

Abstract
Orai1 interacts with transient receptor potential protein of the canonical subfamily (TRPC4) and contributes to calcium selectivity of the endothelial cell store-operated calcium entry current (ISOC). Orai1 silencing increases sodium permeability and decreases membrane-associated calcium, although it is not known whether Orai1 is an important determinant of cytosolic sodium transitions. We test the hypothesis that, upon activation of store-operated calcium entry channels, Orai1 is a critical determinant of cytosolic sodium transitions. Activation of store-operated calcium entry channels transiently increased cytosolic calcium and sodium, characteristic of release from an intracellular store. The sodium response occurred more abruptly and returned to baseline more rapidly than did the transient calcium rise. Extracellular choline substitution for sodium did not inhibit the response, although 2-aminoethoxydiphenyl borate and YM-58483 reduced it by ∼50%. After this transient response, cytosolic sodium continued to increase due to influx through activated store-operated calcium entry channels. The magnitude of this sustained increase in cytosolic sodium was greater when experiments were conducted in low extracellular calcium and when Orai1 expression was silenced; these two interventions were not additive, suggesting a common mechanism. 2-Aminoethoxydiphenyl borate and YM-58483 inhibited the sustained increase in cytosolic sodium, only in the presence of Orai1. These studies demonstrate that sodium permeates activated store-operated calcium entry channels, resulting in an increase in cytosolic sodium; the magnitude of this response is determined by Orai1.
Keywords: transient receptor potential protein of the canonical subfamily, calcium release-activated calcium current, endoplasmic reticulum, calcium release, permeability
endothelium forms a semipermeable barrier that separates blood from the underlying tissue (2, 49). In response to tissue injury, infection, and systemic inflammation, neurohumoral inflammatory mediators act on endothelial cell Gq-coupled receptors and initiate inositol 1,4,5-trisphosphate production (13). Inositol 1,4,5-trisphosphate binds to its receptor on the endoplasmic reticulum, transiently depleting calcium stores and promoting calcium entry through store-operated calcium entry channels. Calcium entry through store-operated calcium entry channels reorganizes the cortical actin rim into stress fibers, which increases centripetally directed tension, and reorganizes junctional complexes at cell-cell and cell-matrix borders (39). Increased tension, along with loss of cell adhesion, promotes interendothelial cell gap formation, which increases water, solute, and macromolecular permeability, leading to tissue edema (39). Tissue edema, especially in the lung, is an important cause of patient morbidity and mortality (57), prompting a search to reveal the molecular anatomy of the endothelial cell store-operated calcium entry signalplex as a potential anti-inflammatory therapeutic target.
Endothelial cells express several transient receptor potential (TRP) proteins of the canonical subfamily, including TRPC1, TRPC3, TRPC4, and TRPC6, all of which have been incriminated in control of endothelial cell barrier integrity (13, 14, 20, 52, 54). However, functional channels are heterotetramers (4, 16, 25, 50). TRPC1 and TRPC4 generate a heterotetramer that forms a channel pore, and this channel appears to provide a calcium source that increases permeability (15, 16, 51, 53, 58). Endoplasmic reticulum calcium store depletion activates the TRPC1/4 channel, generating a small (1–2 pA/pF), calcium (semi)-selective current (reversal potential approximately +40 mV) in endothelial cells, i.e., store-operated calcium entry current (ISOC) (17). Protein 4.1 directly interacts with TRPC4 and links the TRPC1/4 channel to the spectrin membrane skeleton (15, 16). Disruption of the TRPC4-protein 4.1 or the protein 4.1-spectrin interaction abolishes channel activation, whereas disruption of the actin-spectrin interaction is without effect (60). Protein 4.1 interacts directly with few ion channels, but it fulfills a critical role in gating TRPC1/4 (7).
Recently, we found that Orai1 also possesses a protein 4.1-binding domain and, furthermore, that it directly interacts with TRPC4 in the TRPC1/4 signalplex (16). Orai1 is considered to be the molecular basis of the calcium release-activated calcium current (ICRAC), a very small (<0.5 pA/pF) and highly calcium-selective current (reversal potential approximately +80 mV) (42, 43). Endothelial cells possess the ICRAC and the ISOC (1, 61), although the function of a TRPC4-Orai1 interaction remains unclear. We tested whether Orai1 contributes to calcium permeation through the ISOC (16). Orai1 silencing did not abolish the ISOC but shifted the reversal potential to the left and decreased calcium-dependent current inactivation, consistent with the idea that Orai1 contributes to TRPC1/4 channel calcium selectivity. Moreover, loss of Orai1 reduced the anomalous mole fraction effect; in the absence of Orai1, calcium did not effectively inhibit sodium permeation through the ISOC. Under these conditions, activation of store-operated calcium entry did not disrupt the endothelial cell barrier, suggesting an important physiological role for ion selectivity. The nature of the Orai1-TRPC1/4 interaction remains poorly understood. We considered two possibilities: 1) Orai1 forms an independent channel that interacts with TRPC1/4, and 2) Orai1 functions independently as a subunit of the TRPC1/4 channel. Future studies are required to resolve this issue, although work from Trebak and colleagues (1, 61) supports the idea that Orai1 can function as a channel in endothelium. While this prior work established an important role for Orai1 in ion selectivity of store-operated calcium entry channels, it did not evaluate whether sodium entry across the plasma membrane influences cytosolic sodium concentrations. Here we test the hypothesis that, upon activation of store-operated calcium entry channels, Orai1 is a critical determinant of cytosolic sodium transitions.
MATERIALS AND METHODS
Cell isolation and culture.
Rat pulmonary arterial endothelial cells (PAECs) were isolated and cultured (18) in DMEM supplemented with 8% Tet system-approved fetal bovine serum (catalog no. 631101, Clontech) and penicillin-streptomycin (1:100; catalog no. 15140-122, Invitrogen).
Genetic modification for conditional knockdown of Orai1.
Generation of recombinant retro-lentiviruses is described elsewhere (16). PAECs were infected with virus to express the pM2641 construct, which stably expresses Tet-On reverse tetracycline transactivator protein, followed by an internal ribosomal entry site, an enhanced green fluorescent protein (GFP), and a blasticidin resistance gene (16). Infected cells were selected to homogeneity using blasticidin (catalog no. ant-bl-1, InvivoGen; 10 μg/ml); cell homogeneity was confirmed by enhanced GFP fluorescence. The purified cells were infected with a second virus containing the pM2879 construct with tetracycline-operator 7 repeats promoter driven by a doxycycline-reverse tetracycline transactivator protein transcription factor complex, enabling a shRNA expression to knock down Orai1 (16). The doubly infected (PA2879) cells were selected to homogeneity using puromycin and confirmed by mCherry fluorescence. Knockdown of Orai1 was tested by Western blotting from whole cell lysates in a doxycycline (catalog no. 631311, Clontech) dose- and time-dependent manner (16).
Intracellular calcium measurements.
Cells were loaded with 6 μl of fura 2-AM (catalog no. F1221, Invitrogen) and 6 μl of pluronic acid (catalog no. P-6866, Invitrogen) in 2 ml of modified Krebs-Henseleit (Krebs) buffer (catalog no. K3753, Sigma) for 20 min at 37°C (Forma Series II, model 3110, Thermo Scientific) and then switched to Krebs buffer alone for 20 min to allow for dye deesterification. After three rinses with Krebs buffer, coverslips were transferred into the Attofluor chamber (catalog no. A-7816, Invitrogen). Krebs buffer (2 ml) was added to the chamber to measure the background using a Nikon 1X70 fluorescence microscope system with 340- and 380-nm wavelength filters and Felix fluorescence analysis software (Photon Technology International). A field of ∼20 cells were imaged in each experiment, and averaged fluorescence from the cell population was recorded and considered to be equal to 1.
Intracellular sodium measurements.
PA2879 cells were loaded with 10 μl of the intracellular sodium dye Asante NaTRIUM Green-2 (ANG-2/AM; catalog no. 3502, TEFLABS) with 8 μl of pluronic acid in 2 ml of Krebs buffer for 1 h at 37°C. The intracellular sodium signal was measured at 488 nm using a Zeiss fluorescence microscope and AxioVision 4.8 microscope software, which enabled image analysis of single-cell fluorescence within the population of ∼40 cells. Here, in contrast to measurements of cytosolic calcium, both single cell and population responses were evaluated. The sodium signal ratio was calculated by dividing the real-time GFP channel dynamic sodium fluorescence image at each time point by the initial image at 0 s [baseline fluorescence (F0)].
ANG 2 calibration curves were generated in intact PAECs. A sodium solution (110 mM sodium gluconate, 30 mM NaCl, 1.2 mM CaCl2, 0.6 mM MgSO4, and 10 mM sodium-HEPES, adjusted to pH 7.2 using 1 N sodium hydroxide) was prepared, and a similar potassium solution, where sodium ions were replaced with potassium ions, was generated. These solutions were modified from solutions used in previous studies (5, 36). PAECs were grown to near confluence and loaded with ANG 2-AM, as described above. Cells were then rinsed once with the potassium solution (without sodium ions) and loaded with 3 μM gramicidin A in the same solution for 15 min at room temperature. The cover glass was rinsed three times with the potassium solution and assembled into the AutoFluo stainless steel chamber and placed on the microscope stage. After addition of 1 ml of potassium solution to the chamber, images were taken every 5 s to establish baseline fluorescence. After 1 min, extracellular sodium was clamped at ascending concentrations (from 2.5 to 60 mM) by swapping different volumes of the potassium solution with the high-sodium solution. Fluorescence images were recorded for 5 min before the next step-wise increase in extracellular sodium concentration.
Recalcification dose responses.
PA2879 cells were loaded with fura 2-AM and ANG 2-AM dyes, and intracellular calcium and sodium signal ratios, respectively, were measured. Cells were incubated in low extracellular calcium (∼100 nM), and at 200 s, 1 μM thapsigargin (catalog no. T9033, Sigma) was added. At 700 s, calcium chloride (20 μl) was added to increase the final calcium concentration to 10 μM, 100 μM, 500 μM, 1 mM, 5 mM, or 10 mM. At 1,200 s, 60.6 μl of EGTA (catalog no. E4378, Sigma) were added to a final concentration of 5 mM to chelate the extracellular calcium. In separate experiments, cells were incubated with 2-aminoethoxydiphenyl borate (2-APB; catalog no. 42810, Sigma) or YM-58483 (catalog no. Y4895, Sigma).
Statistical analysis.
One-way ANOVA with Bonferroni's post hoc test was used to test differences among treatment groups, as appropriate. Paired t-test was used to compare differences between two groups. Linear regression or two-way ANOVA was used to analyze the time-dependent data. To evaluate the percentage of cells responding to thapsigargin in the presence and absence of Orai1, we used a contingency test, with Cochran-Mantel-Haenszel adjustment, where likelihood ratio and Pearson's P value were calculated. Values are means ± SE, unless specified otherwise. P < 0.05 is considered statistically significant for the comparisons.
RESULTS
Store-operated calcium entry in PAECs.
In our initial studies, we used a recalcification protocol to characterize the thapsigargin-induced store-operated calcium entry response in PAECs (Fig. 1). Thapsigargin (1 μM) was added to PAECs in the presence of low extracellular calcium (∼100 nM) to resolve the endoplasmic reticulum calcium release phase (Fig. 1A). Thapsigargin induced a transient increase in cytosolic calcium that returned to baseline levels within ∼450 s. Extracellular calcium was then replenished over a range of concentrations, from 10 μM to 10 mM, resulting in a concentration-dependent increase in cytosolic calcium (Fig. 1, B and C). Addition of 100 μM extracellular calcium represented the threshold concentration resulting in an increase in cytosolic calcium, whereas 500 μM extracellular calcium represented a concentration near the calculated half-maximal concentration of ∼400 μM calcium. A maximal rise in cytosolic calcium was achieved with addition of 5 mM extracellular calcium.
Fig. 1.
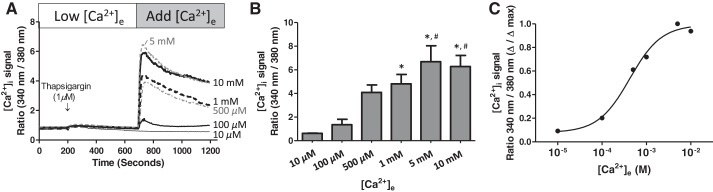
Thapsigargin activates concentration-dependent calcium entry in pulmonary artery endothelial cells (PAECs). A: PAECs were loaded with the ratiometric intracellular calcium indicator fura 2-AM, and cytosolic calcium was continuously monitored in cell populations (average fluorescence from ∼20 cells). Thapsigargin was added to cells in a low-calcium bath solution, then calcium chloride was added to achieve the final concentration of 10 μM (n = 3), 100 μM (n = 8), 500 μM (n = 7), 1 mM (n = 8), 5 mM, (n = 5), and 10 mM (n = 7), respectively. Values are means ± SE. [Ca2+]e, extracellular calcium concentration; [Ca2+]i, intracellular calcium concentration. B: peak store-operated calcium entry level from each calcium dose. Values are means ± SE. Data were analyzed using 1-way ANOVA with Bonferroni's post hoc test. *Significantly different from 10 μM. #Significantly different from 100 μM. C: data in B replotted to illustrate dose-response relationship. Half-maximal effect (EC50) was ∼400 μM.
Orai1 optimizes coupling between calcium store depletion and activation of store-operated calcium entry.
We next sought to determine whether Orai1 impacts endothelial cell store-operated calcium entry in PAECs. PAECs were engineered for conditional expression of Orai1 shRNA using a Tet-On approach, as previously described (16). To silence Orai1, PAECs were treated with doxycycline (3 μg/ml) every 24 h for 3 consecutive days, and Orai1 expression was evaluated by Western blot analysis. Consistent with our previous results (16), Orai1 shRNA expression reduced protein abundance, as assessed by Western blotting of whole cell lysates (Fig. 2A).
Fig. 2.

Orai1 silencing does not significantly reduce the magnitude of the store-operated calcium entry response in PAECs. A: PAECs were engineered for doxycycline-inducible expression of Orai1 shRNA, as described previously (15). Cells were treated with doxycycline (3 μg/ml) every 24 h for a total of 72 h. After doxycycline exposure, cell lysates were collected and subjected to Western blot analysis. Conditional expression of Orai1 shRNA silenced Orai1 protein by 72 h (left), an effect that resulted in an ∼80% decrease in protein as determined by densitometry (middle, n = 20 experiments). Doxycycline treatment induced uniform mCherry expression, consistent with uniform Orai1 shRNA expression (right). B: Orai1 expression was not a determinant of the cytosolic calcium response to thapsigargin in the recalcification protocol following readdition of 100 μM [+Orai1 (n = 4) and −Orai1 (n = 8)], 500 μM [+Orai1 (n = 6) and −Orai1 (n = 10)], and 5 mM [+Orai1 (n = 7) and −Orai1 (n = 9)] calcium, respectively. ns, Not significant. C and D: cytosolic calcium response to 30 nM, 100 nM, and 1 μM thapsigargin in the presence of 2 mM extracellular calcium in the presence and absence of Orai1. C: Orai1 silencing delayed the rise in cytosolic calcium. Pearson P < 0.05 at 30 and 100 nM thapsigargin. *P < 0.05. D: averaged responses following 30 nM [+Orai1 (n = 6) and −Orai1 (n = 7)], 100 nM [+Orai1 (n = 7) and −Orai1 (n = 8)], and 1 μM [+Orai1 (n = 9) and −Orai1 (n = 7)] thapsigargin, respectively.
PAEC store-operated calcium entry was examined using the recalcification protocol in the presence and absence of Orai1 over a range of extracellular calcium concentrations (Fig. 2B). Addition of 1 μM thapsigargin resulted in a transient increase in cytosolic calcium that was independent of Orai1; Orai1 depletion did not influence the amount of calcium released from the endoplasmic reticulum, suggesting that it did not deplete calcium stores. Replenishing extracellular calcium produced a concentration-dependent increase in cytosolic calcium, similar to the results obtained in Fig. 1. We noted that Orai1 silencing did not significantly reduce the global rise in cytosolic calcium upon recalcification at any of the calcium concentrations tested. Orai1 has previously been reported to contribute to the magnitude of store-operated calcium entry, at least in part by optimizing the coupling between stromal interaction molecule 1 (STIM1) on the endoplasmic reticulum and store-operated calcium entry channels (9), although this is not a uniform finding in all cell types. Indeed, our present results are most consistent with a recent report by Sundivakkam and colleagues (51) suggesting that TRPC4, and not Orai1, is the principal store-operated calcium entry mechanism in endothelia. Since calcium release and calcium entry are uncoupled in the recalcification protocol, it is possible that sufficient time is allowed for endoplasmic reticulum-plasma membrane channel coupling, even in the absence of Orai1.
We performed experiments in which thapsigargin was added to PAECs in the presence of physiologically relevant extracellular calcium concentrations, where calcium release and calcium entry phases have not been separated, as in the recalcification studies. Thapsigargin was added to PAECs over a range of concentrations, from 30 nM to 1 μM. We observed that Orai1 was a critical determinant of the probability and the rate at which thapsigargin increased cytosolic calcium, especially at low thapsigargin concentrations (Fig. 2C); 30 nM represented a threshold concentration, where ∼33% of the cell populations failed to respond to thapsigargin with a rise in cytosolic calcium, in the presence and absence of Orai1. In the responding PAEC populations (∼67%), Orai1-expressing cells elicited an acute response to thapsigargin, usually within ∼400 s of its application, whereas Orai1-deficient cells were slow to respond (P < 0.05). A similar trend was observed when 100 nM thapsigargin was assessed, although statistical significance was not achieved (P = not significant). Here, 100% of the Orai1-expressing cells immediately responded to thapsigargin treatment with an increase in cytosolic calcium, whereas only 60% of Orai1-deficient cells responded acutely; no response was recorded in ∼10% of the Orai1-deficient cell studies. When 1 μM thapsigargin was tested, all cells responded acutely with a rise in cytosolic calcium; Orai1 was not a determinant of the store-operated calcium entry response at this maximal thapsigargin concentration. Hence, Orai1 is a critical determinant of thapsigargin-induced store-operated calcium entry in PAECs, increasing coupling efficiency between store depletion and calcium entry, especially at threshold and low thapsigargin concentrations.
We determined whether Orai1 impacts the magnitude of the thapsigargin-induced global cytosolic calcium response. Initially, we averaged responses from cell populations according to time. From these data, it appeared that larger global cytosolic calcium responses were generated by Orai1-expressing than Orai1-deficient cells, especially at lower thapsigargin concentrations. However, since time to peak in the cytosolic calcium response was longer in Orai1-deficient cells, time became a confounding factor in analyzing the data. Data were therefore standardized, so that the cytosolic calcium responses were not aligned according to the time at which thapsigargin was initially delivered but, rather, according to the peak cytosolic calcium response (Fig. 2D). From these data, it became apparent that Orai1 was not a determinant of the magnitude of the thapsigargin-induced rise in cytosolic calcium at any of the concentrations tested. Thus Orai1 primarily regulates the time to activation, and not the magnitude of thapsigargin-induced store-operated calcium entry, in PAECs.
Orai1 and extracellular calcium: determinants of sodium permeability.
Orai1 is generally believed to form a highly calcium-selective store-operated calcium entry channel, in contrast to TRPC channels, which range from being partially calcium-selective (e.g., TRPC1/4 heterotetramers) to calcium- and sodium-nonselective. Recent findings indicate that Orai1 is a determinant of sodium permeation through the TRPC1/4 store-operated calcium entry channel in whole cell patch-clamp studies. On this basis, we examined whether Orai1 contributes to cytosolic sodium concentrations in PAECs.
To begin these experiments, in vivo calibration curves (e.g., calibrations in intact cells) were performed (Fig. 3). PAECs were loaded with the sodium fluorescence indicator ANG 2-AM, and the sodium ionophore gramicidin A (3 μM) was added in the relative absence of extracellular sodium. After a stable baseline fluorescence signal was measured, ascending concentrations of sodium were added to the medium. Sodium dose dependently increased ANG 2 fluorescence; changes in fluorescence were detectable between 2.5 and 30 mM cytosolic sodium, with a half-maximal effect near 11 mM. This sodium-dependent increase in ANG 2 fluorescence was substantially inhibited by choline, demonstrating specificity of the dye for sodium over choline (data not shown). Thus ANG 2 accurately detects low millimolar shifts in cytosolic sodium in intact PAECs, below a maximal concentration of 30 mM.
Fig. 3.

Sodium induces a dose-dependent increase in Asante NaTRIUM Green-2 (ANG 2) fluorescence in intact PAECs. PAECs were loaded with ANG 2-AM. A: baseline fluorescence was measured, and gramicidin A (3 μM) was added in the relative absence of extracellular sodium. After a baseline equilibration period, extracellular sodium was replenished at ascending concentrations. Data represent average fluorescence response from 3 separate experiments. B: raw data in A plotted as a dose-response curve to demonstrate the half-maximal fluorescence signal at ∼11 mM cytosolic sodium. C: representative fluorescence images from a confluent PAEC monolayer exposed to ascending cytosolic sodium concentrations as described in A. [Na+]e, extracellular sodium concentration; [Na+]i, intracellular sodium concentration.
Having established the sensitivity range for ANG 2, cytosolic sodium was measured over a 20-min time course. Basal cytosolic sodium concentrations were stable in the presence of 2 mM extracellular calcium (Fig. 4A). Orai1 depletion revealed a significant increase in cytosolic sodium over the time course studied (Fig. 4B), indicating that Orai1 constitutively limits basal sodium leak. Studies were repeated in low extracellular calcium (Fig. 4). Here, cytosolic sodium was not stable in Orai1-expressing cells but increased steadily over the time course studied. Similar results were obtained in the absence of Orai1, suggesting that extracellular calcium may interact with Orai1 to control basal cytosolic sodium concentrations. Time-lapse fluorescent movies (see Supplemental Movie S1, A–D, in Supplemental Material for this article, available online at the Journal website) illustrate the impact of Orai1 and extracellular calcium on cytosolic sodium; it is apparent that Orai1 and extracellular calcium are necessary to stabilize cytosolic sodium.
Fig. 4.
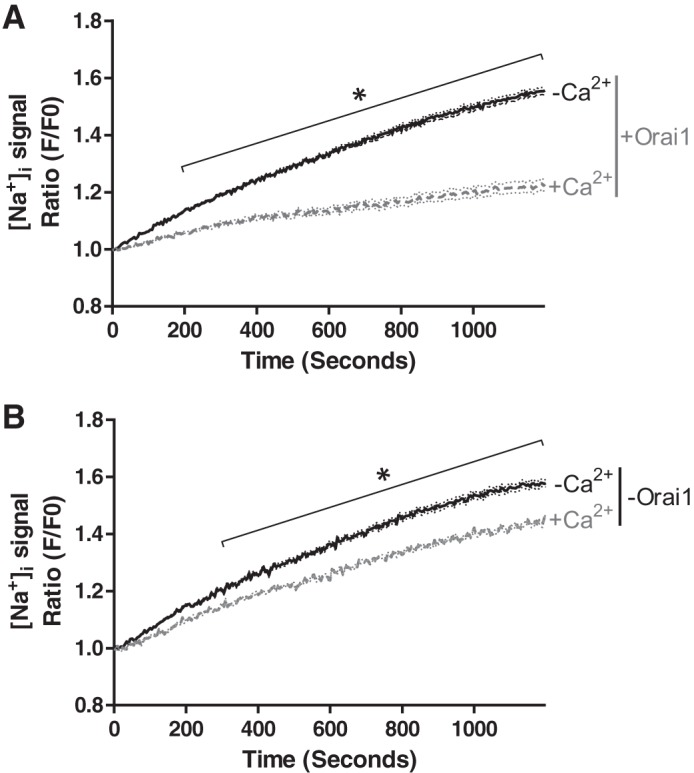
Orai1 suppresses basal sodium leak in the presence of 2 mM extracellular calcium. PAECs were treated with doxycycline to silence Orai1, as described in Fig. 2 legend. Cells were loaded with the intracellular sodium dye ANG 2-AM, and the signal ratio (GFP/F0) was repeatedly measured over time in the presence of 2 mM or low extracellular calcium. Reducing extracellular calcium (A) and silencing Orai1 (B) resulted in an increase in cytosolic sodium, characteristic of increased basal sodium leak. For studies conducted in the presence of both Orai1 and calcium, n = 6 experiments in which 67 cells were analyzed; for studies conducted in the presence of Orai1 and the absence of Ca2+, n = 8 experiments in which 88 cells were analyzed. For studies conducted in the absence of both Orai1 and Ca2+, n = 6 experiments in which 65 cells were analyzed; for studies conducted in the presence of Orai1 and the absence of Ca2+, n = 6 experiments in which 67 cells were analyzed. *P < 0.01.
Sodium influx through store-operated calcium entry channels is controlled by Orai1.
Thapsigargin depletes endoplasmic reticulum calcium and promotes calcium entry through store-operated calcium entry channels, as depicted in Fig. 1. While whole cell electrophysiology experiments reveal that a population of store-operated calcium entry channels conduct calcium and sodium, the influence of sodium influx on cytosolic sodium is unknown. We examined whether thapsigargin increases cytosolic sodium in the presence of 2 mM extracellular calcium and also in the recalcification protocol. Thapsigargin addition transiently increased cytosolic sodium in the presence (Fig. 5A) and absence (Fig. 5B) of extracellular calcium. After this transient increase in cytosolic sodium, sodium continued to increase when cells were incubated in 2 mM extracellular calcium; this steady rise in cytosolic sodium was noted in Orai1-expressing and -deficient cells. By comparing the rate of rise in Figs. 4A, 4B, and 5A, we can see that thapsigargin increased sodium influx in Orai1-expressing cells but had little effect on sodium influx in Orai1-deficient cells. Time-lapse fluorescent movies (see Supplemental Movie S2, A and B) illustrate the thapsigargin-induced rise in cytosolic sodium in low extracellular calcium and demonstrate calcium inhibition of sodium influx following recalcification. Hence, these data suggest that Orai1 expression is necessary for thapsigargin to promote a sustained rise in cytosolic sodium.
Fig. 5.
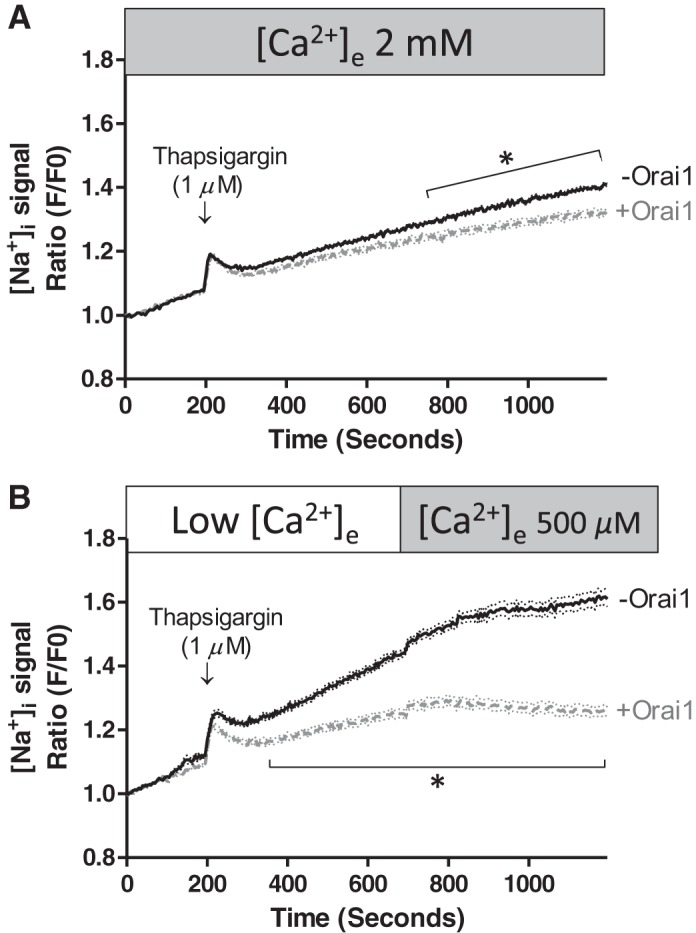
Orai1 suppresses thapsigargin-induced sodium entry through activated store-operated calcium entry channels. PAECs were prepared and treated with doxycycline as described in Fig. 2 legend. Cells were loaded with ANG 2-AM and then imaged for cytosolic sodium. Thapsigargin was applied in the presence (A) and absence (B) of extracellular calcium. A: thapsigargin increased cytosolic sodium in the presence and absence of Orai1. For studies conducted in the presence of Orai1, n = 6 experiments in which 69 cells were analyzed; for studies conducted in the absence of Orai1, n = 10 experiments in which 101 cells were analyzed. B: thapsigargin effect was more pronounced in the absence of extracellular calcium. For studies conducted in the presence of Orai1, n = 5 experiments in which 55 cells were analyzed; for studies conducted in the absence of Orai1, n = 5 experiments in which 55 cells were analyzed. Addition of extracellular calcium in the recalcification protocol resulted in an abrupt reduction in cytosolic sodium. Values are means ± SE. *P < 0.05.
This idea is further substantiated in the recalcification protocol, where a separation in the thapsigargin-induced steady rise in sodium was noted between Orai1-expressing and -deficient cells [significantly greater slope in Orai1-deficient cells (P < 0.01)]. Exposure of Orai1-deficient cells to low extracellular calcium resulted in a steady rise in cytosolic sodium, as was seen in Fig. 4 in the absence of thapsigargin treatment. In Orai1-expressing cells, sodium did not accumulate to a similar degree. Interestingly, in both cases, replenishing extracellular calcium abruptly mitigated sodium entry [significantly lower slope following addition of calcium (P < 0.01)], suggesting that calcium blocks sodium permeation. This inhibitory effect of calcium on cytosolic sodium resembles the anomalous mole fraction effect detected in patch-clamp experiments, where calcium blocks sodium permeation.
Sodium release from an intracellular store has not been detected previously, yet the acute thapsigargin-induced rise in cytosolic sodium resembled the thapsigargin-induced calcium release phase. To specifically compare the temporal nature of the transient thapsigargin-induced increase in cytosolic sodium and calcium, sodium and calcium responses from studies conducted in low extracellular calcium were compared. To interrogate the temporal relationship between sodium- and calcium-release phases, the data for each of the responses were normalized to percent change, where the maximal response for sodium and calcium was set to 1.0, or 100%. As shown in Fig. 6, the thapsigargin-induced rise in sodium was abrupt and occurred prior to the thapsigargin-induced rise in calcium. Cytosolic sodium also returned to baseline concentrations rapidly, whereas cytosolic calcium remained elevated for ∼450 s. The presence (Fig. 6A) or absence (Fig. 6B) of Orai1 was not a determinant of the transient rise in cytosolic sodium or calcium.
Fig. 6.
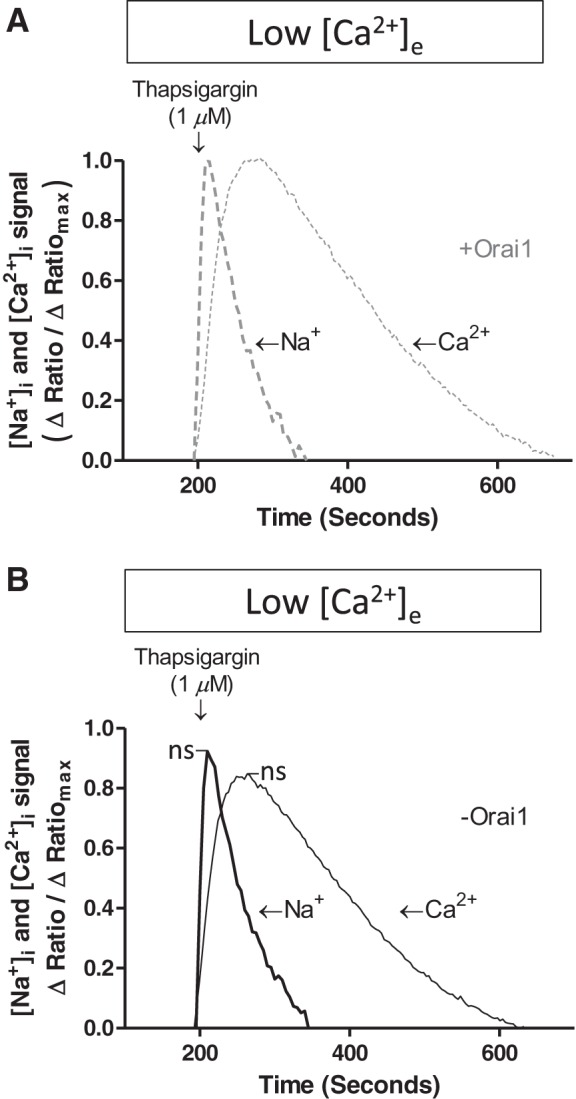
Thapsigargin induced a transient rise in cytosolic sodium prior to endoplasmic reticulum calcium release. PAECs were treated with doxycycline as described in Fig. 2 legend and then loaded with ANG 2-AM or fura 2-AM to measure intracellular sodium or calcium, respectively. Thapsigargin was applied in the absence of extracellular calcium, and cytosolic sodium and calcium responses were evaluated. Raw data were synchronized according to the time at which thapsigargin was added and converted to a maximal response scale (0–100% of maximal response), and sodium and calcium release phases were compared. For studies conducted in the presence of Orai1, [Na+]i data included 17 experiments in which 182 cells were analyzed and [Ca2+]i data included 19 experiments; for studies conducted in the absence of Orai1, [Na+]i data included 15 experiments in which 163 cells were analyzed and [Ca2+]i data included 26 experiments. Data reveal that the sodium peak occurred prior to the calcium peak but was of shorter duration. Similar results were obtained in the presence (A) and absence (B) of Orai1. Mean value from these experiments is shown. Orai1 silencing did not change peak [Na+]i or [Ca2+]i signal (P = not significant).
We considered the possibility that the transient increase in cytosolic sodium was an artifact, due to changes in ANG 2 fluorescence properties after shifts in calcium, potassium, or proton. To test these possibilities, ANG 2 fluorescence was measured in a cuvette with 14 mM sodium (half-maximal ANG 2 fluorescence; Fig. 3A) containing calcium (100 nM, 500 nM, and 1 μM) or potassium (100–140 mM) buffers. Neither calcium nor potassium altered ANG 2 fluorescence (data not shown). We evaluated the impact of pH on ANG 2 excitation and emission spectra within a pH range of 7.0–7.8. Within the low range, 7.0–7.6, pH did not impact the spectra (data not shown). However, pH 7.8 increased the emission signal. Since intracellular pH is in the 7.2–7.5 range, it is unlikely that pH influenced ANG 2 fluorescence. Hence, the thapsigargin-induced transient increase in cytosolic sodium most likely reflects a rapid, transient sodium transition.
Sodium entry occurs through 2-APB- and YM-58483-sensitive channels.
Evidence that Orai1 and extracellular calcium mediate sodium entry into endothelium indicates that pharmacological inhibitors of store-operated calcium entry should similarly reduce sodium influx and cytosolic sodium. To test this idea, basal cytosolic sodium was measured in the presence and absence of Orai1 and extracellular calcium. The change in cytosolic sodium over this time period was measured and compared among treatment groups (Fig. 7A). In low extracellular calcium, Orai1 did not influence cytosolic sodium (P = not significant), as was also seen in Fig. 4. The addition of 2-APB or YM-58483 significantly decreased cytosolic sodium, suggesting that basal sodium permeation occurs through store-operated and/or calcium release-activated calcium-like channels in low extracellular calcium. In the presence of extracellular calcium, Orai1 reduced basal sodium by ∼50%, an effect also observed in Fig. 4. Thus interaction between Orai1 and extracellular calcium is necessary to suppress sodium entry through store-operated calcium entry channels. Here, we also can see that sodium permeation is mainly through the store-operated calcium entry channel, and not the calcium release-activated calcium channel, in the presence of extracellular calcium and sodium, a finding that is consistent with reports that the store-operated calcium entry channel is permeable to calcium and sodium, whereas the calcium release-activated calcium channel is highly calcium-selective.
Fig. 7.

Store-operated calcium entry and calcium release-activated calcium channel inhibitors 2-aminoethoxydiphenyl borate (2-APB) and YM-58483 decrease basal sodium leak and thapsigargin-induced sodium entry only in the presence of Orai1. PAECs were treated with doxycycline as described in Fig. 2 legend and loaded with ANG 2-AM or fura 2-AM. A: baseline sodium. In separate experiments, 2-APB or YM-58483 was added. Orai1 and extracellular calcium (2 mM) together potently limit basal sodium leak. 2-APB (75 μM) and YM-58483 (10 μM) suppress cytosolic sodium, but only in the presence of Orai1. The following studies were conducted in the absence of [Ca2+]e: for studies conducted in the absence of Orai1, data represent 6 experiments in which 67 cells were analyzed; for studies conducted in the presence of Orai1, data represent 8 experiments in which 88 cells were analyzed; for studies conducted in the presence of Orai1 and 2-APB, data represent 7 experiments in which 77 cells were analyzed; for studies conducted in the presence of Orai1 and YM-58483, data represent 5 experiments in which 55 cells were analyzed. The following studies were conducted in the presence of [Ca2+]e: for studies conducted in the absence of Orai1, data represent 6 experiments in which 65 cells were analyzed; for studies conducted in the presence of Orai1, data represent 6 experiments in which 67 cells were analyzed; for studies conducted in the presence of Orai1 and 2-APB, data represent 5 experiments in which 54 cells were analyzed; for studies conducted in the presence of Orai1 and YM-58483, data represent 6 experiments in which 63 cells were analyzed. Data were analyzed using 1-way ANOVA with Bonferroni's post hoc test: *P < 0.0001; #P < 0.01. B–E: after baseline sodium and calcium signals were measured, 2-APB (B and C) or YM-58483 (D and E) was added, followed by thapsigargin in the presence or absence of extracellular calcium. In the presence of 2 mM extracellular calcium, 2-APB (for studies conducted in the presence of Orai1, data represent 5 experiments in which 53 cells were analyzed; for studies conducted in the absence of Orai1, data represent 6 experiments in which 66 cells were analyzed) and YM-58483 (for studies conducted in the presence of Orai1, data represent 6 experiments in which 66 cells were analyzed; for studies conducted in the absence of Orai1, data represent 5 experiments in which 55 cells were analyzed) inhibited thapsigargin-induced sodium entry, but only in the presence of Orai1. In recalcification studies, 2-APB (for studies conducted in the presence of Orai1, data represent 6 experiments in which 66 cells were analyzed; for studies conducted in the absence of Orai1, data represent 5 experiments in which 55 cells were analyzed) and YM-58483 (for studies conducted in the presence of Orai1, data represent 5 experiments in which 55 cells were analyzed; for studies conducted in the absence of Orai1, data represent 7 experiments in which 77 cells were analyzed) again inhibited the thapsigargin-induced increase in cytosolic sodium, only in the presence of Orai1. 2-APB (for studies conducted in the presence of Orai1, data represent 4 experiments; for studies conducted in the absence of Orai1, data represent 4 experiments) and YM-58483 (for studies conducted in the presence of Orai1, data represent 5 experiments; for studies conducted in the absence of Orai1, data represent 7 experiments) decreased the thapsigargin-induced increase in cytosolic calcium, independent of Orai1. Values are means ± SE. F: transient thapsigargin-induced increases in cytosolic sodium and calcium were rescaled as percentage of maximal response. 2-APB and YM-58483 reduced the peak cytosolic sodium response to thapsigargin, whereas only 2-APB decreased the peak cytosolic calcium response to thapsigargin. For [Na+]i peaks: *P < 0.0001; ^P < 0.001; #P < 0.01. For [Ca2+]i peaks, P = not significant. G and H: extracellular sodium was replaced with choline, and the thapsigargin-induced cytosolic sodium response was evaluated. Choline substitution abolished thapsigargin-induced sustained increase in cytosolic sodium (G) but did not alter the transient increase in cytosolic sodium (H).
We next studied the effect of 2-APB and YM-58483 on the thapsigargin-induced increase in cytosolic sodium. 2-APB (Fig. 7, B and C) or YM-58483 (Fig. 7, D and E) was administered prior to thapsigargin in the presence of 2 mM extracellular calcium and in the recalcification protocol. Both agents decreased the transient thapsigargin-induced increase in sodium. 2-APB and YM-58483 also prevented the sustained increase in cytosolic sodium in the presence of Orai1; this effect was absent in Orai1-deficient cells. Hence, as suggested in the studies described in Fig. 7A, 2-APB- and YM-58483-dependent inhibition of sodium influx requires the presence of Orai1.
We examined whether 2-APB and YM-58483 reduced calcium entry through store-operated calcium entry channels. 2-APB (Fig. 7C) and YM-58483 (Fig. 7E) similarly decreased store-operated calcium entry, as determined using the recalcification protocol. In the presence of 2-APB or YM-58483, readdition of 500 μM extracellular calcium in the recalcification protocol increased cytosolic calcium to 340 nm-to-380 nm ratios of ∼2; in contrast, in the absence of pharmacological inhibition, readdition of 500 μM extracellular calcium increased cytosolic calcium to 340 nm-to-380 nm ratios of ∼4 (Fig. 2B). Unlike the effect observed with cytosolic sodium, Orai1 expression was not a determinant of whether 2-APB and YM-58483 reduced calcium influx. Thus, Orai1 is not required for 2-APB or YM-58483 to suppress calcium influx, but it is required for these agents to suppress sodium influx, indicating that Orai1 fulfills a central role in determining the global ion selectivity of store-operated calcium entry channels.
Next, using the approach described in Fig. 6, we quantitatively examined the effect of 2-APB and YM-58483 on the transient thapsigargin-induced increase in sodium (Fig. 7F). First, the transient increases in cytosolic sodium and calcium were evaluated in Orai1-expressing cells. 2-APB and YM-58483 reduced the transient increase in cytosolic sodium, while only 2-APB reduced the cytosolic calcium response. These latter findings are consistent with evidence that 2-APB inhibits the inositol 1,4,5-trisphosphate receptor and plasma membrane store-operated calcium entry channels, whereas YM-58483 only inhibits plasma membrane calcium release-activated calcium channels. Similar results were obtained in Orai1-deficient cells, where both 2-APB and YM-58483 reduced the transient thapsigargin-induced increase in sodium, while only 2-APB reduced the cytosolic calcium response (data not shown). These data suggest that at least a portion of the transient increase in sodium is due to release from an intracellular organelle and, furthermore, that YM-58483 inhibits an Orai1-independent sodium-permeable channel.
To ascertain whether thapsigargin is capable of inducing sodium release from intracellular stores, we replaced extracellular sodium with choline. Thapsigargin treatment induced a transient rise in cytosolic sodium, but it did not cause a sustained increase in cytosolic sodium (Fig. 7G). We then compared the magnitude of this transient increase in cytosolic sodium in the presence and absence of extracellular sodium. In both cases, the temporal nature and the magnitude of the cytosolic sodium signal were nearly identical (Fig. 7H). These results provide compelling evidence that thapsigargin induces sodium release from an intracellular store, at least under these experimental conditions.
DISCUSSION
Store-operated calcium entry is the principal mode of calcium influx in nonexcitable cells, such as endothelium. Activation of store-operated calcium entry in endothelial cells results in gap formation and tissue edema, an effect that is especially important in the lung, where tissue edema results in arterial hypoxemia. TRPC1/4 and Orai1 channel(s) have been incriminated in control of endothelial cell permeability, and recent evidence suggests that Orai1 interacts directly with TRPC4 to control calcium selectivity through store-operated calcium entry pathways. In light of these recent findings, we tested whether activation of store-operated calcium entry channels increases cytosolic sodium and, furthermore, whether Orai1 influences the magnitude of this response. Our results demonstrate that 1) activation of store-operated calcium entry increases cytosolic sodium; 2) Orai1 acts coordinately with extracellular calcium to restrict sodium permeation, under basal conditions and following activation of store-operated calcium entry; 3) Orai1 must be present for 2-APB and YM-58483 to inhibit sodium influx to a significant degree; and 4) thapsigargin induces an apparent transient sodium release from an intracellular store or, alternatively, a transient fluctuation that was not prevented by replacement of extracellular sodium with choline.
Store-operated calcium entry channels conduct sodium; however, under physiological conditions, calcium limits sodium permeability through these channels (42). For example, in the absence of divalent cations, the ICRAC possesses a unitary conductance of sodium that is ∼10-fold greater than that of calcium. Orai1 possesses two glutamate residues in the channel's pore that are responsible for selecting calcium over sodium, E106 and E190; substitution of these residues with aspartate increases sodium permeability (12, 26, 56). The store-operated calcium entry channel is less calcium-selective and more likely to conduct sodium (58). Here, we provide evidence that activation of store-operated calcium entry increases cytosolic calcium and sodium concentrations. Interestingly, Orai1 silencing has little effect on the magnitude of the increase in cytosolic calcium but dramatically increases cytosolic sodium. These findings support the idea that Orai1 interacts with other store-operated calcium entry channels to promote calcium selection, presumably by inhibiting sodium permeation. Future studies are required to determine how Orai1 senses extracellular calcium and restricts sodium permeability through its association with other store-operated calcium entry channels.
The sodium/calcium exchanger modulates intracellular sodium and calcium concentrations upon activation of store-operated calcium entry (6, 21, 22, 33, 37, 41, 46). This exchanger operates in forward and reverse directions. Calcium entry could provide a calcium source that drives sodium/calcium exchange in the forward direction, where cytosolic calcium is extruded from the cell, leading to increased cytosolic sodium. However, prior studies have suggested an alternative possibility: that activation of TRPC3 or TRPC6 channels increases subplasma membrane sodium to a degree sufficient to drive the sodium/calcium exchanger in the reverse direction and increase cytosolic calcium (6, 21, 22, 33, 37, 41, 46). For the sodium/calcium exchanger to operate in reverse, subplasmalemmal sodium concentrations would have to increase ∼10–20 mM (10, 22, 30, 32). In support of this possibility, activation of store-operated calcium entry has been shown to increase cytosolic sodium, albeit in some instances in highly restricted microdomains (6, 22, 41). Reducing extracellular sodium increases the cytosolic calcium response following activation of store-operated calcium entry (46), as does pharmacological blockade of the sodium/calcium exchanger (46). In our studies, the thapsigargin-induced increase in cytosolic sodium was potentiated following Orai1 silencing, an effect that decreases the membrane calcium concentration. Moreover, in Orai1-expressing cells, 2-APB and YM-58483 abolished the thapsigargin-induced sodium influx. These results are therefore more consistent with the idea that thapsigargin increases cytosolic sodium by promoting sodium influx through activated store-operated calcium entry channels, rather than by driving sodium entry through the sodium/calcium exchanger.
An acute transition in sodium across the plasma membrane, based on the electrochemical gradient, is an established mechanism of signal transduction, especially in excitable cells such as neurons and cardiac tissue. In excitable cells, membrane depolarization activates voltage-sensitive sodium channels, which, over millisecond(s) time scales, conduct sodium across the plasma membrane, leading to further depolarization. Aside from these very rapid events, sodium is not a widely accepted second messenger, and in nonexcitable cells, the biological function(s) of a cytosolic sodium transition remains poorly understood.
The best-recognized biological role for sodium transitions in nonexcitable cells pertains to control of membrane potential. In endothelium, thapsigargin and Gq-linked agonists induce a transient hyperpolarization due to activation of calcium-activated potassium channels, followed by a sustained depolarization due to sodium influx, partly attributed to a cyclic nucleotide-gated cation channel and, potentially, other nonselective cation channels (59). This sodium-dependent depolarization limits the electrochemical driving gradient for calcium entry through store-operated channels and provides a negative-feedback mechanism for fine-tuning the global calcium response (55, 59). Along with this feedback, agonist-evoked increases in cytosolic sodium drive the mitochondrial sodium/calcium exchanger, reducing mitochondrial calcium and adjusting the global cytosolic calcium signal (48).
In addition to these mechanisms that regulate a cytosolic calcium signal, a new physiological role for sodium fluxes has recently been revealed in endothelium. Endothelial cells express the epithelial sodium channel ENaC. Sodium influx through aldosterone-responsive ENaC increases cytosolic sodium in endothelial cells (38). The resulting sodium accumulation is accompanied by water uptake through aquaporin channels to produce a physiological “swelling.” In this instance, cell swelling impairs nitric oxide synthesis and represents a mechanism for long-term control of blood pressure. Future studies are needed to determine whether Orai1-dependent sodium influx contributes to cell volume control. In our previous studies, Orai1 silencing reduced gap formation (16). If Orai1 is capable of regulating cell swelling, then sodium transitions leading to cell swelling may impair retraction and serve as a previously unappreciated mechanism for control of endothelial cell barrier integrity.
Increases in cytosolic sodium are not generally thought to control intracellular enzyme activity in ways reminiscent of cytosolic calcium, aside from enzymes such as the sodium-potassium ATPase. However, sodium has been shown to regulate enzyme activity outside the cell; activated protein C (24), thrombin (28, 31, 40), and factor Xa (45) in the blood and/or extracellular space possess a conserved sodium-binding domain that is important for enzymatic activity. This conserved sequence is also found in β-galactosidase near the catalytic pocket, where it regulates enzyme activity inside the cell; in this instance, it is not clear whether acute shifts in cytosolic sodium can control enzyme activation state (29). It will be important to determine whether physiological shifts in cytosolic sodium are capable of transiently regulating the activity of cytosolic enzymes.
We detected a thapsigargin-induced transient rise in cytosolic sodium that was due to an apparent sodium store release. This effect preceded the thapsigargin-induced calcium store release and was of shorter duration. The cytosolic sodium transition was present even after extracellular sodium was replaced with choline, it occurred in the presence and absence of extracellular calcium, it did not require the presence of Orai1, and it was partially inhibited by 2-APB and YM-58483. The source of this intracellular sodium release mechanism is unknown. Intracellular estimates suggest that cytosolic sodium concentrations are ∼5 mM, although concentrations approaching 12 mM have been reported (23, 34, 47, 48). It appears that sodium concentrations vary among intracellular compartments; for example, endoplasmic reticulum, mitochondrial, and nuclear concentrations have been estimated to be ∼27, 10–50, and 108 mM (or higher), respectively (3, 8, 19, 27, 35). From these estimates, it appears that a sodium concentration gradient exists between the luminal contents of organelles and the cytosol. While we consider the source of this transient thapsigargin-induced shift in sodium to be due to release from an intracellular source, our studies have not ruled out other possibilities, including mitochondrial “release” via the sodium/calcium exchanger. Future studies are required to address these possibilities.
Based on this idea, we used parameters established by the Nernst equation {ENa = (RT/zF)ln([Na+]cyt/[Na+]store), where ENa is the reversal potential for sodium, R is the gas constant, T is temperature, z is charge, F is Faraday's constant, and [Na+]cyt is cytosolic sodium concentration} to examine the physiological constraints to sodium release from an intracellular organelle; a similar mathematical approach was used to model calcium (11, 44) and zinc (11, 44) release from the endoplasmic reticulum, respectively.
If the free cytosolic sodium concentration is 5 mM and if the free endoplasmic reticulum luminal sodium concentration is 20 mM, then the equilibrium potential for sodium is −36 mV (Table 1). Although the actual endoplasmic reticulum membrane potential is not known, estimates range from −95 to −74 mV, depending on the cell type tested, where the lumen is negative with respect to the cytosol (11, 44). Under these conditions, sodium release would not be likely, unless thapsigargin depolarizes the organelle membrane potential while activating a channel permeable to sodium. However, if cytosolic sodium is buffered to a free concentration that is <1 mM or if the luminal sodium concentration approaches 100 mM, then sodium release is favorable (Table 1). These findings illustrate the importance of 1) resolving organelle membrane potential and the biophysical properties that establish it, 2) identifying the free sodium concentration in organelles, including the endoplasmic reticulum and nucleus, and 3) determining the actual free vs. buffered cytosolic sodium concentrations in endothelium.
Table 1.
Buffering free cytosolic sodium increases magnitude of the sodium organelle reversal potential to highly negative values
| [Na+]cyt |
||||
|---|---|---|---|---|
| [Na+]store, mM | 10 mM | 5 mM | 2.5 mM | 0.5 mM |
| 5 | 18 | 0 | −18 | −59 |
| 10 | 0 | −18 | −36 | −77 |
| 20 | −18 | −36 | −53 | −95 |
| 50 | −41 | −59 | −77 | −118 |
| 100 | −59 | −77 | −95 | −136 |
Values are reversal potentials, expressed in mV. [Na+]cyt, cytosolic Na+ concentration.
In summary, we provide evidence that activation of store-operated calcium entry in endothelial cells is accompanied by an increase in cytosolic sodium. This rise in cytosolic sodium is most prominent after Orai1 silencing, suggesting an important role for Orai1 in calcium sieving of store-operated calcium entry pathways. We detected thapsigargin-induced sodium release from an intracellular pool, although the origin of the sodium pool has not been established. These results prompt consideration of the physiological role played by acute transitions in sodium through activated store-operated calcium entry channels.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-60024 (T. Stevens and D. L. Cioffi), HL-66299. (T. Stevens, T. C. Rich, and M. Alexeyev), and HL-107778 (D. L. Cioffi) and American Heart Association Predoctoral Fellowship 14PRE18470024 (N. Xu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.X. and T.S. developed the concept and designed the research; N.X., M.A., and T.C.R. performed the experiments; N.X., D.L.C., T.C.R., and T.S. analyzed the data; N.X., T.C.R., and T.S. interpreted the results of the experiments; N.X., T.C.R., and T.S. prepared the figures; N.X. and T.S. drafted the manuscript; N.X., D.L.C., M.A., T.C.R., and T.S. approved the final version of the manuscript; D.L.C., M.A., T.C.R., and T.S. edited and revised the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Eugene Cioffi for helpful discussions pertaining to availability and use of sodium indicators.
REFERENCES
- 1.Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res 103: 1289–1299, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aird WC. Phenotypic heterogeneity of the endothelium. II. Representative vascular beds. Circ Res 100: 174–190, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Allfrey VG, Meudt R, Hopkins JW, Mirsky AE. Sodium-dependent “transport” reactions in the cell nucleus and their role in protein and nucleic acid synthesis. Proc Natl Acad Sci USA 47: 907–932, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amiri H, Schultz G, Schaefer M. FRET-based analysis of TRPC subunit stoichiometry. Cell Calcium 33: 463–470, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Amorino GP, Fox MH. Intracellular Na+ measurements using sodium green tetraacetate with flow cytometry. Cytometry 21: 248–256, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am J Physiol Cell Physiol 278: C163–C173, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Baines AJ, Bennett PM, Carter EW, Terracciano C. Protein 4.1 and the control of ion channels. Blood Cells Mol Dis 42: 211–215, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Baron S, Caplanusi A, van de Ven M, Radu M, Despa S, Lambrichts I, Ameloot M, Steels P, Smets I. Role of mitochondrial Na+ concentration, measured by CoroNa red, in the protection of metabolically inhibited MDCK cells. J Am Soc Nephrol 16: 3490–3497, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Beech DJ. Orai1 calcium channels in the vasculature. Pflügers Arch 463: 635–647, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science 248: 376–378, 1990. [DOI] [PubMed] [Google Scholar]
- 11.Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium 38: 303–310, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Cai X. Molecular evolution and structural analysis of the Ca2+ release-activated Ca2+ channel subunit, Orai. J Mol Biol 368: 1284–1291, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Cioffi DL, Lowe K, Alvarez DF, Barry C, Stevens T. TRPing on the lung endothelium: calcium channels that regulate barrier function. Antioxid Redox Signal 11: 765–776, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cioffi DL, Stevens T. Regulation of endothelial cell barrier function by store-operated calcium entry. Microcirculation 13: 709–723, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Cioffi DL, Wu S, Alexeyev M, Goodman SR, Zhu MX, Stevens T. Activation of the endothelial store-operated ISOC Ca2+ channel requires interaction of protein 4.1 with TRPC4. Circ Res 97: 1164–1172, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, Stevens T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res 110: 1435–1444, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cioffi DL, Wu S, Stevens T. On the endothelial cell ISOC. Cell Calcium 33: 323–336, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Creighton JR, Masada N, Cooper DM, Stevens T. Coordinate regulation of membrane cAMP by Ca2+-inhibited adenylyl cyclase and phosphodiesterase activities. Am J Physiol Lung Cell Mol Physiol 284: L100–L107, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Dick DA. The distribution of sodium, potassium and chloride in the nucleus and cytoplasm of Bufo bufo oocytes measured by electron microprobe analysis. J Physiol 284: 37–53, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietrich A, Kalwa H, Fuchs B, Grimminger F, Weissmann N, Gudermann T. In vivo TRPC functions in the cardiopulmonary vasculature. Cell Calcium 42: 233–244, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Eder P, Poteser M, Romanin C, Groschner K. Na+ entry and modulation of Na+/Ca2+ exchange as a key mechanism of TRPC signaling. Pflügers Arch 451: 99–104, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Fameli N, Kuo KH, van Breemen C. A model for the generation of localized transient [Na+] elevations in vascular smooth muscle. Biochem Biophys Res Commun 389: 461–465, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Harootunian AT, Kao JP, Eckert BK, Tsien RY. Fluorescence ratio imaging of cytosolic free Na+ in individual fibroblasts and lymphocytes. J Biol Chem 264: 19458–19467, 1989. [PubMed] [Google Scholar]
- 24.He X, Rezaie AR. Identification and characterization of the sodium-binding site of activated protein C. J Biol Chem 274: 4970–4976, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogan PG, Rao A. Dissecting ICRAC, a store-operated calcium current. Trends Biochem Sci 32: 235–245, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Hooper G, Dick DA. Nonuniform distribution of sodium in the rat hepatocyte. J Gen Physiol 67: 469–474, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huntington JA. How Na+ activates thrombin—a review of the functional and structural data. Biol Chem 389: 1025–1035, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juers DH, Jacobson RH, Wigley D, Zhang XJ, Huber RE, Tronrud DE, Matthews BW. High resolution refinement of β-galactosidase in a new crystal form reveals multiple metal-binding sites and provides a structural basis for α-complementation. Protein Sci 9: 1685–1699, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science 248: 372–376, 1990. [DOI] [PubMed] [Google Scholar]
- 31.Lechtenberg BC, Freund SM, Huntington JA. An ensemble view of thrombin allostery. Biol Chem 393: 889–898, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science 248: 283, 1990. [DOI] [PubMed] [Google Scholar]
- 33.Lemos VS, Poburko D, Liao CH, Cole WC, van Breemen C. Na+ entry via TRPC6 causes Ca2+ entry via NCX reversal in ATP stimulated smooth muscle cells. Biochem Biophys Res Commun 352: 130–134, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Lidofsky SD, Xie MH, Sostman A, Scharschmidt BF, Fitz JG. Vasopressin increases cytosolic sodium concentration in hepatocytes and activates calcium influx through cation-selective channels. J Biol Chem 268: 14632–14636, 1993. [PubMed] [Google Scholar]
- 35.Moore RD, Morrill GA. A possible mechanism for concentrating sodium and potassium in the cell nucleus. Biophys J 16: 527–533, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Negulescu PA, Harootunian A, Tsien RY, Machen TE. Fluorescence measurements of cytosolic free Na concentration, influx and efflux in gastric cells. Cell Regul 1: 259–268, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nofer JR, Pulawski E, Junker R, Seedorf U, Assmann G, Zidek W, Tepel M. Na+/Ca2+ exchange inhibitors modulate thapsigargin-induced Ca2+ and Na+ influx in human lymphocytes. Int J Clin Lab Res 29: 89–92, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Oberleithner H, Riethmuller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci USA 104: 16281–16286, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ochoa CD, Stevens T. Studies on the cell biology of interendothelial cell gaps. Am J Physiol Lung Cell Mol Physiol 302: L275–L286, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orthner CL, Kosow DP. Evidence that human α-thrombin is a monovalent cation-activated enzyme. Arch Biochem Biophys 202: 63–75, 1980. [DOI] [PubMed] [Google Scholar]
- 41.Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 101: 1030–1038, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Prakriya M. The molecular physiology of CRAC channels. Immunol Rev 231: 88–98, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prakriya M. Store-operated Orai channels: structure and function. Curr Top Membr 71: 1–32, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin Y, Dittmer PJ, Park JG, Jansen KB, Palmer AE. Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci USA 108: 7351–7356, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rezaie AR, He X. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry 39: 1817–1825, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem 279: 13696–13704, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Sage SO, Rink TJ, Mahaut-Smith MP. Resting and ADP-evoked changes in cytosolic free sodium concentration in human platelets loaded with the indicator SBFI. J Physiol 441: 559–573, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sedova M, Blatter LA. Intracellular sodium modulates mitochondrial calcium signaling in vascular endothelial cells. J Biol Chem 275: 35402–35407, 2000. [DOI] [PubMed] [Google Scholar]
- 49.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest 128: 558S–564S, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29: 645–655, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Sundivakkam PC, Freichel M, Singh V, Yuan JP, Vogel SM, Flockerzi V, Malik AB, Tiruppathi C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol Pharmacol 81: 510–526, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Tiruppathi C, Freichel M, Vogel SM, Paria BC, Mehta D, Flockerzi V, Malik AB. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res 91: 70–76, 2002. [DOI] [PubMed] [Google Scholar]
- 54.Townsley MI, King JA, Alvarez DF. Ca2+ channels and pulmonary endothelial permeability: insights from study of intact lung and chronic pulmonary hypertension. Microcirculation 13: 725–739, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Vaca L, Licea A, Possani LD. Modulation of cell membrane potential in cultured vascular endothelium. Am J Physiol Cell Physiol 270: C819–C824, 1996. [DOI] [PubMed] [Google Scholar]
- 56.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol 16: 2073–2079, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ware LB, Matthay MA. Acute pulmonary edema. N Engl J Med 353: 2788–2796, 2005. [DOI] [PubMed] [Google Scholar]
- 58.Wu S, Cioffi EA, Alvarez D, Sayner SL, Chen H, Cioffi DL, King J, Creighton JR, Townsley M, Goodman SR, Stevens T. Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site. Circ Res 96: 856–863, 2005. [DOI] [PubMed] [Google Scholar]
- 59.Wu S, Moore TM, Brough GH, Whitt SR, Chinkers M, Li M, Stevens T. Cyclic nucleotide-gated channels mediate membrane depolarization following activation of store-operated calcium entry in endothelial cells. J Biol Chem 275: 18887–18896, 2000. [DOI] [PubMed] [Google Scholar]
- 60.Wu S, Sangerman J, Li M, Brough GH, Goodman SR, Stevens T. Essential control of an endothelial cell ISOC by the spectrin membrane skeleton. J Cell Biol 154: 1225–1233, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M. Orai1-mediated ICRAC is essential for neointima formation after vascular injury. Circ Res 109: 534–542, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.