

Abstract
Endothelin-1 (ET-1) increases pulmonary vascular tone through direct effects on pulmonary artery smooth muscle cells (PASMC) via membrane-bound ET-1 receptors. Circulating ET-1 contributes to vascular remodeling by promoting SMC proliferation and migration and inhibiting SMC apoptosis. Although endothelial cells (EC) are the primary source of ET-1, whether ET-1 produced by SMC modulates pulmonary vascular tone is unknown. Using transgenic mice created by crossbreeding SM22α-Cre mice with ET-1 flox/flox mice to selectively delete ET-1 in SMC, we tested the hypothesis that PASMC ET-1 gene expression modulates the pulmonary vascular response to hypoxia. ET-1 gene deletion and selective activity of SM22α promoter-driven Cre recombinase were confirmed. Functional assays were performed under normoxic (21% O2) or hypoxic (5% O2) conditions using murine PASMC obtained from ET-1+/+ and ET-1−/− mic and in human PASMC (hPASMC) after silencing of ET-1 using siRNA. Under baseline conditions, there was no difference in right ventricular systolic pressure (RVSP) between SM22α-ET-1−/− and SM22α-ET-1+/+ (control) littermates. After exposure to hypoxia (10% O2, 21–24 days), RVSP was and vascular remodeling were less in SM22α-ET-1−/− mice compared with control littermates (P < 0.01). Loss of ET-1 decreased PASMC proliferation and migration and increased apoptosis under normoxic and hypoxic conditions. Exposure to selective ET-1 receptor antagonists had no effect on either the hypoxia-induced hPASMC proliferative or migratory response. SMC-specific ET-1 deletion attenuates hypoxia-induced increases in pulmonary vascular tone and structural remodeling. The observation that loss of ET-1 inhibited SMC proliferation, survival, and migration represents evidence that ET-1 derived from SMC plays a previously undescribed role in modulating the response of the pulmonary circulation to hypoxia. Thus PASMC ET-1 may modulate vascular tone independently of ET-1 produced by EC.
Keywords: pulmonary hypertension, smooth muscle cells
at all points in organismal development and life, control of pulmonary vascular tone is of critical importance. During fetal life, normal lung development requires closely circumscribed pulmonary flow, while pulmonary vascular resistance (PVR) exceeds systemic vascular resistance. With the onset of air-breathing life, PVR must decrease abruptly to accommodate the increase in pulmonary blood flow that enables gas exchange (3, 4). Early in postnatal life, PVR decreases still further to 20% of systemic vascular resistance, where, under normal conditions (6), it remains throughout the remainder of air-breathing life.
However, in a number of pathological states, pulmonary arterial pressure is increased, leading to profoundly untoward consequences. For example, in the perinatal period, pulmonary vasodilation is biologically imperative. If PVR does not decrease with the onset of air-breathing life, persistent pulmonary hypertension of the newborn results in a substantial cause of neonatal morbidity and mortality (25). During air-breathing life, increases in pulmonary vascular tone possess even more profound untoward consequences.
Pulmonary arterial hypertension (PAH) is a syndrome wherein pulmonary arterial obstruction increases PVR, resulting in right ventricular heart failure. Despite the advent of new biological-based therapies and increased insight into the pathobiology of primary pulmonary hypertension, the actuarial survival of patients has demonstrated only modest improvement, as 50% of all people with primary PAH die within 7 yr of diagnosis (1). No effective preventative or curative treatments are available. When PAH is superimposed on diseases such as bronchopulmonary dysplasia, a chronic lung disease of infancy (26), congenital heart disease, cystic fibrosis, or rheumatological disease (16), the prognosis is dramatically worsened (13). Thus the need to generate new knowledge that might be translated into a therapeutic tool has never been more palpable.
Pulmonary artery smooth muscle cells (PASMC) have the capacity to sense and respond directly to hypoxia (5, 21) and thereby modulate pulmonary vascular tone. In the context of pulmonary hypertension, hypoxia can cause vascular remodeling, which entails PASMC proliferation and migration (28). Molecules produced by PA endothelial cells (EC) modulate vascular tone by direct effects on PASMC. Nitric oxide, for example, activates guanylate cyclase and increases cytosolic cyclic GMP concentration, resulting in vasodilation (7, 11, 24).
Endothelin (ET-1), a 21-amino-acid polypeptide produced primarily by EC, possesses complex effects. ET-1 binds to specific receptors on PASMC to cause an increase in [Ca2+]i and vasoconstriction (40). In many pathological states, including PAH (36), ET-1 production is increased, leading to an increase in vascular tone, smooth muscle cell (SMC) proliferation, migration, and survival (14, 23, 41).
The importance of ET-1 in the pathobiology of PAH (31, 32) provided sound therapeutic rationale for the development of specific ET-1 receptor antagonists. Although ET-1 receptor antagonists are widely used clinically, the therapeutic efficacy has been limited (19). Given the observation that SMC can produce ET-1 (10, 29), we sought to determine whether ET-1 produced by SMC might function in an autocrine manner and thereby contribute to the pathology of PAH independent of ET-1 receptor activation (37, 38). Thus, to test the hypothesis that ET-1 produced by PASMC modulates the PAH response to chronic hypoxia, we generated mice with a SMC-specific deletion of ET-1.
MATERIALS AND METHODS
Generation of SM22α-ET-1−/− mice.
Transgenic mice with selective deletion of ET-1 (SM22α-ET-1−/− mice) in SMC were created by cross breeding SM22α-promoter-driven Cre mice expressing the Cre reporter gene ROSA26-R (kindly provided by Dr. Marlene Rabinovitch, Stanford University) with ET-1flox/flox mice (kindly provided by Dr. Masashi Yanagisawa, UT Southwestern). ET-1 homozygous floxed mice contain the ET-1 exon 2 flanked by LoxP sites as previously described (20). The Institutional Animal Care and Use Committee at Stanford University approved all the procedures and protocols governing the care and use of laboratory animals.
To identify the Cre gene, primers (forward: 5′-CCGGTTATTCAACTTGCACC-3′; reverse: 5′-CTGCATTACCGGTCGATGCAAC-3′) were used to generate a 149-bp PCR product (34). To identify the Cre reporter gene, ROSA26-R, primers [Jackson Laboratories protocol: Gt (ROSA) 26Sortm1sor STD, forward: 5′-AAAGTCGCTCTGAGTTGTTAT-3′; mutant reverse: 5′-GCGAAGAGTTTGTCCTCAACC-3′; wild-type (WT) reverse: 5′-GGAGCGGGAGAAATGGATATG-3′] were used to amplify a 340-bp fragment from mutant and a 550-bp fragment from control mice. To identify the WT ET-1 gene (edn1), primers (WT forward: 5′-GCTGCCCAAAGATTCTGAATTCTG-3′; mutant forward: 5′-CCCAAAGATTCTGAATTGATAACTTCG-3′; reverse: 5′-GATGATGTCCAGGTGGCAG AAG-3′) were used to amplify a 900-bp fragment from mutant (ET-1flox/flox) and a 900-bp fragment from control mice.
X-gal staining of pulmonary tissues.
To identify SM22α-promoter-driven Cre recombinase activity, pulmonary tissues were inflated with optimum cutting temperature (OCT) compound and then stored in OCT at −80°C. Frozen tissues were sectioned, incubated for 15 min in 0.2% glutaraldehyde on ice, stained with 1 mg/ml X-gal (β-Galactosidase Reporter Gene Staining Kit, Sigma-Aldrich) for 3 h at 37°C, and then fixed in 10% formalin solution for 10 min.
Primary mouse PASMC isolation.
Primary mouse PASMC (mPASMC) were isolated from control (Cre-ET-1flox/flox) and SM22α-ET-1−/− mice using a modified elastase/collagenase digestion protocol (18). PA tissue was digested in dispersion medium containing 40 μmol/l CaCl2, 0.5 mg/ml elastase (Worthington Biochemical), 0.5 mg/ml collagenase (Worthington Biochemical), 0.2 mg/ml soybean trypsin inhibitor (Worthington Biochemical), and 2 mg/ml albumin (Sigma-Aldrich) for 20 min at 37°C. After filtration with 100-μm cell strainers, cells were incubated with Dynabeads (Invitrogen) coated with CD31 antibody (BD Biosciences) for 20 min, to deplete endothelial cells expressing CD31. Remaining SMC were collected through centrifugation at 225 g for 5 min at 4°C and cultured in DMEM containing 5% FBS, 1% l-glutamine, and 1% antibiotic-antimycotic solution (Invitrogen/Gibco). To confirm isolation of PASMC, cells were stained for α-smooth muscle actin (α-SMA) (1:400, Sigma) using immunofluorescence technique.
Immunocytochemistry.
mPASMC isolated from control and SM22α-ET1−/− mice were fixed in 3% paraformaldehyde in PBS for 30 min, blocked and permeabilized in blocking solution (0.1% Triton X-100, 15 g/ml glycine, 2.5% FBS in PBS) for 1 h, and then incubated with ET-1 antibody (1:200, Abcam) for 1 h. Cells were then incubated with Alexa Fluor 488 Phalloidin (1:40, Invitrogen/Molecular Probes) and goat α-rabbit Alexa Fluor 568 (1:200, Invitrogen/Molecular Probes) for 30 min, followed by mounting in Vectashield Mounting Medium with DAPI (Vector Laboratories).
Quantitative RT-PCR.
To assay for ET-1 mRNA expression, total RNA was isolated from cultured mPASMC exposed to 24-h hypoxia (5% O2) using the RNeasy Mini Kit (Qiagen). First-strand cDNA was synthesized using SuperScript III Reverse Transcriptase (Life Technologies) and subsequently amplified on the C1000 Thermal Cycler CFX 384 Real-Time System (Bio-Rad) using RT2 SYBR Green qPCR Mastermix (Qiagen). Primer sets are as follows, ET-1 forward: 5′-GTGTCTACTTCTGCCACCTGGACAT-3′ and ET-1 reverse: 5′-GGGCTCGCACTATATAAGGGATGAC-3′; GAPDH forward: 5′-TGCACCACCAACTGCTTAG-3′ and GAPDH reverse: 5′-GGATGCAGGGATGATGT TC-3′. Quantitative RT-PCR was performed using the following cycle: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 60 s, 60°C for 5 min, followed by a dissociation curve analysis. mRNA expression levels were analyzed using the ΔΔCT method.
ET-1 ELISA.
Production of secreted ET-1 was determined by measuring ET-1 levels with a colorimetric immunometric ELISA kit (Enzo), according to the manufacturer's protocol. Briefly, media from transfected human PASMC (hPASMC) [scrambled nontargeted control siRNA (siNTC) and siRNA specific for human ET-1 (siET-1)] and serum from control and SM22α-ET-1−/− mice were plated in duplicate and incubated for 1 h at room temperature. To measure ET-1 protein production in mPASMC isolated from control and SM22α-ET-1−/− mice, cultured mPASMC (ET-1+/+ and ET-1−/−) were exposed to hypoxia (5% O2) for 24 h and then harvested and lysed. Cell lysate samples were normalized to cell number (representing 250,000 cells/well), plated in duplicate, and incubated for 24 h at 4°C. Optical density was measured at 450 nm, with the concentration of ET-1 in samples calculated from a standard curve of recombinant ET-1.
Hemodynamic assessments.
Adult littermates were used in each group. To measure pulmonary arterial pressures, mice were anesthetized with 1.5–2.0% isofluorane, and right ventricular systolic pressure (RVSP) measurements were obtained using a 1.4 F Millar catheter (Millar Instruments) at baseline (normoxia, 21% O2) and after exposure to chronic hypoxia (10% O2, 3 wk) as previously described (17). Left ventricular (LV) fractional shortening, ejection fraction, aortic valve velocity time integral, and heart rate were evaluated by echocardiography using the GE Vivid 7 ultrasound machine with a 13-MHz probe (GE Healthcare). Blood was collected by direct heart venipuncture and assayed for hemoglobin and hematocrit levels. Fulton's index was determined in both experimental groups after hypoxic exposure.
Morphometric analysis.
Assessment of PA muscularization was performed on formalin-fixed and paraffin-embedded lung sections from mice exposed to chronic hypoxia using Movat pentachrome stain. Peripheral PA wall thickness was assessed by measuring Movat-stained vessels less than 100 μm in diameter in 10 fields/mouse at ×400 magnification using Metamorph software and then compared between the two genotypes using the following equation: medial thickness index = [(areaext − areaint)/areaext], where areaext and areaint represent the areas within the external and internal boundaries of the elastic fibers as detected by Movat stain (8). To quantitate the number of distal PA, mean vessel density (number of barium-filled distal arteries <50 μm in diameter per 100 alveoli) was assessed on barium-injected lungs from mice exposed to chronic hypoxia using hematoxylin and eosin stain with six fields/mouse counted at ×400 magnification.
Cell culture.
In vitro studies were performed with hPASMC and mPASMC isolated from control and transgenic mice with selective deletion of ET-1 in SMC (SM22α-ET-1−/−). The hPASMC were purchased from Lonza and grown according to the manufacturer's protocol. Cells from passages 3–8 were used for all experiments.
Immunohistochemistry.
Formalin-fixed and paraffin-embedded lung sections from mice exposed to chronic hypoxia were deparaffinized and rehydrated, incubated with Universal Antigen Retrieval Reagent (R&D Systems) for 30 min at 95°C, permeabilized with 0.25% Triton X-100 in PBS solution for 30 min, incubated with 100 mM glycine solution (pH 7.5) for 20 min to quench autofluorescence, blocked with Sea Block Blocking Solution (Thermo Scientific) for 30 min, blocked with Mouse Detective Reagent (Biocare Medical) for 30 min to block endogenous mouse IgG, and incubated with proliferating cell nuclear antigen (PCNA) (1:150, Abcam) and α-SMA (1:400, Sigma) antibodies overnight at 4°C. Sections were then incubated with goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 568 (1:200, Invitrogen/Molecular Probes) for 1 h, followed by incubation with 1 μg/ml Hoechst solution (Sigma) to visualize nuclei. The relative number of proliferating SMC was assessed as a percentage of total SMC in PA >100 μm and <250 μm in diameter following exposure to chronic hypoxia.
siRNA transfection.
hPASMC, at 50–70% confluency, were transfected using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Briefly, siET-1 (Thermo Scientific Dharmacon) or siNTC (Thermo Scientific Dharmacon) was transfected at a final concentration of 50 nM. Twenty-four hours posttransfection, cells were re-fed with fresh media. After an additional 24 h, the cells were trypsinized and used for cell proliferation, apoptosis, and cell migration studies.
Cell proliferation assays.
SMC proliferation was determined by measuring DNA synthesis with a colorimetric bromodeoxyuridine (BrdU) ELISA kit (Roche Diagnostics), according to the manufacturer's protocol. Briefly, transfected hPASMC (siET-1 and siNTC) and mPASMC (control and ET-1−/−) were seeded in triplicate in a 96-well plate (4,000 cells/well) and incubated in starvation media (0.2% FBS) overnight. Cells were then incubated with BrdU-labeling reagent under normoxic (21% O2) or hypoxic conditions (5% O2) for 24 h in complete media. Cells were fixed and then incubated with BrdU antibody for 90 min. After being washed with PBS, cells were incubated with tetramethylbenzidine for 10 min. Incorporation of BrdU was detected at absorbance 370 nm.
For the assays using endothelin A and B (ETA and ETB) receptor antagonists, hPASMC proliferation was determined as described above with the following exception: 7.5 μM BQ123 (ETA inhibitor) and 0.1 μM BQ788 (ETB inhibitor) were added 30 min before the addition of the BrdU-labeling reagent.
Apoptosis assays.
SMC apoptosis was determined by measuring caspase 3/7 activities using the Caspase-Glo 3/7 Assay (Promega) according to the manufacturer's protocol. Briefly, 3,500 cells/well were seeded in triplicate in a 96-well plate and incubated overnight. Cells were then exposed to normoxia (21% O2) or hypoxia (5% O2) for 24 h in starvation media (0.1% FBS). Caspase 3/7 activities were measured using a GloMax 96-well plate luminometer after the addition of Caspase Glo 3/7 Reagent. As a control for the induction of apoptosis, cells were treated with 0.5 mM H2O2 solution.
For the assays using ETA and ETB receptor antagonists, hPASMC apoptosis was determined as described above with the following exception: 7.5 μM BQ123 (ETA inhibitor) and 0.1 μM BQ788 (ETB inhibitor) were added 30 min before exposure to normoxia or hypoxia for 24 h.
Cell migration assays.
SMC migration was determined by using a modified Boyden Chamber assay (BD BioCoat Matrigel Invasion Chamber, BD Biosciences). Briefly, Matrigel inserts were rehydrated with media at 37°C for 2 h before use. Media (±10 ng/ml PDGF-BB) was added to each well of a 24-well companion plate. Transfected hPASMC (siET-1 and siNTC) and mPASMC (ET-1+/+ and ET-1−/−) were seeded (20,000 cells/well) in duplicate in the top chamber (Matrigel insert). After 24 h of incubation, the nonmigrated cells in the top chamber were carefully removed, and the migrated cells on the lower surface of the membrane were fixed and stained with Diff-Quik kit reagents (Polysciences).
For the assays using ETA and ETB receptor antagonists, hPASMC cell migration was determined as described above with the following exception: 7.5 μM BQ123 (ETA inhibitor) and 0.1 μM BQ788 (ETB inhibitor) were added to the top chamber before exposure to either normoxia or hypoxia for 24 h.
Statistical analysis.
Results are expressed as means ± SE. Statistical significance was assessed with Student's t-test and ANOVA where appropriate. A P value of <0.05 was taken as the threshold level for statistical significance. All experiments were repeated a minimum of three times unless stated otherwise.
RESULTS
Characterization of SM22α-ET-1−/− mice.
Cre recombinase activity and edn1 gene expression in SM22α-ET-1−/− mice was demonstrated using PCR (Fig. 1A). Lung tissues from SM22α-ET-1 mice were stained with X-gal to demonstrate SM22α-promoter-driven Cre recombinase activity in SMC. As shown in Fig. 1B, right, β-gal expression was present in the PA of SM22α-ET-1−/− mice. In contrast, β-gal expression was undetectable in lung tissues from SM22α-ET-1+/+ mice (control, Fig. 1B, left). Both ET-1 mRNA and protein were significantly decreased in PASMC isolated from SM22α-ET-1−/− mice compared with control mice (Fig. 1, C and D). These results confirm the absence of ET-1 in SM22α-expressing cells in the present murine model. Even under hypoxic conditions, ET-1 expression is minimal.
Fig. 1.

Generation of SM22α-ET-1−/− mice. A: Generation of mice with smooth muscle-specific endothelin-1 (ET-1) deficiency. DNA analysis of Cre recombinase and edn1 genes in SM22α-ET-1 mice. Tail DNA isolated from SM22α-ET-1+/+ (control) and SM22α-ET-1−/− mice was subjected to PCR using Cre-specific primers (left) or wild-type (WT) and mutant edn1-specific primers (right). B: smooth muscle cell (SMC)-specific deletion of ET-1 in SM22α-ET-1−/− mice. Pulmonary artery (PA) tissues from control (left) and SM22α-ET-1−/− mice (right) were stained with X-gal to detect β-gal expression. Magnification ×200. C: loss of ET-1 mRNA in pulmonary artery smooth muscle cells (PASMC) isolated from SM22α-ET-1−/− mice. D: loss of ET-1 protein in PASMC isolated from SM22α-ET-1−/− mice. Murine PASMC (mPASMC) were incubated under hypoxic (5% O2) conditions for 24 h. Cell lysates were then assessed for ET-1 levels by ELISA. ***P < 0.001.
Loss of ET-1 in SMC attenuates the pulmonary vascular response to chronic hypoxia.
To characterize the role of PASMC ET-1 production on the pulmonary vascular response to hypoxia, control and SM22α-ET-1−/− mice were evaluated after hypoxic exposure. At baseline, under normoxic conditions, RVSP was 26.9 ± 1.4 mmHg in control and 25.0 ± 1.2 mmHg in SM22α-ET-1−/− mice. After 3 wk of hypoxia (10% O2), RVSP was 41.5 ± 1.2 mmHg in control mice compared with 35.8 ± 1.3 mmHg in SM22α-ET-1−/− mice (Fig. 2, A and B; P < 0.01 vs. control). Fulton's index was determined as the ratio of right ventricular weight to LV and septal weight. (Fig. 2C). Compared with control mice, peripheral PA muscularization was significantly decreased, and the ratio of arteries to alveoli was more well preserved in SM22α-ET-1−/− mice (Fig. 2, D and E). Both groups demonstrated similar increases in hematocrit and heart rate, decreases in body weight, and well-preserved cardiac function after hypoxic exposure (Table 1). After chronic hypoxia, serum ET-1 levels increased similarly in control and SM22α-ET-1−/− mice, supporting the notion that ET-1 production from endothelial cells is the major contributor to circulating ET-1 levels as opposed to ET-1 derived from SMC (Fig. 2F).
Fig. 2.

ET-1 loss in SMC attenuates the pulmonary vascular response to hypoxia. A: representative right ventricular systolic pressure (RVSP) tracings from individual mice exposed to chronic hypoxia. B: RVSP of SM22α-ET-1−/− mice exposed to chronic hypoxia demonstrates a significant reduction in RVSP compared with control mice. Control and SM22α-ET-1−/− littermates were exposed to normoxic (21% O2) or hypoxic (10% O2) conditions for 3 wk (control, n = 7; SM22α-ET-1−/−, n = 9). **P < 0.01, SM22α-ET-1−/− vs. control. C: right ventricular hypertrophy as assessed by Fulton's index (ratio of right ventricular weight to left ventricular + septal weight) in hypoxic mice (control, n = 4; SM22α-ET-1−/−, n = 5). D: representative Movat pentachrome-stained sections show a decrease in muscularization in peripheral PA from SM22α-ET-1−/− mice (right) exposed to chronic hypoxia. Magnification ×400, scale bar = 25 μm. Quantification of peripheral PA muscularization is expressed as the medial thickness index of pulmonary arteries ≤100 μm in diameter. Graph represents the means ± SE; n = 6 per genotype with 10 fields assessed per mouse. **P < 0.01. E: representative histology of barium-injected lungs of control and SM22α-ET-1−/− mice after chronic hypoxia. Magnification ×100, scale bar = 100 μm. Mean vessel density assessed on barium-injected lungs is significantly increased in the lungs of SM22α-ET-1−/− mice. Graph represents the means ± SE of the number of barium-filled distal PA (≤50 μm in external diameter) per 100 alveoli. n = 4 per genotype with 6 fields assessed per mouse. **P < 0.01. F: chronic hypoxia increases serum ET-1 concentration levels in both control and SM22α-ET-1−/− mice. §P < 0.05, §§P < 0.01, hypoxia vs. normoxia.
Table 1.
Hemodynamic assessments of control and SM22α-ET-1−/− mice
| Normoxia |
Chronic Hypoxia |
|||
|---|---|---|---|---|
| Control | SM22α-ET-1−/− | Control | SM22α-ET-1−/− | |
| Hematocrit, % | 44.2 ± 0.8 (6) | 42.0 ± 2.1 (4) | 54.2 ± 0.6† (3) | 56.3 ± 3.2* (3) |
| Heart rate, beats/min | 367 ± 31 (3) | 373 ± 10.7 (4) | 411 ± 16.7* (6) | 419 ± 9.9* (4) |
| Body weight, g | 28.8 ± 1.3 (12) | 30.9 ± 1.2 (10) | 25.3 ± 2.3* (5) | 27.3 ± 2.3* (5) |
| Left ventricular fractional shortening, % | 30.0 ± 3.7 (3) | 30.0 ± 3.9 (4) | 34.5 ± 0.7 (5) | 30.8 ± 1.9 (4) |
| Aortic valve velocity time integral, cm | 2.45 ± 0.3 (3) | 2.84 ± 0.5 (4) | 2.6 ± 0.3 (5) | 2.35 ± 0.3 (4) |
Values are expressed as means ± SE; numbers of mice per group are in parentheses.
P < 0.05,
P < 0.001, hypoxia vs. normoxia. ET-1, endothelin-1.
In both experimental groups, hemoglobin increased after hypoxic exposure. LV function was preserved in both groups after hypoxia. After hypoxia, Fulton's index did not differ between experimental groups (Fig. 2C).
ET-1 depletion inhibits the hypoxia-induced proliferation in PASMC.
To gain insight into the mechanism by which SMC-derived ET-1 may mitigate the pulmonary vascular response to hypoxia, we considered whether ET-1 plays a role in SMC proliferation. Using PASMC isolated from SM22α-ET-1−/− and control mice, we compared proliferation in ET-1+/+ and ET-1−/− PASMC under normoxic and hypoxic conditions. Under normoxic conditions, loss of ET-1 decreased the rate of PASMC proliferation (Fig. 3A). Furthermore, the hypoxia-induced increase in SMC proliferation was significantly attenuated in ET−/− PASMC compared with ET+/+ PASMC. To demonstrate fidelity between in vitro and in vivo findings, we performed PCNA staining on lung tissues. After chronic hypoxia, the percentage of proliferating PASMC was decreased in SM22α-ET-1−/− compared with control mice (Fig. 3B).
Fig. 3.

Loss of SMC ET-1 inhibits the hypoxia-induced proliferative response. A: loss of ET-1 significantly decreases mPASMC proliferation under both normoxic (21% O2) and hypoxic (5% O2) conditions. Cell proliferation was measured by bromodeoxyuridine (BrdU) incorporation assay after 24 h. ***P < 0.001, ET-1−/− vs. ET-1+/+; §§§P < 0.001, hypoxia vs. normoxia. B: representative images of proliferating cell nuclear antigen (PCNA)-expressing cells show a decrease in SMC proliferation in PA of SM22α-ET-1−/− mice exposed to hypoxia (3 wk, 10% O2) (right). PCNA, green; Hoechst (nuclei), blue; α-smooth muscle actin, red. Magnification ×400, scale bar = 50 μm. Percentage of PASMC ≥100 μm in diameter positive for PCNA expression. Graph represents the means ± SE of the number of PCNA-positive SMC/the total number of SMC per PA; n = 3 for each genotype with a minimum of 500 cells counted per mouse. *P < 0.05. C: ET-1 knockdown with siRNA in human PASMC (hPASMC) as assessed by ELISA. ***P < 0.001, siRNA specific for human ET-1 (siET-1) vs. scrambled nontargeted control siRNA (siNTC). D: loss of ET-1 significantly decreases hPASMC proliferation under both normoxic and hypoxic conditions. hPASMC were transfected with siNTC or siET-1. Cell proliferation was measured after 24 h by BrdU incorporation assay. ***P < 0.001, siET-1 vs. siNTC; §§§P < 0.001, hypoxia vs. normoxia.
To buttress the relevance of the findings from the murine model to human biology, hPASMC ET-1 was silenced in hPASMC using siRNA. As shown in Fig. 3C, ET-1 production was significantly diminished in siET-1-transfected cells (86% less than nontargeted control siRNA, siNTC). Consistent with the cell proliferation results from mPASMC, loss of ET-1 decreased proliferation of hPASMC under basal conditions and attenuated the hypoxia-induced proliferative response (Fig. 3D).
ET-1 depletion increases apoptosis in PASMC.
To determine whether SMC-derived ET-1 plays a role in cell survival as well as cell proliferation, we measured the caspase 3/7 activities in ET−/− PASMC. As shown in Fig. 4A, the loss of ET-1 in mPASMC resulted in a significant increase in apoptosis compared with ET-1+/+ SMC under both normoxic and hypoxic conditions. Similarly, knockdown of ET-1 using siRNA in hPASMC resulted in a significant increase in apoptosis compared with siNTC-transfected SMC (Fig. 4B). These results demonstrate that the loss of SMC-derived ET-1 promotes PASMC apoptosis and suggest that SMC-derived ET-1 contributes to PA remodeling in response to hypoxia by promoting SMC survival.
Fig. 4.
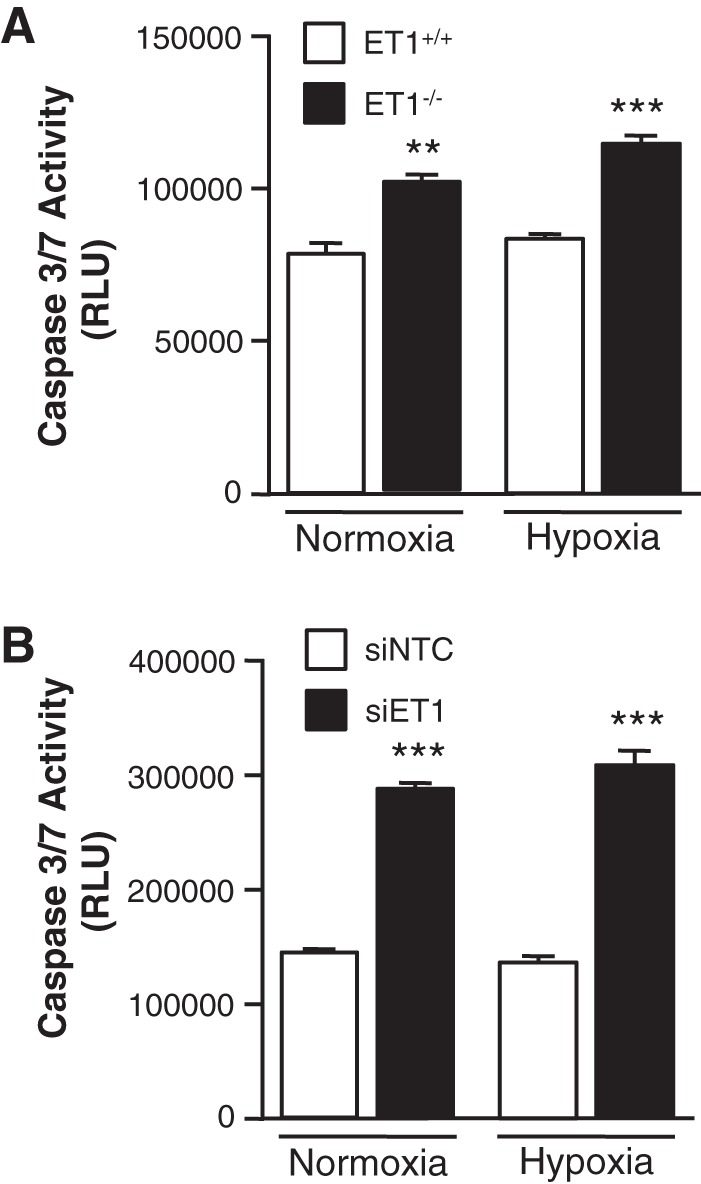
Loss of ET-1 inhibits SMC survival. A: loss of ET-1 significantly increases mPASMC apoptosis under both normoxic and hypoxic conditions. mPASMC apoptosis was measured after 24 h by CaspaseGlo assay to detect caspase 3/7 activities. **P < 0.01, *** P < 0.001, ET-1−/− vs. ET-1+/+. B: loss of ET-1 significantly increases hPASMC apoptosis under both normoxic and hypoxic conditions. hPASMC were transfected with siNTC or siET-1. Apoptosis was measured after 24 h by CaspaseGlo assay to detect caspase 3/7 activities. ***P < 0.001, siET-1 vs. siNTC. RLU, relative light units.
ET-1 depletion inhibits SMC migration in PASMC.
To determine the role of SMC-produced ET-1 in SMC migration, we assessed the effects of ET-1 expression on SMC migration using a modified Boyden Chamber assay. Migration was diminished in ET-1−/− mPASMC compared with ET-1+/+ cells under both normoxic and hypoxic conditions (Fig. 5A). Cell migration was similarly diminished in hPASMC transfected with siET-1 compared with siNTC cells (Fig. 5B). Interestingly, even with PDGF-BB stimulation, a promigratory stimulus for SMC (9, 15), SMC migration was attenuated in ET-1-depleted SMC compared with control cells.
Fig. 5.

Loss of ET-1 inhibits SMC migration. A: mPASMC (ET-1+/+ and ET-1−/−) migration was assessed by modified Boyden Chamber assay. SMC were stimulated with or without 10 ng/ml PDGF-BB for 24 h. Representative images show that the loss of ET-1 (ET1−/−) inhibits SMC migration (right) independent of PDGF-BB stimulation (bottom). Graphs represent the means ± SE of the number of SMC per high-powered field (HPF) from 10 random fields per sample. *P < 0.05, **P < 0.01, ***P < 0.001, ET-1−/− vs. ET-1+/+; §P < 0.05, PDGF-BB vs. untreated. B: hPASMC migration was assessed by modified Boyden Chamber assay. SMC were transfected with siNTC or siET-1 and then stimulated with or without 10 ng/ml PDGF-BB for 24 h. Representative images show that the loss of ET-1 (siET-1) inhibits SMC migration (right) independent of PDGF-BB stimulation (bottom). Graph represents the means ± SE of the number of migrated SMC per HPF from 10 random fields per sample. Magnification ×200. **P < 0.01, ***P < 0.001, siET-1 vs. siNTC; §P < 0.05, §§P < 0.01, PDGF-BB vs. untreated.
Inhibition of ET-1 receptors does not alter SMC proliferation, apoptosis, or migration.
To determine whether the proproliferative effect of SMC ET-1 was contingent on binding to either the ETA or ETB receptors, cell proliferation assays were performed in the presence and absence of specific ET-1 receptor antagonists. Neither BQ123, a specific ETA receptor antagonist, nor BQ788, a specific ETB receptor antagonist (2, 12), had an effect on SMC proliferation under normoxia. Moreover, the hypoxia-induced SMC proliferative response was unaffected by either receptor antagonist (Fig. 6A). To further assess the functional role of SMC ET-1, assays for apoptosis (Fig. 6B) and cell migration (Fig. 6C) were performed in the presence and absence of selective ETA or ETB receptor antagonism. Pharmacological antagonism of either receptor had no effect on either apoptosis or proliferation.
Fig. 6.

ET-1 receptor blockade does not alter the SMC hypoxia-induced proliferative, apoptotic, or migratory responses. A: in hPASMC, hypoxia (5% O2) for 24 h increased proliferation. Inhibition of either the endothelin A or endothelin B (ETA or ETB) receptors had no effect on the proliferative response under normoxic conditions. Moreover, the hypoxia-induced increase in proliferation was not altered by pharmacological blockade of either the ETA receptor with BQ123 or the ETB receptor with BQ788. Cell proliferation was measured (n = 3; for each experimental condition) by BrdU incorporation assay after 24 h. **P < 0.01, hypoxia vs. normoxia. B: ET receptor blockade does not alter hPASMC apoptosis under both normoxic and hypoxic conditions. hPASMC apoptosis was measured after 24 h by CaspaseGlo assay to detect caspase 3/7 activities. Graph represents the means ± SE (n = 6; for each experimental condition) and is represented as a percentage of each respective control (control = 100%). C: ET receptor blockade does not alter hPASMC migration as assessed by modified Boyden Chamber assay. SMC were stimulated with or without 10 ng/ml PDGF-BB for 24 h. Graph represents the means ± SE of the number of SMC per HPF from 5 random fields per sample (n = 3; for each experimental condition). §§§P < 0.001, PDGF-BB vs. untreated.
DISCUSSION
The present series of experiments provides evidence for a previously undescribed role for smooth muscle-derived ET-1 in the pathogenesis of PAH. Compared with controls, RVSP in SM22α-ET-1−/− mice was similar under normoxic conditions but significantly lower after chronic hypoxia. Consistent with these findings, after chronic hypoxia, vascularity was more well preserved and muscularization diminished in SM22α-ET-1−/− mice. To address the potential physiological underpinnings, we evaluated the effect of ET-1 depletion on SMC proliferation, apoptosis, and migration. Loss of ET-1 in SMC compromised both cell survival and migration, suggesting that SMC ET-1 might contribute to pulmonary vascular remodeling by augmenting proliferation and migration, while mitigating apoptosis. Finally, by demonstrating that SMC proliferation, in both hypoxia and normoxia, was not altered by ET-1 receptor blockade, we provide evidence that SMC-derived ET-1 may act independently of ET-1 receptor activation. This observation may, in part, account for the limited therapeutic benefit of ET-1 receptor blockade in the context of PAH characterized by minimal vascular reactivity and structural remodeling (22).
Although ET-1 production by SMC has been previously reported (29, 30), this is the first to demonstrate a physiologically significant role for SMC-derived ET-1 in the pathogenesis of PAH. Whether there is a physiological role for PASMC-derived ET-1 remains unknown. In the present experimental series, ET-1 derived from PASMC modulated proliferation, apoptosis, and migration, even under normoxic conditions. Despite these functional differences, RVSP was similar between the two study groups under normoxic conditions. Differences might become evident in the context of physiological perturbations other than hypoxia, as might be the case in asthma or pneumonia. Further studies are needed to determine the functional significance of PASMC-derived ET-1 under physiological conditions.
Previous studies have demonstrated a role for ET-1 produced by EC in the regulation of peripheral vascular tone (20). Systemic blood pressure is lower in mice with EC-specific deletion of ET-1 with correspondingly lower levels of circulating ET-1. In the present study, under normoxia, there was no difference in RVSP between control and SM22α-ET-1−/− mice. Similarly, after hypoxic exposure for 3 wk, there was no difference in circulating levels of ET-1 between controls and SM22α-ET-1−/− animals. The similarity in circulating levels of ET-1 between genotypes buttresses the notion that ET-1 produced by SMC possesses physiologically significant local effects. Although there was no difference in Fulton's index between the two groups despite divergent RVSP levels after 21 days of hypoxia, right ventricular remodeling might become more apparent only after more sustained hypoxic exposure.
Further support for this construct comes from in vitro experiments. Loss of ET-1 in SMC produced similar results in murine PASMC, wherein ET-1 was never expressed, and in hPASMC, wherein ET-1 expression was silenced using RNA interference. With either approach, PASMC proliferation, migration, and apoptosis were affected, thereby establishing a proproliferative, promigratory, and prosurvival role for SMC-derived ET-1 in the pulmonary circulation. Experiments performed with pharmacological blockers of the ETA and ETB receptors suggest that the effects of ET-1 produced by SMC are not contingent on receptor activation, as antagonism did not affect, whereas ET-1 depletion blocked, the hypoxia-induced increase in proliferation. These results prompt consideration of the notion that ET-1 might play an intracellular role or signal SMC via non-ETA or -ETB receptors.
Overall, although these results are consistent with prior studies detailing ET-1 production from SMC (10, 29, 30), this report points to a prosurvival role for SMC ET-1, which is particularly pronounced in the setting of hypoxia. ET-1 affects SMC consistently irrespective of cellular source, as exogenous ET-1 is proproliferative (41) and promigratory (23) and prevents apoptosis (14). Hypoxia increases ET-1 production (35, 39). The present report adds to these observations by demonstrating that endogenous ET-1 modulates SMC cellular functions that are central to the pathogenesis of pulmonary hypertension. Notwithstanding these observations, how endogenous ET-1 affects SMC function and the signal transduction pathway remains unknown. Endogenous SMC ET-1 may affect migration via ERK 1/2 MAP kinases, as has been previously reported (23). Conclusions that can be drawn from the present series of experiments are limited by the discrete focus. In PASMC, there may be crosstalk between ET-1 and hypoxia-inducible factor-1α, a construct that is unaddressed in the present line of investigation (27). Furthermore, the influence, if any, of endogenous ET-1 on intracellular calcium homeostasis is similarly unaddressed.
The present findings possess important implications for ET-1 signaling in SMC. Even in the presence of pharmacological blockade of ETA and ETB receptors, hypoxia increased SMC proliferation, apoptosis, and migration. These results suggest that ET-1 may possess intracellular effect and may signal through an as yet unknown intracellular route as well as via the well-established and classical membrane-bound receptors. Evidence in support of this construct includes the observation that circulating endothelin levels may not be wholly reflective of tissue levels of endothelin (2, 10, 19, 29, 38). Hence the classical paradigm, namely that endothelin produced by endothelial cells signals predominately through membrane-bound receptors on SMC, may be meaningfully incomplete in a manner that possesses significant physiological implications.
The present findings possess important clinical implications. Although ET-1 receptor blockade is relatively efficacious in less severe PAH, the therapeutic benefit is diminished in patients with more severe disease, structural remodeling, and loss of pulmonary vascular reactivity (33). Whether ET-1 produced by SMC accounts for the constrained therapeutic benefit is unknown. Conceivably, in the presence of already established SMC hypertrophy, ET-1 receptor antagonists might possess limited therapeutic effect owing to SMC-derived ET-1.
Using a murine model of SMC-specific ET-1 deletion, we demonstrate a physiologically significant role for endogenous ET-1 in the pathogenesis of PAH. ET-1 in SMC plays a role in cellular proliferation, survival, and migration, functions that are central to the vascular changes that characterize PAH. These results suggest that in PAH the therapeutic efficacy of ET-1 receptor antagonists might be limited in the presence of marked pulmonary vascular remodeling. Moreover, optimizing the therapeutic benefits of ET-1 receptor antagonists might require a strategy wherein SMC-derived ET-1 production might be limited.
GRANTS
This work was supported by funding from the National Institutes of Health HL060784 and HL0706280 (D. Cornfield).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: F.Y.K., C.M.A., and D.N.C. conception and design of research; F.Y.K., E.A.B., L.Y., C.C., and L.L. performed experiments; F.Y.K., E.A.B., L.Y., L.L., and D.N.C. analyzed data; F.Y.K., L.Y., C.M.A., and D.N.C. interpreted results of experiments; F.Y.K., E.A.B., and D.N.C. drafted manuscript; F.Y.K., E.A.B., L.Y., C.C., L.L., C.M.A., and D.N.C. approved final version of manuscript; E.A.B., C.C., and L.L. prepared Figs.; C.M.A. and D.N.C. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank M. Rabinovitch for the SM22α-promoter-driven Cre/ROSA26 reporter mice and M. Yanigisawa for the ET-1 flox/flox mice.
REFERENCES
- 1.Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 122: 164–172, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Bonvallet ST, Zamora MR, Hasunuma K, Sato K, Hanasato N, Anderson D, Sato K, Stelzner TJ. BQ123, an ETA-receptor antagonist, attenuates hypoxic pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol 266: H1327–H1331, 1994. [DOI] [PubMed] [Google Scholar]
- 3.Cassin S, Dawes GS, Mott JC, Ross BB, Strang LB. The vascular resistance of the fetal and newly ventilated lung of the lamb. J Physiol 171: 61–79, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassin S, Dawes GS, Ross BB. Pulmonary blood flow and vascular resistance in immature fetal lambs. J Physiol 171: 80–89, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornfield DN, Stevens T, McMurtry IF, Abman SH, Rodman DM. Acute hypoxia increases cytosolic calcium in fetal pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 265: L53–L56, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Dawes GS, Mott JC, Widdicombe JG, Wyatt DG. Changes in the lungs of the newborn lamb. J Physiol 121: 141–162, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furchgott RF. Studies on relaxation of rabbit aorta by sodium nitrite: The basis for the proposal that the acid-activatable inhibitory factor from bovine retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide. In: Vasodilatation, edited by Vanhoutte PM. New York, NY: Raven, 1988, p. 410–414. [Google Scholar]
- 8.Girgis RE, Mozammel S, Champion HC, Li D, Peng X, Shimoda L, Tuder RM, Johns RA, Hassoun PM. Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Lung Cell Mol Physiol 292: L1105–L1110, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Grotendorst GR, Seppa HE, Kleinman HK, Martin GR. Attachment of smooth muscle cells to collagen and their migration toward platelet-derived growth factor. Proc Natl Acad Sci USA 78: 3669–3672, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hahn AW, Resink TJ, Scott-Burden T, Powell J, Dohi Y, Buhler FR. Stimulation of endothelin mRNA and secretion in rat vascular smooth muscle cells: A novel autocrine function. Cell Regul 1: 649–659, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 84: 9265–9269, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivy DD, Kinsella JP, Abman SH. Physiologic characterization of endothelin A and B receptor activity in the ovine fetal pulmonary circulation. J Clin Invest 93: 2141–2148, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivy DD, Rosenzweig EB, Lemarie JC, Brand M, Rosenberg D, Barst RJ. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real-world clinical settings. Am J Cardiol 106: 1332–1338, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jankov RP, Kantores C, Belcastro R, Yi M, Tanswell AK. Endothelin-1 inhibits apoptosis of pulmonary arterial smooth muscle in the neonatal rat. Pediatr Res 60: 245–251, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest 89: 507–511, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest 123: 344–350, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Kim YM, Barnes EA, Alvira CM, Ying L, Reddy S, Cornfield DN. Hypoxia-inducible factor-1alpha in pulmonary artery smooth muscle cells lowers vascular tone by decreasing Myosin light chain phosphorylation. Circ Res 112: 1230–1233, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YM, Haghighat L, Spiekerkoetter E, Sawada H, Alvira CM, Wang L, Acharya S, Rodriguez-Colon G, Orton A, Zhao M, Rabinovitch M. Neutrophil elastase is produced by pulmonary artery smooth muscle cells and is linked to neointimal lesions. Am J Pathol 179: 1560–1572, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ. The endothelin system as a therapeutic target in cardiovascular disease: Great expectations or bleak house? Br J Pharmacol 153: 1105–1119, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, Kedzierski RM, Hammer RE, Yanagisawa H, Williams SC, Richardson JA, Suzuki T, Yanagisawa M. Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension 56: 121–128, 2010. [DOI] [PubMed] [Google Scholar]
- 21.Madden JA, Vadula Ms, Kurup VP. Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 263: L384–L393, 1992. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J, American College of Cardiology Foundation Task Force on Expert Consensus Documents, American Heart Association, American College of Chest Physicians, American Thoracic Society, Pulmonary Hypertension Association. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc, and the Pulmonary Hypertension Association. J Am Coll Cardiol 53: 1573–1619, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Meoli DF, White RJ. Endothelin-1 induces pulmonary but not aortic smooth muscle cell migration by activating ERK1/2 MAP kinase. Can J Physiol Pharmacol 88: 830–839, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Moncada S, Higgs A. The L-arginine-nitric oxide pathway. N Engl J Med 329: 2002–2012, 1993. [DOI] [PubMed] [Google Scholar]
- 25.Morin FC 3rd, Stenmark KR. Persistent pulmonary hypertension of the newborn. Am J Respir Crit Care Med 151: 2010–2032, 1995. [DOI] [PubMed] [Google Scholar]
- 26.Mourani PM, Abman SH. Pulmonary vascular disease in bronchopulmonary dysplasia: Pulmonary hypertension and beyond. Curr Opin Pediatr 25: 329–337, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 304: L549–L561, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Resink TJ, Scott-Burden T, Boulanger C, Weber E, Buhler FR. Internalization of endothelin by cultured human vascular smooth muscle cells: characterization and physiological significance. Mol Pharmacol 38: 244–252, 1990. [PubMed] [Google Scholar]
- 30.Resink TJ, Scott-Burden T, Buhler FR. Activation of multiple signal transduction pathways by endothelin in cultured human vascular smooth muscle cells. Eur J Biochem 189: 415–421, 1990. [DOI] [PubMed] [Google Scholar]
- 31.Rubanyi GM, Polokoff MA. Endothelins: Molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev 46: 325–415, 1994. [PubMed] [Google Scholar]
- 32.Rubin LJ. Primary pulmonary hypertension. N Engl J Med 336: 111–117, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 346: 896–903, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem 274: 38071–38082, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Shimoda LA, Sylvester JT, Sham JS. Chronic hypoxia alters effects of endothelin and angiotensin on K+ currents in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 277: L431–L439, 1999. [DOI] [PubMed] [Google Scholar]
- 36.Stelzner TJ, O'Brien RF, Yanagisawa M, Sakurai T, Sato K, Webb S, Zamora M, McMurtry IF, Fisher JH. Increased lung endothelin-1 production in rats with idiopathic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 262: L614–L620, 1992. [DOI] [PubMed] [Google Scholar]
- 37.Wort SJ, Woods M, Warner TD, Evans TW, Mitchell JA. Cyclooxygenase-2 acts as an endogenous brake on endothelin-1 release by human pulmonary artery smooth muscle cells: implications for pulmonary hypertension. Mol Pharmacol 62: 1147–1153, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Wort SJ, Woods M, Warner TD, Evans TW, Mitchell JA. Endogenously released endothelin-1 from human pulmonary artery smooth muscle promotes cellular proliferation: Relevance to pathogenesis of pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol 25: 104–110, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia: Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2 and p300/CBP. J Biol Chem 276: 12645–12653, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415, 1988. [DOI] [PubMed] [Google Scholar]
- 41.Zamora MR, Stelzner TJ, Webb S, Panos RJ, Ruff LJ, Dempsey EC. Overexpression of endothelin-1 and enhanced growth of pulmonary artery smooth muscle cells from fawn-hooded rats. Am J Physiol Lung Cell Mol Physiol 270: L101–L109, 1996. [DOI] [PubMed] [Google Scholar]