

Abstract
Nectins are immunoglobulin-like cell adhesion molecules mainly localized in adherens junctions. The transcription factor p63 is a master regulator of gene expression in stratified epithelia and controls several molecular processes. As mutations in the Pvrl1 and Pvrl4 genes encoding for nectins cause genetic disorders with phenotypes similar to p63-related syndromes, we investigated whether these proteins might be under p63 transcriptional control. Here, we show that in p63-null skin, Pvrl1 gene expression is strongly reduced, whereas Pvrl4 expression is unaffected. In human and mouse primary keratinocytes p63 depletion leads to a specific downregulation of the Pvrl1 gene. Consistent with a direct regulation, chromatin immunoprecipitation experiments (ChIP) indicate that p63 binds to two conserved intronic Pvrl1 enhancer regions. Ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) syndrome is a rare autosomal dominant disorder, caused by mutations in p63 gene, mainly characterized by skin fragility. To test whether nectins may be affected in AEC syndrome, their expression was measured in keratinocytes obtained from patients with AEC or from a conditional mouse model for AEC syndrome. Pvrl1 expression was reduced in AEC keratinocytes, consistent with impaired p63 function. Surprisingly, Pvrl4 expression was similarly affected, in parallel with decreased expression of the transcription factor Irf6. Consistent with the well-characterized role of Irf6 in keratinocyte differentiation and its strong downregulation in AEC syndrome, Irf6 depletion caused reduced expression of Pvrl4 in wild-type keratinocytes. Taken together, our results indicate that Pvrl1 is a bona fide target gene of the transcription factor p63, whereas Pvrl4 regulation is linked to epidermal differentiation and is under Irf6 control.
Keywords: adherens junctions, Irf6, p63, Pvrl1, Pvrl4
Introduction
Adherens junctions (AJ) serve as mechanically adhesive apparatus between neighbouring cells that are associated with actin filaments [for a comprehensive review see (1)]. They consist of two basic adhesive units: the classical calcium-dependent cadherin–catenin complex and the calcium-independent nectin–afadin complex. Epidermal ablation of E-cadherin, the major epidermal transmembrane AJ protein, leads to upregulation of P-cadherin, which is normally restricted to the hair follicle (2). Concomitant ablation of both E-cadherin and P-cadherin in epidermis impairs junction formation and intercellular adhesion, compromising tissue integrity (3).
Nectins are encoded by four different genes PVRL1, PVRL2, PVRL3 and PVRL4 and are capable of forming both homophilic and heterophilic interactions (4,5). Each nectin member is characterized by an extracellular region with three immunoglobulin-like loops, a transmembrane region, and a cytoplasmic intracellular tail that binds afadin, connecting nectin to the actin cytoskeleton. A crucial roles of the nectin-based adherens junctions in embryonic development are revealed by the severe defects detected in afadin (Mllt4) null mice which are characterized by disorganization of the ectoderm, impaired migration of the mesoderm, and loss of somites and other structures derived from both the ectoderm and the mesoderm (6). Depletion of Pvrl1, Pvrl2 or concomitant depletion of Pvrl1 and Pvrl3 do not cause embryonic lethality and lead to relatively mild phenotype in adult mice (7–9), suggesting a functional redundancy among nectins. In human and mouse epidermis, nectin-1 is expressed at cell–cell junctions mainly in the suprabasal layers (10–12). Pvrl1 null newborn mice exhibit a shiny and reddish skin, a phenotype that rapidly disappears, suggesting that compensatory mechanisms may exist in skin, and that Pvrl4, the other nectin gene abundantly expressed in the upper layers of the epidermis (11,13), may compensate for Pvrl1 loss.
Consistent with a crucial and partially overlapping role for PVRL1 and PVRL4, human mutations in these two genes are causative of partially overlapping defects, whereas so far the other nectins have not been linked to human inherited disorders. Mutations in the PVRL1 gene cause cleft lip and/or palate, ectodermal dysplasia syndrome (CLPED1, OMIM 225060), an autosomal recessive disorder mainly characterized by cleft lip/palate, hidrotic ectodermal dysplasia and syndactyly (14–16). The identified homozygous mutations result in protein truncation impairing binding to afadin and abrogating nectin-based cell adhesion. Homozygous or compound heterozygous mutations in the PVRL4 gene are causative of ectodermal dysplasia-syndactyly syndrome (EDSS1, OMIM 613573), an autosomal recessive disorder characterized by cutaneous syndactyly variably involving fingers and toes associated with hair and teeth abnormalities (13,17).
A crucial regulator of epidermal development is p63, a tetrameric transcription factor belonging to the p53 family. p63 null mice die soon after birth with severe defects of the skin and other stratified epithelia, craniofacial abnormalities including cleft lip and palate, as well as impaired limb formation (18–21). The p63 gene is characterized by two independent transcription start sites that give rise to TAp63 and ΔNp63 transcripts (22). The ΔNp63 transcripts are most abundantly expressed in stratified epithelia including those of the skin and the oral cavity and are required for the appropriate development of these structures (22–25). In humans, heterozygous mutations in the p63 gene cause at least six-related syndromes characterized by ectodermal dysplasia, cleft lip/palate and limb abnormalities including ectrodactyly or syndactyly (26). Among them, ankyloblepharon–ectodermal defects–cleft lip/palate (AEC) syndrome (OMIM 106260) is characterized by skin fragility, erosions and blisters (27,28). Consistently, p63 is a crucial regulator of cell–matrix adhesion (25,29–33), and cell–cell adhesion directly regulating a subset of desmosomal genes such as Dsc3, Dsp, Dsg1, Perp (34,35) and the tight junction component claudin-1 (36). Among the adherens junction components, P-cadherin is a direct p63 target gene (37), whereas E-cadherin expression is independent of p63 function.
Given that PVRL1 and PVRL4 associated syndromes have many features in common with those caused by p63 mutations, we investigated the possibility that p63 may regulate Pvrl expression in epidermis. We found that p63 directly controls Pvrl1 transcription, whereas expression of Pvrl4 and of the other Pvrl genes is, at least in part, independent of p63. Pvrl1 expression is inhibited in p63 mutant keratinocytes derived from patients with AEC and in keratinocytes derived from a conditional mouse model of the disorder. Surprisingly, also Pvrl4 expression is impaired in AEC mutant keratinocytes, particularly in differentiating conditions. A crucial regulator of epidermal differentiation is the transcription factor Irf6 (38–40). p63 activates transcription of IRF6, whereas p63 mutant proteins causing ectodermal dysplasias are unable to activate IRF6 transcription (41,42). Accordingly, we found that Irf6 depletion reduces Pvrl4 expression indicating that Pvrl4 may be under Irf6 control in differentiated keratinocytes.
Materials and methods
Mouse models, immunofluorescence and immunoblotting
p63-null mice (B6.129S7-Trp63tm1Brd/J) (18) were obtained from the Jackson Laboratory. Conditional knock-in mice (p63-FLAGL514F/L514F) expressing a clinically relevant missense mutation were obtained crossing a K14-Cre knock-in mouse (43) with p63-FLAGfloxL514F/floxL514F (a complete characterization of the model will be described elsewhere). All mouse experiments were approved by the Italian Ministry of Health.
For immunofluorescence staining, tissue was embedded in O.C.T. compound (Tissue-TekII; Lab-Tek Products) and frozen at −80°C. Cryosections (6 μm) were fixed in 4% PFA and immunofluorescence was performed as previously described (44). Sections were examined using a Zeiss confocal microscope LSM510meta (Zeiss, Oberkoche, Germany) or a Leica DMRB microscope (Leica, Solms, Germany). The following primary antibodies were used: rabbit polyclonal antibodies to nectin-1 (H-62; Santa Cruz Biotechnology (Santa Cruz, CA, USA)) and IRF6 (kindly provided by B. Schutte, Michigan State University), mouse antibodies to p63 (4A4; Santa Cruz Biotechnology), to nectin-4 (HPA016775; Sigma, St. Louise, MO, USA) and to β-actin (Santa Cruz Biotechnology). For immunoblotting, secondary antibodies were donkey anti-rabbit and sheep anti-mouse IgG conjugated to horseradish peroxidase (GE Healthcare, Freiburg, Germany). Immunoblotting was performed using ECL (GE Healthcare, Freiburg, Germany). Fluorescence was revealed using goat anti-mouse biotinylated and goat anti-rabbit biotinylated (Vector Laboratories, Burlingame, CA, USA) in combination with Streptavidin Cy-2 (Vector Laboratories, Burlingame, CA, USA) and sections were counterstained with DAPI.
Cell culture and reporter activity
Primary mouse keratinocytes were isolated from newborn mice and cultured under low calcium conditions (0.05 mm) or treated with 2 mm calcium chloride as previously described (45). Human keratinocytes obtained from patients with AEC Q11X and T533P [kindly provided by J. Zhou, (35,46)], as well as control keratinocytes obtained by unaffected individuals were plated at a density of 104 cells/cm2 and cultured in KBM Gold medium (Lonza) until confluent. Confluent cells were treated with 0.3 mm calcium chloride for subsequent RNA analysis.
Knockdown was achieved by transient transfection of 100 nm small interfering RNA (siRNA) for pan-p63, ΔNp63 (47), Irf6 (Mm_Irf6_2; Qiagen (Valencia, CA, USA) or negative control (Invitrogen, Carlsbad, CA, USA). Mouse Pvrl1 intronic region 2 (p63BS2) was cloned in pGL3-enhancer luciferase reporter plasmid (as described in Supplementary data). Luciferase activity was determined 48 h after transfection with the dual-luciferase reporter assay kit (Promega, Madison, WI, USA). Renilla activity was used to normalize transfection efficiency.
Normal human epidermal keratinocytes (NHEK-Neo, Lonza; Cat. No. 00192907) were plated at a density of 104 cells/cm2 and cultured in KBM Gold medium (Lonza) until confluent. p63 and ΔNp63 knockdown were obtained by transient transfection of 100 nm small interfering RNA (siRNA) for pan-p63 (5′- CAGAACACACAUGGUAUCCAGAUGA-3′; Stealth RNAi Invitrogen), ΔNp63 (47) or negative control (Invitrogen).
Real-time RT-PCR and ChIP analysis
Total RNA from human and mouse keratinocytes were extracted with TRIzol reagent (Invitrogen) and were employed as a template for complementary DNA (cDNA) synthesis performed with SuperScript Vilo (Invitrogen) according to manufacturer's instructions. Two-step real-time reverse transcription RT-PCR was performed using the SYBR Green PCR master mix in an ABI PRISM 7500 (Applied Biosystems, Foster City, CA, USA). Levels of the target genes were quantified with specific oligonucleotide primers and normalized for ß-actin expression in mouse and RPLP0 for human samples. For primer sequences, see Tables S1 and S2. ChIP analysis was performed on primary human keratinocytes and newborn mouse epidermis as previously described (47). For primer sequences, see Tables S3 and S4.
Statistics
All experiments were repeated at least twice, and real-time RT-PCR and ChIP were performed in duplicates. Statistical significance of the gene expression studies was assessed by unpaired two-tailed Student's t test, and P-values are indicated in Figure Legends. All quantitative results are presented as mean ± the standard deviation (SD) as indicated in the Figure Legends and were calculated by Excel software.
Results
Pvrl1 expression is strongly reduced in embryonic skin of p63 deficient mice
In p63 null mice, disorganized and loosely attached epidermal cells are lost at birth. To assess whether p63 may regulates nectin-based adherens junctions, the expression of Pvrl family members was evaluated in p63 null skin and its wild-type counterpart during embryonic development.
In wild-type embryonic skin, both nectin-1 and nectin-4 were expressed in the suprabasal layer of the epidermis at E14.5. In p63 null epidermis, nectin-1 was drastically decreased compared to controls, whereas nectin-4 was still expressed in the single layer of epidermal cells (Fig.1a). Loss of nectin-1 was similarly observed at E16.5 when both wild-type and p63 null skin were characterized by more than one layer of cells, suggesting that loss of nectin-1 expression is unlikely to be due to reduced stratification. Consistent with reduced protein levels, Pvrl1 mRNA was strongly diminished in p63 null mice, whereas an equal amount of the other Pvrl genes, afadin (Mllt4) and E-cadherin (Cdh1) was observed (Fig.1b).
Figure 1.
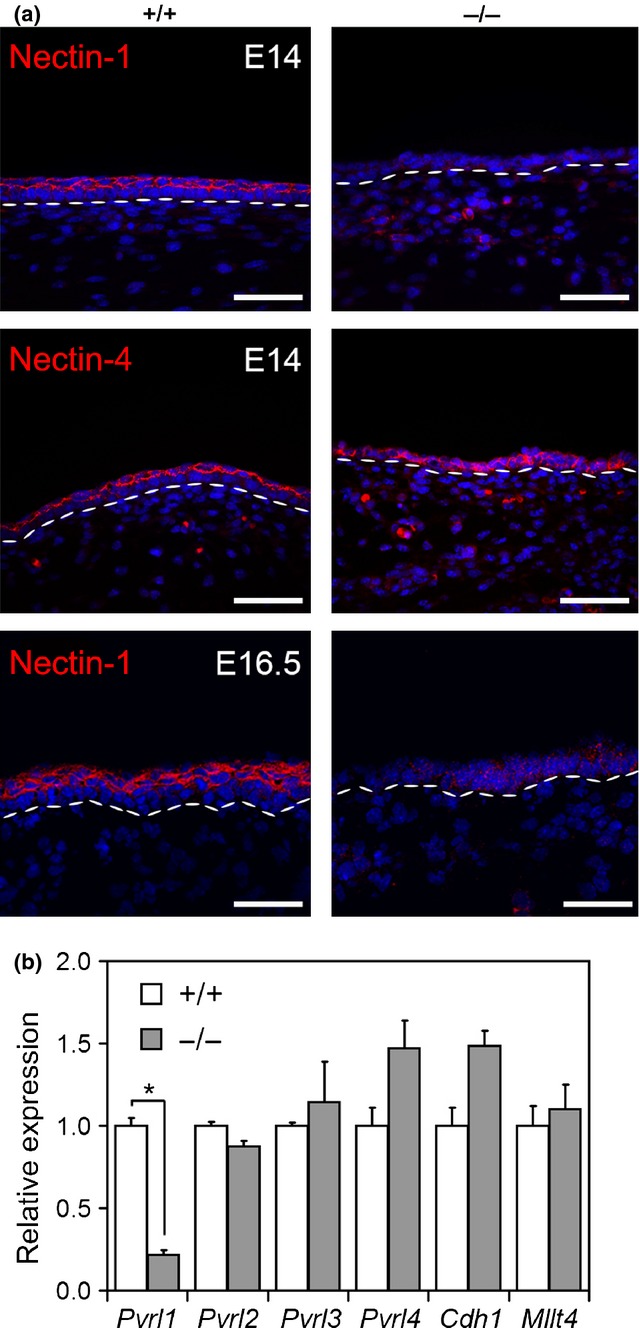
Nectin-1 expression is decreased in p63 null embryos. (a) Immunofluorescence analysis of nectin-1 and nectin-4 on E14 embryo skin derived from p63 null mice (−/−) and controls (+/+) (upper panels). Nectin-1 expression at E16.5 is shown in the lower panel. Dashed white lines indicate the basement membrane. Scale bars, 50 μm. (b) Expression of the indicated genes was performed by real-time RT-PCR in E16.5 p63 null (−/−) and controls (+/+) skin. Pvrl1 mRNA levels were strongly decreased in the skin of p63−/− mice compared to wild type (*P-value=0.007; n = 6). Data are normalized for β-actin mRNA levels and are expressed relative to controls. Error bars denote SD.
We next analysed Pvrl1 expression in primary mouse keratinocytes depleted for p63 or ΔNp63 using specific siRNA (Fig.2a–c). In both p63- and ΔNp63-depleted keratinocytes, Pvrl1 was significantly reduced both at the mRNA and protein levels as compared to controls, whereas Pvrl4 mRNA levels were not affected by p63 knockdown (Fig.2b,c). Similarly, human keratinocytes depleted for p63 or ΔNp63 showed a reduction of PVRL1 compared to the control whereas PVRL4 was not affected by p63 depletion (Fig.2d).
Figure 2.
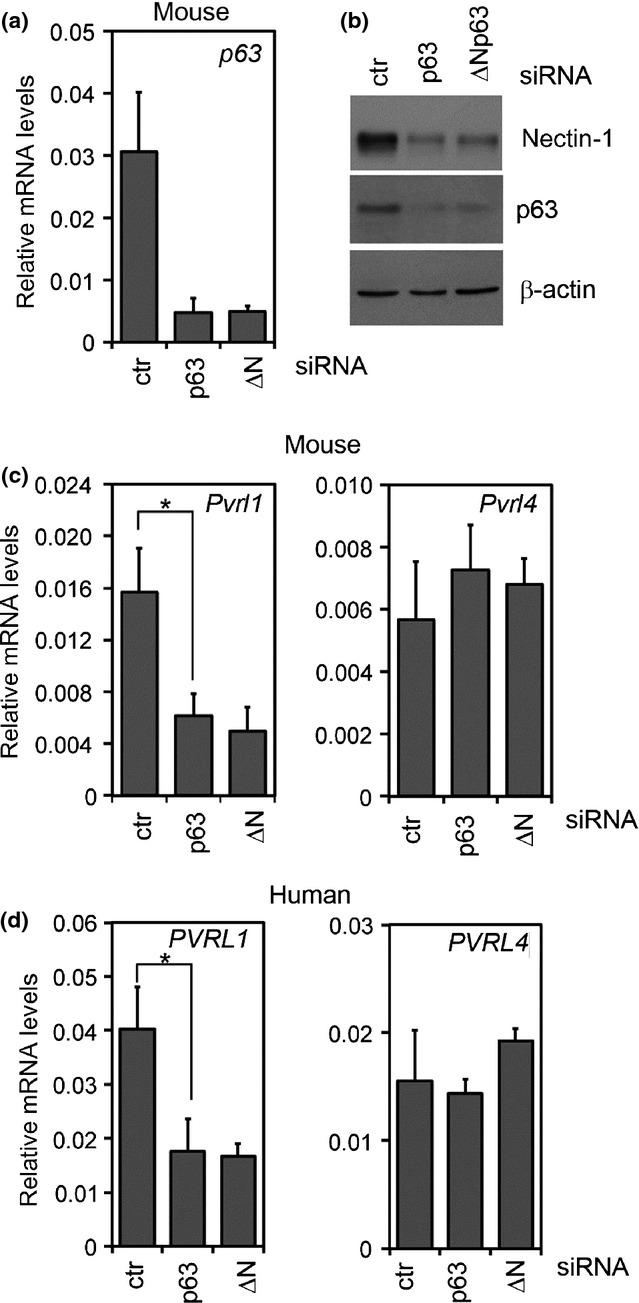
p63 depletion influence Pvrl1 expression in vitro. (a) p63 mRNA expression in control mouse keratinocytes or in keratinocytes depleted of p63 or ΔNp63. Data are normalized for β-actin mRNA levels and are represented as mean of ± SD. (b) Immunoblotting analysis for nectin-1 and p63 in cell extracts depleted of p63 or ΔNp63, or controls (ctr). Data are normalized for β-actin protein expression. (c) Real-time RT-PCR for Pvrl1 in keratinocytes depleted of p63 or ΔNp63. Pvrl1 mRNA expression was strongly downregulated in p63-depleted keratinocytes as compared to controls (*P-value = 0.002; n = 8) whereas Pvrl4 expression is similar to control. Data are normalized for β-actin mRNA levels and are represented as mean ± SD normalized mRNA levels. (d) PVRL1 and PVRL4 mRNA expression in human primary keratinocytes depleted of p63 or ΔNp63. PVRL1 mRNA expression was strongly downregulated in p63-depleted keratinocytes as compared to controls (*P-value = 0.01; n = 6) whereas Pvrl4 expression is similar to control. Data are normalized for RPLP0 mRNA levels and are represented as mean ± SD.
Together these results suggest that p63 specifically regulates the expression of Pvrl1, but not of the other Pvrl genes both in human and mouse.
Pvrl1 is a p63 target gene in skin
To obtain further insights into the regulation of Pvrl1 by p63, we analysed a genome-wide ChIP-seq analysis previously performed in human primary keratinocytes (48) which revealed at least two strong p63-binding regions (p63BS1 and p63BS2) in the first intron of PVRL1 (Figure S1). p63BS1 and p63BS2 were highly conserved in mammals and contained DNAse hypersensitive sites (Figure S1), consistent with their open chromatin state (49,50). In addition p63BS2, and to a lesser extent p63BS1, were enriched for histone H3 acetylation on lysine 27 (H3K27ac) (Figure S1), a chromatin marks known to associate with active regulatory regions (51–53). ChIP-qPCR analysis performed in primary human keratinocytes and mouse epidermis confirmed that p63 efficiently bound both the identified regions and more strongly to p63BS2 in mouse epidermis (Fig.3a), which contains at least two evolutionary conserved p63-binding sites (Figure S1). To gain further insights, we cloned p63BS2 upstream of the luciferase gene. Luciferase assays revealed that the p63BS2 activity was efficiently reduced upon p63 depletion in mouse keratinocytes (Fig.3b). Taken together, these data indicate that Pvrl1 is a bona fide p63 transcriptional target gene.
Figure 3.
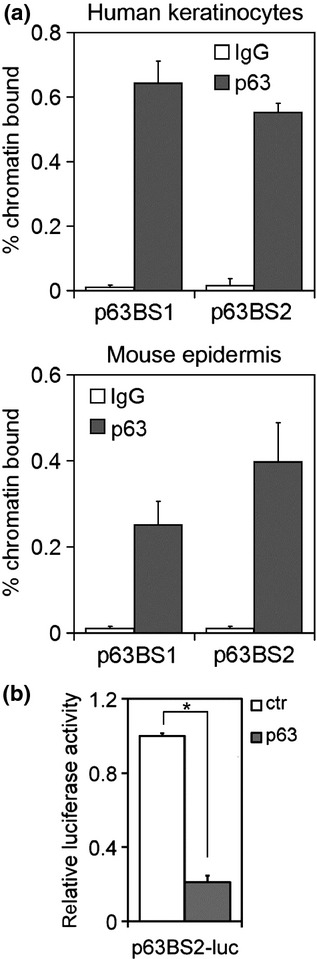
PVRL1 is a target gene of the transcription factor p63. (a) chromatin immunoprecipitation experiments (ChIP)-qPCR of the two identified p63-binding regions (p63BS) in the PVRL1 genomic locus was performed in human primary keratinocytes (upper panel) and in mouse epidermis (lower panel). (b) p63BS2 enhancer activity was analysed by luciferase assay in p63 depleted or control keratinocytes (*P-value = 3.12E-06; n = 6). Data are represented as mean of ± SD of three independent experiments.
PVRL1 and PVRL4 are deregulated in AEC syndrome
PVRL1 and PVRL4 mutations cause syndromic defects partially overlapping with the phenotype of AEC syndrome (13,16,17,27). To test the possibility that PVRL1 and PVRL4 could be impaired in AEC syndrome, we analysed their expression in human keratinocytes derived from individuals affected by this disorder. Surprisingly, both PVRL1 and PVRL4 were strongly reduced (Fig.4a). Similar results were obtained in keratinocytes derived from a conditional mouse model for AEC syndrome (p63-FLAGL514F/L514F), and expressing p63-FLAGL514F to a similar extent of wild-type p63 (Fig.4b). Both Pvrl1 and Pvrl4 were significantly lower in mutant keratinocytes versus controls under basal and differentiating conditions (Fig.4c).
Figure 4.
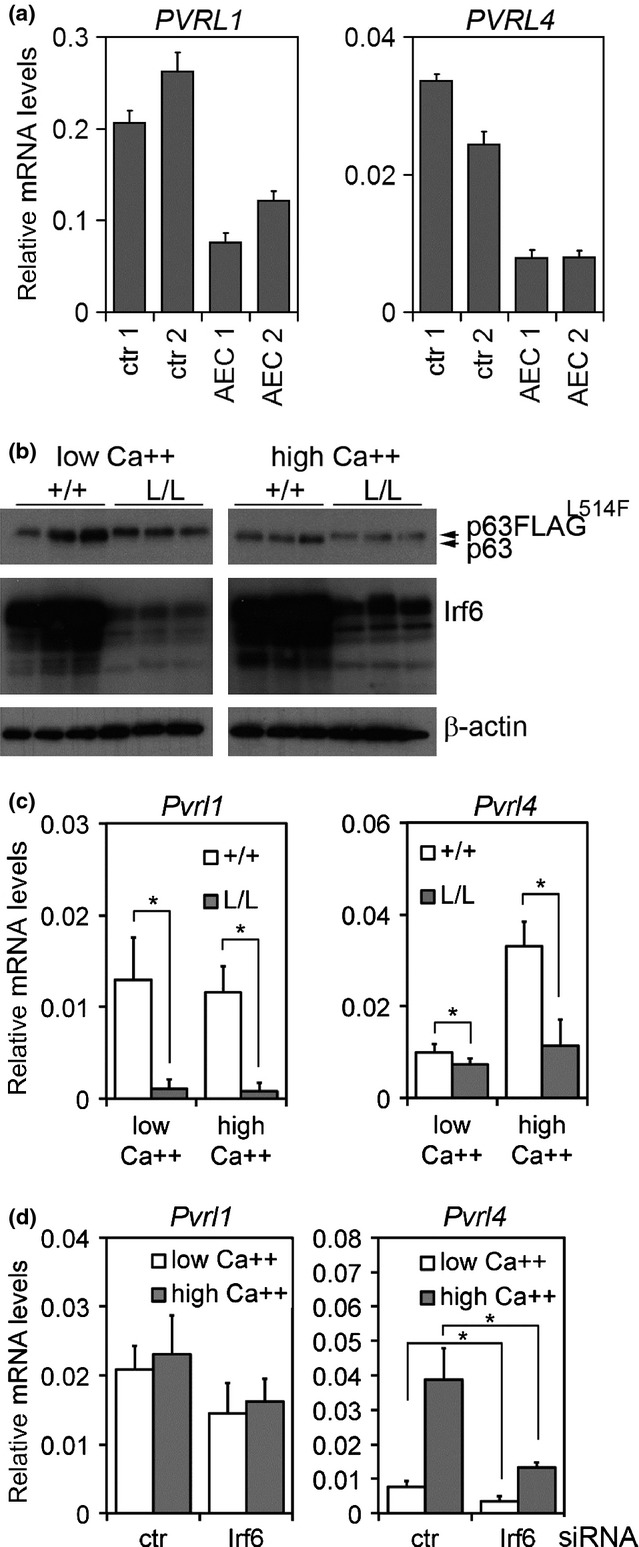
PVRL1 and PVRL4 expression in p63 mutant keratinocytes. (a) PVRL1 and PVRL4 mRNA expression levels in human keratinocytes derived from two affected individuals (AEC1 and AEC2) and controls (ctr1 and ctr2) harvested under differentiating (0.3 mm Ca++) conditions. Data are normalized for RPLP0 mRNA levels and are represented as mean ± SD normalized mRNA levels. (b) Immunoblotting analysis for the indicated proteins in mouse primary keratinocytes derived from p63-FLAGL514F/L514F mice (L/L) and controls (+/+). Arrows indicate the wild-type and FLAG version of the mutant p63 protein. (c) Pvrl1 and Pvrl4 mRNA expression in K14-Cre; p63-FLAGL514F/L514F (L/L) keratinocytes harvested under basal (low Ca++) and differentiating conditions (high Ca++, 2 mm). Pvrl1 mRNA levels were significantly decreased both under basal (*P-value = 8.13E-05; n = 14) and differentiating conditions (*P-value = 9.7E-05; n = 10). Similarly, Pvrl4 mRNA levels were significantly decreased under basal (*P-value = 0.012; n = 14) and differentiating conditions (*P-value = 0.008; n = 6). Data are normalized for β-actin mRNA levels and are represented as mean ± SD. (d) Pvrl1 and Pvrl4 mRNA levels in Irf6-depleted mouse keratinocytes. Pvrl4 mRNA expression is negatively regulated by Irf6 depletion both under basal (*P-value = 0.002; n = 8) and differentiating conditions (*P-value = 0.05; n = 4). Data are normalized for β-actin mRNA levels and are represented as mean ± SD.
Given that Pvrl4 was more highly expressed in differentiated keratinocytes, we tested the possibility that its expression may be regulated by Irf6, a transcription factor involved in epidermal differentiation (40). Irf6 is an established transcriptional target of p63, and p63 mutations causative of AEC syndrome strongly reduce p63 ability to activate IRF6 transcription (41,42). Consistently, we found that p63 mutant mouse keratinocytes exhibited a stronger downregulation of Irf6 (80%) as compared to p63 depleted keratinocytes (40%) or p63 null skin (35–40%) (Fig.4b and Figure S2). To investigate its putative role in the regulation of Pvrl1 and Pvrl4 expression, Irf6 was depleted in primary mouse keratinocytes both under basal and differentiating conditions (Figure S3). Irf6 depletion caused a significant reduction of Pvrl4 expression, whereas Pvrl1 remained unaffected (Fig.4d).
Taken together, these observations suggest that Irf6 controls Pvrl4 expression and that strongly decreased Irf6 expression may be at least in part the cause of reduced Pvrl4 expression in AEC mutant keratinocytes.
Discussion
Among the several functions that have been ascribed to p63 in epidermis, a crucial role played by this transcription factor is to maintain cell–cell adhesion, by controlling several genes including those encoding desmosomal components (34,35,54). Adherens junction significantly contribute to cell adhesion in epidermis and in the hair follicle (55), and among the classical cadherins, the hair follicle expressed P-cadherin is a direct transcriptional target of p63 (37). Here, we demonstrate that p63 regulates the nectin–afadin complex by directly controlling the Pvrl1 gene, encoding for nectin-1. Pvrl1 mRNA and its protein product nectin-1 are strongly reduced in p63 null embryonic skin at E14.5-E16.5, whereas the expression of the other Pvrl family member Pvrl4 is unaffected. Accordingly, Pvrl1 expression is strongly reduced in p63 depleted human and mouse keratinocytes in culture. Consistent with a direct regulation, p63 binds to two intronic bona fide regulatory regions characterized by high levels of H3K27 acetylation and the presence of DNAse hypersensitive sites, both in human keratinocytes and in mouse epidermis. Previous in situ hybridization studies in early embryos (E11-E12) did not show a significant difference in Pvrl1 expression in p63 null surface ectoderm or in oral epithelium (24), possibly due to a relatively low signal to background noise. An alternative possibility that remains to be explored is that Pvrl1 expression may be less dependent on p63 in earlier developmental stages.
The two binding sites for p63 that were characterized in the present study are the strongest ones in the Pvrl1 human genomic region as determined by ChIP-seq (48) and coincide with the putative enhancer regions as determined by H3K27 acetylation (Figure S1) (53). In another genome-wide identification study, a p63-binding region was previously reported in close proximity to the Pvrl1 promoter in human keratinocytes and was found to be responsive to p63γ in heterologous cells (56), suggesting that other p63-binding sites may be functionally relevant for Pvrl1 regulation.
As PVRL1 mutations in humans cause ectodermal dysplasia and cleft lip/palate (14–16), resembling p63-associated syndromes, it has been previously suggested that two genes may be functionally linked (27). Consistent with this hypothesis, we found that Pvrl1 expression is reduced in keratinocytes derived from AEC syndrome patients and from an AEC mouse models. Surprisingly, Pvrl4 expression is also reduced in differentiated AEC keratinocytes, even though its expression is not altered in p63-depleted keratinocytes or epidermis. In searching for a possible mechanism for reduced expression of Pvrl4 in differentiated AEC keratinocytes, we tested the possibility that IRF6, a crucial regulator of keratinocyte terminal differentiation (38–40), may regulate Pvrl4. Consistent with previous observations in human skin (42), we found that Irf6 is strongly reduced in AEC mutant keratinocytes, whereas its expression is more modestly affected in p63-depleted keratinocytes or in p63 null skin. The observed differential Irf6 regulation between AEC keratinocytes and p63 null skin may be explained by the dominant-negative nature of AEC mutations and by the complete abrogation of the entire differentiation program that we observe in AEC keratinocytes, but not in p63-depleted cells (our unpublished data). Taken together these data suggest a model whereby p63 and differentiation-specific transcription factors are required for Irf6 expression (Figure S4). In AEC keratinocytes, loss of p63 and impairment of differentiation lead to dramatic downregulation of Irf6 and Pvrl4. Accordingly, Irf6 depletion results in strong downregulation of Pvrl4, suggesting that reduced Irf6 in AEC mutant keratinocytes may be responsible – at least in part – for Pvrl4 downregulation. Our attempts to identify Irf6-binding sites in the Pvrl4 genomic region have failed (data not shown), thus whether Irf6 controls Pvrl4 directly or indirectly remains to be explored. Finally, reduced nectin-1 and nectin-4 may contribute in part to the skin fragility phenotype observed in AEC syndrome, even though we did not observed strong changes in Pvrl1 and Pvrl4 expression in AEC skin (data not shown), suggesting that compensatory mechanisms may operate in vivo to control nectin expression. Our work provides the first insight into the transcriptional regulation of Pvrl1 and Pvrl4 gene expression in epidermal cells.
Acknowledgments
We thank Dr. Huiqing Zhou for sharing p63 ChIP-seq data and primary keratinocytes derived from patients with AEC. The financial support of Telethon, Italy (GGP09230) (C.M.) and the European ERA-Net Research Program on Rare Disease (E-Rare-2; Skindev) are gratefully acknowledged.
Author contributions
M.R.M and C.M. designed the experiments; M.R.M., D.A., K.M., P.F. performed the research; J.D., A.C., F.B contributed with helpful discussion and essential reagents, F.B. contributed with essential ideas; M.R.M, D.A. and C.M. analysed the data and wrote the paper.
Conflict of interest
The authors have declared no conflicting interests.
Supporting Information
Additional supporting data may be found in the supplementary information of this article.
Figure S1. p63 binding regions in the PVRL1 genomic locus.
Figure S2. Irf6 expression in p63 null epidermis, in p63 depleted keratinocytes, and in AEC mutant keratinocytes.
Figure S3. Irf6 expression in Irf6 depleted mouse keratinocytes.
Figure S4. Scheme illustrating the regulation of Pvrl1 and Pvrl4 by p63.
Table S1. Oligonucleotide primers for Real Time RT-PCR on mouse samples.
Table S2 Oligonucleotide primers for Real Time RT-PCR on human samples.
Table S3. Oligonucleotide primers for ChIP analysis on human genomic DNA.
Table S4. Oligonucleotide primers for ChIP analysis on mouse genomic DNA.
References
- 1.Harris TJ, Tepass U. Nat Rev Mol Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- 2.Tinkle CL, Lechler T, Pasolli HA, et al. Proc Natl Acad Sci USA. 2004;101:552–557. doi: 10.1073/pnas.0307437100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tinkle CL, Pasolli HA, Stokes N, et al. Proc Natl Acad Sci USA. 2008;105:15405–15410. doi: 10.1073/pnas.0807374105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rikitake Y, Mandai K, Takai Y. J Cell Sci. 2012;125:3713–3722. doi: 10.1242/jcs.099572. [DOI] [PubMed] [Google Scholar]
- 5.Meng W, Takeichi M. Cold Spring Harb Perspect Biol. 2009;1:a002899. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda W, Nakanishi H, Miyoshi J, et al. J Cell Biol. 1999;146:1117–1132. doi: 10.1083/jcb.146.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inagaki M, Irie K, Ishizaki H, et al. Development. 2005;132:1525–1537. doi: 10.1242/dev.01697. [DOI] [PubMed] [Google Scholar]
- 8.Barron MJ, Brookes SJ, Draper CE, et al. Hum Mol Genet. 2008;17:3509–3520. doi: 10.1093/hmg/ddn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouchard MJ, Dong Y, McDermott BM, Jr, et al. Mol Cell Biol. 2000;20:2865–2873. doi: 10.1128/mcb.20.8.2865-2873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsushima H, Utani A, Endo H, et al. Br J Dermatol. 2003;148:755–762. doi: 10.1046/j.1365-2133.2003.05225.x. [DOI] [PubMed] [Google Scholar]
- 11.Wakamatsu K, Ogita H, Okabe N, et al. J Biol Chem. 2007;282:18173–18181. doi: 10.1074/jbc.M611159200. [DOI] [PubMed] [Google Scholar]
- 12.Fortugno P, Josselin E, Tsiakas K, et al. J Invest Dermatol. 2014;134:2146–2153. doi: 10.1038/jid.2014.119. [DOI] [PubMed] [Google Scholar]
- 13.Brancati F, Fortugno P, Bottillo I, et al. Am J Hum Genet. 2010;87:265–273. doi: 10.1016/j.ajhg.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zlotogora J, Zilberman Y, Tenenbaum A, et al. J Med Genet. 1987;24:291–293. doi: 10.1136/jmg.24.5.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bustos T, Simosa V, Pinto-Cisternas J, et al. Am J Med Genet. 1991;41:398–404. doi: 10.1002/ajmg.1320410403. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki K, Hu D, Bustos T, et al. Nat Genet. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 17.Jelani M, Chishti MS, Ahmad W. J Hum Genet. 2011;56:352–357. doi: 10.1038/jhg.2011.18. [DOI] [PubMed] [Google Scholar]
- 18.Mills AA, Zheng B, Wang XJ, et al. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 19.Yang A, Schweitzer R, Sun D, et al. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 20.Romano RA, Sinha S. Exp Dermatol. 2014;23:238–239. doi: 10.1111/exd.12356. [DOI] [PubMed] [Google Scholar]
- 21.Missero C, Antonini D. Exp Dermatol. 2014;23:143–146. doi: 10.1111/exd.12320. [DOI] [PubMed] [Google Scholar]
- 22.Yang A, Kaghad M, Wang Y, et al. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 23.Parsa R, Yang A, McKeon F, et al. J Invest Dermatol. 1999;113:1099–1105. doi: 10.1046/j.1523-1747.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- 24.Laurikkala J, Mikkola ML, James M, et al. Development. 2006;133:1553–1563. doi: 10.1242/dev.02325. [DOI] [PubMed] [Google Scholar]
- 25.Romano RA, Smalley K, Magraw C, et al. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinne T, Bolat E, Meijer R, et al. Am J Med Genet A. 2009;149A:1948–1951. doi: 10.1002/ajmg.a.32793. [DOI] [PubMed] [Google Scholar]
- 27.McGrath JA, Duijf PH, Doetsch V, et al. Hum Mol Genet. 2001;10:221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 28.Payne AS, Yan AC, Ilyas E, et al. Arch Dermatol. 2005;141:1567–1573. doi: 10.1001/archderm.141.12.1567. [DOI] [PubMed] [Google Scholar]
- 29.Clements SE, Techanukul T, Lai-Cheong JE, et al. Br J Dermatol. 2012;167:134–144. doi: 10.1111/j.1365-2133.2012.10888.x. [DOI] [PubMed] [Google Scholar]
- 30.Carroll DK, Carroll JS, Leong CO, et al. Nat Cell Biol. 2006;8:551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 31.Kurata S, Okuyama T, Osada M, et al. J Biol Chem. 2004;279:50069–50077. doi: 10.1074/jbc.M406322200. [DOI] [PubMed] [Google Scholar]
- 32.Koster MI, Dai D, Marinari B, et al. Proc Natl Acad Sci USA. 2007;104:3255–3260. doi: 10.1073/pnas.0611376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fenton SE, Denning MF. Exp Dermatol. 2014 doi: 10.1111/exd.12485. Jun 30. doi: 10.1111/exd.12485 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 34.Ihrie RA, Marques MR, Nguyen BT, et al. Cell. 2005;120:843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 35.Ferone G, Mollo MR, Thomason HA, et al. Hum Mol Genet. 2013;22:531–543. doi: 10.1093/hmg/dds464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopardo T, Lo Iacono N, Marinari B, et al. PLoS One. 2008;3:e2715. doi: 10.1371/journal.pone.0002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimomura Y, Wajid M, Shapiro L, et al. Development. 2008;135:743–753. doi: 10.1242/dev.006718. [DOI] [PubMed] [Google Scholar]
- 38.Richardson RJ, Dixon J, Malhotra S, et al. Nat Genet. 2006;38:1329–1334. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 39.Restivo G, Nguyen BC, Dziunycz P, et al. EMBO J. 2011;30:4571–4585. doi: 10.1038/emboj.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingraham CR, Kinoshita A, Kondo S, et al. Nat Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomason HA, Zhou H, Kouwenhoven EN, et al. J Clin Invest. 2010;120:1561–1569. doi: 10.1172/JCI40266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moretti F, Marinari B, Lo Iacono N, et al. J Clin Invest. 2010;120:1570–1577. doi: 10.1172/JCI40267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huelsken J, Vogel R, Erdmann B, et al. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 44.Ferone G, Thomason HA, Antonini D, et al. EMBO Mol Med. 2012;4:192–205. doi: 10.1002/emmm.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antonini D, Russo MT, De Rosa L, et al. J Invest Dermatol. 2010;130:1249–1257. doi: 10.1038/jid.2009.438. [DOI] [PubMed] [Google Scholar]
- 46.Rinne T, Clements SE, Lamme E, et al. Hum Mol Genet. 2008;17:1968–1977. doi: 10.1093/hmg/ddn094. [DOI] [PubMed] [Google Scholar]
- 47.De Rosa L, Antonini D, Ferone G, et al. J Biol Chem. 2009;284:30574–30582. doi: 10.1074/jbc.M109.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kouwenhoven EN, van Heeringen SJ, Tena JJ, et al. PLoS Genet. 2010;6:e1001065. doi: 10.1371/journal.pgen.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nord AS, Blow MJ, Attanasio C, et al. Cell. 2013;155:1521–1531. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford GE, Holt IE, Whittle J, et al. Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rada-Iglesias A, Bajpai R, Swigut T, et al. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Creyghton MP, Cheng AW, Welstead GG, et al. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ernst J, Kheradpour P, Mikkelsen TS, et al. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki D, Senoo M. Exp Dermatol. 2013;22:374–376. doi: 10.1111/exd.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamora C, Fuchs E. Nat Cell Biol. 2002;4:E101–E108. doi: 10.1038/ncb0402-e101. [DOI] [PubMed] [Google Scholar]
- 56.McDade SS, Henry AE, Pivato GP, et al. Nucleic Acids Res. 2012;40:7190–7206. doi: 10.1093/nar/gks389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. p63 binding regions in the PVRL1 genomic locus.
Figure S2. Irf6 expression in p63 null epidermis, in p63 depleted keratinocytes, and in AEC mutant keratinocytes.
Figure S3. Irf6 expression in Irf6 depleted mouse keratinocytes.
Figure S4. Scheme illustrating the regulation of Pvrl1 and Pvrl4 by p63.
Table S1. Oligonucleotide primers for Real Time RT-PCR on mouse samples.
Table S2 Oligonucleotide primers for Real Time RT-PCR on human samples.
Table S3. Oligonucleotide primers for ChIP analysis on human genomic DNA.
Table S4. Oligonucleotide primers for ChIP analysis on mouse genomic DNA.