secondary antibodies used were goat anti-mouse IRDye 680RD (1:15,000, LI-COR Biosciences), goat anti-rabbit IRDye 800CW (1:15,000, LI-COR Biosciences). Bands were quantified using Image Studio (LI-COR Biosciences).
Statistical analysis
For analysis of behavioral data and westerns, multiple groups were compared for significant differences over time using repeated measures two-way ANOVA or between groups using a standard two-way ANOVA followed by Holm-Sidak multiple comparison correction. Area-under-the-curve (AUC) was calculated using the trapezoidal method and analyzed using a one-way ANOVA with Holm-Sidak multiple comparison correction or an unpaired, two-tailed t-test. Ipsilateral versus contralateral (paired) or pioglitazone versus saline (unpaired) comparisons of GFAP immunohistochemistry ROIs (6 total analyses) or GFAP western quadrants were analyzed using a two-tailed t-test. An alpha value of α=0.05 was used to determine statistical significance. All data were analyzed and graphed using Prism 6.0 (GraphPad, La Jolla, CA) and are presented as mean ± SEM.
RESULTS
Systemic pioglitazone reduces mechanical and cold hypersensitivity
To determine whether PPARγ activation reduces hyperalgesia, we evaluated behavioral indices of neuropathic pain after a single i.p. injection of pioglitazone. Spared nerve injury (SNI) decreased mechanical [time; F (1, 37) = 1453; P < 0.0001] and cold [time; F (1, 37) = 86; P < 0.0001] sensitivities at 14 d after injury. As illustrated in Fig 1A–B, systemic pioglitazone attenuated mechanical [dose × time; F (21, 259) = 5.756; P < 0.0001] and cold hypersensitivity [F (21, 259) = 3.485; P < 0.0001]. Area under the curve (AUC) analyses in Fig 1C–D indicate that pioglitazone dose-dependently attenuated mechanical [dose; F (3, 37) = 6.747; P = 0.001] and cold hypersensitivity [F (3, 37) = 3.575; P = 0.023].
Figure 1. A single systemic injection of pioglitazone (Pio) dose-dependently reduced behavioral signs of neuropathic pain.

Spared nerve injury (SNI, arrow) resulted in pain-like hypersensitivity. Intraperitoneal (i.p.) pioglitazone dose-dependently reversed (A) mechanical and (B) cold hypersensitivity 14d after SNI. Pain-related behavioral responses to plantar application of von Frey and acetone stimuli were measured before (0d) and after (14d) SNI and at 15, 30, 60, 90, 120, 180, and 240 min after intraperitoneal (i.p.) injection of pioglitazone (0, 10, 30, 100 mg/kg) in saline vehicle. Area under the curve (AUC) is shown for (C) von Frey and (D) acetone pain-related behaviors. ★ “i.p. Pio 100 mg/kg” significantly different from “i.p. Saline”. † “i.p. Pio 30 mg/kg” and “i.p. Pio 100 mg/kg” significantly different from “i.p. Saline”. “n” are shown in parentheses.
PPARγ in the spinal cord mediates the anti-hyperalgesic actions of systemic pioglitazone
Churi et al. reported that a single intrathecal injection of the PPARγ agonist rosiglitazone produced anti-hyperalgesia within one hour [13]. However, the authors did not test for PPARγ-dependency. To address this gap and test the hypothesis that PPARγ mediates the acute anti-hyperalgesic effects of i.p. pioglitazone, we pretreated SNI rats with GW9662, an irreversible PPARγ antagonist [12;45]. As illustrated in Fig 2A–D, systemic GW9662 (i.p.) prevented pioglitazone reductions in mechanical [AUC; F (2, 25) = 13.14; P = 0.0001] and cold [AUC; F (2, 27) = 3.992; P = 0.03] hypersensitivity.
Next we sought to determine whether the anti-hyperalgesic effects of systemic pioglitazone are mediated through activation of spinal PPARγ. As illustrated in Fig 2E–H, intrathecal (i.t.) GW9662 attenuated pioglitazone reduction of mechanical [AUC; F (3, 23) = 18.55; P < 0.0001] and cold [AUC; F (3, 23) =12.85; P < 0.0001] hypersensitivity. Both systemic and spinal administration of GW9226 attenuated the reduction of mechanical and cold hypersensitivity by administration of systemic pioglitazone.
PPARγ in the spinal cord mediates the anti-hyperalgesic actions of intrathecal pioglitazone
To support the PPARγ antagonist experiment above in identifying the spinal cord as a key site of PPARγ anti-hyperalgesic action, we administered pioglitazone and/or GW9662 by the intrathecal or intracerebroventricular route. Fig 3A demonstrates that i.t. pioglitazone attenuated mechanical hyperalgesia [dose × time; F (18, 162) = 2.951; P = 0.0001] as rapid as 30 min after injection in a dose-dependent manner. To account for possible supraspinal activation of PPARγ after i.t. pioglitazone via the cerebrospinal fluid circulation, we injected the same doses via the intracerebroventricular (i.c.v.) route. We found no anti-hyperalgesic effect of pioglitazone at these doses [F (3, 21) = 1.959; P = 0.15]. Fig 3B illustrates that co-administration of the PPARγ antagonist GW9662 completely blocked the anti-hyperalgesic actions of i.t. pioglitazone [drug × time; F (18, 198) = 16.26; P < 0.0001].
Figure 3. Spinal PPARγ mediates pioglitazone anti-hyperalgesia.

(A) Intrathecal (i.t.) pioglitazone (Pio) dose-dependently reduced mechanical hyperalgesia at doses that have no effect when administered by the intracerebroventricular (i.c.v.) route. (B) Intrathecal co-administration of the PPARγ antagonist GW9662 (GW) completely reversed pioglitazone anti-hyperalgesia. † high and medium ‡ high, medium, low dose Pio significantly different from “i.t. Saline”. ★ significantly different from “i.t. Saline + i.t. Vehicle”. ‘n’ are shown in parentheses.
PPARγ activation does not produce motor deficits or analgesia
We determined whether the anti-hyperalgesic actions of pioglitazone might be confounded by off-target effects on motor systems or transient reflexive pain [84]. Fig 4A–B illustrates that a relatively high dose of pioglitazone did not change rotarod performance after i.p. [p = 0.81] or i.t. [p = 0.35] injection. Because von Frey mechanical withdraw thresholds are maximal in uninjured rats (e.g. 15 g), we assessed response latencies to noxious heat to determine if pioglitazone altered acute nociception in sham rats. Intrathecal pioglitazone did not change heat response latencies [p = 0.5] suggesting that the effects of PPARγ activation are not analgesic. Together with our previous reports [13;59;60], we conclude that reduction of mechanical hyperalgesia by PPARγ agonists is not secondary to adverse effects on motor coordination or normal sensory thresholds.
Figure 4. Pioglitazone did not produce ataxia or changes in transient nociception.

Motor coordination was assessed on an accelerating rotarod before and at 1, 2, and 3 h after high dose pioglitazone (Pio) administration by (A) systemic (i.p. 100 mg/kg) or (B) spinal (i.t. 1000 μg) routes. Pioglitazone did not change time spent on the accelerating rotarod. The analgesic effects of pioglitazone were tested using the Hargreaves assay before and after intrathecal saline or pioglitazone administration in sham rats (C). Pioglitazone did not change paw withdraw latencies to noxious heat. n = 8–10 per group.
PPARγ agonists rapidly reduce nociceptive and neuropathic pain-like behavior
The above studies illustrate that i.t. administration of pioglitazone reduces tactile hypersensitivity within 30 min of administration in SNI rats. Consistent with this finding, intrathecal pioglitazone, compared to saline, reduced nociceptive behavior when given 20 min prior to intraplantar capsaicin (nociceptive responses; 16.3 ± 2.3 vs. 62.2 ± 16.1; n = 5–6) [p = 0.013]. To test the hypothesis that PPARγ anti-hyperalgesic mechanisms occur rapidly after traumatic nerve injury, we extended our temporal analysis of pain-like behavior in the SNI model to earlier time points. Fig 5A–B demonstrates that the PPARγ agonists rosiglitazone [F (1, 9) = 5.942; P = 0.0375] and pioglitazone [F (1, 8) = 28.24; P = 0.0007] attenuated SNI-associated mechanical hypersensitivity within 5 min of injection. We repeated the study with 7.5 min as the first behavioral timepoint to allow full recovery from isoflurane anesthesia, as evidenced by the slight increase in von Frey thresholds in the saline control group in Fig 5B. Fig 5C illustrates a robust anti-hyperalgesic effect of pioglitazone [F (1, 14) =26.62; P = 0.0001] and at the 7.5 minute timepoint [p < 0.0001] in the absence of a residual anesthetic effect [p = 0.9998]. Our findings that rosiglitazone and pioglitazone produce anti-hyperalgesia and anti-nociception within 5–20 min of administration suggest that non-genomic PPARγ mechanisms mediate rapid reduction of pain-like behavior.
Figure 5. PPARγ agonists rapidly reduce mechanical hyperalgesia.

Intrathecal injection of the PPARγ ligands (A) rosiglitazone (Rosi) or (B) pioglitazone (Pio) rapidly (within 5 min) reduced mechanical hypersensitivity 14d after SNI. Intrathecal injections were performed under isoflurane anesthesia, which resulted in a small anti-hyperalgesic effect at 5 min. Therefore, we repeated the experiment with 7.5 min the earliest timepoint tested. (C) Pioglitazone rapidly attenuated mechanical hypersensitivity that lasted from 7.5 to 90 min with no anesthesia effect in the saline group. ★ significantly different from “i.t. Saline”. ‘n’ are shown in parentheses.
In vivo blockade of protein translation in the dorsal horn by intrathecal anisomycin
To examine whether a translation-independent (i.e. non-genomic) mechanism mediates the rapid anti-hyperalgesic effects of PPARγ, we administered intrathecal anisomycin to block protein synthesis in the dorsal horn in vivo at a dose that did not produce confounding alterations in nociception [43]. To achieve this, we evaluated the effect of multiple doses of anisomycin, based on previously used doses of 25 to 125 μg [2;7;69;76], on capsaicin-induced protein (i.e. Fos) expression. Capsaicin activates TRPV1-positive primary afferent nociceptors [8] resulting in spontaneous, dose-dependent pain in humans [79]. Intraplantar injection of capsaicin in rodents elicits spontaneous nociceptive behavior [80] and induces Fos expression in the superficial dorsal horn within 30 to 90 min [3;35], providing a suitable tool for evaluating effects of anisomycin on new protein synthesis.
As depicted in Fig 6A, we administered intrathecal pioglitazone and/or anisomycin twenty minutes prior to evaluation of capsaicin-evoked nociceptive behavior. Sixty minutes after capsaicin injection, we euthanized animals for quantification of Fos expression in the L4–L5 dorsal horn. As illustrated in Fig 6B, combinations of capsaicin, pioglitazone, and anisomycin administration significantly altered Fos expression in laminae I-V [group; F (7, 17) = 9.38; P < 0.0001]. Compared to vehicle injection, intraplantar capsaicin increased Fos expression [p = 0.007], an effect that was dose-dependently inhibited by pretreatment with 100–200 μg doses of anisomycin [p < 0.05] but not 50 μg [p = 0.064]. Anisomycin (200 μg) also reduced Fos expression when co-administered with pioglitazone [p = 0.0006]. Pioglitazone did not alter capsaicin-evoked Fos [p = 0.74] when administered alone.
Figure 6. Anisomycin reduces translation of capsaicin-evoked Fos without blocking the rapid anti-nociceptive actions of pioglitazone.

(A) Experimental timeline to test the hypothesis that capsaicin evoked Fos translation is inhibited by intrathecal anisomycin pretreatment. (B) Vehicle (0μg Cap), pioglitazone (Pio), and/or anisomycin (Ani) were co-administered i.t. 20 min prior to i.pl. capsaicin (50μg). Capsaicin-induced spontaneous licking, lifting, and flinching nociceptive responses from 0–2 min after injection were almost completely abolished by pioglitazone (300μg) alone or in combination with anisomycin (200μg) pretreatment. Anisomycin (50–200μg) alone did not alter capsaicin nociception. (C) Capsaicin-evoked Fos expression was dose-dependently inhibited by anisomycin and unaltered by pioglitazone. (D) Sham injection of vehicle produced moderate Fos expression when compared to (E) i.t. vehicle injections followed by i.pl. capsaicin. (F) 50μg, (G) 100μg, (H) 150μg, and (I) 200μg anisomycin dose-dependently reduced Fos. (J) Pioglitazone alone had no effect on Fos expression while (K) anisomycin completely abolished Fos when co-administered with pioglitazone. ★ significantly different from capsaicin only (black bars). n = 3–9 for behavioral and n = 3–4 for Fos analyses.
We offer two explanations for the lack of effect of pioglitazone on spinal Fos expression even though nociceptive behavior was reduced after intraplantar capsaicin injection. First, Fos and behavior do not always positively correlate: drug-induced reduction of pain-like behavior can occur in the absence of [14;26;33] or with increases in [65] Fos expression. Second, the target of pioglitazone, PPARγ, directly activates c-fos transcription [71]. If pioglitazone directly increases Fos expression in inhibitory interneurons, which can account for a significant population of total cells expressing Fos [87], then this would confound the ability of our Fos method to reflect noxious stimulus-evoked spinal neuron activation.
Anisomycin does not alter the anti-nociceptive effects of intrathecal pioglitazone
Having validated that anisomycin reduces protein translation (i.e. Fos) in the dorsal horn, we used this method to explore the anti-nociceptive and translation-independent actions of PPARγ activation in the capsaicin model. As illustrated in Fig 6C, combinations of capsaicin, pioglitazone, and anisomycin administration significantly altered nociceptive behavior [group; F (7, 35) = 5.476; P = 0.0003]. Intraplantar capsaicin markedly increased nociceptive behavior when compared to vehicle injection [p = 0.0055]. Anisomycin alone (50–200 μg) did not alter capsaicin-evoked nociceptive behavior [p > 0.05]. Pioglitazone reduced nociceptive behavior when administered alone [p = 0.0029] or in combination with the 200 μg dose of anisomycin [p = 0.0029]. These results indicate that anisomycin blocks genomic activity (i.e. Fos expression) in the dorsal horn without altering nociceptive behavior or pioglitazone anti-nociception, providing proof of principle for this method.
Spinal anisomycin does not change the rapid anti-hyperalgesic effects of pioglitazone
Next we extended spinal anisomycin blockade of translation in the dorsal horn to the SNI model of neuropathic pain. We began with a dose of anisomycin that, when administered into the brain, inhibits conditioned taste aversion [72], auditory fear conditioning [68;69;76], and incorporation of radioactive methionine into nascent proteins [69;72]. We found that the 200 μg dose of anisomycin abolished capsaicin-evoked Fos, but also produced a confounding attenuation of SNI-induced mechanical hypersensitivity. By lowering the anisomycin dose to 100 μg, we obtained a significant reduction in Fos without altering SNI-induced hypersensitivity. As illustrated in Fig 7A, combinations of intrathecal vehicle, pioglitazone, or anisomycin changed mechanical sensitivity in SNI rats when analyzed across the entire 240 min time course [drug × time; F (24, 168) = 3.416; P < 0.0001]. Further analysis of the 7.5 – 120 min timepoints revealed that pioglitazone attenuated mechanical hypersensitivity when injected alone [vs. Vehicle + Saline; F (1, 9) = 29.41; P = 0.004] or with 100 μg anisomycin [vs. Vehicle + Saline; F (1, 11) = 5.098; P = 0.045]. At this dose, anisomycin alone did not inhibit mechanical hypersensitivity [p = 0.54]. To compare the effect of anisomycin on pioglitazone anti-hyperalgesia at early versus delayed timepoints, we analyzed the data at each of four timepoints (Fig 7B): before intrathecal injections (pre-drug), during early pioglitazone anti-hyperalgesia (7.5 min), during late pioglitazone anti-hyperalgesia (60 min), and after anti-hyperalgesia resolved (180 min). Compared to intrathecal vehicle, pioglitazone [p = 0.013] and pioglitazone plus anisomycin [p = 0.026] attenuated hypersensitivity at 7.5 min when compared to intrathecal vehicle only [drug; F (3, 20) = 5.173; P = 0.008]. By contrast, the delayed anti-hyperalgesic actions of pioglitazone at 60 min [p = 0.003 vs. Vehicle + Saline] were significantly reduced by anisomycin [p = 0.021 vs. Vehicle + Pio].
Figure 7. Early but not late pioglitazone anti-hyperalgesia is independent of translation.

To test whether immediate PPARγ anti-hyperalgesia is dependent on canonical genomic activity, we co-administered pioglitazone with an anisomycin dose that blocks translation but not nociception (100μg; see discussion for details). (A) Timecourse of mechanical thresholds after i.t. injection of pioglitazone (Pio) or anisomycin (Ani). (B) Analysis of mechanical thresholds at 7.5, 60 and 180 min timepoints. Anisomycin did not change the anti-hyperalgesic effects of pioglitazone at the early 7.5 min period, but reduced pioglitazone anti-hyperalgesia at 60 min. These results suggest that pioglitazone produces its anti-hyperalgesic effects by both translation-independent (7.5 min) and translational-dependent (60 min) mechanisms. ★ significantly different. ‘n’ are shown in parentheses.
Acute pioglitazone reduces expression of GFAP after nerve injury
Neuropathic pain is associated with astrocyte activation after nerve injury [94]. We previously reported that repeated administration of pioglitazone reduced not only established neuropathic pain, but also astrocyte activation in the dorsal horn of SNI rats [60]. To determine whether a single administration of pioglitazone also reduces astrocyte activation after SNI, we evaluated the protein expression of GFAP in the lumbar dorsal horn using both immunohistochemistry and western blot as soon as 60 min.
Similar to previous reports [94], Fig 8A–B illustrates that SNI produced a unilateral increase in GFAP expression in the ipsilateral dorsal horn 14d after injury [ipsi vs. contra; F (1, 12) = 8.55; P = 0.0127]. This occurred in both injured (tibial and common peroneal) and uninjured (sural) innervation territories [t-test; p < 0.05]. Fig 8C–D illustrates that pioglitazone reduced GFAP expression in the contralateral and ipsilateral dorsal horn within 60 min of injection [drug; F (1, 8) = 74.93; P < 0.0001].
Figure 8. Acute PPARγ activation reduces GFAP expression.

(A) Spared nerve injury produced an increase in GFAP expression on the injured (ipsilateral) side when compared to the uninjured (contralateral) side of the lumbar dorsal horn. (B) SNI-induced a GFAP increase in injured (tibial and common peroneal) and uninjured (sural) innervation territories. (C–D) Pioglitazone (100 mg/kg i.p.; 1 h prior to perfusion) reduced GFAP expression on both the contralateral and ipsilateral dorsal horn. Each region of interest was analyzed separately using a two-tailed t-test. # significantly different from “contralateral” in the “Saline” group (paired). ★ significantly different from “Saline” (unpaired). n = 3 per group.
To confirm that the acute reduction of GFAP expression elicited by pioglitazone (Fig 8) did not result from structural changes that mask the GFAP antibody epitope in fixed tissue, we performed denaturing western blots. At 14 d after sham or SNI surgery, we administered i.p. pioglitazone, measured behavioral anti-hyperalgesia, and harvested L4-5 spinal cord quadrants 90 min later. As illustrated in Fig 9A, pioglitazone but not saline reduced mechanical hyperalgesia in SNI animals [drug; F (1, 10) = 11.17; P = 0.0075] at 60 [p = 0.023] and 90 [p < 0.0001] min after administration. Western blot analysis of the four lumbar quadrants is reported in Fig 9B (dorsal) and Fig 9C (ventral). Neither injury [p = 0.58] nor drug [p = 0.33] treatment changed GFAP expression in the contralateral dorsal horn. By contrast, we found a significant injury × drug interaction in the ipsilateral dorsal horn [F (1, 20) = 11.39; P = 0.003]. Post-hoc tests revealed that SNI increased GFAP expression when compared to sham animals treated with saline [p = 0.0014]. This increase was reduced by pioglitazone in the ipsilateral [p = 0.0026] but not contralateral [p = 0.54] dorsal horn of SNI animals (Fig 9B). In the contralateral ventral horn, neither injury [p = 0.14] nor pioglitazone [p = 0.40] changed GFAP expression. Injury [p = 0.41] or pioglitazone [p = 0.84] did not change GFAP expression in the ipsilateral ventral horn.
Figure 9. SNI is required for pioglitazone reduction of pain and astrocyte activation.
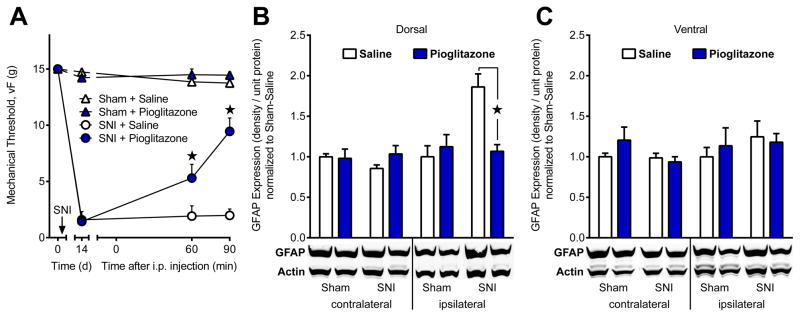
To determine whether pioglitazone is toxic to astrocytes and to rule out conformational changes in the GFAP antibody epitope during immunohistochemical analysis we performed a denaturing western blot. Spinal cord quadrants (L4–5) were harvested 90 min after i.p. pioglitazone administration at d14 after sham or spared nerve injury (SNI) surgery. (A) Pioglitazone attenuated mechanical hypersensitivity in SNI, but not sham, animals. (B) Dorsal and (C) Ventral integrated densities normalized to Sham + Saline are shown. There was no effect of injury or drug treatment in the ventral horn or in the contralateral dorsal horn segments. GFAP expression was increased in the ipsilateral dorsal horn of SNI + Saline animals when compared to all other groups. Pioglitazone significantly reduced ipsilateral dorsal horn GFAP expression in SNI, but not sham, animals. This suggests that the anti-hyperalgesic effects of pioglitazone are associated with decreased astrocyte activation after nerve injury. ★ significantly different from “SNI + Saline”. n=6–7 per group.
DISCUSSION
Anisomycin to assess translation-independent activity of nuclear receptors in the spinal cord
Here we used in vivo administration of anisomycin to dissect genomic versus non-genomic mechanisms of a nuclear receptor in the spinal cord. Previous studies using anisomycin suggested that protein translation is necessary for late phase LTP in the brain [25;44], hyperalgesic priming in the peripheral nervous system [2], ongoing pain transmission in the spinal cord [43], and reduction of established hyperalgesia during pain memory reconsolidation in the spinal cord [7]. As these studies did not confirm inhibition of translation, we found it important to demonstrate that intrathecal anisomycin inhibits spinal protein expression. We found that anisomycin, at a dose of 100 μg, decreased expression of capsaicin-evoked Fos without changing SNI-induced hyperalgesia. This provides proof of principle for using this approach to determine the contribution of non-genomic PPARγ mechanisms to the rapid and delayed phases of pioglitazone anti-hyperalgesia.
Non-genomic PPARγ activity mediates the early anti-hyperalgesic effect of pioglitazone
Based on our finding that the anti-hyperalgesic effect of pioglitazone occurred very rapidly (7.5 min) and this was maintained in the presence of 100 μg anisomycin, we suggest the involvement of a non-genomic PPARγ mechanism. Non-genomic mechanisms in the dorsal horn might also explain the rapid anti-hyperalgesic effects observed after interruption of other nuclear receptors including estrogen and PPARα receptors. For example, in vivo administration of 17β-estradiol rapidly (within 15 min) enhanced bradykinin-induced hyperalgesia [73] and inhibited opioid receptor-like 1 anti-nociception [81]: these were unaffected by pretreatment with anisomycin or conjugation of 17β-estradiol to membrane-impermeable BSA [73;81] suggesting that membrane estrogen receptors contribute to rapid pain modulation. Also, the inhibition of neuropathic pain (within 30 min) by the PPARα agonists PEA and GW7647 [16] was abrogated in PPARα knockout mice or by pharmacological blockade of calcium-activated potassium channels [16;47;48] suggesting both rapid and PPARα-dependent analgesic actions. We speculate that, in a similar manner, the rapid anti-hyperalgesic effect of pioglitazone is mediated by membrane PPARγ that is linked to ion channel activity, neuronal excitability, and/or central sensitization mechanisms.
Genomic PPARγ activity mediates the late anti-hyperalgesic effects of pioglitazone
We found that anisomycin significantly reduced the anti-hyperalgesic effects of pioglitazone at later time points (60 minutes), supporting a contribution of classical genomic mechanisms involving transcription and translation. This is consistent with several reports indicating that repeated pioglitazone administration produced sustained anti-hyperalgesic actions persisting for days to weeks after cessation of drug administration [60;83], beyond the time necessary for drug clearance [55]. Similarly, our current results and previous studies [13;59;67] indicate that a single TZD injection produces anti-hyperalgesia lasting for several hours, much longer than the half-life of pioglitazone [55]. In summary, our results suggest pioglitazone anti-hyperalgesia transitions from a rapid, anisomycin–resistant mechanism to a delayed, anisomycin-sensitive mechanism within approximately 60 minutes.
The anti-hyperalgesic effect of pioglitazone is mediated by spinal PPARγ
PPARγ agonists reduce tactile hypersensitivity after nerve injury, but the site(s) of action and selectivity of these agents remain elusive. Here, systemic or spinal pioglitazone decreased nerve injury-induced tactile hypersensitivity, and this effect was lost when given in the presence of intrathecal GW9662. We conclude that PPARγ in the dorsal horn of the spinal cord contributes to the anti-hyperalgesic effects of pioglitazone. This is consistent with previous studies showing that: 1) PPARγ is expressed in the lumbar spinal cord [13;53]; 2) hyperalgesia after SNI was exacerbated by intrathecal administration of the PPARγ antagonist BADGE [22]; and 3) intrathecal GW9662 inhibited the anti-hyperalgesic effect of both repeated systemic or single intrathecal administration of rosiglitazone or 15d-PGJ2 [13;60]. It is unlikely that brain PPARγ mediates anti-hyperalgesia after intrathecal pioglitazone, because i.c.v. administration of comparable doses had no effect; this is consistent with previous results showing that low i.c.v. doses of rosiglitazone or 15d-PGJ2 [13] did not change tactile hypersensitivity.
Our studies do not rule out “off-target” effects that might contribute to rapid pioglitazone anti-hyperalgesia. Gras et al (2009) reported that rosiglitazone-stimulated calcium mobilization was lost after GPR40 siRNA knockdown, suggesting that TZDs (i.e. PPARγ agonists) may activate GPR40 [28]. Activation of GPR40, which is expressed in brain [51;52] and spinal cord neurons [61], by endogenous (DHA) and exogenous (GW9508) ligands reduced inflammatory pain [61;62]. Thus GPR40 is an intriguing target for future studies.
Pioglitazone acutely inhibits astrocyte activation
We previously reported that repeated pioglitazone administration spanning several weeks reduced GFAP in the dorsal horn [60]. Here we observed the same result in a timeframe of just 60 minutes following a single dose of pioglitazone, leading us to speculate that astrocytes contribute to rapid anti-hyperalgesia mechanisms. This might be PPARγ-dependent, since markers of astrocyte activation are reduced by other PPARγ agonists [31;82].
Our results are not the first to describe that an acute intervention (i.e. pioglitazone) can alter astrocyte function. For example, increases in GFAP expression occur as soon as 30 min after traumatic nerve injury [96], electrical stimulation of primary afferents [89], and intraplantar injection of CFA [89] or snake venom toxin sPLA2-Asp49 [9]. Second, the astrocyte toxins l-α-aminoadipate and fluorocitrate reduced neuropathic pain and GFAP expression within 30 to 60 min of administration [91;97]. Third, fluorocitrate prevented the early phase of TNFα-induced long term potentiation (LTP) in the ex vivo spinal cord [30]. Therefore, based on the current finding that pioglitazone reduced GFAP in the dorsal horn, we propose that pioglitazone inhibits the astrocytic release of neuron-sensitizing molecules that contribute to LTP and, ultimately, the maintenance of chronic neuropathic pain. Indeed, pioglitazone reduced the release of TNFα from astrocytes in culture [82].
Our results do not rule out a supraspinal component to the anti-hyperalgesic actions of systemically-administered pioglitazone. This hypothesis is suggested by the findings that: 1) in models of CNS injury, pioglitazone reduced mitochondrial dysfunction [36;37;75;93] and promoted glucose metabolism in astrocytes [19;66]; 2) pioglitazone reduced Aβ-induced astrocyte activation in the hippocampus [34]; 3) PPARγ is expressed in cortical astrocytes [17;58]; 4) astrocytes in the anterior cingulate cortex facilitate pain sensitization and inhibition [11;27;38;92]. This provides for the possibility that systemic pioglitazone administration produces anti-hyperalgesia through cortical, in addition to spinal, astrocytic mechanisms.
Investigations of nerve injury-induced activation of astrocytes in areas of the dorsal horn innervated by injured versus uninjured afferents are sparse [4;15;94]. Here, our immunohistochemical quantification method [15] revealed that nerve injury increased GFAP expression in regions of the ipsilateral dorsal horn innervated by both injured tibial afferents (medial) and uninjured sural afferents (lateral). This is consistent with qualitative studies showing that astrocyte activation extends beyond injured territories [4], and suggests that astrocytes induce hyperalgesia by sensitizing intact nociceptive pathways.
Validation of immunohistochemical results with a complementary approach is essential because IHC assays can be misinterpreted due to drug or injury-induced cytotoxic changes in the conformation and/or the formalin sensitivity of the GFAP epitope (i.e. epitope masking) [5;21]. We chose an approach involving a denaturing western blot, and the results indicate that pioglitazone does not change GFAP expression in sham animals, nor on the contralateral side in SNI animals, nor in the ventral horn in sham or SNI animals. This not only suggests that astrocyte viability is preserved after pioglitazone injection, but also that epitope masking does not confound our conclusion that a single injection of pioglitazone exerts anti-hyperalgesic actions in part by reducing astrocyte activation.
Conclusions
Our results indicate that pioglitazone acts at spinal PPARγ to inhibit astrocyte activation and to produce fast-acting, dose-dependent, and translation-independent inhibition of pain-like hypersensitivity after traumatic nerve injury. The specific mechanisms of rapid pain reduction by PPARγ and the involvement of astrocytes warrant further investigation. Our studies do not rule out actions of PPARγ agonists in the peripheral nervous system [53;83] that would reduce spinal sensitization, but do illustrate an important behavioral and neurobiological role for non-genomic inhibition of pain in the spinal cord by a nuclear receptor.
SUMMARY.
Pioglitazone activation of spinal PPARγ reduces GFAP and rapidly attenuates hyperalgesia after traumatic nerve injury. PPARγ pain reduction occurs via both non-genomic and genomic mechanisms.
Acknowledgments
Supported by: 5R01NS062306 and R01NS045954 to Bradley K. Taylor; T32NS077889 and F31NS083292 to Ryan B. Griggs; NIH CTSA UL1TR000117; NHMRC 1054091 and AAA Sir Keith Murdoch Fellowship to Peter M. Grace.
Footnotes
The authors declare no competing financial conflict of interests.
Keith A Strand helped run western blots in Fig 9B–C, Andrew New helped collect behavioral pharmacology data in Fig 3A, Chris Meriweather helped quantify immunohistochemistry in Fig 8, Heather Scuderi-Porter assisted with behavioral pharmacology experiments in Fig 2A–D, Colleen Garrett helped collect behavioral pharmacology data in Fig 1A–D, Danielle Lyons assisted with behavioral pharmacology experiments in Fig 2E–H, Greg Corder provided consultation on experimental design, Marion Coe critically red the manuscript.
References
- 1.Andersson DA, Gentry C, Light E, Vastani N, Vallortigara J, Bierhaus A, Fleming T, Bevan S. Methylglyoxal Evokes Pain by Stimulating TRPA1. PLoS One. 2013;8(10):e77986. doi: 10.1371/journal.pone.0077986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asiedu MN, Tillu DV, Melemedjian OK, Shy A, Sanoja R, Bodell B, Ghosh S, Porreca F, Price TJ. Spinal protein kinase M zeta underlies the maintenance mechanism of persistent nociceptive sensitization. J Neurosci. 2011;31(18):6646–6653. doi: 10.1523/JNEUROSCI.6286-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaudry H, Dubois D, Gendron L. Activation of spinal mu- and delta-opioid receptors potently inhibits substance P release induced by peripheral noxious stimuli. J Neurosci. 2011;31(37):13068–13077. doi: 10.1523/JNEUROSCI.1817-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beggs S, Salter MW. Stereological and somatotopic analysis of the spinal microglial response to peripheral nerve injury. Brain, Behavior, and Immunity. 2007;21(5):624–633. doi: 10.1016/j.bbi.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell PB, Rundquist I, Svensson I, Collins VP. Formaldehyde sensitivity of a GFAP epitope, removed by extraction of the cytoskeleton with high salt. Journal of Histochemistry & Cytochemistry. 1987;35(12):1375–1380. doi: 10.1177/35.12.2445810. [DOI] [PubMed] [Google Scholar]
- 6.Bernardo A, Minghetti L. Regulation of Glial Cell Functions by PPAR-gamma Natural and Synthetic Agonists. PPAR research. 2008;2008:864140. doi: 10.1155/2008/864140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonin RP, De Koninck Y. A spinal analog of memory reconsolidation enables reversal of hyperalgesia. Nat Neurosci. 2014;17(8):1043–1045. doi: 10.1038/nn.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol. 2013;716(1–3):61–76. doi: 10.1016/j.ejphar.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Chacur M, Gutiérrez JM, Milligan ED, Wieseler-Frank J, Britto LRG, Maier SF, Watkins LR, Cury Y. Snake venom components enhance pain upon subcutaneous injection: an initial examination of spinal cord mediators. Pain. 2004;111(1–2):65–76. doi: 10.1016/j.pain.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of Neuroscience Methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 11.Chen FL, Dong YL, Zhang ZJ, Cao DL, Xu J, Hui J, Zhu L, Gao YJ. Activation of astrocytes in the anterior cingulate cortex contributes to the affective component of pain in an inflammatory pain model. Brain Res Bull. 2012;87(1):60–66. doi: 10.1016/j.brainresbull.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 12.Chen Q, Chen J, Sun T, Shen J, Shen X, Jiang H. A yeast two-hybrid technology-based system for the discovery of PPARγ agonist and antagonist. Analytical Biochemistry. 2004;335(2):253–259. doi: 10.1016/j.ab.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Churi SB, Abdel-Aleem OS, Tumber KK, Scuderi-Porter H, Taylor BK. Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J Pain. 2008;9:639–649. doi: 10.1016/j.jpain.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coggeshall RE. Fos, nociception and the dorsal horn. Progress in Neurobiology. 2005;77(5):299–352. doi: 10.1016/j.pneurobio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Corder G, Siegel A, Intondi AB, Zhang X, Zadina JE, Taylor BK. A Novel Method to Quantify Histochemical Changes Throughout the Mediolateral Axis of the Substantia Gelatinosa After Spared Nerve Injury: Characterization with TRPV1 and Substance P. The Journal of Pain. 2010;11(4):388–398. doi: 10.1016/j.jpain.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain. 2008;139(3):541–550. doi: 10.1016/j.pain.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Cristiano L, Bernardo A, Ceru MP. Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J Neurocytol. 2001;30(8):671–683. doi: 10.1023/a:1016525716209. [DOI] [PubMed] [Google Scholar]
- 18.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87(2):149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 19.Dello Russo C, Gavrilyuk V, Weinberg G, Almeida A, Bolanos JP, Palmer J, Pelligrino D, Galea E, Feinstein DL. Peroxisome Proliferator-activated Receptor γ Thiazolidinedione Agonists Increase Glucose Metabolism in Astrocytes. Journal of Biological Chemistry. 2003;278(8):5828–5836. doi: 10.1074/jbc.M208132200. [DOI] [PubMed] [Google Scholar]
- 20.Dixon WJ. Efficient analysis of experimental observations. Annual review of pharmacology and toxicology. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 21.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochemical research. 2000;25(9–10):1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 22.Fehrenbacher JC, LoVerme J, Clarke W, Hargreaves KM, Piomelli D, Taylor BK. Rapid pain modulation with nuclear receptor ligands. Brain Research Reviews. 2009;60(1):114–124. doi: 10.1016/j.brainresrev.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feinstein DL. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes technology & therapeutics. 2003;5(1):67–73. doi: 10.1089/152091503763816481. [DOI] [PubMed] [Google Scholar]
- 24.Filardo EJ, Quinn JA, Frackelton AR, Bland KI. Estrogen Action Via the G Protein-Coupled Receptor, GPR30: Stimulation of Adenylyl Cyclase and cAMP-Mediated Attenuation of the Epidermal Growth Factor Receptor-to-MAPK Signaling Axis. Molecular Endocrinology. 2002;16(1):70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 25.Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain research. 1988;452(1–2):57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- 26.Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? The open pain journal. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gras D, Chanez P, Urbach V, Vachier I, Godard P, Bonnans C. Thiazolidinediones induce proliferation of human bronchial epithelial cells through the GPR40 receptor. Am J Physiol Lung Cell Mol Physiol. 2009;296(6):L970–978. doi: 10.1152/ajplung.90219.2008. [DOI] [PubMed] [Google Scholar]
- 29.Grollman AP, Walsh Wttao M. Inhibitors of Protein Biosynthesis: II. MODE OF ACTION OF ANISOMYCIN. Journal of Biological Chemistry. 1967;242(13):3226–3233. [PubMed] [Google Scholar]
- 30.Gruber-Schoffnegger D, Drdla-Schutting R, Honigsperger C, Wunderbaldinger G, Gassner M, Sandkuhler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci. 2013;33(15):6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurley C, Nichols J, Liu S, Phulwani NK, Esen N, Kielian T. Microglia and Astrocyte Activation by Toll-Like Receptor Ligands: Modulation by PPAR-gamma Agonists. PPAR Res. 2008;2008:453120. doi: 10.1155/2008/453120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 33.Harris JA. Using c-fos as a Neural Marker of Pain. Brain Research Bulletin. 1998;45(1):1–8. doi: 10.1016/s0361-9230(97)00277-3. [DOI] [PubMed] [Google Scholar]
- 34.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPAR{gamma} agonist pioglitazone and ibuprofen reduces glial inflammation and A{beta}1-42 levels in APPV717I transgenic mice. Brain. 2005;128(6):1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 35.Hossaini M, Duraku LS, Saraç Ç, Jongen JLM, Holstege JC. Differential distribution of activated spinal neurons containing glycine and/or GABA and expressing c-fos in acute and chronic pain models. Pain. 2010;151(2):356–365. doi: 10.1016/j.pain.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 36.Hunter RL, Bing G. Agonism of Peroxisome Proliferator Receptor-Gamma may have Therapeutic Potential for Neuroinflammation and Parkinson’s Disease. Curr Neuropharmacol. 2007;5(1):35–46. doi: 10.2174/157015907780077123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter RL, Choi D-Y, Ross SA, Bing G. Protective properties afforded by pioglitazone against intrastriatal LPS in Sprague–Dawley rats. Neuroscience Letters. 2008;432(3):198–201. doi: 10.1016/j.neulet.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikeda H, Mochizuki K, Murase K. Astrocytes are involved in long-term facilitation of neuronal excitation in the anterior cingulate cortex of mice with inflammatory pain. Pain. 2013;154(12):2836–2843. doi: 10.1016/j.pain.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 39.Jia H-b, Wang X-m, Qiu L-l, Liu X-y, Shen J-c, Ji Q, Yang J-j. Spinal neuroimmune activation inhibited by repeated administration of pioglitazone in rats after L5 spinal nerve transection. Neuroscience Letters. 2013 doi: 10.1016/j.neulet.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 40.Jia H, Zhu S, Ji Q, Hui K, Duan M, Xu J, Li W. Repeated administration of pioglitazone attenuates development of hyperalgesia in a rat model of neuropathic pain. Exp Clin Psychopharmacol. 2010;18(4):359–365. doi: 10.1037/a0020181. [DOI] [PubMed] [Google Scholar]
- 41.Joels M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev. 2012;64(4):901–938. doi: 10.1124/pr.112.005892. [DOI] [PubMed] [Google Scholar]
- 42.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SJ, Thomas KS, Calejesan AA, Zhuo M. Macromolecular synthesis contributes to nociceptive response to subcutaneous formalin injection in mice. Neuropharmacology. 1998;37(8):1091–1093. doi: 10.1016/s0028-3908(98)00099-9. [DOI] [PubMed] [Google Scholar]
- 44.Krug M, Lössner B, Ott T. Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Research Bulletin. 1984;13(1):39–42. doi: 10.1016/0361-9230(84)90005-4. [DOI] [PubMed] [Google Scholar]
- 45.Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG. Functional Consequences of Cysteine Modification in the Ligand Binding Sites of Peroxisome Proliferator Activated Receptors by GW9662. Biochemistry. 2002;41(21):6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 46.Levin ER. Rapid signaling by steroid receptors. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1425–1430. doi: 10.1152/ajpregu.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67(1):15–19. doi: 10.1124/mol.104.006353. Epub 2004 Oct 2001. [DOI] [PubMed] [Google Scholar]
- 48.LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi: 10.1124/jpet.106.111385. Epub 2006 Sep 1022. [DOI] [PubMed] [Google Scholar]
- 49.Lu Y-C, Chen C-W, Wang S-Y, Wu F-S. 17{beta}-Estradiol Mediates the Sex Difference in Capsaicin-Induced Nociception in Rats. J Pharmacol Exp Ther. 2009 doi: 10.1124/jpet.109.158402. jpet.109.158402. [DOI] [PubMed] [Google Scholar]
- 50.Luconi M, Cantini G, Serio M. Peroxisome proliferator-activated receptor gamma (PPARgamma): Is the genomic activity the only answer? Steroids. 2010;75(8–9):585–594. doi: 10.1016/j.steroids.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 51.Ma D, Lu L, Boneva NB, Warashina S, Kaplamadzhiev DB, Mori Y, Nakaya M-a, Kikuchi M, Tonchev AB, Okano H, Yamashima T. Expression of free fatty acid receptor GPR40 in the neurogenic niche of adult monkey hippocampus. Hippocampus. 2008;18(3):326–333. doi: 10.1002/hipo.20393. [DOI] [PubMed] [Google Scholar]
- 52.Ma D, Tao B, Warashina S, Kotani S, Lu L, Kaplamadzhiev DB, Mori Y, Tonchev AB, Yamashima T. Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neuroscience Research. 2007;58(4):394–401. doi: 10.1016/j.neures.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 53.Maeda T, Kiguchi N, Kobayashi Y, Ozaki M, Kishioka S. Pioglitazone attenuates tactile allodynia and thermal hyperalgesia in mice subjected to peripheral nerve injury. J Pharmacol Sci. 2008;108(3):341–347. doi: 10.1254/jphs.08207fp. [DOI] [PubMed] [Google Scholar]
- 54.Maeda T, Kishioka S. Chapter 13 PPAR and Pain. In: Bagetta GCMTST, Sakurada S, editors. International Review of Neurobiology. Vol. 85. Academic Press; 2009. pp. 165–177. [DOI] [PubMed] [Google Scholar]
- 55.Maeshiba Y, Kiyota Y, Yamashita K, Yoshimura Y, Motohashi M, Tanayama S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittel-Forschung. 1997;47(1):29–35. [PubMed] [Google Scholar]
- 56.Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. 2010;204(2):105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- 57.Mestre C, Pelissier T, Fialip J, Wilcox G, Eschalier A. A method to perform direct transcutaneous intrathecal injection in rats. Journal of pharmacological and toxicological methods. 1994;32(4):197–200. doi: 10.1016/1056-8719(94)90087-6. [DOI] [PubMed] [Google Scholar]
- 58.Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid × receptors in the adult rat CNS. Neuroscience. 2004;123(1):131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- 59.Morgenweck J, Abdel-aleem OS, McNamara KC, Donahue RR, Badr MZ, Taylor BK. Activation of peroxisome proliferator-activated receptor [gamma] in brain inhibits inflammatory pain, dorsal horn expression of Fos, and local edema. Neuropharmacology. 2010;58(2):337–345. doi: 10.1016/j.neuropharm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morgenweck J, Griggs RB, Donahue RR, Zadina JE, Taylor BK. PPARγ activation blocks development and reduces established neuropathic pain in rats. Neuropharmacology. 2013 doi: 10.1016/j.neuropharm.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakamoto K, Nishinaka T, Matsumoto K, Kasuya F, Mankura M, Koyama Y, Tokuyama S. Involvement of the long-chain fatty acid receptor GPR40 as a novel pain regulatory system. Brain research. 2012;1432:74–83. doi: 10.1016/j.brainres.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 62.Nakamoto K, Nishinaka T, Sato N, Mankura M, Koyama Y, Kasuya F, Tokuyama S. Hypothalamic GPR40 Signaling Activated by Free Long Chain Fatty Acids Suppresses CFA-Induced Inflammatory Chronic Pain. PLoS One. 2013;8(12):e81563. doi: 10.1371/journal.pone.0081563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 2004;29(10):1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- 64.Oliveira ACP, Bertollo CM, Rocha LTS, Nascimento JEB, Costa KA, Coelho MM. Antinociceptive and antiedematogenic activities of fenofibrate, an agonist of PPAR alpha, and pioglitazone, an agonist of PPAR gamma. European Journal of Pharmacology. 2007;561:194–201. doi: 10.1016/j.ejphar.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 65.Orii R, Ohashi Y, Guo T, Nelson LE, Hashimoto T, Maze M, Fujinaga M. Evidence for the involvement of spinal cord alpha1 adrenoceptors in nitrous oxide-induced antinociceptive effects in Fischer rats. Anesthesiology. 2002;97(6):1458–1465. doi: 10.1097/00000542-200212000-00018. [DOI] [PubMed] [Google Scholar]
- 66.Pancani T, Anderson KL, Porter NM, Thibault O. Imaging of a glucose analog, calcium and NADH in neurons and astrocytes: dynamic responses to depolarization and sensitivity to pioglitazone. Cell Calcium. 2011;50(6):548–558. doi: 10.1016/j.ceca.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park SW, Yi JH, Miranpuri G, Satriotomo I, Bowen K, Resnick DK, Vemuganti R. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther. 2007;320(3):1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 68.Parsons RG, Gafford GM, Baruch DE, Riedner BA, Helmstetter FJ. Long-term stability of fear memory depends on the synthesis of protein but not mRNA in the amygdala. Eur J Neurosci. 2006;23(7):1853–1859. doi: 10.1111/j.1460-9568.2006.04723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parsons RG, Riedner BA, Gafford GM, Helmstetter FJ. The formation of auditory fear memory requires the synthesis of protein and mRNA in the auditory thalamus. Neuroscience. 2006;141(3):1163–1170. doi: 10.1016/j.neuroscience.2006.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press, Inc; 1997. [Google Scholar]
- 71.Rogue A, Spire C, Brun M, Claude N, Guillouzo A. Gene Expression Changes Induced by PPAR Gamma Agonists in Animal and Human Liver. PPAR Res. 2010;2010:325183. doi: 10.1155/2010/325183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenblum K, Meiri N, Dudai Y. Taste memory: The role of protein synthesis in gustatory cortex. Behavioral and Neural Biology. 1993;59(1):49–56. doi: 10.1016/0163-1047(93)91145-d. [DOI] [PubMed] [Google Scholar]
- 73.Rowan MP, Berg KA, Milam SB, Jeske NA, Roberts JL, Hargreaves KM, Clarke WP. 17β-Estradiol Rapidly Enhances Bradykinin Signaling in Primary Sensory Neurons In Vitro and In Vivo. Journal of Pharmacology and Experimental Therapeutics. 2010;335(1):190–196. doi: 10.1124/jpet.110.167445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Saltiel AR, Olefsky JM. Thiazolidinediones in the Treatment of Insulin Resistance and Type II Diabetes. Diabetes. 1996;45(12):1661–1669. doi: 10.2337/diab.45.12.1661. [DOI] [PubMed] [Google Scholar]
- 75.Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G, Sullivan PG. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Experimental Neurology. 2011;227(1):128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20(18):RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10(11):1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 78.Seabrook GR, Sutton KG, Jarolimek W, Hollingworth GJ, Teague S, Webb J, Clark N, Boyce S, Kerby J, Ali Z, Chou M, Middleton R, Kaczorowski G, Jones AB. Functional Properties of the High-Affinity TRPV1 (VR1) Vanilloid Receptor Antagonist (4-Hydroxy-5-iodo-3-methoxyphenylacetate ester) Iodo-Resiniferatoxin. Journal of Pharmacology and Experimental Therapeutics. 2002;303(3):1052–1060. doi: 10.1124/jpet.102.040394. [DOI] [PubMed] [Google Scholar]
- 79.Simone DA, Baumann TK, LaMotte RH. Dose-dependent pain and mechanical hyperalgesia in humans after intradermal injection of capsaicin. Pain. 1989;38(1):99–107. doi: 10.1016/0304-3959(89)90079-1. [DOI] [PubMed] [Google Scholar]
- 80.Simone DA, Ngeow JY, Putterman GJ, LaMotte RH. Hyperalgesia to heat after intradermal injection of capsaicin. Brain research. 1987;418(1):201–203. doi: 10.1016/0006-8993(87)90982-6. [DOI] [PubMed] [Google Scholar]
- 81.Small KM, Nag S, Mokha SS. Activation of membrane estrogen receptors attenuates opioid receptor-like1 receptor-mediated antinociception via an ERK-dependent non-genomic mechanism. Neuroscience. 2013;255(0):177–190. doi: 10.1016/j.neuroscience.2013.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Storer PD, Xu J, Chavis JA, Drew PD. Cyclopentenone prostaglandins PGA2 and 15-deoxy-delta12,14 PGJ2 suppress activation of murine microglia and astrocytes: implications for multiple sclerosis. J Neurosci Res. 2005;80(1):66–74. doi: 10.1002/jnr.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi Y, Hasegawa-Moriyama M, Sakurai T, Inada E. The Macrophage-Mediated Effects of the Peroxisome Proliferator-Activated Receptor-Gamma Agonist Rosiglitazone Attenuate Tactile Allodynia in the Early Phase of Neuropathic Pain Development. Anesth Analg. 2011 doi: 10.1213/ANE.0b013e31821b220c. [DOI] [PubMed] [Google Scholar]
- 84.Taylor BK. Pathophysiologic mechanisms of neuropathic pain. Curr Pain Headache Rep. 2001;5(2):151–161. doi: 10.1007/s11916-001-0083-1. [DOI] [PubMed] [Google Scholar]
- 85.Taylor BK, Abhyankar SS, Vo NT, Kriedt CL, Churi SB, Urban JH. Neuropeptide Y acts at Y1 receptors in the rostral ventral medulla to inhibit neuropathic pain. Pain. 2007;131(1–2):83–95. doi: 10.1016/j.pain.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor BK, Holloway D, Printz MP. A unique central cholinergic deficit in the spontaneously hypertensive rat: physostigmine reveals a bradycardia associated with sensory stimulation. Journal of Pharmacology and Experimental Therapeutics. 1994;268(3):1081–1090. [PubMed] [Google Scholar]
- 87.Todd AJ, Spike RC, Brodbelt AR, Price RF, Shehab SAS. Some inhibitory neurons in the spinal cord develop c-fos-immunoreactivity after noxious stimulation. Neuroscience. 1994;63(3):805–816. doi: 10.1016/0306-4522(94)90525-8. [DOI] [PubMed] [Google Scholar]
- 88.Vega-Avelaira D, Moss A, Fitzgerald M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain, Behavior, and Immunity. 2007;21(5):617–623. doi: 10.1016/j.bbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 89.Wang H, Guo W, Yang K, Wei F, Dubner R, Ren K. Contribution of Primary Afferent Input to Trigeminal Astroglial Hyperactivity, Cytokine Induction and NMDA Receptor Phosphorylation. The open pain journal. 2010;2010(3):144–152. doi: 10.2174/1876386301003010144]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends in neurosciences. 2001;24(8):450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 91.Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28(42):10482–10495. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamashita A, Hamada A, Suhara Y, Kawabe R, Yanase M, Kuzumaki N, Narita M, Matsui R, Okano H, Narita M. Astrocytic activation in the anterior cingulate cortex is critical for sleep disorder under neuropathic pain. Synapse. 2014;68(6):235–247. doi: 10.1002/syn.21733. [DOI] [PubMed] [Google Scholar]
- 93.Yonutas HM, Sullivan PG. Targeting PPAR isoforms following CNS injury. Current drug targets. 2013;14(7):733–742. doi: 10.2174/1389450111314070003. [DOI] [PubMed] [Google Scholar]
- 94.Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem. 2006;97(3):772–783. doi: 10.1111/j.1471-4159.2006.03746.x. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Lu N, Zhao ZQ, Zhang YQ. Involvement of estrogen in rapid pain modulation in the rat spinal cord. Neurochemical research. 2012;37(12):2697–2705. doi: 10.1007/s11064-012-0859-1. [DOI] [PubMed] [Google Scholar]
- 96.Zhuang Z-Y, Kawasaki Y, Tan P-H, Wen Y-R, Huang J, Ji R-R. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain, behavior, and immunity. 2007;21(5):642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26(13):3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]