

Abstract
Polyunsaturated fatty acid (PUFA) intake has increased over the last 100 yr, contributing to the current obesogenic environment. Obesity and aging are prominent risk factors for myocardial infarction (MI). How obesity interacts with aging to alter the post-MI response, however, is unclear. We tested the hypothesis that obesity in aging mice would impair the resolution of post-MI inflammation. PUFA diet (PUFA aging group) feeding to 12-mo-old C57BL/6J mice for 5 mo showed higher fat mass compared with standard lab chow (LC)-fed young (LC young group; 3–5 mo old) or aging alone control mice (LC aging group). LC young, LC aging, and PUFA aging mice were subjected to coronary artery ligation to induce MI. Despite similar infarct areas post-MI, plasma proteomic profiling revealed higher VCAM-1 in the PUFA aging group compared with LC young and LC aging groups, leading to increased neutrophil infiltration in the PUFA aging group (P < 0.05). Macrophage inflammatory protein-1γ and CD40 were also increased at day 1, and myeloperoxidase remained elevated at day 5, an observation consistent with delayed wound healing in the PUFA aging group. Lipidomic analysis showed higher levels of arachidonic acid and 12(S)-hydroxyeicosatetraenoic acid at day 1 post-MI in the PUFA aging group compared with the LC aging group (all P < 0.05), thereby mediating neutrophil extravasation in the PUFA aging group. The inflammation-resolving enzymes 5-lipoxygenase, cyclooxygenase-2, and heme oxyegnase-1 were altered to delay wound healing post-MI in the PUFA aging group compared with LC young and LC aging groups. PUFA aging magnifies the post-MI inflammatory response and impairs the healing response by stimulating prolonged neutrophil trafficking and proinflammatory lipid mediators.
Keywords: inflammation, lipid mediators, proteomics, lipidomics, neutrophils, polyunsaturated fatty acids
after myocardial infarction (MI), a poorly controlled age-associated inflammation primes the left ventricle (LV) for the development of congestive heart failure (24, 36). Moreover, body mass index alone is a dominant risk factor that increases the risk of MI and ischemic heart disease in overweight and obese subjects (43). Cardiovascular disease is responsible for 30% of deaths worldwide, exceeding all other diseases, including cancer (31). The global economy costs $863 billion each year for the treatment of cardiovascular disease (4). Sedentary lifestyle and obesogenic diet increase the risk of cardiovascular disease, particularly pleiotropic actions of obesity and insulin resistance-mediated inflammation alter the metabolic sequelae (32). To control unresolved inflammation post-MI and to reduce heart failure rates, the identification of novel targets for the treatment of post-MI remodeling is needed to treat elderly heart failure patients.
A previous report by Stanley et al. (42) suggested that the source of the fat [either from polyunsaturated fatty acid (PUFA) (n-6) or saturated fat] impacts inflammation, healing, and the post-MI progression to heart failure. Surprisingly, the heart failure rate was reduced in saturated fat-fed hamsters compared with high n-6 fatty acid (linoleate)-fed hamsters in the male δ-sarcoglycan-null cardiomyopathic heart failure model (14), whereas pre- or post-MI saturated fat diet intake did not worsen the LV remodeling outcome in young rodents. At present, there is no consensus on the effects of n-6 fatty acids or saturated fat intake in the post-MI setting. Progressive heart failure is commonly observed in the aging population post-MI, and obesity itself remodels the LV and predisposes the heart to failure. The prevalence of heart failure increases with age, in part due to increased incidences of obesity (39, 40).
The increased intake of n-6 enriched fatty acids leads to insulin resistance and hyperglycemia (5, 8). Our previous studies (17, 19, 20) showed that chronic intake of n-6 fatty acids reduces the lifespan of lupus-prone mice, increases bone marrow adiposity, and alters the bone marrow microenvironment in obese and aging mice. Of note, both ω-3 and ω-6 fatty acids are essential for cell growth and signaling and cell membrane dynamics and integrity, with optimal ratios occurring at 1:1 or 2:1. In the setting of obesity, this ratio can reach as high as 30:1 (10).
The present study was designed to answer the following fundamental question: “How does the LV respond to post-MI remodeling in the setting of obesity and aging?” Several groups have used young rodent models of morbid obesity, using 45% or 60% kcal high-fat diets. In actuality, ∼3% of the United States population is morbidly obese (with body mass index ≥ 40), and the number of overweight or obese Americans (with body mass index ≥ 25) is 69% (25, 33). Therefore, models that show moderate rather than gigantic increases in body weight would be more closely applicable to the obese and overweight population. To mimic this adult overweight or obese population, we selected a model of obesity that uses 22% kcal from a safflower oil-enriched diet.
Our study rationales were as follows: first, MI and heart failure are quite frequently studied in young or adult healthy mice subjected to coronary ligation; however, young and healthy humans rarely develop MI. For this reason, a model of obesity in the setting of aging was used in this study. Second, although n-6 fatty acids are considered safe, a recent meta-analysis (35) indicated that linoleic acid-enriched n-6 fatty acids increase the risk of death from coronary artery disease and cardiovascular disease. Finally, an epidemiological study (2) has indicates that n-6 fatty acid consumption has dramatically increased in the 20th century, significantly contributing to increased energy intake and an obesogenic environment. Thus, a linoleic acid-enriched n-6 PUFA was used to induce obesity in aging mice. Our results indicate that n-6 PUFA-induced obesity drove the post-MI acute inflammatory response in aging. These findings underscore a delayed post-MI resolution of inflammation in obese and aging mice.
MATERIALS AND METHODS
Mice.
All animal procedures were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th ed., 2011) and were approved by the Institutional Animal Care and Use Committees of the University of Texas Health Science Center (San Antonio, TX) and University of Alabama at Birmingham (Birmingham, AL). Male C57BL/6J mice of 9 mo of age were acquired from the National Institute of Aging colony and maintained on a standard diet for 3 mo. At 12 mo of age, mice began a diet supplemented with n-6 PUFA [10% (wt/wt)] to induce obesity, and they continued on this diet for 5 mo (PUFA aging group). A group of standard lab chow (LC) diet-fed mice was maintained as age-matched controls (LC aging group). Diet composition details are shown in Table 1 for every 100 g of diet. The young mouse group (3–5 mo old, n = 30) was maintained for the comparative analysis of body weight, fat mass, and post-MI echocardiographic measurements (LC young group). Both LC aging (n = 21) and PUFA aging (n = 21) mice were subjected to coronary ligation at 17 mo of age, whereas LC young (n = 30) mice underwent surgery at 3–5 mo of age, as previously described (18), and were evaluated at days 1 or 5 post-MI (Fig. 1A, study design). Due to the high perioperative mortality attributed to aging, we collected samples from 16 LC aging and 13 PUFA aging mice, whereas 24 LC young mice with no MI were used as the respective group (day 0 control group). The MI surgery procedure was minimally invasive; we did not cut and cauterize the ribs and did not open the chest along its full length. Because of this difference from earlier surgery procedures, the need for a sham control group was replaced with the need for a time 0 control group. Sample sizes for analyses were as follows: day 1 (n = 4–9 mice/group), day 5 (n = 6–9 mice/group), and day 0 (n = 3–7 mice/group) in the LC young, LC aging, and PUFA aging groups. In brief, mice were anesthetized with 2% isoflurane, and the left anterior descending coronary artery was permanently ligated using minimally invasive surgery. To reduce post-MI surgical pain, buprenorphine (0.1 mg/kg ip) was given immediately after the ligation (22).
Table 1.
Diet composition with a major emphasis on fatty acids
| LC Young Group | LC Aging Group | PUFA Aging Group | |
|---|---|---|---|
| Dietary ingredients, g% | |||
| Protein | 14 | 14 | 15 |
| Carbohydrate | 73 | 73 | 66 |
| Fat | 4 | 4 | 10 |
| ω-3 Fatty acids | 0.011 | 0.011 | 0.015 |
| ω-6 Fatty acids | 3.1 | 3.1 | 6.63 |
| ω-9 Fatty acids | 0.9 | 0.9 | 1.34 |
| kcal/g | 3.8 | 3.8 | 4.2 |
Fatty acid composition in the diets is shown, including protein, carbohydrates, and fat, which were enriched with polyunsaturated fatty acids (PUFAs; ω-6 fatty acids) and the minimum amount of ω-3 fatty acids. The following group of mice are shown: lab control diet-fed young (LC young), lab control diet-fed aging (LC aging group), and PUFA diet-fed aging (PUFA aging group) C57BL/6J mice.
Fig. 1.

A: study design illustrating the three groups and post-myocardial infarction (post-MI) time points in young and aging mice. The group of mice are shown: lab control diet-fed young (LC young; 3–5 mo, n = 30), lab control diet-fed aging (LC aging group; 17 mo, n = 21), and polyunsaturated fatty acid (PUFA) diet-fed aging (PUFA aging group; 17 mo, n = 21) C57BL/6J mice. B: age-associated changes in the body weight of LC young, LC aging, and PUFA aging C57BL/6J mice. PUFA [10% (wt/wt)] supplementation increased body weight and fat mass in aging mice independent of lean mass. B–E: body weights (B), fat mass (C), lean mass (D), fasting glucose (5 h; E) in LC young, LC aging, and PUFA aging mice. Numbers of mice per group are shown in columns. *P < 0.05 vs. the LC young group; ¥P < 0.05 vs. LC young and LC aging groups (analyzed by ANOVA).
Measurements of fat and lean mass using quantitative MRI.
LC young, LC aging, and PUFA aging mice were subjected to the whole body composition measurements using quantitative MRI (QMRI instrument, Echo Medical System). This equipment uses nuclear magnetic resonance to measure the physical state of lean and fat mass; thus, quantitative MRI provides an accurate measurement of total body fat, lean mass, and free water (18).
Echocardiographic measurements.
For the echocardiography analysis, 0.8–1.0% isoflurane in an oxygen mix was used to anesthetize mice. Electrocardiograms and heart rates were monitored using a surface electrocardiogram. Images were acquired using the in vivo imaging system (Vevo 2100 high-resolution system equipped with MS-400 transducer, 30MHz, Visual Sonics) at heart rates of >400 beats/min to achieve physiologically relevant measurements. Before the acquisition of images, each mouse was allowed to rest for 5–7 min on the echocardiographic platform. Measurements were taken from two-dimensional parasternal long-axis and short-axis (M-mode) recordings from the midpapillary region. Echocardiographic experiments were performed before euthanization at day 0 in LC young, LC aging, and PUFA aging groups and at days 1 or 5 post-MI. An independent analyzer blinded to the groups measured three images from consecutive cardiac cycles and averaged the results (22).
Necropsy of day 0 control and post-MI surviving mice.
Mice were anaesthetized using 2% isoflurane, and heparin was injected (4 IU/g body wt ip). Blood was collected from the carotid artery and centrifuged for 5 min to collect plasma. Plasma aliquots were stored in −80°C for plasma analysis. The LV was perfused with cardioplegic solution to arrest hearts in diastole and then cut into three sections. The remote region [LV control (LVC)] and infarct region [LV infarct (LVI)] were separated from apex and base sections and used for gene expression measurements and immunoblot analysis, whereas the midcavity LV section was fixed in 10% formalin and paraffin embedded for histology and immunohistochemistry. As a naïve control, the midcavity was processed similar to post-MI samples, and the remaining LV was frozen as the LVC region and processed for gene expression and immunoblot measurements.
Plasma proteomic profiling.
Heparinized plasma was used for the measurement of 59 analytes in Rodent MAP (version 3.0), available from a Clinical Laboratory Improvement Amendments certified biomarker testing laboratory (Myriad RBM). This laboratory used multiplexed immunoassay-based technology that allowed us to get reproducible and quantitative measurements of 59 analytes in an 80-μl sample. Analyte levels below the limit of detection were excluded from the results (18).
LV RNA isolation, reverse transcription, and real-time PCR of an inflammatory gene array.
Postnecropsy, frozen samples of LVC or LVI regions were processed separately for RNA extraction. A total of 4–8 mg of LV tissue was homogenized with a sonic dismembrator (Fisher Scientific, at amplitude between 10 and 100 Hz) in 500 μl TRIzol (Invitrogen). The PureLink RNA isolation kit (Invitrogen) was used to isolate RNA. Before cDNA synthesis, the RNA concentration was determined using ND1000 Nanodrop (Thermo Scientific). The RT2 first-strand kit (Qiagen) was used to synthesis cDNA. Each sample was prepared on a RT2-PCR plate (Inflammatory Cytokine and Receptor, Qiagen PAMM-011E) and run on an ABI 7900HT. Threshold cycle (Ct) values were converted into 2−ΔCt values and normalized to hypoxanthine phosphoribosyltransferase 1 as a control.
Mass spectrometric analysis of plasma lipid mediators.
Plasma lipids were extracted and analyzed by liquid chromotography tandem mass spectroscopy (LC-MS/MS) using an AB/Sciex API-4000 Q TRAP mass spectrometer. Aliquots of plasma (100 μl) were acidified by the addition of 850 μl of 6 N HCl (pH 2.3), vortexed briefly, and incubated on ice for 15 min. Deuterated standards for each measured hydroxyloctadecadienoic (HODE) and hydroperoxyeicosatetraenoic acid (HETE; 20 ng of each, Cayman Chemical) were spiked into samples before extraction. Samples were subsequently added to solid phase extraction columns (Oasis HLB 30 mg 1 ml) while using a slow vacuum, so that each sample took ∼2 min to pass through. After solid phase extraction columns were washed with 5% methanol, lipids were eluted with 1 ml of 100% methanol, dried under argon, and resuspended in 100 μl of 80% methanol. Samples (50 μl) were injected onto a Luna C18(2)-HST reverse-phase LC column (2 × 100 mm, 2.5 μl, Phenomenex) using a Shimadzu auto-sampler with gradient elution at 0.3 ml/min (mobile phase A: 0.1% formic acid in H2O and mobile phase B: 0.1% formic acid in acetonitrile). Gradient elution was as follows: from 0 to 2 min, hold at 50% B; from 2 to 3 min, increase to 60% B; from 3 to 15 min, increase to 65% B; from 15 to 17 min, hold at 65% B; from 17 to 19 min, increase to 100% B; from 19 to 21 min, hold at 100% B; and from 21 to 23 min, decrease back to 50% B and hold for the system to equilibrate to initial conditions until 27 min. A standard curve (0.1, 1, 2.5, 5, 7.5, 10, 25, 50, 75, 100, 250, 500, 750, and 1,000 ng/ml) of a mixture of unlabeled HODEs and HETEs (13-HODE, 9-HODE, 13-HpODE, 5-HETE, 15-HETE, 20-HETE, and 12-HETE) were also run for quantification of individual HODE/HETE species. Each point of this standard curve was spiked with deuterated HODE/HETE standards exactly as described above for samples (20 ng of each deuterated HODE/HETE per standard curve point). The transitions monitored were the following mass-to-charge ratios: 295.1→194.8 for 13-HODE, 295.0→171.0 for 9-HODE, 319.1→115.0 for 5-HETE, 319.1→219.0 for 15-HETE, 319.1→179.0 for 12-HETE, 299.0→197.9 for 13(S)-HODE-d4, 299.1→172.0 for 9(S)-HODE-d4, 327.1→116.0 for 5(S)-HETE-d8, 327.1→226.1 for 15(S)-HETE-d8, and 327.1→184.0 for 12(S)-HETE-d8.
Immunohistochemistry.
LV midcavity sections were fixed, paraffin embedded, and sectioned at 5 μm. Unstained sections were deparaffinized in citrisolv (Thermo Fisher Scientific), rehydrated through graded ethanol, and subsequently exposed to antigen epitopes (Target Retrieval Solution, Dako S1699). Furthermore, sections were blocked with normal rabbit antibody and incubated with rat anti-mouse neutrophil antibody (CL 8993AP, clone 7 1:50, Cedarlane). The Vectastain Elite ABC kit (Vector) was used to stain positive cells. Slides were mounted using permount, and five to six images were acquired for each slide. The percent stained area in the infarct region was quantified.
LV protein extraction for immunoblot analysis.
Each LVC or LVI sample was weighed and placed into a 1.5-ml tube. For every milligram of tissue, 16 μl of 1× PBS (without calcium, Invitrogen) and with 1× proteinase inhibitor (Roche Diagnostics) were added to the tube. On ice, samples were dismembrated in short (5 s) intervals with up to 100 A using a sonic dismembrator. Dismembration continued until the sample was completely homogenous. Sets of eight samples were completed at a time to prevent samples from overheating. Samples were centrifuged at the maximum speed (14,000 rpm) for 5 min at 4°C. The supernatant was transferred to a fresh tube and snap frozen in liquid nitrogen. This was used as the soluble protein fraction. For the insoluble pellet, the pellet was washed three times with PBS and centrifuged at maximum speed (5 min) after each wash. Using 16 μl for every milligram or original tissue, reagent B (Reagent 4 from Sigma with 1× PI) was added to the tube containing the pellet, and the pellet was dismembrated using the same method as the A fraction. The new homogenous solution was snap frozen and used as the insoluble protein fraction. Total protein was determined using the Bradford assay, with the insoluble fraction diluted 1:40 to account for the high urea concentration.
LV protein immunoblot analysis.
Immunoblot analysis was used to quantify the protein levels of homogenized LV samples. Each immunoblot was run on a criterion XT bis-Tris 4–12% 18-lane gel in either MES buffer or MOPS buffer (Bio-Rad) depending on the molecular weight of the protein. The Kaleidoscope precision plus standard (Bio-Rad) was used to determine the molecular weight of the protein. Samples were prepared with water, 5× loading buffer, and 10 μg protein. All samples were denatured by incubation for 5 min at 95°C. Once samples were loaded, the gel was run for 10 min at 80 V and then 150 V until completion. The protein was transferred to a nitrocellulose membrane (Bio-Rad) at 65 V for 1 h. Total protein was stained using reversible protein stain (Pierce). After a rinse with water, the membrane was blocked at room temperature for 1 h using 5% nonfat milk powder (Bio-Rad) and probed with primary antibodies [cyclooxygenase (COX)-2, 1:1,000; 5-lipoxygenase (5-LOX), 1:500; and heme oxygenase (HO)-1, 1:1,000] overnight at 4°C. The membrane was placed in horseradish peroxidase-conjugated secondary antibody solution in 5% milk for 1 h at room temperature. Using pico or femto substrate (Thermo Scientific), the membrane was incubated for 5 min at room temperature and exposed to Hawkins X-ray films. Densitometry was performed using ImageJ software, and each immunoblot was normalized to the respective total protein amount (in arbitrary units) for that lane.
Statistical analysis.
Data are expressed as means ± SE. Statistical analyses were performed using Graphpad Prism 5. ANOVA followed by a Student Newman-Keuls post hoc test was used for multiple comparisons of LC young, LC aging, and PUFA aging groups for day 1 or 5 post-MI samples compared with day 0. Two-way analyses were applied to determine the effects of obesity and aging on post-MI LV remodeling followed by a Bonferroni posttest. For two-group comparisons, Student's t-test (unpaired) was applied. P values of <0.05 were considered significant.
RESULTS
Excessive PUFA intake in aging led to obesity but did not alter LV function pre-MI.
To determine the impact of obesity and aging on LV function, control C57BL/6J mice were maintained for 17 mo on a standard diet (LC aging group) and developed age-associated obesity, which was indicated by a 25% increase in body weight compared with the LC young group (3- to 5-mo-old mice, P < 0.05). The linoleic acid-enriched PUFA aging group showed a 41% increase in body weight compared with the LC young group and a 12% increase in body weight compared with the LC aging group (all P < 0.05; Fig. 1B). As expected, PUFA aging mice showed a 172% increase in fat mass compared with LC young mice (P < 0.05). PUFA aging mice showed a 43% increase in fat mass compared with age-matched LC aging mice, indicating a significant contribution of the PUFA diet to fat mass (P < 0.05; Fig. 1C). In humans, a body mass index of 24.9 is normal and 30.0 is considered obese, indicating a 20.5% increase from normal to obese. Based on these criteria, LC aging mice and PUFA aging mice can both be considered obese models. Fat mass increased 89% in LC aging mice compared with LC young mice (P < 0.05). Lean mass remained unchanged between LC young, LC aging, and PUFA aging mice (all P = not significant; Fig. 1D). Fasting 5-h glucose levels were increased in PUFA aging mice, who developed hyperglycemia compared with LC young and LC aging mice (both P < 0.05; Fig. 1E). Thus, our results suggest that the increased body weight was due to an increase in fat mass in LC aging and PUFA aging mice, indicating age-related obesity, and PUFA intake amplified weight weight gain and fat mass. To test if PUFA diet-induced obesity impacted on LV function pre-MI, echocardiography measurements before MI did not show any functional abnormalities due to the PUFA diet.
PUFA-induced obesity increased mortality and led to early LV dysfunction post-MI.
To define the PUFA effect on LV function and post-MI mortality in aging, a total of 30 LC young mice and 21 mice each of the LC aging and PUFA aging groups were enrolled in the MI experiment. Perioperative mortality (mice that died within 24 h of surgery) was 9% for LC young mice, 20% for LC aging mice, and 33% for PUFA aging mice (P < 0.05 for PUFA aging vs. LC young mice). Higher postoperative (after 24 h) mortality was also observed in PUFA aging mice by day 5 post-MI (19% for LC young mice, 28% for LC aging mice, and 44% for PUFA aging mice, P < 0.05 for PUFA aging mice vs. LC young and aging mice). To gain insight into the impact of PUFA-induced obesity on LV post-MI, we performed echocardiography in LC young, LC aging, and PUFA aging post-MI mice. PUFA aging mice developed post-MI LV dysfunction earlier compared with LC young and LC aging mice (Fig. 2, A and B). All three groups showed significantly increased end-diastolic volume and end-systolic volume compared with the day 0 control group (all P < 0.05). PUFA aging mice showed a significant increase in end-diastolic volume compared with LC young and LC aging mice at day 1 post-MI, indicating early LV dysfunction (P < 0.05). Of note, by day 5, LC aging mice caught up and showed similar LV dysfunction compared with PUFA aging mice. The ejection fraction was decreased at day 1 post-MI in PUFA aging mice compared with LC young and LC aging mice (Fig. 2C). Post-MI wall thinning was observed to a similar extent in all three groups as measured by echocardiography (Fig. 2D). In summary, the PUFA aging led to accelerated LV dilation post-MI.
Fig. 2.

PUFA-induced obesity increased end-diastolic volume (EDV) and reduced ejection fraction in PUFA aging mice at days 1 and 5 post-MI. A: EDV. B: end-systolic volume (ESV). C: ejection fraction. Ejection fraction = [(EDV − ESV)/EDV] × 100. D: infarcted wall. n = 3–8 mice/group per time point. Numbers of mice per group are shown in columns. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. the LC young group at the respective day (analyzed by ANOVA).
Necropsy data revealed that post-MI LV mass was higher in PUFA aging and LC aging groups at day 5 post-MI compared with the day 0 control group, indicating a dilated cardiomyopathy phenotype (P < 0.05; Table 1). Lung wet mass was increased at day 5 post-MI in PUFA aging and LC aging groups compared with the day 0 control group (P < 0.05; Table 2). These results suggest that MI leads to increased LV mass and volume overload-induced pulmonary edema and that the magnitude of the response was not altered by diet but the timing was accelerated in the PUFA aging group.
Table 2.
Necropsy data in LC young, LC aging, and PUFA aging mice at baseline and at days 1 and 5 post-MI
|
Day 0 Control Group |
Day 1 Post-MI Group |
Day 5 Post-MI Group |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| LC young | LC aging | PUFA aging | LC young | LC aging | PUFA aging | LC young | LC aging | PUFA aging | |
| n | 9 | 3 | 3 | 9 | 4 | 4 | 8 | 9 | 6 |
| LV mass, mg | 90 ± 2 | 101 ± 2 | 88 ± 9 | 104 ± 4 | 104 ± 7 | 102 ± 5* | 108 ± 3* | 114 ± 3* | 122 ± 4* |
| Tibia length, mm | 18 ± 0.3 | 17 ± 0.1 | 17 ± 0.1 | 18 ± 0.1 | 17 ± 0.1 | 17 ± 0.7 | 18 ± 0.2 | 18 ± 0.2 | 17 ± 0.2 |
| LV mass/tibia length | 5 ± 0.3 | 5 ± 0.1 | 5 ± 0.3 | 5 ± 0.1 | 6 ± 0.3 | 6 ± 0.3 | 6 ± 0.2 | 6 ± 0.1 | 7 ± 0.1*† |
| RV mass, mg | 18 ± 1 | 20 ± 3 | 21 ± 3 | 19 ± 1 | 20 ± 2 | 24 ± 1*† | 20 ± 1 | 26 ± 1* | 28 ± 2*† |
| RV/BW, mg/g | 0.6 ± 0.3 | 0.6 ± 0.1 | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.8 ± 0.01* | 0.9 ± 0.03* | 0.8 ± 0.1* |
| Wet lung mass, mg | 140 ± 25 | 157 ± 47 | 204 ± 10 | 160 ± 13 | 174 ± 17 | 204 ± 26*† | 196 ± 13* | 320 ± 30* | 265 ± 26* |
| Wet lung mass/BW, mg/g | 5 ± 2 | 5 ± 1 | 6 ± 1 | 5 ± 1 | 6 ± 1 | 6 ± 1 | 8 ± 1* | 11 ± 1* | 8 ± 1* |
Data are means ± SE; n, number of mice/group.
LV, left ventricular; RV, right ventricular; BW, body weight.
P < 0.05 vs. the day 0 control group;
P < 0.05 vs. the LC aging group at the respective time point.
PUFA aging intensified inflammatory plasma analyte levels at day 1 post-MI.
Proteomic profiling of plasma revealed that of 59 analytes, 8 proinflammatory analytes and 1 anti-inflammatory analyte were significantly higher in PUFA aging mice compared with LC aging mice before MI, indicating a basal increase in the proinflammatory status. These proinflammatory analytes were CD40 ligand, interferon-γ-induced protein 10, lymphotactin, macrophage-derived chemokine, matrix metalloproteinase-9, macrophage chemotactic protein (MCP)-1, MCP-5, and VCAM-1, whereas anti-inflammatory analyte tissue inhibitor of metalloproteinase-1 was increased compared with the age-related LC aging group (all P < 0.05; Supplemental Table S1 in the Supplemental Material).1
At day 1 post-MI, PUFA aging mice developed earlier impairment in LV function compared with LC young and LC aging mice, which correlated with increased macrophage inflammatory protein (MIP)-1γ and CD40 levels (all P < 0.05). Interestingly, LC young mice showed a robust increase in MCP-3 at day 1 but a rapid decrease by day 5 post-MI, whereas PUFA aging and LC aging mice maintained higher levels by day 5, suggesting an impaired healing response in aging. Furthermore, myeloperoxidase was significantly increased in PUFA aging mice at day 5 post-MI compared with LC aging mice (all P < 0.05; Fig. 3, A–D). These results revealed that excessive PUFA intake during aging delays the resolving and healing process by exacerbating acute inflammation post-MI.
Fig. 3.

PUFA-induced aging amplified proinflammatory analytes compared with LC young and LC aging groups post-MI. A–D: plasma levels of macrophage inflammatory protein (MIP)-1γ (A), CD40 at day 1 (B), and myeloperoxidase (MPO) at day 5 (C) were increased, whereas macrophage chemotactic protein (MCP)-3 (D) was decreased at day 1 post-MI in PUFA aging mice. n = 3–8 mice/group per time point. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. LC young and LC aging groups; #P < 0.05, PUFA aging group vs. LC aging group at the respective day (analyzed by ANOVA).
PUFA-induced obesity impaired post-MI acute inflammatory responses in aging mice.
We observed a robust increase in the post-MI inflammatory status due to PUFA diet, leading us to investigate whether PUFA intake altered leukocyte numbers in aging compared with LC young and LC aging groups. No neutrophils were observed in LC young, LC aging, and PUFA aging mice at day 0, indicating an absence of neutrophils in myocardia without MI injury (Fig. 4, A and B). At day 1 post-MI, there were greater numbers of neutrophils in the infarct area of PUFA aging mice compared with LC aging mice (P < 0.05). Of note, VCAM-1 (a primary mediator of neutrophil endothelial adhesion and extravasation) was higher in the plasma of PUFA aging mice than LC aging mice before MI (Fig. 4C). VCAM-1 levels plummeted post-MI but remained higher in PUFA aging mice compared with LC aging mice post-MI (P < 0.05). The increased VCAM-1 levels in PUFA aging mice could explain the higher recruitment of neutrophils compared with LC aging mice. To minimize the variation within groups, we included mouse samples with an infarct area above 40% (Fig. 4D). Thus, the increased neutrophil density at day 1 post-MI in the PUFA aging group explains the earlier LV dysfunction and higher levels of plasma proinflammatory analytes.
Fig. 4.

A: MI-induced left ventricular (LV) neutrophil density was increased in PUFA aging mice compared with LC young and LC aging mice. A: LV tissue stained with anti-mouse neutrophils (Cedarlane). Top, no-MI day 0 control group; bottom, day 1 post-MI (24 h) group. Infiltrated neutrophils are circled. B: quantitation of neutrophil density. n = 5–6 slides/mouse per group. ¥P < 0.05 vs. LC young and LC aging groups. C: plasma VCAM-1 levels. D: post-MI day 1 infarct area with representative LV midcavities stained with 1% 2,3,5-triphenyltetrazolium chloride. n = 3–8 mice/group per time point. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. the LC young group at the respective day (analyzed by ANOVA).
PUFA intake fueled post-MI inflammatory responses in aging as measured by gene array.
Because plasma analyses indicated increased inflammatory analytes post-MI, we measured LV inflammatory cytokine and receptor gene expression of LC young, LC aging, and PUFA aging mice at day 0 and post-MI at days 1 and 5 in both LVC and LVI areas (n = 3–4 mice/group per day time point).
At day 0, LC aging mice had increased IL-10 receptor-β expression, whereas complement component 3 and small inducible cytokine subfamily E member 1 were increased in PUFA aging mice compared with LC young mice (all P < 0.05). Of 84 genes, we noticed that chemokine (C-C motif) ligand (Ccl)19 was increased at day 1 and that β2-integrin, Ccl2, Ccl7, Ccl12, and Ccl19 (all P < 0.05) were increased in the infarcted area of PUFA aging mice at day 5 compared with LC aging mice. The increased β2-integrin mRNA expression reinforced neutrophil extravasation and recruitment in PUFA aging mice compared with LC young and LC aging mice. Of 84 genes, 18 genes were increased in PUFA aging mice, and, particularly, secreted phosphoprotein 1 and transforming growth factor-β1 (P < 0.05) were increased in response to MI in all three groups (P < 0.05; Supplemental Tables S2 and S3). Thus, the gene expression data indicated that the post-MI acute inflammatory response was amplified in the PUFA aging group.
PUFA-induced obesity increased proinflammatory lipid mediators in aging mice.
Because analyses of plasma and the LV array showed intensified inflammation in PUFA aging mice compared with LC young and LC aging mice, we determined the role of PUFA-derived lipid mediators in LV dysfunction. Plasma from day 0 control and day 1 and 5 post-MI mice were subjected to LC-MS/MS-based lipidomic analysis. As expected, PUFA aging mice showed elevated levels of arachidonic acid (AA) in plasma at all three time points: before MI and at days 1 and 5 post-MI. In addition, levels of the AA metabolite 12(S)-HETE were significantly higher in PUFA aging mice at day 1 post-MI compared with LC aging mice (both P < 0.05; Fig. 5, A–D). An increase in an eicosanoid metabolite derived from AA could explain the altered proinflammatory environment in PUFA aging mice compared with LC aging mice. Thus, a diet enriched in ω-6 fatty acids stimulated a proinflammatory milieu after MI in the PUFA aging group.
Fig. 5.

Liquid chromotography tandem mass spectroscopy-based lipidomic analysis revealed higher levels of plasma arachidonic acid (AA) and 12(S)-HETE at day 1 post-MI in PUFA aging mice. A and B: quantitative measurements of AA (A) and 12(S)-HETE (B). C and D: representative chromatograms of AA at day 0 and 12(S)-HETE at day 1 post-MI in LC aging (C) and PUFA aging (D) groups. n = 3–8 mice/group per time point. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. the LC aging group at the respective day (analyzed by ANOVA).
PUFA-induced obesity impaired the resolution of inflammation post-MI.
MI initiates a rapid inflammatory response that begins to resolve around day 5 post-MI. We examined whether PUFA aging impacted the resolution of post-MI inflammation. COX-2 mediates pro- and anti-inflammatory lipid mediators, whereas 5-LOX stimulates the resolution of overactive inflammation. Therefore, we measured COX-2 and 5-LOX expression. PUFA aging mice showed a robust increase of COX-2 at day 1 post-MI that returned to the baseline level by day 5 post-MI. In comparison, COX-2 levels were elevated in LC young and LC aging mice at day 5 post-MI (P < 0.05; Fig. 6, A and B). To counterbalance and stimulate resolution of increased post-MI inflammation, 5-LOX was consistently increased at days 1 and 5 post-MI in both non-PUFA-fed LC young and LC aging groups but was relatively lower in the PUFA aging group (Fig. 6, C and D). Furthermore, expression of the inflammation-resolving enzyme HO-1 was lowered in PUFA aging mice compared with LC young and LC aging mice at day 5 post-MI (P < 0.05; Fig. 7, A and B). Decreased LV expression of HO-1, confirmed using LV immunohistochemistry, suggests that excessive intake of PUFA in aging impairs stimulation of HO-1 (Fig. 7C). Thus, the imbalance of COX-2, 5-LOX, and HO-1 enzymes in the infarcted area in part contributed to the overactive inflammation and impaired post-MI resolution of inflammation in the PUFA aging group.
Fig. 6.

MI-induced cyclooxygenase (COX)-2 and 5-lipoxygenase (5-LOX) resolved inflammation in LC young and LC aging mice but not in PUFA aging mice. A–D: COX-2 (A and B) and 5-LOX (C and D) immunoblot and densitometry analyses. The densitometry data were normalized to total protein. n = 2 mice in the day 0 control group and 3 mice in the day 1 and 5 post-MI groups. AU, arbitrary units. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. the LC aging group (analyzed by ANOVA).
Fig. 7.

PUFA aging mice showed reduced inflammation-resolving enzyme heme oxygenase (HO)-1 levels compared with age-matched LC aging and LC young mice post-MI. A and B: HO-1 immunoblot (A) and densitometry (B) measurements. Densitometry data were normalized to total protein. n = 2 mice in the day 0 control group and 3 mice in the day 1 and 5 post-MI groups. *P < 0.05 vs. the day 0 control group; ¥P < 0.05 vs. the LC young group (analyzed by ANOVA). C: LV HO-1 immunohistochemistry in the day 0 control, day 1 post-MI, and day 5 post-MI groups.
DISCUSSION
In the present study, PUFA-induced obesity superimposed on aging led to the development of post-MI LV dysfunction earlier and impaired resolution of inflammation and delayed LV healing. The major outcomes in PUFA-fed aging mice showed 1) increased proinflammatory analytes before MI and that this increase was exacerbated by MI, 2) increased neutrophil density into the infarcted area, 3) elevated plasma AA and 12(S)-HETE in the acute inflammatory phase, and 4) imbalanced resolution of inflammation by altering resolving enzymes (COX-2, 5-LOX, and HO-1) post-MI to favor unresolved inflammation. Thus, our results indicate that increased PUFA intake during aging delays the post-MI resolution of inflammation.
The post-MI innate inflammatory response has been well studied in young and lean animal models; however, the resolution of inflammation or the role of lipid mediators after MI in the setting of obesity and aging requires additional investigation. Therefore, our study not only ratifies post-MI inflammation in PUFA aging but also focuses on plasma proteomic profiling, measurement of lipid mediators, and neutrophil trafficking post-MI. PUFA intake during aging stimulated eight proinflammatory analytes before MI, including MIP-1γ, MCP-3, and VCAM-1. Post-MI, increased levels of MCP-1 have been well studied; MCP-1-deficient mice exhibit lower macrophage recruitment in the infarcted heart, delayed phagocytosis of dead cardiomyocytes, and reduced cardiac remodeling post-MI (45). Levels of MIP-1γ and CD40 were significantly peaked, whereas MCP-3 decreased, at day 1 in PUFA aging mice, indicated that prior elevation of proinflammatory analytes impacted the healing and delayed containment of inflammation. Furthermore, VCAM-1 plays a role in the adhesion, recruitment, and transmigration process of neutrophils from the circulation to the site of injury (37). Increased expression of β2-integrin and VCAM-1 levels assisted the lingering of neutrophils into the LV and promoted an overactive inflammatory response in PUFA aging mice compared with young mice. In aging, VCAM-1 is known to activate endothelial cells that promote leukocyte adhesion to the endothelial lining. Proinflammatory lipid mediators, such as 12-HETE, may activated the endothelium, leading to increased VCAM-1, which could potentially lead to the development of endothelial dysfunction (26). Thus, VCAM-1-mediated increased endothelial dysfunction may serve as the trigger to increase neutrophil trafficking and their recruitment in the infarcted area. Of note, post-MI neutrophils trafficking is essential to curb inflammation by phagocytizing necrotic myocytes, but their excessive stay in the infarcted area may lead to nonresolving inflammation or delay the tissue regeneration process in the PUFA aging group. In a population-based study (29), patients with increased blood neutrophil numbers were prone to MI, and death underscores unresolved post-MI inflammation (29).
Post-MI inflammation has been well described as an innate inflammatory response and after reparative response (11). Now with a better understanding of post-MI neutrophil and macrophage kinetics, it is known that post-MI inflammation involves initiation, amplification, and resolution. The levels of eicosanoids, docosanoids, and oxidized linoleic acid metabolites not only coordinate the innate inflammatory response but also modify immune cell plasticity and the pain response (16). Eicosanoids are known to accelerate inflammation; therefore, increased 12(S)-HETE may ease vascular permeability to extravsate neutrophil endothelial transmigration in PUFA-fed aging mice. Targeted analysis of ω-6 eicosanoids in MI patients revealed higher 12-HETE than in individuals with stable angina (12). One of the noteworthy features of obesity-triggered metainflammation was that PUFA-enriched diet increased neutrophils in PUFA aging mice at day 1 post-MI. It would be an oversimplification to state that n-6 fatty acids are responsible for the extended stay of neutrophils in the infarcted area, but it is quite possible that these mice had higher neutrophils in their body or that these mice were prone to an exacerbated inflammatory response (34). Gosh et al. (15) showed that aging mice supplemented with a ω-6 fatty acid diet lead to increased neutrophils in the gut and the development of dysbiosis in aging. This may be a spillover of neutrophils coming from the gut that are recruited at the site of severe injury. However, future precise immune cell tracking studies in rodents and humans are warranted.
The incorporation of n-6 and n-3 PUFAs in myocardial membranes has been well established, suggesting that increased consumption of n-3 PUFA increases myocardial n-3 PUFA levels (41); however, the way in which these precursor classic fatty acids lead to the formation of lipid mediators in the post-MI setting is unclear. An imbalance of n-6 and n-3 fatty acids leads to excessive and extensive metainflammation and cardiovascular disease in aging (34). n-3 fatty acid (particularly eicosapentaenoic and docosahexaenoic acid) intake is known to reduce the risk of congestive heart failure and mortality in the aging population (30). Despite the widely touted cardioprotective effects of ω-3 supplements, it is evident that high doses of n-3 fatty acids merely reduce triglycerides in elderly patients (6). We noticed that excessive intake of n-6 fatty acids deteriorated post-MI repair in aging. By analogy, if the car oil goes bad, we drain the old oil and replace with fresh oil. In the human body, how can we drain excessive unused fatty acids during aging? It is possible that exercise will help to burn or reduce lipotoxicity in obesity. This analogy gives us a hint to the reason why ω-3 products are not effective in unresolved inflammation or in obesity. A possibility is that the preexistence of excessive n-6 fatty acids dominates competition and requires strong inflammation resolvents to contain the post-MI inflammation. Thus, our results surfaced a long-debated question: “Do we need to limit n-6 fatty acid intake? As such, the American Heart Association recommendation for n-6 intake is 17 g for men and 12 g for women (21). However, with a current scenario of reduced energy expenditure present in a sedentary lifestyle during aging, should it be advised to reduce n-6 fatty acids levels to control post-MI defective resolution of inflammation in obese aging? The feasibility of this concept was recently tested in obese individuals and demonstrated that a reduction of energy intake from 4.6% to 2% by reducing linoleic intake increased n-3 fatty acid levels (44).
Gas chromatographic analysis revealed that total n-3 fatty acids were 0.015% and linoleic acid content was 6.6% in PUFA diet, which mirrored in plasma AA levels in PUFA aging mice (Fig. 5, A–D). In elderly humans, linoleic acid-enriched diet shows higher body weight gain as evidenced by storage in adipose tissue (9). Linoleic acid intake is not only relevant to obesity but also high intake can lead to transgenerational obesity in mice (28). A recent randomized double-blind overfeeding LIPOGAIN trial (7 wk) in young lean healthy individuals indicated that PUFA or saturated fat intake not only increased weight gain by >2% but also increased endothelial dysfunction markers such as VCAM-1, ICAM-1, and E-selectin (23). Thus, our findings have significant implications for the overactive inflammatory response post-MI in the setting of obesity and aging that delays healing. Investigation of the substrates that reduce proinflammatory milieu and promote the resolution of inflammation in aging will be an area of active investigation in obese aging. A recently published, comprehensive report on high-fat diet-induced obesity using either milk fat or lard fat (60% kcal) indicated that saturated fat alone is insufficient to induce murine cardiac dysfunction (1, 7). In agreement with these results, excessive intake of PUFA-induced obesity did not alter pre-MI LV function. In the post-MI setting, PUFA-fed aging mice developed post-MI LV dysfunction earlier than LC young and LC aging mice. At the translational level, the saturated fat does not contribute to heart dysfunction (27); however, excessive nutrient intake of either saturated or PUFA fat can lead to insulin resistance, nonalcoholic fatty liver disease, and hyperlipidemia (13). Research on daily energy input (quantity and quality of fat), bioenergetics, and the production of lipid mediators that maintain inflammation-resolving homeostasis will provide additional evidence on the fat intake dilemma.
To explain post-MI inflammation and resolution of the inflammation axis after MI in the setting of PUFA aging, we measured inflammation-inducible and proresolving enzymes. COX-2 is induced in response to post-MI inflammation. We noticed higher expression in the LVI area in PUFA-fed aging mice at day 1, which returned to baseline before day 5 compared with LC aging and LC young mice. Of note, COX-2 is essential for cardiovascular physiology; however, selective pharmacological inhibition of COX-2 (e.g., celecoxib and valdecoxib) impedes inflammation and induced MI events that led to the postmarketing withdrawal of selective inhibitors (38). Taken together, our results provide evidence that while maintaining caution with the COX-2 inhibition strategy, estimation of resolving enzymes is necessary in the post-MI setting. Counterregulating, pro-resolving enzymes, such as 5-LOX and HO-1, were lower in PUFA aging infarcted areas at day 1, indicating a dampened post-MI-resolving mechanism compared with the LC young group. 5-LOX and HO-1 are major inflammation-resolving enzymes and promote post-MI ventricular repair and attenuate cardiac remodeling (3, 46). Future studies with a major focus on COX and LOX coordination in the resolution of post-MI inflammation will be an area of active investigation to determine role of resolvins, lipoxins, protectins, and maresins (24).
Thus, our results suggest that excess PUFA intake during aging drives the unresolved inflammatory response by increasing inflammatory mediators, promoting neutrophil trafficking and leading to impaired wound healing post-MI (Fig. 8). Future studies are essential to shed light on the containment of inflammation in obesity and aging and how attempts can be made to resolve or limit inflammatory cell trafficking. The reduction of inflammatory cells lingering in the infarcted area will reduce the uncontrolled inflammation post-MI and delay the progression toward heart failure.
Fig. 8.
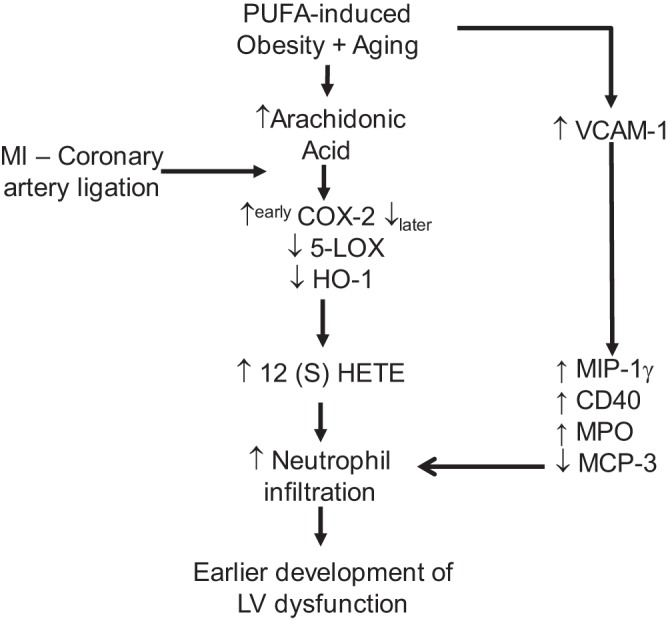
Schematic based on major findings. Higher PUFA intake in aging leads to obesity with a higher load of proinflammatory lipid mediators in plasma that aggravates inflammation mediated by increased neutrophil trafficking, thereby impairing the resolution of inflammation in the LV.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R00-AT-006704 (to G. V. Halade) and HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center as well as NIH Grants R01-HL-075360 and HL051971 and 5I01BX000505 (to M. L. Lindsey). The authors acknowledge funding support for the mass spectrometry analysis from the University of Alabama at Birmingham (UAB)-Health Services Foundation General Endowment Fund and the UAB-University of California-San Diego O'Brien Acute Kidney Injury Center (NIH Grant P30-DK-079337) (to Dr. A. Agarwal, UAB).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank to Doyle Ray Moore II for technical assistance.
Footnotes
Supplemental Material for this article is available at the American Journal of Physiology-Heart and Circulatory Physiology website.
REFERENCES
- 1.Aurich AC, Niemann B, Pan R, Gruenler S, Issa H, Silber RE, Rohrbach S. Age-dependent effects of high fat-diet on murine left ventricles: role of palmitate. Basic Res Cardiol 108: 369, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr 93: 950–962, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, Frey A, Bayer B, Vogel B, Ertl G, Bauersachs J, Frantz S. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol 108: 367, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bloom DE, Cafiero ET, Jané-Llopis E, Abrahams-Gessel S, Bloom LR, Fathima S, Feigl AB, Gaziano T, Mowafi M, Pandya A, Prettner K, Rosenberg L, Seligman B, Stein AZ, &, Weinstein C. The Global Economic Burden of Noncommunicable Diseases. Geneva: World Economic Forum, 2011. [Google Scholar]
- 5.Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 18: 139–143, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonds DE, Harrington M, Worrall BB, Bertoni AG, Eaton CB, Hsia J, Robinson J, Clemons TE, Fine LJ, Chew EY. Effect of long-chain ω-3 fatty acids and lutein + zeaxanthin supplements on cardiovascular outcomes: results of the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA Intern Med 174: 763–771, 2014. [DOI] [PubMed] [Google Scholar]
- 7.Brainard RE, Watson LJ, Demartino AM, Brittian KR, Readnower RD, Boakye AA, Zhang D, Hoetker JD, Bhatnagar A, Baba SP, Jones SP. High fat feeding in mice is insufficient to induce cardiac dysfunction and does not exacerbate heart failure. PloS One 8: e83174, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canete R, Gil-Campos M, Aguilera CM, Gil A. Development of insulin resistance and its relation to diet in the obese child. Eur J Nutr 46: 181–187, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Dayton S, Hashimoto S, Dixon W, Pearce ML. Composition of lipids in human serum and adipose tissue during prolonged feeding of a diet high in unsaturated fat. J Lipid Res 7: 103–111, 1966. [PubMed] [Google Scholar]
- 10.DeFilippis AP, Sperling LS. Understanding omega-3's. Am Heart J 151: 564–570, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48: 504–511, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez Peralbo MA, Priego-Capote F, Galache-Osuna JG, Luque de Castro MD. Targeted analysis of omega-6-derived eicosanoids in human serum by SPE-LC-MS/MS for evaluation of coronary artery disease. Electrophoresis 34: 2901–2909, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Galbo T, Perry RJ, Jurczak MJ, Camporez JP, Alves TC, Kahn M, Guigni BA, Serr J, Zhang D, Bhanot S, Samuel VT, Shulman GI. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci USA 110: 12780–12785, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galvao TF, Brown BH, Hecker PA, O'Connell KA, O'Shea KM, Sabbah HN, Rastogi S, Daneault C, Des Rosiers C, Stanley WC. High intake of saturated fat, but not polyunsaturated fat, improves survival in heart failure despite persistent mitochondrial defects. Cardiovasc Res 93: 24–32, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Molcan E, DeCoffe D, Dai C, Gibson DL. Diets rich in n-6 PUFA induce intestinal microbial dysbiosis in aged mice. The Br J Nutr 110: 515–523, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Green DP, Ruparel S, Roman L, Henry MA, Hargreaves KM. Role of endogenous TRPV1 agonists in a postburn pain model of partial-thickness injury. Pain 154: 2512–2520, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halade GV, El Jamali A, Williams PJ, Fajardo RJ, Fernandes G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp Gerontol 46: 43–52, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halade GV, Ma Y, Ramirez TA, Zhang J, Dai Q, Hensler JG, Lopez EF, Ghasemi O, Jin YF, Lindsey ML. Reduced BDNF attenuates inflammation and angiogenesis to improve survival and cardiac function following myocardial infarction in mice. Am J Physiol Heart Circ Physiol 305: H1830–H1842, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halade GV, Rahman MM, Bhattacharya A, Barnes JL, Chandrasekar B, Fernandes G. Docosahexaenoic acid-enriched fish oil attenuates kidney disease and prolongs median and maximal life span of autoimmune lupus-prone mice. J Immunol 184: 5280–5286, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halade GV, Rahman MM, Williams PJ, Fernandes G. High fat diet-induced animal model of age-associated obesity and osteoporosis. J Nutr Biochem 21: 1162–1169, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris WS, Mozaffarian D, Rimm E, Kris-Etherton P, Rudel LL, Appel LJ, Engler MM, Engler MB, Sacks F. Omega-6 fatty acids and risk for cardiovascular disease: a science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 119: 902–907, 2009. [DOI] [PubMed] [Google Scholar]
- 22.Heaberlin JR, Ma Y, Zhang J, Ahuja SS, Lindsey ML, Halade GV. Obese and diabetic KKAy mice show increased mortality but improved cardiac function following myocardial infarction. Cardiovasc Pathol 22: 481–487, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iggman D, Rosqvist F, Larsson A, Arnlov J, Beckman L, Rudling M, Riserus U. Role of dietary fats in modulating cardiometabolic risk during moderate weight gain: a randomized double-blind overfeeding trial (LIPOGAIN study). J Am Heart Assoc 3: e001095, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kain V, Prabhu SD, Halade GV. Inflammation revisited: inflammation versus resolution of inflammation following myocardial infarction. Basic Res Cardiol 109: 444, 2014. [DOI] [PubMed] [Google Scholar]
- 25.Lavie CJ, De Schutter A, Milani RV. Healthy obese versus unhealthy lean: the obesity paradox. Nat Rev Endocrinol; doi: 10.1038/nrendo.2014.165. [DOI] [PubMed] [Google Scholar]
- 26.Liao JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest 123: 540–541, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malhotra A. Saturated fat is not the major issue. BMJ 347: f6340, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Massiera F, Barbry P, Guesnet P, Joly A, Luquet S, Moreilhon-Brest C, Mohsen-Kanson T, Amri EZ, Ailhaud G. A Western-like fat diet is sufficient to induce a gradual enhancement in fat mass over generations. J Lipid Res 51: 2352–2361, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner J, Irfan A, Twerenbold R, Mueller S, Reiter M, Haaf P, Reichlin T, Schaub N, Winkler K, Pfister O, Heinisch C, Mueller C. Use of neutrophil count in early diagnosis and risk stratification of AMI. Am J Med 124: 534–542, 2011. [DOI] [PubMed] [Google Scholar]
- 30.Mozaffarian D, Lemaitre RN, King IB, Song X, Spiegelman D, Sacks FM, Rimm EB, Siscovick DS. Circulating long-chain ω-3 fatty acids and incidence of congestive heart failure in older adults: the cardiovascular health study: a cohort study. Ann Intern Med 155: 160–170, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med 366: 54–63, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339: 172–177, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 311: 806–814, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patterson E, Wall R, Fitzgerald GF, Ross RP, Stanton C. Health implications of high dietary ω-6 polyunsaturated Fatty acids. J Nutr Metab 2012: 539426, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramsden CE, Zamora D, Leelarthaepin B, Majchrzak-Hong SF, Faurot KR, Suchindran CM, Ringel A, Davis JM, Hibbeln JR. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ 346: e8707, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reina-Couto M, Carvalho J, Valente MJ, Vale L, Afonso J, Carvalho F, Bettencourt P, Sousa T, Albino-Teixeira A. Impaired resolution of inflammation in human chronic heart failure. Eur J Clin Invest 44: 527–538, 2014. [DOI] [PubMed] [Google Scholar]
- 37.Ross EA, Douglas MR, Wong SH, Ross EJ, Curnow SJ, Nash GB, Rainger E, Scheel-Toellner D, Lord JM, Salmon M, Buckley CD. Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood 107: 1178–1183, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sellers RS, Radi ZA, Khan NK. Pathophysiology of cyclooxygenases in cardiovascular homeostasis. Vet Pathol 47: 601–613, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol 57: 9–17, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shioi T, Inuzuka Y. Aging as a substrate of heart failure. J Cardiol 60: 423–428, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Slee EL, McLennan PL, Owen AJ, Theiss ML. Low dietary fish-oil threshold for myocardial membrane n-3 PUFA enrichment independent of n-6 PUFA intake in rats. J Lipid Res 51: 1841–1848, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stanley WC, Dabkowski ER, Ribeiro RF Jr, O'Connell KA. Dietary fat and heart failure: moving from lipotoxicity to lipoprotection. Circ Res 110: 764–776, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomsen M, Nordestgaard BG. Myocardial infarction and ischemic heart disease in overweight and obesity with and without metabolic syndrome. JAMA Intern Med 174: 15–22, 2014. [DOI] [PubMed] [Google Scholar]
- 44.Wood KE, Lau A, Mantzioris E, Gibson RA, Ramsden CE, Muhlhausler BS. A low ω-6 polyunsaturated fatty acid (n-6 PUFA) diet increases ω-3 (n-3) long chain PUFA status in plasma phospholipids in humans. Prostaglandins Leukot Essent Fatty Acids 90: 133–138, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia Y, Frangogiannis NG. MCP-1/CCL2 as a therapeutic target in myocardial infarction and ischemic cardiomyopathy. Inflamm Allergy Drug Targets 6: 101–107, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, Dzau VJ, Lee ME, Perrella MA. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ Res 89: 168–173, 2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.