

Abstract
Monoamines and neuropeptides modulate neuronal excitability and synaptic strengths, shaping circuit activity to optimize behavioral output. In C. elegans, a pair of bipolar polymodal nociceptors, the ASHs, sense 1-octanol to initiate escape responses. In the present study, 1-octanol stimulated large increases in ASH Ca2+, mediated by L-type voltage-gated Ca2+ channels (VGCCs) in the cell soma and L-plus P/Q-type VGCCs in the axon, which were further amplified by Ca2+ released from intracellular stores. Importantly, 1-octanol-dependent aversive responses were not inhibited by reducing ASH L-VGCC activity genetically or pharmacologically. Serotonin, an enhancer of 1-octanol avoidance, potentiated 1-octanol-dependent ASH depolarization measured electrophysiologically, but surprisingly, decreased the ASH somal Ca2+ transients. These results suggest that ASH somal Ca2+ transient amplitudes may not always be predictive of neuronal depolarization and synaptic output. Therefore, although increases in steady-state Ca2+ can reliably indicate when neurons become active, quantitative relationships between Ca2+ transient amplitudes and neuronal activity may not be as straightforward as previously anticipated.
Keywords: C. elegans, ASH, 1-octanol, neuromodulation, Ca2+ imaging, nociception, electrophysiology, Ca2+ dynamics, 5-HT
the potential outputs of a neural circuit are specified by the chemical and electrical synapses that interconnect the neurons. However, the actual output is largely determined by the concentrations of neuromodulatory monoamines and neuropeptides present at a given moment, which can drastically reconfigure circuits by modulating intrinsic cell excitability and synaptic strengths (Briggman and Kristan 2008; Komuniecki et al. 2014; Marder and Bucher 2007). Therefore, to understand animal behavior in terms of individual neurons, we need both an anatomical map of their neuronal connectivity and an understanding of the prevalent neuromodulatory states and their functional consequences (Bargmann 2012; Bargmann and Marder 2013). To achieve this goal, it is necessary to quantitatively record neuronal activity states during circuit operation, compare activity patterns under different modulatory conditions, and relate the observed activity patterns to overall behavior. The complexity of this task grows exponentially as nervous system size increases. Not surprisingly, the most detailed insights thus far have come from studies of small discrete circuits with well-defined outputs, such as the crustacean somatogastric ganglion. These circuits can orchestrate multiple distinct patterns of muscle contraction, depending on which monoamines and neuropeptides are present, to select behaviorally appropriate rhythmic outputs (Blitz and Nusbaum 2011; Marder and Bucher 2007). Similarly, neuromodulators can reconfigure more complex mammalian circuits to modulate sensory function and behavior (Gleason 2012; Hermans et al. 2011; Pena-Ortega 2012), suggesting neuromodulatory strategies are highly conserved.
An important current goal in neuroscience is to extend these studies to the whole animal, to explain complex behaviors in terms of neuronal activity patterns and understand how monoamines and neuropeptides alter behavioral states. The C. elegans hermaphrodite provides a useful model system, with only 302 neurons that are interconnected by about 7,000 chemical and about 1,000 electrical synapses, fully mapped in a wiring diagram based on serial-section electron microscopy (White et al. 1986). Importantly, C. elegans is transparent, so nervous system-wide neuronal activity patterns can be recorded from immobilized (glued to a substrate or held in microfluidics devices), or freely moving worms using Ca2+ imaging techniques. These approaches have been very fruitful in elucidating the circuitry that controls sensory function and locomotion and have begun to unravel the mechanisms by which neuromodulators control behavior (Chalasani et al. 2007; Ezcurra et al. 2011; Hilliard et al. 2005; Macosko et al. 2009; Piggott et al. 2011; Suzuki et al. 2008). Ca2+ imaging has the advantage of being completely noninvasive and may be used to monitor many neurons simultaneously, and in C. elegans, perhaps even the entire nervous system (Chen et al. 2011; Grienberger and Konnerth 2012; Schrodel et al. 2013; Seelig and Jayaraman 2013). However, Ca2+ influx is an indirect measure of neuronal activity, since the Ca2+ enters the cytoplasm through voltage-gated Ca2+ channels (VGCCs) that become activated upon depolarization. Ca2+ influxes may underestimate depolarization where VGCCs are relatively scarce or become downregulated through intracellular signaling cascades (Budde et al. 2002). Conversely, Ca2+ influxes may overestimate depolarization if the VGCCs are coupled to inositol tris-phosphate receptors (IP3Rs) or ryanodine receptors (RYRs), which can amplify Ca2+ signals by Ca2+-induced Ca2+ release from internal stores (Busch et al. 2012; Goto and Mikoshiba 2011; Kato et al. 2012; Simpson et al. 1995). Importantly, C. elegans neurons are isopotential and do not fire action potentials (Goodman et al. 1998), so membrane depolarization can spread the length of the neuron independently of voltage-gated channels. For these reasons, care must be taken to properly interpret changes in Ca2+ dynamics caused by monoamines and neuropeptides because they can potentially reflect direct modulation of the VGCCs or their coupling to IP3Rs or RYRs rather than altered neuronal excitation per se.
We are studying neuromodulation in an aversive olfactory circuit mediated by the C. elegans nociceptive ASH sensory neurons, focusing particularly on the functional significance of ASH Ca2+ dynamics. The ciliated endings of the ASHs sense the noxious odorant 1-octanol through the amphid opening at the tip of the nose, and stimulate backwards locomotion escape behavior. This response is modulated at multiple levels in the sensorimotor circuitry by 5-HT, octopamine, tyramine, dopamine, and several neuropeptides (Chao et al. 2004; Ezak and Ferkey 2010; Hapiak et al. 2013; Mills et al. 2012; Wragg et al. 2007). Worms off food reverse around 10 s after exposure to 30% 1-octanol, but reverse in around 5 s when preincubated on 5-HT containing plates (Chao et al. 2004; Harris et al. 2009). 5-HT acts through three distinct receptors to potentiate aversive behavior, including SER-5 acting in the ASHs (Harris et al. 2009, 2010, 2011). 1-Octanol induces large Ca2+ transients in the soma of the ASHs (Mills et al. 2012). Here, we dissected the Ca2+ signal genetically and pharmacologically and compared it to behavioral responses and ASH depolarization measured directly. The magnitude of the ASH somal Ca2+ transient did not predict the depolarization strength or synaptic output as inferred from the initiation of aversive responses, and, in fact, ASH somal Ca2+ transients varied inversely with depolarization strength during 5-HT modulation. These results highlight the potential for nonlinearity in the relationship between Ca2+ signal amplitude and neuronal depolarization strength.
MATERIALS AND METHODS
Strains and constructs.
Strains were maintained on nematode growth media (NGM) agar plates seeded with OP50 bacteria according to standard protocols. The following strains were used: N2, CX10979 kyEx2865[Psra-6::GCaMP3], FY907 grIs17 [Psra-6::GCaMP3], FY867 ser-5(tm2654) I; kyEx2865[Psra-6::GCaMP3], FY882 itr-1(sa73) IV; grIs17[Psra-6::GCaMP3], FY933 egl-19(n582) IV; grIs17[Psra-6::GCaMP3], FY934 egl-30(n686sd) I; grIs17[Psra-6::GCaMP3], FY935 unc-68(e540) V; grIs17[Psra-6::GCaMP3]. Neuron-specific RNA interference (RNAi) transgenes were created as previously described (Esposito et al. 2007), and coinjected with Punc-122::RFP at a concentration of 50 ng/μl. Animals were analyzed within four generations of original injection. The primers used for Psra-6::egl-19RNAi transgene construction were as follows: sra-6F: 5′-CTTTTCATCTCGACCAGACGGTG-3′; sra-6F*: 5′-CAATGTCCACTGATGTACCTTTCTATC-3′; egl-19 Tf: 5′-GTTCCGTGTGATGCGTCTCGTG-3′; egl-19 Tf*: 5′-GCGTCTCGTGAAGCTGCTTTC-3′; egl-19 Tr: 5′-CTTGCCAGGCTTCTCCAGTTG-3′; egl-19 Tr*: 5′-CAGGCTTCTCCAGTTGCTGATC-3′; Psra-6::egl-19 Prs: 5′-CACGAGACGCATCACACGGAACGGCAAAATCTGAAATAAATAAATATTAAATTCTGCG-3′; Psra-6::egl-19 Pra: 5′-CAACTGGAGAAGCCTGGCAAGGGCAAAATCTGAAATAATAAATATTAAATTCTGC-3′.
Calcium imaging.
Calcium imaging experiments were performed essentially as previously described (Mills et al. 2012). Animals were glued to 1.5-mm round coverslips coated with Sylgard (Dow Corning, Midland, MI), immersed in electrophysiology external solution (see below) using WormGlu cyanoacrylate glue (GluStitch, Delta, Canada). Coverslips were placed in a laminar flow chamber (Warner RC26G, Warner Instruments, Hamden, CT) and perfused continuously with fresh external solution. Saturated 1-octanol solution (∼2.37 μM in electrophysiological external) or 1 mM dihydrocaffeic acid was delivered under gravity feed through solenoid valves using a Perfusion Pencil (AutoMate Scientific, Berkeley, CA) or homemade equivalent. All odorant solutions also contained the fluorescent tracer sulforhodamine 101 (SR101, 1 μM), which stains animals on contact. After exposure, animals were visually examined for staining to confirm successful application. No response was observed in ASHs exposed to 1 μM SR101 alone or in the ASI neurons (which are not required for 1-octanol sensitivity, but express the Psra-6::GCaMP3 reporter transgene) and osm-9 mutants [lacking the transient receptor potential (TRP) channel required downstream of ODR-3 in the ASH chemosensory signaling pathway (Colbert et al. 1997)], demonstrating specificity in the ASH 1-octanol responses (not shown). To control for the possibility that high Ca2+ levels produced GCaMP3 saturation in these experiments that could obscure the kinetics of the off responses, we applied 150 mM K+-containing external solution to dissected ASHs expressing GCaMP3 to artificially depolarize them, obtaining change in fluorescence over original fluorescence intensity (ΔF/Fo) values comparable to the 1-octanol-evoked responses. These transients activated and deactivated very quickly, relative to 1-octanol responses (not shown), suggesting that kinetic difference between 1-octanol responses of wild-type and itr-1 mutants were not artifacts of GCaMP3 saturation by relatively high ASH Ca2+ levels in the wild type. Recordings were performed on an Axioskop 2 FS Plus upright compound microscope (×40 Achroplan water immersion objective, GFP filter set no. 38), fitted with an Orca ER CCD camera (Hamamatsu, Skokie, IL) and an automated shutter (Uniblitz, Vincent Associates, Rochester, NY). Minimal illumination intensity was used to minimize GCaMP3 photobleaching, and we did not observe differential photobleaching rates between different genotypes and treatment groups. 5-HT exposure was performed by incubating animals on NGM agar plates containing 4 mM 5-HT for 30 min before recording. Nemadipine-A exposure was performed by incubating animals on NGM agar plates containing 5 μM nemadipine-A spread across the top of the plate for 30 min, and recordings were performed with nemadipine-A in the bath perfusion. 5-HT and nemadipine-A plates were prepared fresh each day. Animals had typically been removed from food for 10–20 min prior to recording, so were considered to be in the “off food” state. Fluorescent images were acquired using MetaVue 7.6.5 (MDS Analytical Technologies, Sunnyvale, CA), and analyzed with Jmalyze software (Rex Kerr). Exposure times were 50 ms with 4 × binning. For quantifying soma fluorescence changes, square regions of interest were drawn centered on the starting position of the soma, corresponding to ∼100 × soma area. For axons, roughly equal size regions of interest were chosen, excluding the soma and dendrite. We routinely compared baseline fluorescence values between mutant or drug-treated worms and corresponding controls and observed no significant differences.
Electrophysiology.
For patch-clamp analyses, animals were glued and placed in the recording chamber as described above. ASH cell bodies (identified by GCaMP3 expression) were exposed for whole-cell recordings by slitting the cuticle with a glass patch pipette (TW150-3, World Precision Instruments, Sarasota, FL) that had been melted and drawn to a fine point on a Narishige MF-83 microforge (Narishige, Setagaya-ku, Tokyo, Japan), and mounted on a micromanipulator (Sutter MP285, Sutter Instruments, Novato, CA). Whole-cell recordings were performed using pressure-polished patch pipettes [15–30 MΩ resistance (Johnson et al. 2008)]. Internal solution contained 15 mM KCl, 115 mM potassium-gluconate, 10 mM HEPES, 5 mM MgCl2, 0.25 mM CaCl2, 5 mM EGTA, 20 mM sucrose, 5 mM MgATP, 0.25 mM NaGTP; pH 7.20, 315 mOsm. External solution contained 150 mM NaCl, 5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 15 mM HEPES; pH 7.30, 327–333 mOsm. 1-Octanol solution was delivered using the Perfusion Pencil as described above, mounted on a Warner SF77B Perfusion Fast Step device (step size 100 μm; Warner Instruments) to permit precise computer control of pipette position. A second patch pipette (20–30 μm opening, driven by a syringe pump; KD Scientific, Holliston, MA) was placed in the bath to provide a shielding stream of external solution that could be used to precisely steer the odorant stream (see Fig. 5). The shielding stream diverted the 1-octanol solution away from the animal's nose prior to and after application and prevented 1-octanol from contacting the exposed ASH neuron during application, thus ensuring precise timing, and preventing direct contact of the odorant solution with the exposed cell body. After termination, the animal was inspected, and any sample with visible SR101 staining of the exposed neuron due to faulty laminar flow or pipette positioning error was discarded. Signals were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) in current clamp mode (0-pA injected current; 10-kHz sampling, 2-kHz filtering), digitized with a Digidata 1440A digitizer, and analyzed using pCLAMP 10 software (Molecular Devices). 5-HT treatment was performed as described above.
Fig. 5.

5-HT potentiates ASH depolarization in response to 1-octanol. A: diagram of recording setup. Left: arrangement of pipettes before exposure, as whole cell recording is being established. At bottom right, the recording pipette and protruding neuronal cell bodies are shown. At top left, flow pipette providing 1-octanol solution and a fluorescent dye to monitor flow (shaded plume) is shown; at bottom left, flow pipette providing a stream of buffer to shield exposed neuron is shown. Exposure is initiated by moving 1-octanol pipette to preset position closer to nose (middle), and terminated by returning 1-octanol pipette to original position (right). B: representative traces of ASH depolarization in response to 1-octanol exposure in control (left) or 5-HT-treated worms (right). C: 5-HT effect on ASH 1-octanol-induced depolarization. ΔVm, change in membrane potential. *P < 0.0005. D: 1-octanol responses of ASH neurons dissected for electrophysiology, but analyzed by Ca2+ imaging, showing that 5-HT signaling to reduce Ca2+ signals remained intact through the dissection process. *P < 0.05. Values are means ± SE; nos. within/above bars indicate n.
Behavioral assays.
Behavioral responses to 1-octanol were assayed as previously described (Chao et al. 2004; Harris et al. 2009). L4 animals were picked the night before assaying to fresh plates seeded with OP50. 5-HT-containing NGM plates were prepared 2 h prior to assays by adding 4 mM 5-HT creatinine sulfate monohydrate to molten solid media (∼50°C) before pouring. A glass capillary or paintbrush hair was dipped in 30% 1-octanol (dissolved in 100% ethanol, vol/vol) and placed in front of a forward-moving animal. For experiments in the absence of 5-HT, animals were transferred to an intermediate (food-free) plate for 1 min and then transferred to assay plates and tested after 10 min. In contrast, for assays in the presence of 4 mM 5-HT, worms were transferred to an intermediate plate for 1 min, but transferred to the 5-HT-containing plate (food-free) and assayed after 30 min.
All reagents were obtained from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich (St. Louis, MO). Statistical analysis was performed using unpaired, two-tailed Student's t-tests, unless otherwise stated, using Prism software with data presented as means ± SE (GraphPad, La Jolla, CA).
RESULTS
Distinct ASH Ca2+ pools mediate ASH-driven aversive responses.
The two ASH nociceptive neurons are necessary and sufficient for aversive responses to dilute 1-octanol; i.e., laser ablation of the ASHs or the ASH-selective RNAi knockdown of the EAT-4 vesicular glutamate transporter, essential for glutamatergic signaling, abolish the aversive response (Chao et al. 2004; Harris et al. 2010). Transgenic animals expressing the Ca2+ reporter GCaMP3 under the control of the ASH-selective sra-6 promoter (Psra-6::GCaMP3) display large increases in ASH Ca2+ when exposed to buffer saturated with 1-octanol (2.4 μM) (Mills et al. 2012), making them a useful strain to study the modulation of 1-octanol-dependent aversive behavior. To begin probing the significance of these increases in ASH Ca2+, we identified the underlying Ca2+ channels using genetic and pharmacological approaches. 1-Octanol-dependent increases in ASH Ca2+ were observed in the soma, dendrite, and axon (Fig. 1A). Somal signals were strongly reduced in the presence of nemadipine-A, a specific inhibitor of the C. elegans EGL-19 L-type VGCC (Kwok et al. 2006), and in egl-19 loss-of-function mutants, but unaffected in mutants lacking the UNC-2 P/Q-type VGCC, indicating that L-type VGCCs predominate in the soma (Fig. 1, A and B; dendritic signals behaved the same as somal signals). Axonal signals were much less sensitive to nemadipine-A, significantly reduced by loss of UNC-2, and nearly abolished by nemadipine-A plus unc-2 mutation, suggesting both channels are important in axons (Fig. 1, A and C).
Fig. 1.
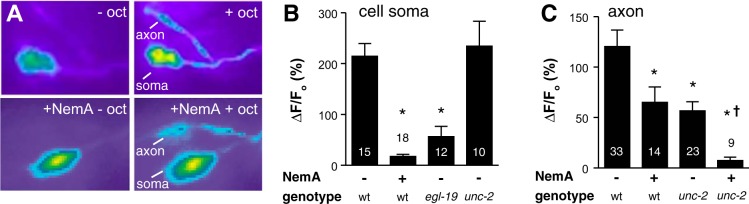
Ca2+ channels mediating 1-octanol-evoked Ca2+ influx into the ASH soma and axon. A: 1-octanol (oct) stimulates Ca2+ transients in the soma and axon of ASHs; nemadipine-A (NemA) preferentially inhibited somal Ca2+ signals. B: loss or block of the L-type (EGL-19), but not the P/Q-type (UNC-2) Ca2+ channels strongly decreased the somal Ca2+ signal. C: axonal Ca2+ signals required both L-type and P/Q-type channels. ΔF/Fo, change in fluorescence over original fluorescence intensity. Values are means ± SE; nos. within/above bars indicate n. *P < 0.05 compared with wild type (wt)/untreated. †P < 0.05 compared with unc-2 untreated.
Ca2+ entering the cytoplasm from outside the cell can stimulate Ca2+ release from internal stores through the IP3R (encoded by itr-1) and the RYR (encoded by unc-68) (Baker et al. 2013). This Ca2+-induced Ca2+ release appears to operate in both the soma and axon of ASHs, dependent on IP3Rs and RYRs in the soma, but only IP3Rs in the axon (Fig. 2). For example, loss of either channel reduced 1-octanol-dependent increases in ASH peak Ca2+ fluorescence by roughly one-half in the soma (Fig. 2, A and B), suggesting that much of the observed Ca2+ signal was not the direct result of voltage-gated processes. Loss of IP3Rs also altered the kinetics of the 1-octanol-induced Ca2+ signal, increasing the rise time (Fig. 2, A and E) and changing the decay time course. In wild-type animals, the Ca2+ signal peaked and began to desensitize within 10 s, while 1-octanol was still present, then continued decreasing at the same rate after 1-octanol withdrawal, indicating the ASH somal Ca2+ pool was no longer detectably responsive to the presence or absence of stimulus. In contrast, in IP3R mutants, the Ca2+ signal desensitized more slowly and responded to 1-octanol withdrawal with an accelerated return to baseline (Fig. 2, A and F). Together, these results suggest IP3Rs first amplify the somal Ca2+ signal initiated by depolarization/EGL-19 VGCC activation, then release additional Ca2+ driven by positive feedback and eventually desensitize due to high cytoplasmic Ca2+ levels (Keizer et al. 1995), resulting in a persistent somal Ca2+ pool that eventually decays independent of stimulus withdrawal. Similar trends are observed in axons (comparing IP3R mutants to wild type), with decreased amplitudes (Fig. 2, C and D) increased rise times (Fig. 2G), and decreased desensitization rates (Fig. 2H). In contrast, loss of RYRs had no effect on Ca2+ kinetics in either the somas or axons (Fig. 2, A, C, E–H). In summary, 1-octanol depolarizes ASH neurons at the amphid (Roayaie et al. 1998), activating L-type VGCCs in the soma and a combination of L- and P/Q-type VGCCs in the axon, and stimulates significant Ca2+ release from internal stores through IP3Rs and RYRs; amplification of Ca2+ signals in ASHs by IP3Rs adds a layer of kinetic complexity that obscures the relationship between ASH stimulation and optically-measurable Ca2+ signals.
Fig. 2.

Ca2+ release from intracellular stores amplifies and shapes Ca2+ dynamics in ASH neurons. A and C: time courses of 1-octanol-evoked Ca2+ transients in ASH somata (A) and axons (C) in wild-type and inositol tris-phosphate receptor (IP3R) mutants (left) or ryanodine receptor (RYR) mutants (right; itr-1 and unc-68, respectively). 1-Octanol was applied between 0 and 20 s (gray box). B and D: peak amplitudes of somal (B) and axonal (D) Ca2+ transients. E–H: kinetic analysis of somal (E and F) and axonal (G and H) Ca2+ transients: signal rise times (E and G), decay slopes (F and H). Black bars in F and H indicate desensitization in the continued presence of 1-octanol; white bars indicate decay rate after 1-octanol removal (rates measured at the intervals denoted by the black and white boxes in A and C, respectively). Values are means ± SE; nos. within bars indicate n. n = 13 wild type, 12 itr-1, and 7 unc-68 (A and F); n = 6 wild type, 8 itr-1, and 6 unc-68 (C and H). *P < 0.05 compared with corresponding wild-type measurement. †P < 0.05 compared with itr-1 desensitization rate.
Surprisingly, the ASH RNAi knockdown of EGL-19 L-type VGCC had no effect on ASH-mediated aversive responses to 30% 1-octanol off food, with wild-type and transgenic animals initiating backward locomotion in about 10 s after 1-octanol exposure, even though ASH EGL-19 RNAi dramatically reduced 1-octanol dependent ASH somal Ca2+ transients (Fig. 3). These data strongly suggest that large increases in ASH somal Ca2+ are not required for ASH-mediated aversive responses to 1-octanol off food.
Fig. 3.
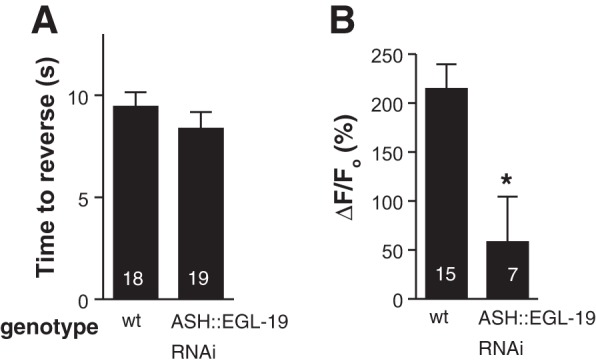
Aversive responses not affected by reduction of Ca2+ influx through EGL-19 L-type voltage-gated Ca2+ channel (VGCC). ASH-selective RNA interference (RNAi) knockdown of the EGL-19 L-type channel (using the sra-6 promoter) did not alter 1-octanol response times (P > 0.05; A), even though Ca2+ signals in the ASH soma in response to 1-octanol exposure were strongly reduced (B). Values are means ± SE; nos. within bars indicate n. *P < 0.05 compared with wild type.
5-HT decreases 1-octanol-dependent ASH somal Ca2+ transients, but increases ASH depolarization.
Food and 5-HT significantly decrease the time taken to initiate backward locomotion in response to dilute 1-octanol, i.e., they increase aversive responses, through a complex extrasynaptic modulatory circuit involving three distinct 5-HT receptors, including the Gαq-coupled SER-5 in the ASHs (Harris et al. 2009). Therefore, we examined the role of 5-HT on 1-octanol-dependent Ca2+ dynamics. To confirm that ASH expression of Psra-6::GCaMP3 did not perturb 1-octanol avoidance, we showed 5-HT still potentiated 1-octanol aversive responses off food (from about 10 s to about 5 s) in the Psra-6::GCaMP3 transgenics (Fig. 4A); these responses did not differ significantly from wild type (Fig. 4F, P > 0.05). Surprisingly, although 5-HT increased aversive responses, it dramatically inhibited 1-octanol-dependent increases in ASH somal Ca2+ (Fig. 4, B and C). As predicted (Harris et al. 2009), both SER-5 and EGL-30 Gαq were required for the 5-HT inhibition of the 1-octanol-dependent somal Ca2+ signal (Fig. 4C). Interestingly, axonal Ca2+ signals were not significantly reduced by 5-HT treatment (Fig. 4D), reinforcing the earlier observation that the Ca2+ pools in the ASH soma and axon are independent. To test whether this 5-HT effect was general or 1-octanol specific, we compared responses to 1 mM dihydrocaffeic acid, another ASH-sensed soluble ligand (Aoki et al. 2011; Kato et al. 2014), and found a similar 5-HT-dependent decrease in ASH somal Ca2+ (Fig. 4E). This result suggests that the 5-HT sensitization of ASH-mediated aversive responses occurs, at least in part, through decreased, not increased, ASH somal Ca2+ transients. In animals with the EGL-19 L-VGCC knocked down in the ASHs using RNAi, basal aversive responses were wild type, but 5-HT failed to potentiate, and even slightly inhibited, aversive responses (Fig. 4F). To eliminate the possibility that the ASI neurons were the relevant site of EGL-19 L-VGCC knockdown (i.e., Psra-6 is active in ASHs and ASIs), we performed a control using the srb-6 promoter (active in ASHs, ADLs, PHAs and PHBs, but not ASIs), and observed the same result. The abolition of 5-HT modulation by EGL-19 L-VGCC knockdown reinforces a role for ASH somal Ca2+ transients in modulation. That the decrease in ASH Ca2+ transients in ASH-selective EGL-19 RNAi knockdown animals was not sufficient to potentiate aversive responses in the absence of 5-HT (Figs. 3B and 4F) suggests that other aspects of 5-HT signaling are also required for the more rapid initiation of aversive responses.
Fig. 4.

5-HT modulation of ASH chemosensory responses. A: aversive behavioral responses to 1-octanol in Psra-6::GCaMP3 worms. Basal responses and 5-HT stimulation were normal, indicating that GCaMP3 expression in ASHs does not interfere with ASH function or 5-HT modulation. B: 5-HT treatment reduced Ca2+ signals in ASHs stimulated by 1-octanol (saturated aqueous solution); n = 7 and 13 for untreated and +5-HT, respectively, dashed lines are SEM, gray box indicates 1-octanol exposure. C: 5-HT modulation of ASH Ca2+ transients required the 5-HT receptor SER-5 and the Gαq subunit EGL-30. D: axonal Ca2+ signals were not significantly reduced by 5-HT. E: ASH responses to 1 mM dihydrocaffeic acid (DHCA) were also reduced by 5-HT treatment. F: ASH-selective RNAi knockdown of EGL-19 prevented 5-HT potentiation of 1-octanol aversive responses; instead, 5-HT slightly inhibited them. Two different ASH-selective promoters were used, sra-6 and srb-6, as indicated in each bar. Values are means ± SE; nos. within bars indicate n. *P < 0.05, **P < 0.0001 compared with untreated.
To better understand the relationship between the 1-octanol-dependent ASH somal Ca2+ signal and ASH depolarization, we turned to electrophysiology. It was necessary to modify standard C. elegans neuronal patch-clamping protocols by adding a second flow pipette to deflect the stream of 1-octanol-saturated external solution away from the exposed neuron during recordings, since long-chain alcohols can directly partition into cell membranes and affect ion channel function (Fig. 5A, materials and methods). Using this system, we measured robust 1-octanol-evoked depolarizations that developed slowly [relative to touch responses (Geffeney et al. 2011)] and returned to baseline once 1-octanol was removed (Fig. 5B). These depolarizations, measured at the cell soma, are likely to reflect membrane potential changes throughout the ASH neuron, due to the presumed isopotentiality of C. elegans neurons (Goodman et al. 1998). Surprisingly, preexposure of the animals to 5-HT (using the same protocol as in behavioral and Ca2+ imaging experiments) led to a significant potentiation of 5-HT-evoked depolarization amplitudes (Fig. 5, B and C), even though 5-HT appeared to decrease 1-octanol-dependent increases in somal Ca2+ (Fig. 4, B and C). To ensure that dissection and exposure of ASHs to the bath solution did not disrupt 5-HT signaling, we also measured Ca2+ transients in dissected ASHs that were not further processed for patch-clamp recording. As observed in intact animals, 5-HT pretreatment also decreased 1-octanol-evoked Ca2+ signals in the somas of these dissected neurons (Fig. 5D). Together, these data demonstrate that 5-HT preexposure reduces the 1-octanol-dependent increase in ASH somal Ca2+, but increases ASH depolarization; i.e., the somal Ca2+ transient varies inversely with depolarization. Therefore, although increases in steady-state Ca2+ can reliably indicate when neurons become active, the quantitative relationship between steady-state Ca2+ and neuronal activity may not be as straightforward as previously anticipated.
DISCUSSION
The two ASH sensory neurons are necessary and sufficient for aversive responses to dilute 1-octanol. In the present study, we have recorded 1-octanol-dependent ASH somal and axonal calcium transients, using the genetically-encoded Ca2+ indicator GCaMP3, and ASH depolarization by direct electrophysiological measurement, and shown that 1) the amplitude of the ASH somal Ca2+ transient does not accurately predict the strength of aversive response off food, as ASH-selective RNAi knockdown of EGL-19 dramatically decreases the 1-octanol-dependent ASH somal Ca2+ signal, but has no effect on aversive responses (Fig. 3); and 2) 5-HT pretreatment potentiates aversive responses and increases 1-octanol-dependent ASH depolarization, but dramatically depresses 1-octanol-dependent Ca2+ transients in the ASH soma (Figs. 4C and 5). These data show that the magnitude of the ASH somal Ca2+ transient does not necessarily predict the depolarization strength or synaptic output and suggest that receptor potentials generated at the amphid may be sufficient to activate VGCCs and synaptic vesicle release in the axons. Consistent with this interpretation, input resistances of C. elegans neurons are very high (Goodman et al. 1998), so for a neuron the size of ASH, depolarization can spread passively from distal dendrite to axon with little loss of strength; however, this does not exclude the possibility that small rises in Ca2+ are required to initiate the behavior. In contrast, ASH somal Ca2+ clearly plays a role in 5-HT modulation, since ASH-selective EGL-19 RNAi knockdown prevents the 5-HT potentiation of aversive behavior (Fig. 3). However, while the depression of ASH Ca2+ influx is probably necessary, it is not sufficient for 5-HT modulation, as the ASH RNAi knockdown of EGL-19 does not stimulate aversive responses in and of itself (Fig. 3). These observations raise two questions: 1) how does decreasing 1-octanol-dependent ASH somal Ca2+ signal increase ASH depolarization; and 2) what additional aspects of serotonergic signaling are required for the 5-HT sensitization of aversive responses?
A Ca2+-dependent negative feedback loop controlling ASH excitability would provide a parsimonious explanation for how reduced Ca2+ influx could lead to increased depolarization amplitude (Fig. 6). 1-Octanol, activating an as-yet-unidentified sensory transduction cascade, causes depolarization of the ASH plasma membrane [based on other ASH stimuli, this cascade may involve the ODR-3 G protein, and polyunsaturated fatty acid signaling to activate the cationic OSM-9/OCR-2 TRP channels (Bargmann 2006; Colbert et al. 1997; Kahn-Kirby et al. 2004; Roayaie et al. 1998)]. The depolarization spreads passively with little attenuation through the soma to the axon, where it activates VGCCs in the soma (EGL-19 L-type VGCCs), and the axon (EGL-19 plus UNC-2 P/Q-type VGCCs). Ca2+ influx then stimulates Ca2+ release from internal stores through IP3Rs and RYRs, thus amplifying and shaping the kinetics of the Ca2+ transient (Fig. 2). This Ca2+ pool could then activate a number of different hyperpolarizing conductances, including the SLO-1 Ca2+-activated K+ channel, or the anoctamin or bestrophin Cl− channels (Francois et al. 2012; Hogan 2007; Jeon et al. 2013; Liu et al. 2013), to potentially brake the 1-octanol-induced depolarization. Direct inhibition of the Ca2+ influx by 5-HT through SER-5 and Gαq could disinhibit ASHs by reducing negative feedback, and, in fact, Gαq-dependent pathways for inhibition of neuronal L-type VGCCs have been described in mammals (Olson et al. 2005).
Fig. 6.
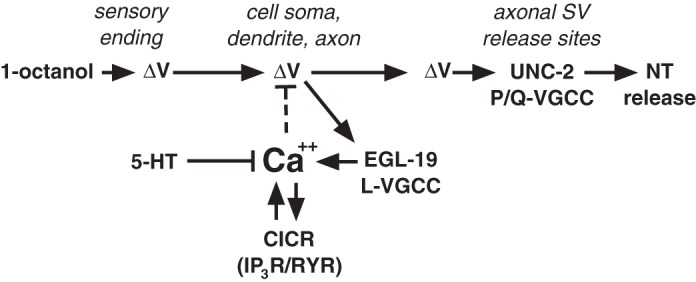
Model of a hypothetical Ca2+-driven negative feedback loop controlling ASH excitability. The activation of the ASH sensory neuron by exposure to 1-octanol involves an as-yet-unidentified amphidial signal transduction cascade, resulting in a membrane depolarization (ΔV), which spreads down the ASH dendrite to the soma and axon, probably passively, given the apparent isopotentiality of C. elegans neurons (Goodman et al. 1998). The amphidial ASH signal transduction cascade activated by other stimuli includes the ODR-3 G protein and polyunsaturated fatty acid signaling to activate the cationic OSM-9/OCR-2 transient receptor potential channels (Bargmann 2006; Colbert et al. 1997; Kahn-Kirby et al. 2004; Roayaie et al. 1998), and these components are also probably involved in activation by 1-octanol. The stimulus-dependent ASH depolarization activates L-type VGCCs (EGL-19) in dendritic, somal, and axonal membranes, and P/Q-type VGCCs (UNC-2) at synaptic vesicle (SV) release sites in the axon, leading to glutamate release. Ca2+ influx through EGL-19 leads to a rise in cytoplasmic Ca2+ that is further amplified by the Ca2+-dependent release of Ca2+ from endogenous stores through both the RYR (UNC-68) and IP3R (ITR-1). This cytoplasmic Ca2+ pool can potentially drive neuronal repolarization by activating Ca2+-dependent inhibitory ion channels (Ca2+-activated K+ channels, anoctamins, or bestrophins; dashed line). Importantly, ASH 5-HT signaling through the SER-5 receptor has the potential to regulate ASH excitability at many levels, including the modulation of ASH Ca2+ dynamics. CICR, calcium-induced calcium release; NT, neurotransmitter.
Besides reducing ASH somal Ca2+ transients and potentiating ASH depolarization, 5-HT activates at least two other pathways relevant to potentiation of 1-octanol aversive behavior. First, Gαq signaling (i.e., downstream of SER-5) regulates synaptic vesicle fusion at C. elegans neuromuscular junctions (Miller et al. 1999; Perez-Mansilla and Nurrish 2009), and potentially also in ASHs (Esposito et al. 2010). Second, 5-HT potentiation of 1-octanol avoidance requires neuropeptide release from ASHs, specifically NLP-3-encoded neuropeptides (Harris et al. 2010). Maximal 5-HT potentiation of aversive responses may require all of these pathways simultaneously, whereas the ASH-selective EGL-19 RNAi knockdown may only activate one pathway (i.e., increased ASH excitability) while failing to activate (or even inhibiting) others. Finally, the potential role of intrinsic homeostatic adaptation in the ASHs to maintain normal electrical excitability must be considered. In many types of neurons, loss of one inhibitory conductance is compensated for by upregulation of another (Bergquist et al. 2010; Misonou 2010; Nerbonne et al. 2008; Turrigiano 2011). For example, the transient K+ current (IA) K+ conductance is upregulated after loss of a Ca2+-activated K+ conductance following blockade of L-type VGCCs with Cd2+ in the cardiac ganglion of the crab Cancer borealis (Ransdell et al. 2012). By analogy, the replacement of the SLO-1 Ca2+-activated K+ conductance by a Ca2+-insensitive K+ conductance (such as the transient K+ current K+ channel KVS-1) in response to ASH-selective EGL-19 RNAi knockdown could explain both the failure to observe fast (5 s) basal 1-octanol responses, and the loss of 5-HT potentiation in the Ca2+-dependent negative feedback model described above.
The relationship between 5-HT signaling, Ca2+ influx and electrical excitability in ASH nociceptors bears a striking similarity to inflammation-induced changes in mammalian somatic nociceptors associated with chronic pain. At sites of injury, the “inflammatory soup” of signaling molecules, including 5-HT, initiates signaling cascades that chronically hypersensitize nociceptors (Basbaum et al. 2009; Reichling et al. 2013). In several rodent models of nerve injury and inflammation, hyperalgesia is observed coincident with nociceptor hyperexcitability, reduced Ca2+ influxes through N- and L-type VGCCs, and a loss of Ca2+-activated K+ conductances in dorsal root ganglion neurons (Hendrich et al. 2012; Hogan 2007; McCallum et al. 2011; Zhang et al. 2012). However, to definitively show that these events are causally related, it will be necessary to demonstrate that inflammatory signaling leads to increased excitability by decreasing Ca2+ influx and reducing Ca2+-activated K+ currents, all within a single cell, which is difficult in rodents because each dorsal root ganglion contains thousands of sensory neuron cell bodies representing multiple nociceptive and nonnociceptive classes (Hogan 2010), and absolute cell identities cannot be assigned between animals. The C. elegans system importantly complements the mammalian model, because its neuroanatomy is stereotyped and relatively simple. With only two ASH neurons (left and right), we can be certain that correlated changes in multiple parameters are taking place within the same cells. Future studies of this conserved nociceptive signaling pathway in C. elegans should provide important insights into the initiation and progression of chronic pain in humans.
Our results add to a growing body of observations highlighting the diversity of roles for somal Ca2+ in the ASH nociceptors. Several studies support the conventional view, that Ca2+ transient amplitudes are predictive of the strength of ASH output. First, six out of six ASH-sensed noxious stimuli produced ASH somal Ca2+ transients, while nonnoxious stimuli did not (Hilliard et al. 2005). In this study, the Ca2+ signals were EGL-19 dependent, and Ca2+ transients elicited by exposure to Cu2+ ions (an ASH-sensed chemorepellent) were diminished by 5-HT treatment (Hilliard et al. 2005). Second, dopamine positively modulates Cu2+-dependent avoidance and ASH Ca2+ transients in parallel, through the DOP-4 receptor (Ezcurra et al. 2011). Third, ASH-mediated avoidance of high pH shows strong correlation between stimulus intensity, ASH Ca2+ signal peak amplitudes, and induction of avoidance behavior (Sassa et al. 2013), and fourth, egl–4 mutation affects ASH Ca2+ signal intensity and avoidance behavior in parallel for the ASH-sensed repellent quinine (Krzyzanowski et al. 2013). Other studies, however, reveal alternative roles for ASH somal Ca2+. For example, loss of RGS-3 proteins that dampen G protein signaling abolish aversive responses to 100% 1-octanol, but sensitivity is restored when Ca2+ buffering proteins are expressed in ASHs, implying that excessive ASH Ca2+ levels inhibit activation (Ferkey et al. 2007). Additionally, loss of function egl-19 mutation does not affect aversive responses to high osmotic strength glycerol, despite reducing ASH Ca2+ transient peak amplitude, but does result in a failure to adapt (i.e., downregulate) during repeated stimulation (Kato et al. 2014). Similarly, a point mutation in the OSM-9 TRP vanilloid channel subunit reduces somal ASH Ca2+ transients below the level of detectability by GCaMP3, but does not prevent osmotic avoidance (Lindy et al. 2014). Interestingly, these animals also showed defects in adaptation and modulation of osmotic avoidance by monoamines (Lindy et al. 2014). These studies support our conclusion that amplitudes of the somal Ca2+ transients in ASHs are not always predictive of response strength and confirm our observation that ASH somal Ca2+ transients are involved in response plasticity.
These findings have important implications for neural circuit analysis. Circuits in both vertebrates and invertebrates switch among several different “output states,” dependent on the intrinsic excitability of the neurons and the synaptic strengths of their connections, subject to modulation by neuropeptides and monoamines (Bargmann 2012; Bargmann and Marder 2013; Briggman and Kristan 2008; Gleason 2012; Komuniecki et al. 2014; Pena-Ortega 2012). Therefore, quantitative measures of neuronal excitation are crucial for understanding how neuromodulators select one circuit output pattern over another. In vertebrates, brain-wide Ca2+ imaging will continue to be a powerful tool (Chen et al. 2013), and even though vertebrate neurons use all-or-none action potentials, Ca2+ transient amplitudes still provide important information about action potential frequency and subthreshold excitation events (Chen et al. 2013; Glickfeld et al. 2013; Sasaki et al. 2008; Vogelstein et al. 2009). Accounting for modulation of Ca2+ entry dynamics by neuromodulatory signaling cascades will be important for the correct interpretation of these data sets. In C. elegans, appreciating the relationship between depolarization and Ca2+ influx may be even more important, because neurons signal through graded potentials, whose amplitudes probably encode analog information about excitation strength. Ca2+ signals are usually equated with membrane potential changes, but, as we show here, somal Ca2+ signals can become uncoupled from, or even vary inversely with, depolarization amplitude and behavioral output. Consistent with our observations, an independent study showed Ca2+ signals can vary inversely with synaptic vesicle release [measured optically using a synaptobrevin-pHluorin fusion reporter in C. elegans salt-sensing neurons (Oda et al. 2011)]. So while Ca2+ influxes can reliably indicate when neurons become active, they are potentially error prone for reporting the degree of activation, a critical parameter for comparing circuit configurations under different modulatory conditions. This potential nonlinearity in the input-output relationships in Ca2+ imaging approaches may be a significant source of error, and direct measurement of depolarization amplitude and/or neurotransmitter release should be performed in parallel to confirm Ca2+ imaging results where possible.
GRANTS
This work was supported by a grant from the Whitehall Foundation to B. A. Bamber, and National Institute of Allergy and Infectious Diseases Grant AI-072644 to R. W. Komuniecki.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.A.Z., R.W.K., and B.A.B. conception and design of research; J.A.Z., P.D.E.W., P.J.S., and B.A.B. performed experiments; J.A.Z., P.D.E.W., P.J.S., and B.A.B. analyzed data; J.A.Z., R.W.K., and B.A.B. interpreted results of experiments; J.A.Z., P.D.E.W., P.J.S., and B.A.B. prepared figures; J.A.Z., R.W.K., and B.A.B. edited and revised manuscript; B.A.B. drafted manuscript; B.A.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the C. elegans Genetics Center for strains; Yifan Xu and C. Bargmann for the kind gift of the ASH::GCaMP3 marker; T. Lindsay, S. Lockery, and M. Goodman for assistance with electrophysiology; A. Korchnak for technical assistance; and members of the Komuniecki and Bamber laboratories for helpful discussion.
REFERENCES
- Aoki R, Yagami T, Sasakura H, Ogura K, Kajihara Y, Ibi M, Miyamae T, Nakamura F, Asakura T, Kanai Y, Misu Y, Iino Y, Ezcurra M, Schafer WR, Mori I, Goshima Y. A seven-transmembrane receptor that mediates avoidance response to dihydrocaffeic acid, a water-soluble repellent in Caenorhabditis elegans. J Neurosci 31: 16603–16610, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KD, Edwards TM, Rickard NS. The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci Biobehav Rev 37: 1211–1239, 2013. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Beyond the connectome: how neuromodulators shape neural circuits. Bioessays 34: 458–465, 2012. [DOI] [PubMed] [Google Scholar]
- Bargmann CI. Chemosensation in C. elegans. WormBook Oct 25: 1–29, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI, Marder E. From the connectome to brain function. Nat Methods 10: 483–490, 2013. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 139: 267–284, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist S, Dickman DK, Davis GW. A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron 66: 220–234, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitz DM, Nusbaum MP. Neural circuit flexibility in a small sensorimotor system. Curr Opin Neurobiol 21: 544–552, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggman KL, Kristan WB. Multifunctional pattern-generating circuits. Annu Rev Neurosci 31: 271–294, 2008. [DOI] [PubMed] [Google Scholar]
- Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci 3: 873–883, 2002. [DOI] [PubMed] [Google Scholar]
- Busch KE, Laurent P, Soltesz Z, Murphy RJ, Faivre O, Hedwig B, Thomas M, Smith HL, de Bono M. Tonic signaling from O(2) sensors sets neural circuit activity and behavioral state. Nat Neurosci 15: 581–591, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, Bargmann CI. Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature 450: 63–70, 2007. [DOI] [PubMed] [Google Scholar]
- Chao MY, Komatsu H, Fukuto HS, Dionne HM, Hart AC. Feeding status and serotonin rapidly and reversibly modulate a Caenorhabditis elegans chemosensory circuit. Proc Nat Acad Sci U S A 101: 15512–15517, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Andermann ML, Keck T, Xu NL, Ziv Y. Imaging neuronal populations in behaving rodents: paradigms for studying neural circuits underlying behavior in the mammalian cortex. J Neurosci 33: 17631–17640, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Leischner U, Rochefort NL, Nelken I, Konnerth A. Functional mapping of single spines in cortical neurons in vivo. Nature 475: 501–505, 2011. [DOI] [PubMed] [Google Scholar]
- Colbert HA, Smith TL, Bargmann CI. OSM-9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans. J Neurosci 17: 8259–8269, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Amoroso MR, Bergamasco C, Di Schiavi E, Bazzicalupo P. The G protein regulators EGL-10 and EAT-16, the Gialpha GOA-1 and the G(q)alpha EGL-30 modulate the response of the C. elegans ASH polymodal nociceptive sensory neurons to repellents. BMC Biol 8: 138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Di Schiavi E, Bergamasco C, Bazzicalupo P. Efficient and cell specific knock-down of gene function in targeted C. elegans neurons. Gene 395: 170–176, 2007. [DOI] [PubMed] [Google Scholar]
- Ezak MJ, Ferkey DM. The C. elegans D2-like dopamine receptor DOP-3 decreases behavioral sensitivity to the olfactory stimulus 1-octanol. PLos One 5: e9487, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezcurra M, Tanizawa Y, Swoboda P, Schafer WR. Food sensitizes C. elegans avoidance behaviours through acute dopamine signalling. EMBO J 30: 1110–1122, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferkey DM, Hyde R, Haspel G, Dionne HM, Hess HA, Suzuki H, Schafer WR, Koelle MR, Hart AC. C. elegans G protein regulator RGS-3 controls sensitivity to sensory stimuli. Neuron 53: 39–52, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A, Grauso M, Demondion E, Bozzolan F, Debernard S, Lucas P. Bestrophin-encoded Ca(2)(+)-activated Cl(-) channels underlie a current with properties similar to the native current in the moth Spodoptera littoralis olfactory receptor neurons. PLos One 7: e52691, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffeney SL, Cueva JG, Glauser DA, Doll JC, Lee TH, Montoya M, Karania S, Garakani AM, Pruitt BL, Goodman MB. DEG/ENaC but not TRP channels are the major mechanoelectrical transduction channels in a C. elegans nociceptor. Neuron 71: 845–857, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason E. The influences of metabotropic receptor activation on cellular signaling and synaptic function in amacrine cells. Vis Neurosci 29: 31–39, 2012. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Andermann ML, Bonin V, Reid RC. Cortico-cortical projections in mouse visual cortex are functionally target specific. Nat Neurosci 16: 219–226, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MB, Hall DH, Avery L, Lockery SR. Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 20: 763–772, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto J, Mikoshiba K. Inositol 1,4,5-trisphosphate receptor-mediated calcium release in Purkinje cells: from molecular mechanism to behavior. Cerebellum 10: 820–833, 2011. [DOI] [PubMed] [Google Scholar]
- Grienberger C, Konnerth A. Imaging calcium in neurons. Neuron 73: 862–885, 2012. [DOI] [PubMed] [Google Scholar]
- Hapiak V, Summers P, Ortega A, Law WJ, Stein A, Komuniecki R. Neuropeptides amplify and focus the monoaminergic inhibition of nociception in Caenorhabditis elegans. J Neurosci 33: 14107–14116, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris G, Korchnak A, Summers P, Hapiak V, Law WJ, Stein AM, Komuniecki P, Komuniecki R. Dissecting the serotonergic food signal stimulating sensory-mediated aversive behavior in C. elegans. PLos One 6: e21897, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris G, Mills H, Wragg R, Hapiak V, Castelletto M, Korchnak A, Komuniecki RW. The monoaminergic modulation of sensory-mediated aversive responses in Caenorhabditis elegans requires glutamatergic/peptidergic cotransmission. J Neurosci 30: 7889–7899, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris GP, Hapiak VM, Wragg RT, Miller SB, Hughes LJ, Hobson RJ, Steven R, Bamber B, Komuniecki RW. Three distinct amine receptors operating at different levels within the locomotory circuit are each essential for the serotonergic modulation of chemosensation in Caenorhabditis elegans. J Neurosci 29: 1446–1456, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich J, Alvarez P, Chen X, Levine JD. GDNF induces mechanical hyperalgesia in muscle by reducing I(BK) in isolectin B4-positive nociceptors. Neuroscience 219: 204–213, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans EJ, van Marle HJ, Ossewaarde L, Henckens MJ, Qin S, van Kesteren MT, Schoots VC, Cousijn H, Rijpkema M, Oostenveld R, Fernandez G. Stress-related noradrenergic activity prompts large-scale neural network reconfiguration. Science 334: 1151–1153, 2011. [DOI] [PubMed] [Google Scholar]
- Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, Schafer WR. In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J 24: 63–72, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan QH. Labat lecture: the primary sensory neuron: where it is, what it does, and why it matters. Reg Anesth Pain Med 35: 306–311, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan QH. Role of decreased sensory neuron membrane calcium currents in the genesis of neuropathic pain. Croat Med J 48: 9–21, 2007. [PMC free article] [PubMed] [Google Scholar]
- Jeon JH, Paik SS, Chun MH, Oh U, Kim IB. Presynaptic localization and possible function of calcium-activated chloride channel anoctamin 1 in the mammalian retina. PLos One 8: e67989, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BE, Brown AL, Goodman MB. Pressure-polishing pipettes for improved patch-clamp recording. J Vis Exp 20: 964, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn-Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, Bargmann CI, Watts JL. Specific polyunsaturated fatty acids drive TRPV-dependent sensory signaling in vivo. Cell 119: 889–900, 2004. [DOI] [PubMed] [Google Scholar]
- Kato HK, Kassai H, Watabe AM, Aiba A, Manabe T. Functional coupling of the metabotropic glutamate receptor, InsP3 receptor and L-type Ca2+ channel in mouse CA1 pyramidal cells. J Physiol 590: 3019–3034, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Xu Y, Cho CE, Abbott LF, Bargmann CI. Temporal Responses of C. elegans chemosensory neurons are preserved in behavioral dynamics. Neuron 81: 616–628, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keizer J, Li YX, Stojilkovic S, Rinzel J. InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Mol Biol Cell 6: 945–951, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuniecki R, Hapiak V, Harris G, Bamber B. Context-dependent modulation reconfigures interactive sensory-mediated microcircuits in Caenorhabditis elegans. Curr Op Neurobiol 29C: 17–24, 2014. [DOI] [PubMed] [Google Scholar]
- Krzyzanowski MC, Brueggemann C, Ezak MJ, Wood JF, Michaels KL, Jackson CA, Juang BT, Collins KD, Yu MC, L'Etoile ND, Ferkey DM. The C. elegans cGMP-dependent protein kinase EGL-4 regulates nociceptive behavioral sensitivity. PLoS Genet 9: e1003619, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok TC, Ricker N, Fraser R, Chan AW, Burns A, Stanley EF, McCourt P, Cutler SR, Roy PJ. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature 441: 91–95, 2006. [DOI] [PubMed] [Google Scholar]
- Lindy AS, Parekh PK, Zhu R, Kanju P, Chintapalli SV, Tsvilovskyy V, Patterson RL, Anishkin A, van Rossum DB, Liedtke WB. TRPV channel-mediated calcium transients in nociceptor neurons are dispensable for avoidance behaviour. Nat Commun 5: 4734, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Chen B, Wang ZW. Postsynaptic current bursts instruct action potential firing at a graded synapse. Nat Commun 4: 1911, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Pokala N, Feinberg EH, Chalasani SH, Butcher RA, Clardy J, Bargmann CI. A hub-and-spoke circuit drives pheromone attraction and social behaviour in C. elegans. Nature 458: 1171–1175, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D. Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu Rev Physiol 69: 291–316, 2007. [DOI] [PubMed] [Google Scholar]
- McCallum JB, Wu HE, Tang Q, Kwok WM, Hogan QH. Subtype-specific reduction of voltage-gated calcium current in medium-sized dorsal root ganglion neurons after painful peripheral nerve injury. Neuroscience 179: 244–255, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB. Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron 24: 323–333, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills H, Wragg R, Hapiak V, Castelletto M, Zahratka J, Harris G, Summers P, Korchnak A, Law W, Bamber B, Komuniecki R. Monoamines and neuropeptides interact to inhibit aversive behaviour in Caenorhabditis elegans. EMBO J 31: 667–678, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H. Homeostatic regulation of neuronal excitability by K(+) channels in normal and diseased brains. Neuroscientist 16: 51–64, 2010. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Gerber BR, Norris A, Burkhalter A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J Physiol 586: 1565–1579, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda S, Tomioka M, Iino Y. Neuronal plasticity regulated by the insulin-like signaling pathway underlies salt chemotaxis learning in Caenorhabditis elegans. J Neurophysiol 106: 301–308, 2011. [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci 25: 1050–1062, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Ortega F. Tonic neuromodulation of the inspiratory rhythm generator. Front Physiol 3: 253, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Mansilla B, Nurrish S. A network of G-protein signaling pathways control neuronal activity in C. elegans. Adv Genet 65: 145–192, 2009. [DOI] [PubMed] [Google Scholar]
- Piggott BJ, Liu J, Feng Z, Wescott SA, Xu XZ. The neural circuits and synaptic mechanisms underlying motor initiation in C. elegans. Cell 147: 922–933, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransdell JL, Nair SS, Schulz DJ. Rapid homeostatic plasticity of intrinsic excitability in a central pattern generator network stabilizes functional neural network output. J Neurosci 32: 9649–9658, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, Green PG, Levine JD. The fundamental unit of pain is the cell. Pain 154, Suppl 1: 10–1016., 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roayaie K, Crump JG, Sagasti A, Bargmann CI. The G alpha protein ODR-3 mediates olfactory and nociceptive function and controls cilium morphogenesis in C. elegans olfactory neurons. Neuron 20: 55–67, 1998. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takahashi N, Matsuki N, Ikegaya Y. Fast and accurate detection of action potentials from somatic calcium fluctuations. J Neurophysiol 100: 1668–1676, 2008. [DOI] [PubMed] [Google Scholar]
- Sassa T, Murayama T, Maruyama IN. Strongly alkaline pH avoidance mediated by ASH sensory neurons in C. elegans. Neurosci Lett 555: 248–252, 2013. [DOI] [PubMed] [Google Scholar]
- Schrodel T, Prevedel R, Aumayr K, Zimmer M, Vaziri A. Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat Methods 10: 1013–1020, 2013. [DOI] [PubMed] [Google Scholar]
- Seelig JD, Jayaraman V. Feature detection and orientation tuning in the Drosophila central complex. Nature 503: 262–266, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson PB, Challiss RA, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends Neurosci 18: 299–306, 1995. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Thiele TR, Faumont S, Ezcurra M, Lockery SR, Schafer WR. Functional asymmetry in Caenorhabditis elegans taste neurons and its computational role in chemotaxis. Nature 454: 114–117, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34: 89–103, 2011. [DOI] [PubMed] [Google Scholar]
- Vogelstein JT, Watson BO, Packer AM, Yuste R, Jedynak B, Paninski L. Spike inference from calcium imaging using sequential Monte Carlo methods. Biophys J 97: 636–655, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314: 1–340, 1986. [DOI] [PubMed] [Google Scholar]
- Wragg RT, Hapiak V, Miller SB, Harris GP, Gray J, Komuniecki PR, Komuniecki RW. Tyramine and octopamine independently inhibit serotonin-stimulated aversive behaviors in Caenorhabditis elegans through two novel amine receptors. J Neurosci 27: 13402–13412, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XL, Mok LP, Lee KY, Charbonnet M, Gold MS. Inflammation-induced changes in BK(Ca) currents in cutaneous dorsal root ganglion neurons from the adult rat. Mol Pain 8: 37, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]