

Abstract
Chemosensitive neurons in the retrotrapezoid nucleus (RTN) provide a CO2/H+-dependent drive to breathe and function as an integration center for the respiratory network, including serotonergic raphe neurons. We recently showed that serotonergic modulation of RTN chemoreceptors involved inhibition of KCNQ channels and activation of an unknown inward current. Hyperpolarization-activated cyclic-nucleotide-gated (HCN) channels are the molecular correlate of the hyperpolarization-activated inward current (Ih) and have a high propensity for modulation by serotonin. To investigate whether HCN channels contribute to basal activity and serotonergic modulation of RTN chemoreceptors, we characterize resting activity and the effects of serotonin on RTN chemoreceptors in vitro and on respiratory activity of anesthetized rats in the presence or absence of blockers of KCNQ (XE991) and/or HCN (ZD7288, Cs+) channels. We found in vivo that bilateral RTN injections of ZD7288 increased respiratory activity and in vitro HCN channel blockade increased activity of RTN chemoreceptors under control conditions, but this was blunted by KCNQ channel inhibition. Furthermore, in vivo unilateral RTN injection of XE991 plus ZD7288 eliminated the serotonin response, and in vitro serotonin sensitivity was eliminated by application of XE991 and ZD7288 or SQ22536 (adenylate cyclase blocker). Serotonin-mediated activation of RTN chemoreceptors was blocked by a 5-HT7-receptor blocker and mimicked by a 5-HT7-receptor agonist. In addition, serotonin caused a depolarizing shift in the voltage-dependent activation of Ih. These results suggest that HCN channels contribute to resting chemoreceptor activity and that serotonin activates RTN chemoreceptors and breathing in part by a 5-HT7 receptor-dependent mechanism and downstream activation of Ih.
Keywords: retrotrapezoid nucleus, serotonin, 5-HT7, cAMP, Gs-coupled receptor
central chemoreception is the mechanism by which the brain regulates breathing in response to changes in CO2/H+. A region of the brainstem called the retrotrapezoid nucleus (RTN) is an important site of chemoreception. Neurons (Wang et al. 2013) and astrocytes (Gourine et al. 2010) in this region sense changes in CO2/H+ to produce an integrated CO2/H+-dependent drive to breathe. Chemosensitive RTN neurons also integrate respiratory drive from other regions of the respiratory circuit such as serotonergic raphe neurons (Mulkey et al. 2007a). Serotonin is a potent modulator of breathing (Richerson 2004; Corcoran et al. 2014), including at the level of the RTN, where serotonin has been shown to stimulate chemoreceptor activity in vitro and increase breathing in conscious and anesthetized animals (Hawryluk et al. 2012). In addition, disruption of serotonergic signaling has been shown to decrease the respiratory chemoreflex (Hodges et al. 2008; Ray et al. 2011) and may contribute to respiratory failure associated with sudden unexpected death in epilepsy (SUDEP) (Massey et al 2014; Buchanan et al. 2014). Despite this critical physiological role, the mechanisms by which serotonin activates RTN chemoreceptors have yet to be fully elucidated.
Serotonergic signaling is mediated through seven receptor families (5-HT1–7), including Gi (5-HT1)-, Gq (5HT2)-, and Gs (5HT4,7)-coupled receptors (Richter et al. 2003). In the RTN, serotonin sensitivity can be blocked with ketanserin (Mulkey et al. 2007a), an antagonist for both 5-HT2 and 5-HT7 receptors (Beique et al. 2004; Jasper et al. 1997). Evidence also indicates that serotonergic modulation of RTN neurons is determined, in part, by inhibition of KCNQ channels (Hawryluk et al. 2012), most likely by a 5-HT2 Gq-coupled receptor mechanism (Mulkey et al. 2007a,b; Delmas and Brown 2005). However, RTN chemoreceptors retain approximately half their serotonin sensitivity when KCNQ channels are blocked, suggesting involvement of other channels. Preliminary data suggest that when KCNQ channels are blocked, serotonin sensitivity of RTN neurons can be eliminated by inhibiting hyperpolarization-activated cyclic-nucleotide-gated (HCN) channels (Hawryluk et al. 2012), which produce a hyperpolarization-activated inward current (Ih). HCN channels are known to be activated by cAMP (DiFrancesco and Totora 1991) and Gs-coupled receptor signaling (Ulens and Tytgat 2001). Therefore, we hypothesize that HCN channels contribute to serotonergic modulation of RTN chemoreceptors.
Here, we demonstrate the functional presence of both KCNQ and HCN channels in RTN chemoreceptors neurons. We show that inhibition of HCN channels increases ventilation in vivo and the firing rate of RTN chemoreceptors under control conditions but not when Ca2+ channels or other Cd2+-sensitive currents are blocked (Abbruzzese et al. 2010), suggesting that HCN channels dampen basal chemoreceptor excitability by a Cd+2-sensitive mechanism that may involve voltage-dependent Ca2+ channels, as described in other brain regions (Tsay et al. 2007). Inhibition of HCN channels also blunted serotonergic modulation of RTN neurons in vitro and attenuated the ventilatory response to serotonin in vivo. During HCN and KCNQ blockade, serotonin failed to stimulate chemoreceptor activity or breathing, suggesting that coordinated activity of these channels is entirely responsible for serotonergic modulation of RTN chemoreceptors. Furthermore, inhibition of adenylate cyclase blocked the Ih component of the serotonergic response in RTN chemoreceptors, suggesting involvement of a GS-coupled receptor signaling. Consistent with this, serotonin-mediated activation of RTN chemoreceptors was blocked with a 5-HT7-receptor blocker and mimicked by a 5-HT7-receptor activator. We also find that serotonin shifted the voltage dependence of Ih activation to more depolarized potentials. These results build on our understanding of the mechanisms by which serotonin regulates breathing by identifying a novel role of HCN channels and 5-HT7 receptors in the serotonergic modulation of RTN chemoreceptors. Furthermore, considering that HCN and KCNQ are epilepsy-associated ion channels and disruption of serotoninergic signaling may contribute to SUDEP, these results suggest that HCN and KCNQ channels are common substrates for respiratory dysfunction and epilepsy.
METHODS
Animals.
Animal use was in accordance with guidelines approved by the Universities of Connecticut and São Paulo Institutional Animal Care and Use Committees. The in vivo experiments were performed on male Wistar rats weighing 250–300 g (8–10 mo old). In vitro experiments were performed on brain slices isolated from rat pups (7–12 days postnatal).
In vivo preparation.
The surgical procedures and experimental protocols were done as described previously (Hawryluk et al. 2012). Briefly, general anesthesia was induced with 5% halothane in 100% O2. Artificial ventilation with 1.4–1.5% halothane in 100% O2 was maintained throughout surgery. Standard surgical procedures (bilateral vagotomy, arterial cannulation, phrenic nerve dissection, and dorsal transcerebellar access to the ventrolateral medulla oblongata) were used. After surgery, halothane was gradually replaced by urethane (1.2 g/kg, administered intravenous over 20 min). This initial dose was supplemented hourly and at least twice with additional urethane injections of 0.1 g/kg. After a total of 1.4–1.5 g/kg, the level of anesthesia was stable for the duration of the experiment (up to 4 h after initial anesthetic crossover). Rats were ventilated with 100% O2 throughout the experiment, and muscle relaxation was performed with pancuronium (1 mg/kg iv). Rectal temperature was maintained at 37°C, and end-tidal CO2 was monitored with a microcapnometer. The adequacy of anesthesia was continually monitored by testing for the absence of arterial pressure or phrenic nerve discharge (PND) responses to firm toe or tail pinch. After these criteria were satisfied, the muscle relaxant pancuronium was administered intravenously at an initial dose of 1 mg/kg, and the adequacy of the anesthesia was thereafter gauged by the lack of cardiorespiratory responses to a firm toe pinch.
In vivo recordings of physiological parameters.
As described (Mulkey et al. 2004), mean arterial pressure (MAP) and PND were digitized with a Micro 1401 (Cambridge Electronic Design), stored on a computer, and processed offline with Spike 2 software. Integrated PND (iPND) was obtained after rectification and smoothing (τ = 0.015 s) of the original signal, which was acquired with a 30- to 300-Hz bandpass filter. PND amplitude, frequency, and the product of PND frequency and amplitude (i.e., the neural equivalent of minute ventilation) were expressed for each animal on a scale ranging from 0 (value during apnea) to 100 (value while breathing 10% CO2).
Hypercapnia was produced by addition of pure CO2 to the 100% O2 supplied by artificial ventilation to increase the maximum end-expiratory CO2 to 9.5–10%. End-expiratory CO2 was maintained for 5 min, and CO2 was removed. The level of CO2 required to stimulate breathing was identified by increasing inspired CO2 in a stepwise fashion (Δ1%) from 2 to 3% up to 9–10% (5 min per step), and PND was measured during the last 30 s of each step when end-expiratory CO2 and PND appeared to have reached equilibrium. The CO2 PND threshold was determined as the percent CO2 that first elicited PND. End-expiratory CO2 was measured by averaging the maximum values recorded from 50 consecutive breaths at the midpoint of the time interval sampled. One arbitrary unit represents the highest value of PND measured at steady state with end-expiratory CO2 set at 9.5–10%.
Serotonin was injected by pressure (40–60 psi, 4-ms pulses, 50 nl in 3–5 s) through glass pipettes (20-μm outside diameter) filled with 1 mm serotonin creatinine sulfate in a pH 7.3 normal saline solution containing 1% (vol/vol) fluorescent microbeads (for histological verification of injection sites). The concentration of serotonin used in these experiments was based on the EC50 response elicited by serotonin injections into the hypoglossal nucleus of anesthetized rats (Fenik and Veasey 2003). The pipette tip was placed 200 μm ventral to the caudal edge of the facial motor nucleus under electrophysiological guidance as described below for drug injections. In all cases, correct injection was verified by postmortem histological inspection of the location of fluorescent microbeads (Lumafluor, New City, NY).
Histology.
At the end of each in vivo experiment, rats were deeply anesthetized with halothane and perfused through the heart with PBS, pH 7.4, followed by paraformaldehyde (4% in 0.1 m phosphate buffer, pH 7.4). The brains were removed, and the medulla was cut into 40-μm-thick coronal sections (Vibratome 1000S Plus). We confirmed that injection sites were located within the RTN by fluorescent visualization using an Axioskop 2 microscope (Carl Zeiss, Thornwood, NY). Sections from different brains were aligned with respect to a reference section, which was the most caudal section containing an identifiable cluster of facial motor neurons. This reference section was assigned a value of 11.6 mm caudal to bregma (bregma: −11.6 mm; Paxinos and Watson 1989).
Brain slice preparation and slice-patch electrophysiology.
As described (Mulkey et al. 2004; Wenker et al. 2012b), neonatal rats (P7–12 days postnatal) were decapitated under ketamine/xylazine anesthesia, and transverse brainstem slices (300 μm) were cut using a microslicer (DSK 1500E; Dosaka) in ice-cold substituted Ringer solution containing the following (in mm): 260 sucrose, 3 KCl, 5 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, and 1 kynurenic acid. Slices were incubated for ∼30 min at 37°C and subsequently at room temperature in normal Ringer solution containing the following (in mm): 130 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. All solutions were equilibrated with 95% O2-5% CO2, extracellular pH 7.35.
Individual slices containing the RTN were transferred to a recording chamber mounted on a fixed-stage microscope (Zeiss Axioskop FS) and perfused continuously (∼2 ml min−1) with normal Ringer solution bubbled with 95% O2-5% CO2. Hypercapnic solution was made by equilibrating normal Ringer solution with 15% CO2 balance O2 (pHo ∼6.90). All recordings were made from neurons located within 100 μm of the ventral surface and below the caudal end of the facial motor nucleus using an Axopatch 200B patch-clamp amplifier, digitized with a Digidata 1322A A/D converter, and recorded using pCLAMP 10.0 software (Molecular Devices, Sunnyvale, CA). Recordings were obtained at room temperature (∼22°C) with electrodes pulled from borosilicate glass capillaries (Harvard Apparatus, Hamden, CT) on a two-stage puller (P89; Sutter Instrument) to a DC resistance of 5–7 MΩ when filled with an internal solution containing the following (in mm): 120 KCH3SO3, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 HEPES, 10 EGTA, 3 Mg-ATP, and 0.3 GTP-Tris (pH 7.2), and electrode tips were coated with Sylgard 184 (Dow Corning, Midland, MI). All recordings of the firing rate were performed using the cell-attached configuration, and firing rate histograms were generated by integrating action potential discharge in 10-s bins and plotted using Spike 5.0 software. Whole cell current-clamp recordings were made using internal solution containing the following (in mM): 125 potassium gluconate, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 4 Mg-ATP, 0.3 Na-GTP, 0.1 EGTA, 10 2-Tris-phosphocreatine, and 0.05% biocytin (pH 7.3). Cells were held at −60 mV, and depolarizing steps from −100 pA were applied (Δ +10 mV). Input resistance (Rin) was calculated from the voltage responses to hyperpolarizing steps. Initial spike frequency was measured following a 50-pA current injection, and the rebound spike latency was taken as the mean time to first action potential following hyperpolarizing steps (−100 to −70 pA). The sag ratio was expressed as [(1 − ΔVss/ΔVmin) × 100%], where ΔVss = resting membrane potential (RMP) − Vss, ΔVmin = RMP − Vmin, RMP is the resting membrane potential, Vs is the steady-state potential, and Vmin is the initial minimum potential. Voltage-clamp recordings were made at a holding potential of −60 mV and in the presence of tetrodotoxin (TTX; 0.5 μM; Alomone Laboratories) to block action potentials and barium (1 mM) to block leak K+ channels. Holding current, conductance, and current-voltage (I–V) relationships were determined by applying hyperpolarizing voltage steps from −60 to −150 mV for 1,000 ms (Δ −10 mV). The maximal amplitude of Ih was quantified as the size of the time-dependent current during a step to −150 mV. The voltage dependence of Ih activation was obtained from tail currents measured at −70 mV following a series of hyperpolarizing steps; those data were normalized, plotted as a function of the initial step potential, and fitted to a Boltzmann function for calculation of voltage for half-maximal activation (V50) of Ih. For whole cell recordings series resistance (Ra) was typically <20 MΩ and was compensated by 65–70%. A liquid junction potential of 10 mV was corrected offline. Recordings were discarded if Ra varied >10% during an experiment.
Drugs.
All drugs were purchased from Tocris Bioscience unless otherwise stated. For in vivo experiments, the KCNQ channel blocker XE991 (Abcam Biochemicals, Cambridge, MA) and the HCN channel blocker ZD7288 (Sigma-Aldrich) were diluted to 50 μM in sterile saline (pH 7.4) and injected into the RTN using a single-barrel glass pipette (tip diameter of 20 μm) connected to a pressure injector (Picospritzer III; Parker Hannifin, Cleveland, OH). Since the RTN is located bilaterally and both structures contribute to the chemoreflex, to test for contributions of KCNQ and HCN channels to the CO2/H+-drive to breathe we injected XE991 or ZD7288 bilaterally. However, since unilateral RTN injection of serotonin can elicit a ventilatory response, we also tested contributions of KCNQ and HCN to this response by making same side injections of the blockers. For each injection we delivered a volume of 50 nl over a period of 5 s. These glass pipettes also allowed for recordings of field-potential properties that were used to help direct the electrode tip to the desired site. Injections in the RTN region were guided by recordings of the facial field potential (Brown and Guyenet 1985) and were placed 250 μm below the lower edge of the field, 1.7 mm lateral to the midline, and 200 μm rostral to the caudal end of the field. Recordings were made on one side only; the second injection was made 1–2 min later at the same level on the contralateral side. We included a 2% dilution of fluorescent latex microbeads with all drug applications to mark the injection sites and verify the spread of the injections (Takaura and Moreira 2011; Takakura et al. 2011). For in vitro experiments, we bath-applied serotonin hydrochloride (5 μM; Sigma-Aldrich), XE991 (10 μM; Abcam Biochemicals) to block KCNQ channels, ZD7288 (50 μM), or CsCl (2 mM; Sigma-Aldrich) to block HCN channels, CdCl2 (100 μM; Sigma-Aldrich) to block voltage-gated calcium channels, synaptic block perfusate contained kynurenic acid (1 mM; Sigma-Aldrich) to block ionotropic glutamate receptors, bicuculline (10 μM; Sigma-Aldrich) to block GABAA receptors and strychnine (20 μM; Sigma-Aldrich) to block glycine receptors, SQ22536 (100 μM) to inhibit adenylate cyclase activity, SB258719 (10 μM) to block 5-HT7 receptors, 5-carboxamidotryptamine (5-CT, 5 μM) to activate 5-HT7 as well as several Gi-coupled receptors (i.e., 5-HT1A/1B/1D/5), and WAY100635 (0.1 μM) to block 5-HT1A receptors and the specific 5-HT7 agonist LP-44 (2 uM).
Statistical analysis.
Data are reported as means ± SE and illustrated as bar graphs with individual data points. Statistical analysis was performed using GraphPad Prism version 3.0 software. Data were assessed for normality, and the Student's t-test or one-way ANOVA followed by Newman-Keuls multiple-comparisons test were used as appropriate. The relevant values used for statistical analysis are provided in results.
RESULTS
This study consists of three sets of experiments designed to address the mechanistic underpinnings of serotonin-induced respiratory activity in the RTN. First, to determine if serotonin-mediated cardiorespiratory control within the RTN involves HCN channels, we examined breathing and blood pressure in anesthetized rats injected with serotonin into the RTN, either alone or after injection of KCNQ and/or HCN channel blockers. KCNQ channels were blocked in a number of experiments to reveal the KCNQ-independent component to the serotonin response. Second, to determine whether HCN channels regulate excitability and serotonergic modulation of RTN chemoreceptors we tested effects of blocking HCN channels, alone and during blockade of KCNQ channels, on RTN neuron activity (basal firing rate and serotonin-sensitivity) in the brain slice preparation. Third, to identify mechanism(s) underlying serotonergic modulation of HCN channels in RTN chemoreceptors, we assessed serotonin responses when adenylate cyclase activity is blocked and tested the effects of subtype-specific serotonin receptor modulators on RTN chemoreceptor activity. Furthermore, in voltage-clamp experiments, we determined the effects of serotonin on amplitude and voltage-dependent activation of Ih.
Here we focus on chemosensitive RTN neurons, which have been shown to express the transcription factor Phox2b (Lazarenko et al. 2009; Stornetta et al. 2006). However, Phox2b is also expressed by other cells in relatively close proximity to the RTN, including catecholaminergic neurons (C1 and A5), facial motor neurons, and the superior salivatory nucleus (Kang et al. 2007). Therefore, we chose to functionally identify CO2/H+-sensitive RTN neurons based on their characteristic firing rate response to CO2/H+. Neurons were considered chemosensitive if they increased the firing rate by ≥1.5 Hz following exposure to 15% CO2. This level of CO2/H+ response is similar to what we, and others, have reported for chemosensitive RTN neurons in vitro (Mulkey et al. 2006; Ritucci et al. 2005; Wenker et al. 2010). RTN neurons that did not exhibit this minimum response were considered nonchemosensitive and excluded from this study.
HCN channels in the RTN contribute to basal activity and serotonergic modulation of respiratory drive.
Previous evidence indicates that KCNQ channels regulate activity and serotonergic modulation of RTN neurons (Hawryluk et al. 2012). Preliminary in vitro evidence also suggested that HCN channels are expressed in RTN neurons and contribute to serotonin responses (Hawryluk et al. 2012). To establish the functional significance of HCN channels in controlling breathing, we tested whether RTN injection of an HCN channel blocker (ZD7288) affects resting respiratory motor output of anesthetized rats. We then tested whether the ventilatory response to serotonin was altered by RTN injection of ZD7288. All injections (uni- or bilateral) were placed 250 μm below the facial motor nucleus and 200 μm rostral to the caudal end of this nucleus to target the region containing the highest density of CO2-sensitive RTN neurons (Mulkey et al. 2004; Takakura and Moreira 2011; Takakura et al. 2011) (Figs. 1E and 2B).
Fig. 1.
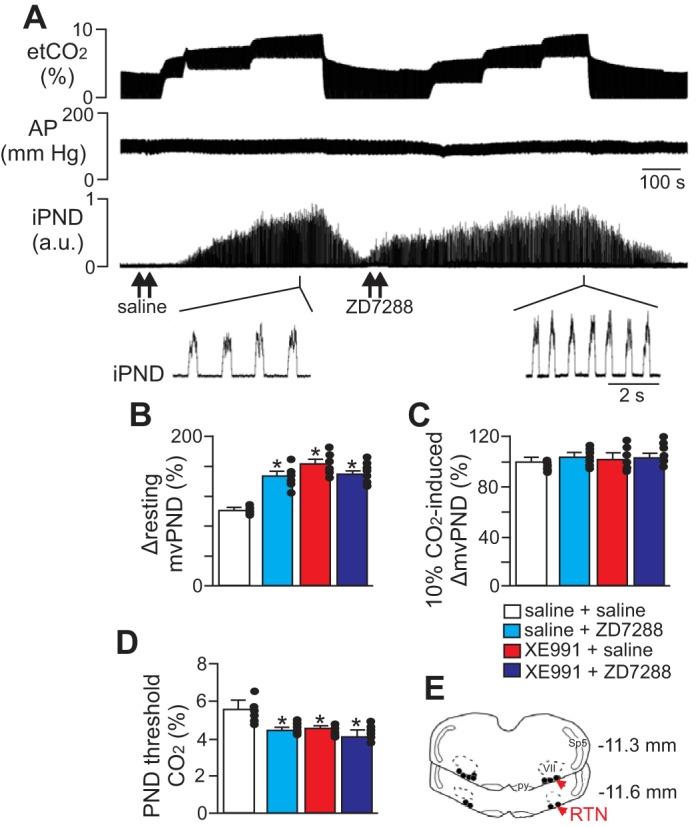
Hyperpolarization-activated cyclic-nucleotide-gated (HCN) channels in the retrotrapezoid nucleus (RTN) regulate resting breathing activity and the ventilatory response to CO2 in anesthetized rats. A: traces of end expiratory CO2 (etCO2), arterial pressure (AP), and integrated phrenic nerve discharge (iPND) show the respiratory response to bilateral injections (arrows) of saline or HCN channel inhibitor ZD7288 in the RTN. Under control conditions, injections of ZD7288 (50 μM, 50 nl each side) increased respiratory activity with an increase in mvPND (product of PND amplitude and frequency) and significantly lowered PND CO2 threshold (D). However, CO2 responsiveness was otherwise unaffected by application of ZD7288 in the RTN; lowering etCO2 from to 3 to 4% inhibited respiratory output, and graded increases etCO2 up to 9–10% increased mvPND by an amount similar to saline (control). B: summary data plotted as change in mvPND shows the effect of ZD7288 alone (50 μM) or in combination with KCNQ channel inhibitor XE991 (50 μM) on resting respiratory activity (n = 7 animals). C: summary data showing CO2-induced changes in mvPND under control conditions, after injections of ZD7288 or XE991 or after injections of ZD7288 + XE991 (n = 7 animals). D: summary data showing that ZD7288 alone or in combination with XE991 decreased the level of CO2 required to stimulate PND activity (n = 7 animals). E: computer-assisted plots of the center of the injection sites (coronal projection on the plane Bregma: −11.6 and −11.3 mm; Paxinos and Watson 1989). Note that all injections were made in the caudal aspect of the RTN where there is the highest density of chemosensitive RTN neurons. Sp5, spinal trigeminal tract; py, pyramids; VII, facial motor nucleus; au, arbitrary units. *P < 0.01.
Fig. 2.

HCN channels in the RTN contribute to serotonergic modulation of respiratory drive in anesthetized rats. A: traces of iPND and AP show effects of unilateral RTN injections of serotonin (5-HT; 1 mM) on breathing and blood pressure under control conditions, after RTN injection of XE991 (50 μM; A1) or ZD7288 (50 μM; A2) alone, and after injections of both XE991 and the HCN channel blocker ZD7288 (A1). Inset: traces of iPND plotted on an expanded time scale. Under control conditions RTN injection of serotonin increased PND amplitude and decreased mean arterial pressure (MAP). Unilateral RTN injection of XE991 or ZD7288 decreased effects of serotonin on PND amplitude by ∼40% and coadministration of ZD7288 with XE991 eliminated the serotonin response. Note the effect of serotonin on PND fully recovered after 60 min. B: computer-assisted plot shows the locations of injection sites within the RTN (Bregma: −11.6 mm). C: summary data (n = 6 animals) showing the effects of serotonin on PND amplitude under control conditions, XE991 alone, ZD7288 alone, and XE991 plus ZD7288. RPa, raphe pallidus. *P < 0.05 vs. control.
In anesthetized animals, we found that bilateral RTN injections of ZD7288 (50 μM, 50 nl each side) increased resting respiratory output as measured by a change in PND. As described previously, bilateral injections of XE991 into the RTN increased the neural equivalent of minute ventilation (mvPND, product of PND amplitude and frequency; Fig. 1, B and E). Bilateral injections of ZD7288 to inhibit HCN channels also increased respiratory activity by 48 ± 7% [F(3,56) = 144.12, P < 0.01; Fig. 1, A and B]. Bilateral blockade of both KCNQ and HCN within the RTN did not increase respiratory activity further than injection of either blocker alone (Fig. 1, A and B). We also found that the ventilatory response to a maximum stimulus of 10% CO2 was unaffected by application of XE991 and/or ZD7288 into the RTN [Fig. 1, A and C; F(3,56) = 0.0254, P > 0.05]. However, bilateral application of XE991 and/or ZD7288 shifted the CO2 threshold (i.e., the level of CO2 required to stimulate ventilatory output) from 5.3 ± 0.09 to 4.5 ± 0.1% [F(3,56) = 87.44, P < 0.01; Fig. 1D]. These data suggest that inhibition of KCNQ and HCN channels increases excitability of RTN chemoreceptors and consequently the respiratory system responses to changes in CO2.
In agreement with previous studies (Hawryluk et al. 2012; Mulkey et al. 2007a), unilateral RTN injection of serotonin (1 mM) increased PND amplitude by 37 ± 5% above baseline [F(3,29) = 129.83, P < 0.01; Fig. 2, A and C]. After respiratory activity returned to control levels, unilateral RTN injection of XE991 (50 μm, 50 nl) or the HCN blocker ZD7288 (50 μM, 50 nl) had no discernible effect on resting PND [F(3,29) = 0.011, P > 0.05; Fig. 2A]. However, injection with either channel blocker significantly blunted, and together essentially eliminated, serotonin modulation of respiratory activity (Fig. 2, A and C). For example, injection of serotonin after application of either XE991 (Fig. 2A1) or ZD7288 (Fig. 2A2) only increased PND amplitude 22 ± 2 and 29 ± 3% above baseline, respectively. This reflects a 40 ± 3 and 21 ± 2% decrease in serotonin responsiveness, respectively, relative to control conditions [F(3,29) = 167.74, P < 0.01; Fig. 2, A and C]. After coapplication of both XE991 and ZD7288 into the RTN, subsequent injection of serotonin only increased PND amplitude by 5 ± 2% (Fig. 2C). Serotonin injection into this region also decreased MAP by 15 ± 4 mmHg, and this hypotensive response was unaffected by XE991 or ZD7288 [F(3,29) = 0.0012, P > 0.05; Fig. 2A; summary data not shown], suggesting that KCNQ and HCN channels are not involved in cardiovascular regulation at this level of the rostral ventrolateral medulla. These results demonstrate that KCNQ channels and HCN channels within the RTN serve as functional determinants of serotonergic modulation of respiratory drive.
HCN channels contribute to both resting firing behavior and serotonergic modulation of RTN chemoreceptors in vitro.
To examine the contribution of HCN channels to activity of RTN chemoreceptors, we first characterized membrane potential responses to hyperpolarizing and depolarizing current steps under control conditions and during HCN channel blockade. In the whole cell current-clamp configuration, chemosensitive RTN neurons have an Rin of 910 ± 83 MΩ and exhibit a sag ratio of 32.3 ± 4.6% in response to a −100-pA hyperpolarizing voltage step (Fig. 3A). At the end of the hyperpolarizing step, chemosensitive RTN neurons produce a rebound spike with a latency of 109 ± 19 ms (Fig. 3A4). Bath application of ZD7288 (50 μM) increased Rin to 1196.0 ± 64.5 MΩ, eliminated the sag [to a sag ratio of only 3.1 ± 4.5%; T(5) = 5.96, P = 0.0019; Fig. 3A2], and increased the rebound spike latency [T(5) = 3.68, P = 0.0142; Fig. 3A4]. We also found that the firing rate response to a depolarizing current injection (20 pA) was unaffected by ZD7288 (Fig. 3A3). This is not surprising considering that HCN channels are inhibited by depolarization. Furthermore, blocking KCNQ channels with XE991 (10 μM) caused a modest increase in Rin [T(4) = 4.71, P = 0.0093] but with no effect on the sag potential or rebound spike latency (Fig. 3B). In addition, XE991 also increased the instantaneous spike frequency elicited by 20-pA current injections (Fig. 3B4). These results indicate that HCN and KCNQ channels regulate intrinsic electrical properties of chemosensitive RTN neurons.
Fig. 3.
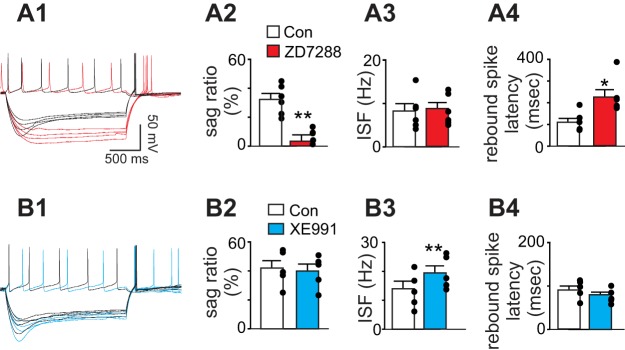
Contributions of HCN and KCNQ channels to repetitive firing behavior of chemosensitive RTN neurons. A1: membrane potential responses to a depolarizing step (+20 pA) and hyperpolarizing current steps (−70 to −100 pA, Δ10 pA) under control conditions and in ZD9288 (50 μM, red). A2–A4: summary data (n = 6) showing sag ratio (A2), instantaneous spike frequency (ISF; A3) and rebound spike latency (A4) under control conditions and in ZD7288. Note the characteristic HCN channel-dependent depolarizing sag and short latency rebound spike. B1: voltage responses to a depolarizing step (+20 pA) and hyperpolarizing current steps (−70 to −100 pA, Δ10 pA) under control conditions and in XE991 (10 μM, blue). B2-B4: summary data (n = 5) showing sag ratio (B2), ISF (B3), and rebound spike latency (B4) under control conditions and in XE991 (10 μM, blue). Blocking KCNQ channels increased ISF but did not affect the sag ratio or rebound spike latency. *P < 0.05, **P < 0.01.
Next, to determine whether HCN channels regulate excitability of RTN chemoreceptors, we tested the effects of HCN channel blockers (ZD7288 or Cs+) on basal activity in the rat brainstem slice preparation. We also used HCN channel blockers alone and in conjunction with XE991 to test for contributions of HCN channels to the serotonin response of RTN chemoreceptors. We found that under control conditions inhibition of HCN channels with ZD7288 (50 μM) or Cs+ (2 mM) increased chemoreceptor activity by 1.01 ± 0.17 Hz [T(16) = 5.93, P < 0.0001; Fig. 4, A and B], suggesting that HCN channels can limit basal activity of RTN chemoreceptors. Note that there was no difference in the effects of ZD7288 and Cs+ on chemoreceptor activity [T(15) = 1.497, P = 0.1551] so data were pooled. Based on evidence from other brain regions, HCN channels may inhibit neural activity by shunting synaptic potentials (Stuart and Spruston 1998) or by influencing activity of other voltage-dependent channels including KCNQ (George et al. 2009) and Ca2+ channels (Tsay et al. 2007). To determine which of these possibilities may contribute to the inhibitory effects of HCN channel on resting activity of RTN chemoreceptors, we characterized effects of HCN channel blockade 1) when excitatory and inhibitory receptors are blocked with a cocktail containing kynurenic acid (1 mM), bicuculline (10 μM), and strychnine (20 μM) (i.e., synaptic blockade); 2) when KCNQ channels were blocked with XE991; and 3) when voltage-gated Ca2+ channels were blocked with Cd2+ (100 μM). We found that the firing rate response of RTN chemoreceptors to HCN channel blockade (i.e., bath application of ZD7288 or Cs+) was unaffected by synaptic blockade but was reduced to a change of 0.38 ± 0.16 Hz in XE991 and eliminated (0.08 ± 0.06 Hz) in Cd2+ [F(3,39) = 7.13, P = 0.0006; Fig. 4, A and B], suggesting that HCN channels limit basal activity of RTN chemoreceptors indirectly by mechanisms involving KCNQ and Cd+2-sensitive channels including Ca2+ channels.
Fig. 4.

HCN channels contribute to resting firing behavior and serotonergic modulation of RTN chemoreceptors in vitro. A1: representative trace of firing rate from a chemosensitive RTN neuron. Under control conditions exposure to serotonin (5-HT; 5 μM) increased firing rate in a CO2-sensitive neuron. After serotonin was washed out and returned to a basal level of activity, bath application of XE991 (10 μM) also increased the firing rate. In the continued presence of XE991 (with baseline activity adjusted to near control levels by DC current injection), a second exposure to serotonin only increased the firing rate by approximately half the control response. Subsequent bath application of ZD7288 (50 μM) also increased firing rate by ∼1 Hz. In the presence of XE991 plus ZD7288 (with baseline activity adjusted to near control levels by DC current injection), a third exposure to serotonin had no discernible effect on firing rate. A2: firing rate trace shows the response of an RTN chemoreceptor to 5-HT and Cs+ under control conditions and in the presence of Cd2+ (100 μM). Note that Cd2+ eliminated the inhibitory effect of HCN channel blockade (reflected as an increase in the firing rate) but did not affect the serotonin response. B: summary data show the firing rate response of RTN chemoreceptors to HCN channel inhibition by ZD7288 or Cs+ (2 mM) under control conditions (n = 17 cells), in XE991 to block KCNQ channels (n = 12 cells), in synaptic blockers (n = 7 cells), and in Cd2+ to block channels including voltage-gated Ca2+ channels (n = 7 cells). Both XE991 and Cd2+ significantly reduced the responses to HCN channel inhibition. C: summary data show the firing rate response to serotonin under control conditions (n = 16 cells), in XE991 alone (n = 13 cells), in ZD7288 alone (n = 4 cells), and in XE991 plus ZD7288 (n = 7 cells) or Cs2+ (2 mM; n = 6 cells). D: firing rate trace shows that exposure to 15% CO2 increased activity of a chemosensitive RTN neuron by similar amounts under control conditions and in the presence of ZD7288 (50 μM). E: summary data (n = 6 cells) showing that CO2/H+ sensitivity of RTN neurons is fully retained when HCN channels are blocked by ZD7288. F: summary data (N = 7 cells) shows that serotonin responses were also retained during Ca2+ channel blockade. // Marks 10-min time breaks and ↓ indicates DC current injection. *P < 0.05, **P < 0.01, ***P < 0.001.
As expected, bath application of serotonin (5 μM) increased activity of these cells by 1.18 ± 0.13 Hz [paired T(14) = 9.00, P < 0.001; Fig. 4, A and C] and it did so in a reversible and repeatable manner (ratio of the third serotonin response divided by the second response was 0.9 ± 0.1; Hawryluk et al. 2012). Bath application of XE991 (10 μM) to block KCNQ channels increased baseline firing by 1.04 ± 0.43 Hz [paired T(10) = 5.42, P = 0.0003] and blunted serotonin-responsiveness by 54.3 ± 11.9% [F(3,38) = 20.06, P < 0.0001; Fig. 4, A and C], as previously reported (Hawryluk et al. 2012). Application of ZD7288 (50 μM) to block HCN channels (with DC current injection to adjust baseline to control levels) reduced subsequent serotonin response to an increase of only 0.57 ± 0.09 Hz (Fig. 4, A and C). Notably, when both KCNQ and HCN channels are blocked with XE991 and ZD7288 or Cs+ (with baseline activity adjusted by DC current injection to near control levels), serotonin increased firing rate less than under control conditions (Fig. 4, A and C), i.e., serotonin increased activity by only 0.11 ± 0.04 Hz in XE991 plus ZD7288 and only 0.07 ± 0.17 Hz in XE991 plus Cs+. When the inhibitory contribution of HCN channel activity was blocked by Cd2+, exposure to serotonin stimulated chemoreceptor activity by an amount similar to that of control conditions [T(5) = 0.66; P = 0.5407; Fig. 4, A2 and F], as expected for activation of an inward depolarizing current. It should be noted that CO2/H+-sensitivity of RTN neurons was retained when HCN channels were blocked with ZD7288 or Cs+ (Fig. 4, D and E). These results suggest that both KCNQ and HCN channels are essential components of serotonergic modulation in RTN chemoreceptor neurons. Collectively, these results also suggest that the net contribution of HCN channels to RTN chemoreceptor basal activity is to limit excitability indirectly by influencing activity of KCNQ and Ca2+ channels. However, targeted activation of presumably a select subset of HCN channels by serotonin can increase chemoreceptor activity and contribute to the excitatory effects of serotonin on breathing.
Serotonin activates Ih in RTN neurons by a Gs-coupled receptor and cAMP-dependent mechanism.
It is unlikely that a single serotonin receptor subtype mediates these divergent effects on KCNQ and HCN channels. HCN channels are known to be activated by Gs-coupled receptors (Ulens and Tygat 2001) but inhibited by Gq-coupled receptor signaling (Liu et al. 2003), indicating that HCN channel activity may be regulated by diverse signal transduction pathways. Gs-coupled receptor signaling involves activation of adenylate cyclase and increased cAMP production (Wettschureck and Offermanns 2005), and it is well known that cAMP can directly bind HCN channels to facilitate Ih (DiFrancesco et al. 1991). We therefore tested the possibility that serotonin activates HCN channels by Gs-coupled receptor signaling.
We first sought to determine if adenylate cyclase activity is required for serotonin-mediated activation of Ih. To study serotonin modulation of HCN channels in relative isolation, we performed these experiments in the presence of XE991 (10 μM) to block KCNQ channels. As before, KCNQ channel blockade decreased the firing rate response to serotonin [control, 1.34 ± 0.21 Hz; XE991, 0.81 ± 0.22 Hz; F(2,4) = 11.38, P = 0.0046; Fig. 5, A and C]. During KCNQ channel blockade (and after baseline activity was adjusted by DC current injection to near control levels), application of the adenylate cyclase inhibitor SQ22536 (100 μM) increased the firing rate of RTN chemoreceptors by 0.80 ± 0.70 Hz [T(4) = 6.43, P = 0.0030; Fig. 5, A and B]. However, when HCN channels are blocked with Cs+ (2 mM), bath application of SQ22536 still increases in the chemoreceptor activity by an amount similar to that under control conditions (n = 3; data not shown), suggesting cAMP can modulate activity of RTN chemoreceptors by mechanisms in addition to activation of HCN. Furthermore, in the presence of SQ22536 inhibition of HCN with Cs+ increased RTN chemoreceptor activity by an amount similar to that under control conditions (n = 4; data not shown), suggesting that cAMP is not required for the maintenance of HCN channel activity under basal conditions. This latter finding is consistent with the possibility that the inhibitory contribution of HCN to basal activity of RTN neurons is determined indirectly by the voltage-dependent modulation of KCNQ and Ca2+ channels. Nevertheless, in the presence of XE991 and SQ22536, exposure to serotonin increased the firing rate by only 0.25 ± 0.06 Hz [F(2,4) = 11.38, P = 0.0046; Fig. 5, A and C]. These findings suggest that adenylate cyclase activity is required for the Ih component of serotonergic modulation of chemosensitive RTN neurons.
Fig. 5.
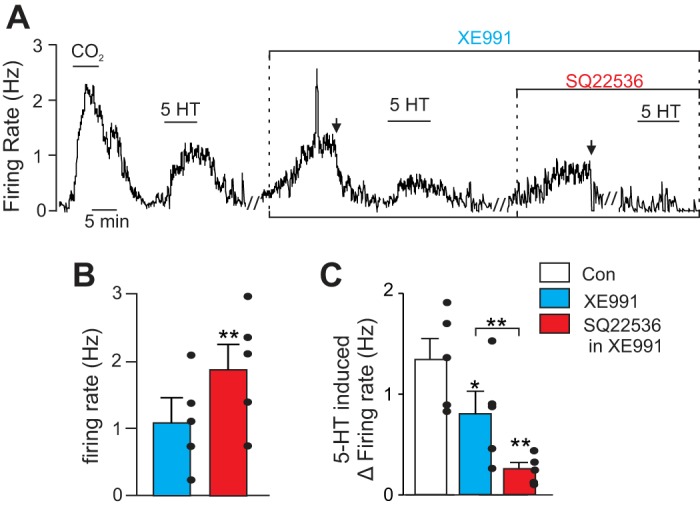
Serotonin activates HCN channels by a cAMP-dependent mechanism. A: representative trace of firing rate from a chemosensitive RTN neuron. Under control conditions exposure to serotonin (5-HT; 5 μM) increased firing in a CO2 sensitive neuron by ∼1 Hz. After washout of serotonin, bath application of XE991 (10 μM) also increased firing rate by ∼1.2 Hz. In the continued presence of XE991 (with baseline activity adjusted to near control levels by DC current injection), a second exposure to serotonin only increased firing rate by ∼0.5 Hz. Subsequent bath application of the adenylate cyclase inhibitor SQ22536 (100 μM) increased firing rate by ∼0.8 Hz. In the presence of XE991 plus SQ22536 (with baseline activity adjusted to near control levels by DC current injection), a third exposure to serotonin no longer led to increased firing. B: summary data (n = 5 cells) of firing rate responses to SQ22536 when KCNQ channels are blocked with XE991. C: summary data (n = 5 cells) of firing rate responses to serotonin under control conditions, in XE991 alone, and in XE991 plus SQ22536. // Marks 10-min time breaks and ↓ indicates DC current injection. *P < 0.05, **P < 0.01.
In other brain regions, cAMP-mediated activation of HCN channels results from a depolarizing shift in voltage-dependent activation of Ih (DiFrancesco and Totora 1991). To confirm this is the case for RTN chemoreceptors, we characterized effects of serotonin on the maximum amplitude and voltage-dependent activation of Ih. Maximum Ih amplitude was defined as the difference between instantaneous and steady-state currents during a −150-mV step (represented in Fig. 6A, by dashed lines). In the whole cell voltage-clamp configuration (holding potential of −60 mV and in TTX [0.5 μM] to block action potentials), Ih was detected by its characteristic time-dependent activation during hyperpolarizing voltage steps. The maximum amplitude of Ih in RTN chemoreceptors was 64.55 ± 14.53 pA under control conditions and 57.1 ± 12.5 pA during exposure to serotonin (Fig. 6B). In addition, bath application of ZD7288 largely eliminated Ih [F(2,21) = 5.21, P = 0.0146; Fig. 6B]. These results suggest that serotonin does not affect maximum Ih in these cells.
Fig. 6.
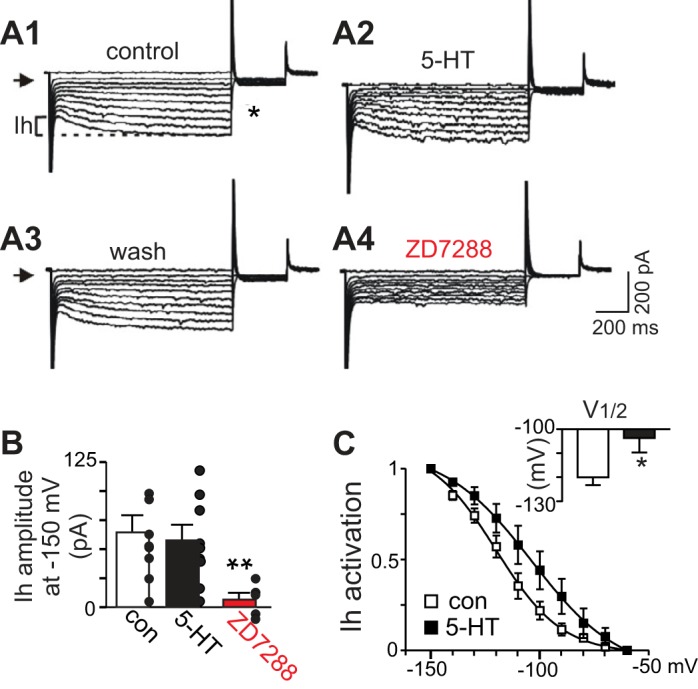
Serotonin causes a depolarizing shift in the voltage-dependent activation of hyperpolarization-activated inward current (Ih) in RTN chemoreceptors. A: current responses to hyperpolarizing voltage steps recorded under control conditions (A1), in serotonin (5 μM; A2), after being washed serotonin for 5 min (A3), and in ZD7288 (50 μM). Note that serotonin caused a modest decrease in holding current (arrows indicate zero current levels). B: summary data (n = 6 cells/animals) showing that serotonin did not increase maximal Ih amplitude, measured as the difference between steady-state and instantaneous currents at −150 mV (illustrated in A1). C: Ih activation curves where tail currents were measured during a fixed step to −70 mV that followed each test pulse (* in A) and normalized data were fitted using the Boltzmann equation, under control conditions and in serotonin. Serotonin causes a depolarizing shift in the voltage-dependent activation of Ih (V1/2 values inset, n = 7). *P < 0.05, **P < 0.01.
To determine the effects of serotonin on the voltage dependency of Ih activation, we measured tail currents at a fixed potential of −70 mV under control conditions and after activating Ih with a series of hyperpolarizing voltage steps. Normalized Ih tail currents were plotted as a function of membrane potential during the initial hyperpolarizing steps and fitted with a Boltzmann function to determine the half-activation voltage (V1/2) (Sirois et al. 2002; Wenker et al. 2012a). The V1/2 for Ih activation averaged −120.1 ± 3.2 mV in control conditions and −103.7 ± 6.0 mV in serotonin [T(4) = 2.35, P = 0.0394; Fig. 6C], indicating that serotonin caused a depolarizing shift of ∼16 mV in the voltage-dependent activation of Ih. These results demonstrate Ih within RTN chemoreceptor neurons and strongly suggest that serotonin activates HCN channels in the RTN by a Gs- and cAMP-dependent mechanism.
Several members of the serotonin receptor family are Gs-coupled including 5-HT4, 5-HT6, and 5-HT7. Of these, 5-HT7 receptors in particular are inhibited by ketanserin (Lesage et al. 1998; Shen et al. 1993). Since ketanserin was shown to block the serotonin responses of chemosensitive RTN neurons (Mulkey et al. 2007a), we considered the possibility that 5-HT7 receptors mediate the effects of serotonin on Ih. To test for involvement of 5-HT7 receptors, we used 5-CT, which activates 5-HT7 as well as several Gi-coupled receptors (i.e., 5-HT1A/1B/1D/5), and WAY100635, which selectively blocks 5-HT1A receptors (Beique et al. 2004). If exposure to 5-CT mimicked effects of serotonin on chemoreceptor activity under control and WAY100635-exposed conditions by a Ih-dependent mechanism, this would suggest that 5-HT7 receptors contribute to serotonin-mediated activation of HCN channels in RTN neurons. Finally, we tested this by using the 5-HT7-specific antagonist SB258719, along with the 5-HT7 receptor agonist LP-44.
We found that bath application of 5-CT (5 μM) increased chemoreceptor activity from 0.97 ± 0.37 to 1.48 ± 0.41 Hz [T(12) = 7.02, P < 0.0001; Fig. 7, A and B]. This change in activity (0.52 ± 0.07 Hz) was similar in magnitude to the serotonin response when KCNQ channels were blocked (Fig. 4C). A second exposure to 5-CT, this time when HCN channels were blocked with ZD7288, failed to significantly increase the firing rate [with a rise of only 0.16 ± 0.05 Hz; F(4,27) = 7.83, P = 0.0003; Fig. 7, A and B]. Bath application of WAY100635 had no effect on baseline activity [T(6) = 1.67, P = 0.1452] or firing-rate response to 5-CT [F(4,27) = 7.83, P = 0.0003; Fig. 7B], suggesting that 5-HT1A receptors do not exert direct or indirect control (e.g., modulation of inhibitory input) over chemosensitive RTN neurons in vitro. Application of a 5-HT7 receptor blocker (SB258719, 10 μM) reduced the firing rate response of RTN chemoreceptors to serotonin by approximately half [T(3) = 6.98, P = 0.006; data not illustrated] and eliminated the 5-CT response [T(4) = 3.70, P = 0.0208; Fig. 7C]. Furthermore, the 5-HT7 agonist LP-44 (2 μM) induced a 0.77 ± 0.16 Hz increase in the chemoreceptor firing rate and this response was blocked by a 5-HT7 antagonist [SB258719, 10 μM; T(3) = 5.56, P = 0.0309; Fig. 7D]. Together, these results suggest that 5-HT7 receptors mediate the effects of serotonin on HCN channels in RTN neurons.
Fig. 7.
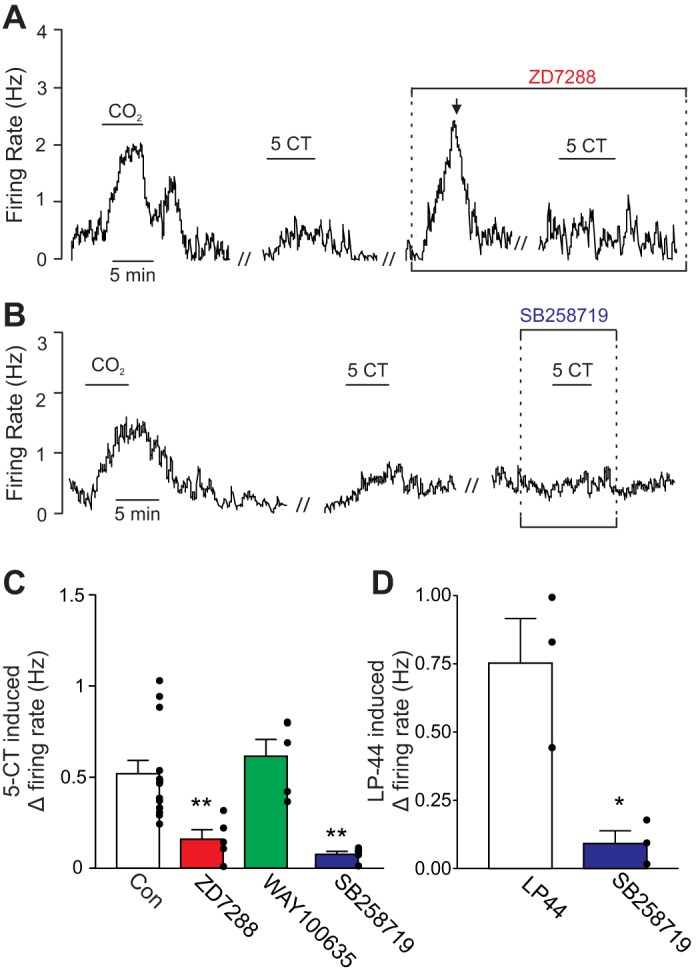
5-HT7 receptor activation mediates effects of serotonin on RTN chemoreceptors. A: representative trace of firing rate from a chemosensitive RTN neuron shows that under control conditions exposure to 5-carboxamidotryptamine (5-CT; 5 μM) increased firing by ∼0.4 Hz. As before, bath application of ZD7288 (50 μM) also increased firing rate. A second exposure to 5-CT, this time in the presence of ZD7288 (with baseline activity adjusted to near control levels by DC current injection), had no effect on firing rate. B: trace of firing rate shows the response of an RTN chemoreceptor to 5-CT was blocked by 10-min incubation in SB258719 (10 μM), a 5-HT7 receptor blocker. C: summary data (n ≥ 4 cells) showing the firing rate response to 5-CT under control conditions and in the presence of ZD7288 alone, the 5-HT1A receptor antagonist WAY100635 (0.1 μM) alone, or the 5-HT7 receptor antagonist SB258719. Note that WAY100635 did not affect the response to 5-CT, suggesting 5-HT1A receptors do not contribute to this response whereas 5-HT7 inhibition significantly reduced the serotonin response. D: average data (n = 3 cells) showing the firing rate response of RTN chemoreceptors the 5-HT7 receptor agonist LP-44 (2 μM) under control conditions and in the presence of the 5-HT7 receptor antagonist SB258719. // Marks 10-min time breaks and ↓ indicates DC current injection. *P < 0.05, **P < 0.01.
DISCUSSION
The RTN is an important locus of respiratory control, as neurons in this region provide a CO2/H+-dependent drive to breathe (Mulkey et al. 2004; Wang et al. 2013) and serve as a point of convergence for other respiratory centers including the medullary raphe (Hawryluk et al. 2012; Mulkey et al. 2007a). Despite the importance of RTN chemoreceptors in breathing, we are only just beginning to understand the ionic mechanisms that control their resting membrane potential and response to neurotransmitters. Recent evidence indicates that KCNQ channels in RTN neurons are key determinants of resting firing behavior and serotonergic modulation of breathing (Hawryluk et al. 2012). Here we show that HCN channels are expressed in RTN chemoreceptor neurons and are a second essential regulator of serotonin-mediated chemoreceptor function. Specifically, we show that HCN channels influence repetitive firing behavior of RTN chemoreceptors by depolarizing membrane potential during hyperpolarization and shortening the rebound spike latency. We also show that inhibition of HCN channels increased spontaneous activity, blunted serotonergic modulation of RTN neurons in vitro, and attenuated the ventilatory response to serotonin in vivo. The mechanism underlying serotonergic activation of HCN channels in RTN neurons appears to involve adenylate cyclase activity and can be mimicked or blunted by drugs that activate or block 5-HT7 receptors. Together with previous data, these results suggest that serotonin regulates the activity of RTN neurons by Gs-mediated activation of HCN channels and Gq-mediated inhibition of KCNQ channels.
A limitation of our experimental approach is that our in vivo and in vitro conditions differ in terms of animal age, temperature, and CO2/H+ stimulus intensity. Age and temperature can influence many physiological parameters including adenylate cyclase activity and HCN channel kinetics, both of which are increased by warming from room to body temperature. Therefore, it is possible that we underestimate the extent to which these channels contribute to chemoreceptor activity. However, we have shown previously that chemosensitive RTN neurons in slices from rat pups (recorded at room temperature) respond to raphe neurotransmitters (e.g., serotonin) in a manner similar to chemosensitive RTN neurons in anesthetized adult rats (Mulkey et al. 2007a). Therefore, it is unlikely that the fundamental mechanisms underlying neurotransmitter modulation of these neurons (i.e., HCN channel modulation) differ significantly with age or temperature. Although ZD7822 is a commonly used HCN channel blocker, there is some evidence that it can also block Na+ and Ca2+ channels (Wu et al. 2012). Therefore, to minimize potential off target effects, we also used a second HCN channel block (Cs+) that is chemically distinct and unlikely to produce common off target effects. It should also be noted that Cd+2, in addition to blocking Ca2+ channels, can also block certain voltage-gated K+ channels (Abbruzzese et al. 2010). Therefore, additional experiments are required to confirm involvement of Ca2+ channels. Lastly, we also recognize that the level of CO2 used here to identify chemosensitive RTN neurons is high. However, we consider this a minor issue because cells identified in vitro as chemoreceptors using 15% CO2 appear to be of the same population of cells identified in vivo as RTN chemoreceptors, i.e., located in the same region, are glutamatergic and express Phox2B (Mulkey et al. 2004; Mulkey et al. 2007b).
HCN channels generate an inward current (Ih) at subthreshold potentials that can have opposing influences on neuronal excitability. For example, activation of Ih has been shown to increase neuronal excitability by depolarizing membrane potential (Wenker et al. 2012a). However, activation of Ih also inhibits neuronal activity by decreasing input resistance, as well as by modulating activity of other voltage-dependent channels, e.g., Ih-mediated depolarization can increase activity of KCNQ channels (George et al. 2009) and prolong inactivation of Ca2+ channels (Tsay et al. 2007). Therefore, both activation and inhibition of HCN channels may cause similar effects on neural activity. Furthermore, the net contribution of Ih to RTN chemoreceptor activity likely depends on several factors including subcellular HCN channel distribution and proximity to other voltage-gated ion channels and/or neurotransmitter receptors. Here, we show in vitro that blocking HCN channels increased the firing rate of RTN neurons under control conditions. Likewise, bilateral RTN injections of ZD7288 increased basal respiratory activity in anesthetized rats. We also found in vitro that the firing rate response of RTN chemoreceptors to HCN channel blockade was blunted by XE991 and eliminated by Cd2+, suggesting that under resting conditions HCN channels limit chemoreceptor activity by mechanisms involving KCNQ and Cd+2-sensitive channels including Ca2+ channels. Consistent with a contribution of KCNQ channels to the inhibitory effects of HCN channel blockade, we found in vivo that bilateral RTN injections of ZD7288 and XE991 increased respiratory output by an amount similar to either drug alone (i.e., the responses were not additive). These results are consistent with evidence from other brain regions that showed HCN-mediated depolarization can enhance activity of voltage-dependent KCNQ channels and in doing so produced a net inhibitory influence on neuronal excitability (George et al. 2009). An additional inhibitory effect of HCN channel activation has been shown to result from prolonging Ca2+ channel inactivation (Tsay et al. 2007). Importantly, the excitatory effect of serotonin on chemoreceptor activity was retained in Cd2+, as expected for activation of an inward current. These results suggest that the excitatory and inhibitory effects of HCN channel activity can coexist in the same cells and can contribute to RTN chemoreceptor activity by different mechanisms.
Serotonergic modulation of RTN chemoreceptor neurons appears to be intrinsic. For example, previous (Mulkey et al. 2007a) and present results suggest that in the presence of TTX (to block action potentials) RTN chemoreceptors respond to serotonin with a modest decrease in holding current. Furthermore, we show here that RTN chemoreceptors exhibit a characteristic Ih that was activated by serotonin, suggesting a direct effect of serotonin on HCN channels in RTN chemoreceptors. Although RTN astrocytes are also known to contribute to the mechanism of chemoreception (Gourine et al. 2010; Huckstepp et al. 2010; Wenker et al. 2012), presumably by a TTX-independent mechanism, preliminary results suggest that CO2/H+-sensitive RTN astrocytes do not exhibit a serotonin-sensitive current (data not shown). However, there is some evidence that astrocytes in other brain regions express 5-HT receptors (Hirst et al. 1997; Miyazaki et al. 2013) and so it remains possible that astrocytes influence serotonergic modulation of chemoreception at other levels of the respiratory network.
The role we are attributing to HCN channels in chemosensitive RTN neurons differs from that previously described for CO2-inhibited medullary neurons isolated from fetal rats, where inhibition of HCN channels decreased resting activity and blocked CO2/H+ sensitivity (Wellner-Kienitz and Shams 1998). The divergent roles of Ih in these cell types may be due to developmental changes in the expression of HCN channel subunits as described in other brainstem regions (Bayliss et al. 1994); however, this possibility requires further investigation.
Our working model for serotonergic modulation of RTN chemoreceptor activity is summarized in Fig. 8. Ketanserin has been shown to block serotonin-stimulated activity of RTN neurons (Mulkey et al. 2007a), suggesting involvement of 5-HT2 and/or 5-HT7 receptors. It is well known that 5-HT2 receptors are coupled to the phosphoinositol (i.e., Gq-coupled) second messenger system, which can modulate ion channel function by depleting plasma membrane levels of phosphatidylinositol 4,5-bisphosphate (PIP2) and by formation of inositol 1,4,5-trisphosphate and diacylglycerol (Suh and Hille 2002). Although not tested directly in this study, it is likely that activation of 5-HT2 receptors and subsequent PIP2 depletion are the likely mechanisms by which serotonin inhibits KCNQ channels in RTN neurons (Hawryluk et al. 2012; Delmas and Brown 2005). It is also well established that 5-HT7 receptors are coupled to trimeric Gs proteins and when activated stimulate adenylate cyclase production of cAMP followed by activation of protein kinase A (PKA) (Matthys et al. 2011). We show that activation of HCN channels via this signaling pathway also contributes to serotonergic modulation of RTN chemoreceptors. Although activation of each of these pathways can independently increase neuronal activity, simultaneous activation of both may antagonize the downstream effects on KCNQ and HCN channels. For example, Gq-mediated PIP2 depletion (Pian et. al. 2006) and PKC activation (Liu et al. 2003) have been shown to inhibit Ih whereas Gs-mediated activation of PKA may activate certain KCNQ channels (Schroeder et al. 1998). It is possible that coordinated activation of both Gs and Gq cascades ensures a robust serotonin response, while cross talk between pathways prevents overactivation.
Fig. 8.
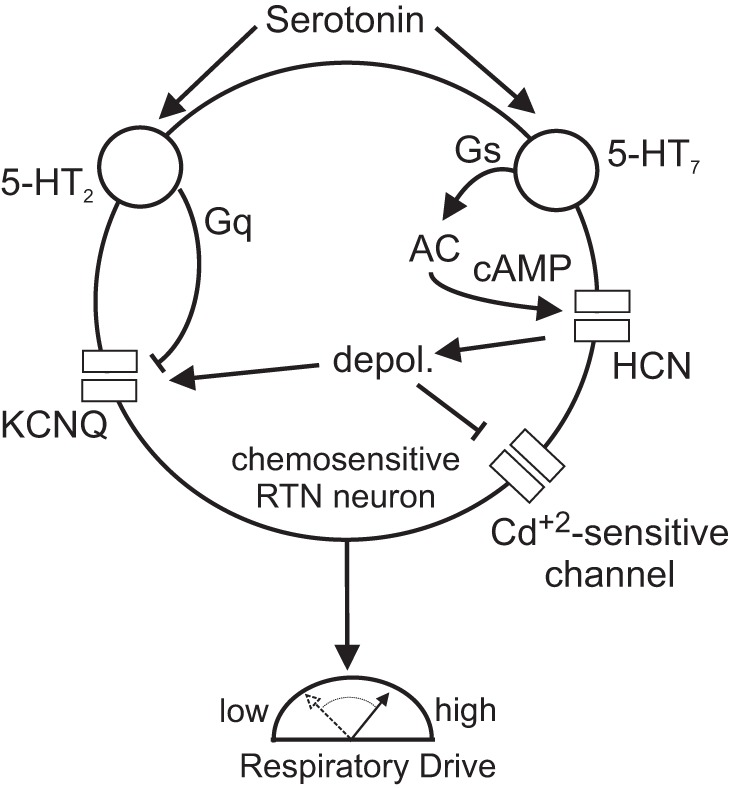
Model depicting the molecular basis for serotonergic modulation of RTN chemoreceptors. Previous (Hawryluk et al. 2012) and present results show that serotonin stimulates activity of RTN chemoreceptors by 2 independent but coordinated signaling pathways: 5-HT2-mediated inhibition of KCNQ channels most likely by Gq signaling, and 5-HT7-mediated activation of HCN channels by a mechanism involving activation of adenylate cyclase- and cAMP-mediated depolarizing shift in the voltage-dependent activation of Ih. Our evidence suggests that each of these signaling pathways can independently stimulate activity of RTN chemoreceptors and together their coordinated activity ensures a robust response to serotonin. Note that our evidence also suggests that under basal conditions HCN channel activity may indirectly modulate activity of voltage-dependent KCNQ and Cd+2-sensitive channels including Ca2+ channels.
In summary, our data identify HCN channels as key determinants of both excitability and serotonergic modulation of RTN chemoreceptor neurons and breathing. The mechanism by which serotonin activates HCN channels in RTN chemoreceptor neurons involves Gs-coupled receptor signaling via 5-HT7 receptors.
GRANTS
This work was supported by funds from National Institutes of Health Grants HL-104101 (to D. K. Mulkey) and NS-073981 (to A. V. Tzingounis) and public funding from São Paulo Research Foundation (FAPESP) Grants 09/54888-7, 13/10573-8 (to T. S. Moreira), and 10/09776-3 (to A. C. Takakura).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: V.E.H., A.V.T., and D.K.M. conception and design of research; V.E.H., J.M.H., A.C.T., and T.S.M. performed experiments; V.E.H., A.C.T., and T.S.M. analyzed data; V.E.H. and D.K.M. interpreted results of experiments; V.E.H. prepared figures; V.E.H., J.M.H., A.C.T., A.V.T., T.S.M., and D.K.M. approved final version of manuscript; A.C.T., A.V.T., T.S.M., and D.K.M. edited and revised manuscript; D.K.M. drafted manuscript.
REFERENCES
- Abbruzzese J, Sachse FB, Tristani-Firouzi M, Sanguinetti MC. Modification of hERG1 channel gating by Cd2+. J Gen Physiol 136: 203–224, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss DA, Viana F, Bellingham MC, Berger AJ. Characteristics and postnatal development of a hyperpolarization-activated inward current in rat hypoglossal motoneurons in vitro. J Neurophysiol 71: 119–128, 1994. [DOI] [PubMed] [Google Scholar]
- Beique JC, Campbell B, Perring P, Hamblin MW, Walker P, Mladenovic L, Andrade R. Serotonergic regulation of membrane potential in developing rat prefrontal cortex: coordinated expression of 5-hydroxytryptamine (5-HT)1A, 5-HT2A, and 5-HT7 receptors. J Neurosci 24: 4807–4817, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DL, Guyenet PG. Electrophysiological study of cardiovascular neurons in the rostral ventrolateral medulla in rats. Circ Res 56, 359–369, 1985. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Murray NM, Hajek MA, Richerson GB. Serotonin neurons have anti-convulsant effects and reduce seizure-induced mortality. J Physiol 592: 4395–4410, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran AE, Commons KG, Wu Y, Smith JC, Harris MB, Richerson GB. Dual effects of 5-HT(1a) receptor activation on breathing in neonatal mice. J Neurosci 34: 51–59, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6: 850–862, 2005. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 351: 145–147, 1991. [DOI] [PubMed] [Google Scholar]
- Fenik P, Veasey SC. Pharmacological characterization of serotonergic receptor activity in the hypoglossal nucleus. Am J Respir Crit Care Med 167: 563–569, 2003. [DOI] [PubMed] [Google Scholar]
- George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+) channels. Nat Neurosci 12: 577–584, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K, Kasparov S. Astrocytes control breathing through pH-dependent release of ATP. Science 329: 571–575, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk JM, Moreira TS, Takakura AC, Wenker IC, Tzingounis AV, Mulkey DK. KCNQ channels determine serotonergic modulation of ventral surface chemoreceptors and respiratory drive. J Neurosci 32: 16943–16952, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst WD, Price GW, Rattray M, Wilkin GP. Identification of 5-hydroxytryptamine receptors positively coupled to adenylyl cyclase in rat cultured astrocytes. Br J Pharmacol 120: 509–515, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, McEvoy SD, Richerson DN, Deneris ES, Johnson RL, Chen ZF, Richerson GB. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci 28: 2495–2505, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, id Bihi R, Eason R, Spyer KM, Dicke N, Willecke K, Marina N, Gourine AV, Dale N. Connexin hemichannel-mediated CO2-dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J Physiol 588: 3901–3920, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper JR, Kosaka A, To ZP, Chang DJ, Eglen RM. Cloning, expression and pharmacology of a truncated splice variant of the human 5-HT7 receptor (h5-HT7b). Br J Pharmacol 122: 126–132, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BJ, Chang DA, Mackay DD, West GH, Moreira TS, Takakura AC, Gwilt JM, Guyenet PG, Stornetta RL. Central nervous system distribution of the transcription factor Phox2b in the adult rat. J Comp Neurol 503: 627–641, 2007. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Milner TA, Depuy SD, Stornetta RL, West GH, Kievits JA, Bayliss DA, Guyenet PG. Acid sensitivity and ultrastructure of the retrotrapezoid nucleus in Phox2b-EGFP transgenic mice. J Comp Neurol 517: 69–86, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage AS, Wouters R, Van Gompel P, Heylen L, Vanhoenacker P, Haegeman G, Luyten WH, Leysen JE. Agonistic properties of alniditan, sumatriptan and dihydroergotamine on human 5-HT1B and 5-HT1D receptors expressed in various mammalian cell lines. Br J Pharmacol 123: 1655–1665, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Bunney EB, Appel SB, Brodie MS. Serotonin reduces the hyperpolarization-activated current (Ih) in ventral tegmental area dopamine neurons: involvement of 5-HT2 receptors and protein kinase C. J Neurophysiol 90: 3201–3212, 2003. [DOI] [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol 10: 271–282, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthys A, Haegeman G, Van CK, Vanhoenacker P. Role of the 5-HT7 receptor in the central nervous system: from current status to future perspectives. Mol Neurobiol 43: 228–253, 2011. [DOI] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Murakami S, Takeshima M, Torigoe N, Kitamura Y, Miyoshi K. Targeting 5-HT(1A) receptors in astrocytes to protect dopaminergic neurons in Parkinsonian models. Neurobiol Dis 59: 244–256, 2013. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Mistry AM, Guyenet PG, Bayliss DA. Purinergic P2 receptors modulate excitability but do not mediate pH sensitivity of RTN respiratory chemoreceptors. J Neurosci 26: 7230–7233, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Rosin DL, West G, Takakura AC, Moreira TS, Bayliss DA, Guyenet PG. Serotonergic neurons activate chemosensitive retrotrapezoid nucleus neurons by a pH-independent mechanism. J Neurosci 27: 14128–14138, 2007a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Talley EM, Stornetta RL, Siegel AR, West GH, Chen X, Sen N, Mistry AM, Guyenet PG, Bayliss DA. TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci 27: 14049–14058, 2007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA, Guyenet PG. Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci 7: 1360–1369, 2004. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates (4th ed). San Diego, CA: Academic, 1989. [Google Scholar]
- Pian P, Bucchi A, Robinson RB, Siegelbaum SA. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J Gen Physiol 128: 593–604, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RS, Corcoran AE, Brust RD, Kim JC, Richerson GB, Nattie E, Dymecki SM. Impaired respiratory and body temperature control upon acute serotonergic neuron inhibition. Science 333: 637–642, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci 5: 449–461, 2004. [DOI] [PubMed] [Google Scholar]
- Richter DW, Manzke T, Wilken B, Ponimaskin E. Serotonin receptors: guardians of stable breathing. Trends Mol Med 9: 542–548, 2003. [DOI] [PubMed] [Google Scholar]
- Ritucci NA, Erlichman JS, Leiter JC, Putnam RW. Response of membrane potential and intracellular pH to hypercapnia in neurons and astrocytes from rat retrotrapezoid nucleus. Am J Physiol Regul Integr Comp Physiol 289: R851–R861, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396: 687–690, 1998. [DOI] [PubMed] [Google Scholar]
- Shen Y, Monsma FJ, Metcalf MA, Jose PA, Hamblin MW, Sibley DR. Molecular cloning and expression of 5-hydroxytryptamine7 serotonin receptors subtype. J Biol Chem 268: 18200–204, 1993. [PubMed] [Google Scholar]
- Sirois JE, Lynch C III, Bayliss DA. Convergent and reciprocal modulation of a leak K+ current and I(h) by an inhalational anaesthetic and neurotransmitters in rat brainstem motoneurones. J Physiol 541: 717–729, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci 26: 10305–10314, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N. Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci 18: 3501–3510, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507–520, 2002. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Colombari E, Menani JV, Moreira TS. Ventrolateral medulla mechanisms involved in cardiorespiratory responses to central chemoreceptor activation in rats. Am J Physiol Regul Integr Comp Physiol 300: R501–R510, 2011. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS. Contribution of excitatory amino acid receptors of the retrotrapezoid nucleus to the sympathetic chemoreflex in rats. Exp Physiol 96: 989–999, 2011. [DOI] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56: 1076–1089, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulens C, Tytgat J. Gi- and Gs-coupled receptors up-regulate the cAMP cascade to modulate HCN2, but not HCN1 pacemaker channels. Pflügers Arch 442: 928–942, 2001. [DOI] [PubMed] [Google Scholar]
- Wang S, Shi Y, Shu S, Guyenet PG, Bayliss DA. Phox2b-expressing retrotrapezoid neurons are intrinsically responsive to H+ and CO2. J Neurosci 33: 7756–7761, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner-Kienitz MC, Shams H. Hyperpolarization-activated inward currents contribute to spontaneous electrical activity and CO2/H+ sensitivity of cultivated neurons of fetal rat medulla. Neuroscience 87: 109–121, 1998. [DOI] [PubMed] [Google Scholar]
- Wenker IC, Benoit JP, Chen X, Liu H, Horner RL, Mulkey DK. Nitric oxide activates hypoglossal motoneurons by cGMP-dependent inhibition of TASK channels and cGMP-independent activation of HCN channels. J Neurophysiol 107: 1489–1499, 2012a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Kreneisz O, Nishiyama A, Mulkey DK. Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1-Kir51-like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol 104: 3042–3052, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Sobrinho CR, Takakura AC, Moreira TS, Mulkey DK. Regulation of ventral surface CO2/H+-sensitive neurons by purinergic signaling. J Physiol 590: 2137–2150, 2012b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev 85: 1159–1204, 2005. [DOI] [PubMed] [Google Scholar]
- Wu X, Liao L, Liu X, Luo F, Yang T, Li C. Is ZD7288 a selective blocker of hyperpolarization-activated cyclic nucleotide-gated channel currents? Channels 6: 438–442, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]