

Abstract
Lipid accumulation in the heart is associated with obesity and diabetes and may play an important role in the pathogenesis of heart failure. The renin-angiotensin system is also thought to contribute to cardiovascular morbidity in obese and diabetic patients. We hypothesized that the presence of lipid within the myocyte might potentiate the cardiomyopathic effects of ANG II in the cardiac diacylglycerol acyl transferase 1 (DGAT1) transgenic mouse model of myocyte steatosis. Treatment with ANG II resulted in a similar increase in blood pressure in both nontransgenic and DGAT1 transgenic mice. However, ANG II in DGAT1 transgenic mice resulted in a marked increase in interstitial fibrosis and a reduction in systolic function compared with nontransgenic littermates. Lipidomic analysis revealed that >20% of lipid species were significantly altered between nontransgenic and DGAT1 transgenic animals, whereas 3% were responsive to ANG II administration. ROS were also increased by ANG II in DGAT1 transgenic hearts. ANG II treatment resulted in increased expression of transforming growth factor (TGF)-β2 and the type I TGF-β receptor as well as increased phosphorylation of Smad2 in DGAT1 transgenic hearts. Injection of neutralizing antibodies to TGF-β resulted in a reduction in fibrosis in DGAT1 transgenic hearts treated with ANG II. These results suggest that myocyte steatosis amplifies the fibrotic effects of ANG II through mechanisms that involve activation of TGF-β signaling and increased production of ROS.
Keywords: angiotensin, diacylglycerol acyl transferase 1, fibrosis, lipotoxixity, transforming growth factor-β
obese subjects have twice the risk for the development of heart failure compared with nonobese patients after adjustment for hypertension, diabetes, and dyslipidemia (24). The mechanism(s) underlying this effect remains incompletely understood; however, both obesity and diabetes are associated with cardiac steatosis, a state of abnormal triglyceride (TG) accumulation within cardiac myocytes (35, 37, 46). A growing body of work suggests that intracellular lipids and lipid metabolites may have direct toxic effects on the heart (i.e., lipotoxicity) (18, 57, 61, 62).
Several mouse models of obesity and diabetes mellitus result in both increased deposition of TG within myocytes and impaired heart function (9, 38, 39, 54, 69). Studies carried out using transgenic (Tg) mice that selectively promote fatty acid uptake in the cardiac myocyte (7, 8, 64) resulted in TG accumulation and cardiac dysfunction. Deletion of adipose triglyceride lipase (ATGL), the enzyme that catalyzes the initial step in TG hydrolysis, results in marked TG accumulation in cardiac myocytes and significant cardiomyopathy (21, 22). In contrast, mice expressing a cardiac-selective ATGL transgene have reduced cardiac TG levels and appear to be protected from pressure-induced cardiomyopathy (27). Similarly, cardiac overexpression of hormone-sensitive lipase, which also facilitates TG hydrolysis, results in a reduction of cardiac TG and diminished fibrosis in streptozotocin-treated mice (56). We have previously demonstrated that cardiac myocyte-selective expression, using the α-myosin heavy chain (αMHC) promoter, of diacylglycerol acyltransferase 1 (MHC-DGAT1), an enzyme that catalyzes the final step in the TG synthesis pathway, results in increased TG accumulation, increased fibrosis, and cardiac dysfunction (17). This occurred in the absence of differences in blood pressure, body weight, fasting glucose, insulin sensitivity, or serum lipid levels (17). However, severe cardiomyopathy does not occur in this model until mice are 52–60 wk of age (17).
Activation of the renin-angiotensin system has been shown to be linked to the pathogenesis of many forms of heart disease (43), including those due to obesity and diabetes mellitus (4), and blockade of this system has proven to be an effective strategy for managing cardiac disease. We hypothesized that ANG II might synergize mechanistically with steatosis-dependent alterations in myocyte physiology to accelerate the development of cardiomyopathic changes in the heart. Using our murine model of isolated myocyte steatosis, we examined the interaction between ANG II infusion and elevated myocyte TG levels in promoting cardiac dysfunction. Exposure to ANG II led to a moderate increase in blood pressure in both non-Tg (NTg) control and MHC-DGAT1 Tg mice. However, ANG II in MHC-DGAT1 Tg mice resulted in a marked increase in heart size and cardiac fibrosis compared with NTg littermates. These results suggest that myocyte steatosis creates an abnormal metabolic environment that amplifies the profibrotic effects of ANG II.
METHODS
Animal experiments.
All experiments were approved by the Institutional Animal Care and Use Committee of the University of California (San Francisco, CA) and complied with guidelines for the care of laboratory animals published by National Institutes of Health (NIH Pub. No. 85-23, Revised 1996). Mice were fed standard chow diet (PicoLab Mouse Diet 20 5058). Cardiac myocyte-selective DGAT1 expression was achieved using a mouse transgene containing the α-MHC promoter linked to the FLAG-tagged, murine DGAT1 coding sequence in the DBA/2J background as previously described (17). Male and female mice (12–14 wk of age) were used in the study to guarantee adequate numbers of mice from the same generations to perform the analyses. There was no major difference in the manner in which male versus female mice reacted to genetic or pharmacological stimuli. NTg littermates were used as controls. Mice were anesthetized with isoflurane (4%) followed by cervical dislocation before the collection of tissues.
Osmotic pump placement.
Mice were anesthetized with 3.5% isoflurane. The intrascapular region was shaved and prepped with betadine antiseptic solution before the administration of bupivacaine (0.1 mg/kg) and buprenorphine (0.1 mg/kg). An incision was made, and an Alzet osmotic minipump (catalog no. 1002) containing either saline or 500 ng·kg−1·min−1 ANG II (EMD, Millipore, Billerica, MA) was surgically implanted. Pumps were left in place for 14 days.
Blood pressure measurement.
Blood pressure was measured by the tail-cuff plethysmography method in unanesthetized mice using a Hatteras Instruments SC1000 Blood Pressure Analysis System as previously described (17). Measurements were made on 3 consecutive days before osmotic pump implantation and on days 12 and 13 after implantation.
Echocardiography.
Mice were anesthetized with 1.5% isoflurane, and echocardiography was carried out using a Vevo 660 system (VisualSonics, Toronto, ON, Canada) equipped with a 30-MHz real-time microvisualization scan head according to the method of Zhang et al. (68). Measurements were taken at day 14 after osmotic pump implantation.
Transforming growth factor-β-neutralizing antibody treatment.
The effect of transforming growth factor (TGF)-β-neutralizing antibody (NAb) was assessed in NTg and MHC-DGAT1 Tg mice in the presence and absence of ANG II (see above) as previously described by Teekakirikul et al. (53). TGF-β NAb (catalog no. AB-100NA, R&D Systems, Minneapolis, MN) or isotype IgG control in saline (catalog no. AB-105-C, R&D Systems) was administered by intraperitoneal injection (5 mg/kg body wt) 1 day before placement of the osmotic pump containing saline or ANG II and then every third day (5 injections total) for 14 days.
RNA isolation and quantitative PCR.
Left ventricular (LV) tissue, preserved in RNAlater (Life Technologies, Grand Island, NY), was used to isolate total RNA with the RNeasy kit (Qiagen, Valencia, CA) followed by cDNA synthesis from 500–1,000 ng total RNA using Superscript III (Life Technologies). Quantitative PCR was carried out and normalized to GAPDH as an internal control using the following Taqman primer sets (Life Technologies): atrial natriuretic peptide (Mm01255748_g1), NADPH oxidase (Nox)1 (Mm00549170_m1), neutrophil cytosolic factor 1 (Mm00447921_m1), Nox4 (Mm00479246_m1), cytochrome b-α (Mm00514478_m1), cytochrome b-β (Mm01287743_m1), collagen type 1A (Mm00801666_g1), and collagen type 3A (Mm01254476_m1). SYBR Green detection agent (Applied Biosystems) was used for detection of the following genes: Tgfb1 (sense: 5′-GTGGAGCAACATGTGGAACTCTAC-3′ and antisense 5′-GTCAGCAGCCGGTTACCAA-3′), Tgfb2 (sense: 5′-CCAGCCGGCGGAAGA-3′ and antisense 5′-GCGAAGGCAGCAATTATCCT-3′), Tgfb3 (sense: 5′-CCGCAGCGCAGACACA-3′ and antisense 5′-AGACGCCTCTGGGTTCAGG-3′), TGF-β receptor (TGFBR)1 (sense: 5′-CCACCGTGTGCCAAATGAAG-3′ and antisense 5′-TGGTGCCCTCTGAAATGAAAG-3′), and TGFBR2 (sense: 5′-AAAAGACCAAGGAATAGCATCGC-3′ and antisense 5′-TGAATATGGCCGAAGTGTTCA-3′).
Western blot analysis.
Total LV lysates or membrane protein preparations were prepared in lysis buffer containing 20 mmol/l Tris (pH 8.0), 150 mmol/l NaCl, 1% Triton X-100, Complete protease inhibitor and PhosSTOP phosphatase inhibitor was used as directed by the manufacturer (Roche Applied Science, Indianapolis, IN) as previously described (17). Protein was quantified using colorimetric Coomassie assay reagent (Thermo Scientific). Protein (total: 10–40 μg) was resolved on SDS-PAGE and transferred to ImmunBlot polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were probed with antibodies as indicated. Blots were stripped and reprobed with anti-GAPDH or anti-β-tubulin, which served as loading controls. The following antibodies were used: anti-GAPDH, anti-TGFBR1, anti-β-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phosphorylated (p-)Smad2, and anti-Smad2 (Cell Signaling Technologies, Danvers, MA). Detection of immunoreactive bands was achieved using horseradish peroxidase-conjugated secondary antibody followed by incubation with SuperSignal West Femto Chemiluminescent Substrate (Pierce, Rockford, IL) and exposure to film. Quantification of band intensity was performed using ImageJ software.
Histology and immunofluorescence.
Briefly, paraffin-embedded LV tissue was cut on a microtome to yield 5-μm-thick transverse sections and transferred to glass slides. Samples were rehydrated in xylene and ethanol before hematoxylin and eosin and Masson's trichrome staining, which were carried out according to the manufacturer's instructions (Sigma-Aldrich, St. Louis, MO.). Hematoxylin and eosin staining was used to evaluate cardiac structure. Masson's trichrome staining was used to detect fibrosis, which was shown as blue-green. Oil red O staining was carried out to assess lipid droplet content in ventricular sections according to Koopman et al. (28) and visualized under fluorescent microscopy.
Fibrotic area.
Analysis of the fibrotic area fraction was performed with 5-μm-thick transverse cardiac cross-sections stained with Masson's trichrome. Four to five photomicrographs were obtained to cover the entire midventricular section at ×200 magnification. Connective tissue and muscle areas were measured using Metamorph analysis software (Molecular Devices, Sunnyvale, CA) excluding perivascular and epicardial fibrosis. Fibrotic area was then calculated for the heart as the sum of connective tissue areas divided by the sum of connective tissue plus muscle areas in all fields.
Lipidomic analysis.
A global unbiased characterization of cardiac lipids was conducted using ultra-performance liquid chromatography quadrupole-time-of-flight mass spectrometry (UPLC/Q-TOF-MS; Agilent 6530). Cardiac lipids were extracted from homogenized tissue using a modified protocol (41a) consisting of a biphasic solvent system of cold methanol, methyl tert-butyl, and water. Dried extracts were resuspended in methanol-toluene (9:1), and lipids were separated using a CSH C18 (100 × 2.1,m; 1.7 μm) column with a 15-min gradient. Mass spectra were acquired on the Q/TOF-MS (Agilent 6530) operated both in positive ESI(+) and negative ESI(−) modes (65–1,700 m/z). Lipids were identified based on their MS/MS fragmentation profiles using in-house software (LipidBlast). A total of 734 mass spectral features were identified, including glycerophospholipids, glycerolysophospholipids, glycerolipids, sphingolipids, cholesterol esters, ceramides, and fatty acids. Lipidomic measurements were analyzed using univariate statistics, multivariate modeling, and correlation network analysis. Analyses were implemented in the R statistical programming environment (41a), imDEV (version 1.4) (19), and Cytoscape (45). Two-way ANOVA was used to test for significant differences in lipids due to genotype (NTg vs. DGAT1), treatment (ANG II vs. control), and genotype-treatment interactions. Probability levels for test statics were adjusted for the multiple hypotheses tested (3) to allow for a maximum 5% probability (q = 0.05) of false positive detection. Orthogonal partial least-squares discriminant analysis (55) was used to develop a multivariate classification model to concomitantly discriminate between genotype and treatment effects. Models were fit to autoscaled measurements, and the latent variable number was determined using leave-one-out cross-validation. Model validation was conducted through the comparison of performance statistics (Q2 and root mean squared error of prediction) for 100 randomly determined test/training cross-validations to that of a permuted class label models. Correlation-based networks were used to analyze variable interrelationships in the context of statistical and multivariate model results. Hierarchical cluster analysis of Spearman's correlations was used to group all parameters into seven major correlated clusters. Variable cluster identities, statistical significance, and multivariate importance were projected into a network of lipids connected based on significant correlations (Spearmans's ρ, P < 0.0001, q < 0.1). Separate networks were used to map genotype and treatment effects.
Measurement of superoxide, lipid peroxide, and oxidative DNA damage.
Samples of the LV were embedded in optimum cutting temperature reagent (Tissue-Tek, Fisher Scientific), and 5-μm-thick sections were cut and mounted on glass slides. Unfixed frozen sections were then incubated with 5 μM dihydroethidium (Sigma-Aldrich) for 25 min at 37°C followed by three washes in PBS. Images were obtained using the Leica TCS SP5 confocal microscope and analyzed using ImageJ.
For other ROS assays, heart tissue was quickly harvested and snap frozen in liquid nitrogen until the time of the assays. Frozen tissue was weighed, and 4-hydroxynonenal histidine protein adducts were measured using the OxiSelect HNE-His Adduct ELISA Kit (Cell Biolabs, San Diego, CA) according to the manufacturer's instructions. Oxidative damage to DNA was assessed by measurements of 8-hydroxydeoxyguanosine using the OxiSelect Oxidative DNA Damage ELISA kit (Cell Biolabs).
Statistical analysis.
Results are presented as means ± SD. Fold changes and SD for quantitative PCR were calculated as previously described by Livak and Schmittgenn (33). Data were analyzed using two-way ANOVA using GraphPad Prism 5 statistical software. P values are reported for the main effects of ANG II and MHC-DGAT1 Tg genotype and the interaction between ANG II and MHC-DGAT1 Tg genotype in the four experimental groups (NTg, NTg + ANG II, MHC-DGAT1 Tg, and MHC-DGAT1 Tg + ANG II). Statistical differences were considered significant when P values were <0.05.
RESULTS
At 12 wk of age, the MHC-DGAT1 Tg mouse displays activation of the hypertrophic gene program as well as evidence of diastolic dysfunction, but systolic dysfunction is preserved (17). We asked whether ANG II infusion would lower the threshold for the development of cardiomyopathy in MHC-DGAT1 Tg mice. NTg and MHC-DGAT1 Tg mice (12–14 wk of age) were infused with either saline (sham) or ANG II (500 ng·kg−1·min−1) for 14 days. The dose of ANG II was selected to provide a submaximal pressor response (6, 50). The magnitude of the increase in blood pressure was similar in NTg and MHC-DGAT1 Tg mice 12 days after ANG II infusion (Fig. 1, A–C), and there was no statistically significant interaction between ANG II and genotype.
Fig. 1.
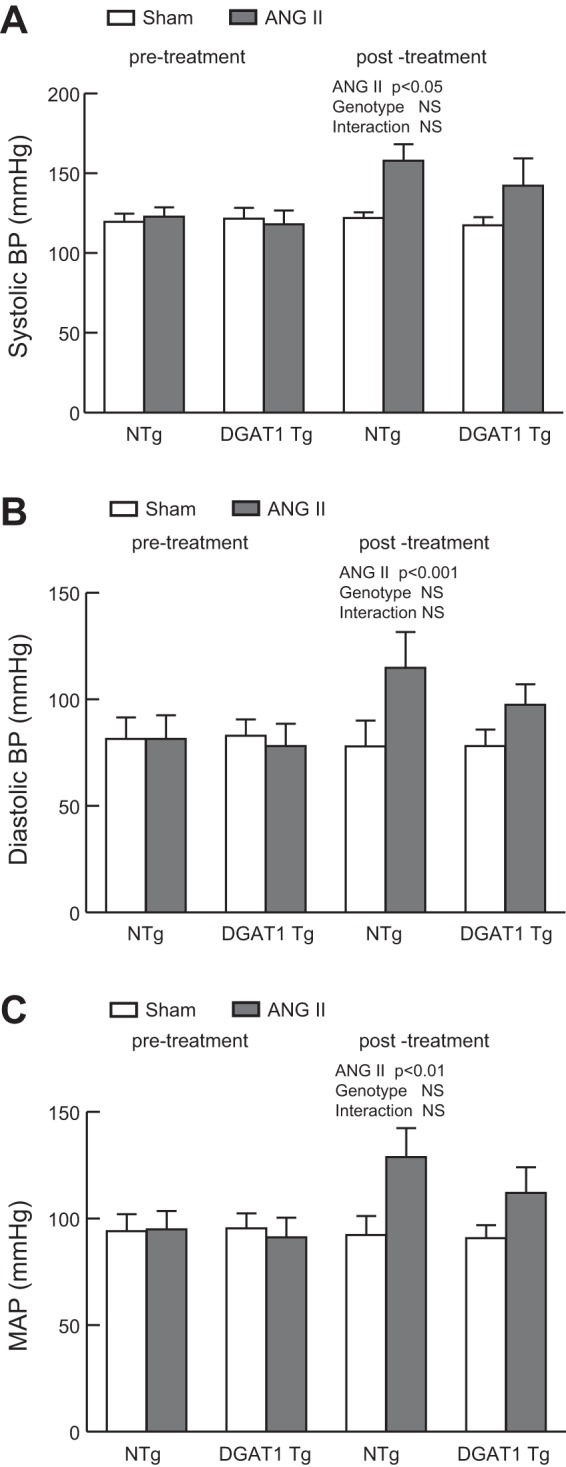
Blood pressure in non-transgenic (NTg) and α-myosin heavy chain promoter of diacylglycerol acyltransferase 1 (MHC-DGAT1) transgenic (Tg) mice. Mice were treated with either saline (sham) or ANG II (500 ng·kg−1·min−1) delivered by osmotic minipump for 14 days. A–C: systolic blood pressure (BP; A), diastolic BP (B), and mean arterial pressure (MAP; C) were assessed in NTg and MHC-DGAT1 Tg mice before treatment and on day 13 postsurgery. n = 6 NTg, NTg + ANG II, and MHC-DGAT1 Tg mice and 9 MHC-DGAT1 Tg + ANG II mice. Significance is indicated. NS, not significant.
Histological analysis revealed disruption of the normal cardiac architecture, including diffuse mononuclear cell infiltrates and the accumulation of fibrosis (Fig. 2A), in MHC-DGAT1 Tg mice treated with ANG II, which was not apparent in control mice. Lipid accumulation, as assessed by oil red O staining, demonstrated an increase in MHC-DGAT1 Tg ventricles relative to NTg ventricles (Fig. 2B). Serum TG, on the other hand, did not differ significantly in NTg and MHC-DGAT1 Tg mice at baseline or with ANG II treatment (data not shown). Both lipid accumulation and ANG II treatment have independently been shown to result in cardiac fibrosis (5, 17, 65, 66, 69). Treatment of MHC-DGAT1 Tg mice with ANG II resulted in a marked increase in fibrosis, quantified as fibrotic area (Fig. 2, C and G), compared with MHC-DGAT1 Tg mice treated with saline or NTg mice treated with ANG II. Analysis of heart size revealed a significant increase in heart weight, normalized to body weight, in MHC-DGAT1 Tg mice treated with ANG II versus saline (Fig. 2D). There was a reduction in body weight noted in both NTg and MHC-DGAT1 Tg mice treated with ANG II, although this was not statistically significant (data not shown). Therefore, heart weight was also normalized to tibial length, which showed a similar increase heart size in MHC-DGAT1 Tg mice treated with ANG II (Fig. 2E). Atrial natriuretic peptide, a sensitive molecular marker of hypertrophy, was significantly increased in MHC-DGAT1 Tg versus NTg mice treated with ANG II (Fig. 2F). Expression of collagen types 1A and 3A1 (34) were also increased in MHC-DGAT1 Tg mice treated with ANG II (Fig. 2, H and I), although the interaction was not significant. Collectively, these results demonstrate an enhanced response to ANG II in the MHC-DGAT1 Tg heart.
Fig. 2.

Cardiac hypertrophy and fibrosis in MHC-DGAT1 Tg mice treated with ANG II. A–C: representative left ventricular (LV) micrographs in sham and ANG II-treated NTg and MHC-DGAT1 Tg mice stained with hematoxylin and eosin (A), oil red O (B), where red fluorescence indicates lipid accumulation, and Masson's trichrome stain (C), where blue-green stain indicates fibrosis. Bars = 50 μm. D and E: calculated heart weight-to-body weight ratios (HW/BW; D) and heart weight-to tibia-length ratios (HW/TL; E). F: atrial natriuretic peptide (ANP) gene expression normalized to GAPDH expression and compared with the NTg sham group as assessed by quantitative PCR. G: quantification of fibrotic area, expressed as a percentage of tissue area. H and I: gene expression analysis of collagen type 1A1 (Col1a1; H) and type 3A1 (Col3A1; I) normalized to GAPDH expression. n = 6 NTg, NTg + ANG II, and MHC-DGAT1 Tg mice and 9 MHC-DGAT1 Tg + ANG II mice.
Cardiac function was evaluated by echocardiography at the end of the 14-day treatment with either saline or ANG II (Table 1). Representative Doppler and M-mode examples are shown in Fig. 3, A and B. Diastolic function was assessed by Doppler analysis of the early and late ventricular filling velocities and measurement of the deceleration time of early filling velocity. We found evidence of diastolic dysfunction in MHC-DGAT1 Tg mice treated with saline, as previously reported (17), and ANG II (Fig. 3, C and D) and in NTg mice treated with ANG II (Fig. 3D), similar to a previous report (23). However, a marked reduction in LV ejection fraction and fractional shortening was seen in MHC-DGAT1 Tg mice exposed to ANG II compared with NTg mice in the presence or absence of ANG II or with MHC-DGAT1 Tg mice at baseline (Fig. 3, E and F). This suggests that ANG II exposure in the setting of cardiac lipid accumulation can result in significant systolic dysfunction.
Table 1.
Cardiac function evaluated by echocardiography at the end of the 14-day treatment with either saline or ANG II
| NTg Mice |
MHC-DGAT1 Tg Mice |
P Values |
|||||
|---|---|---|---|---|---|---|---|
| Sham | ANG II | Sham | ANG II | ANG II | Genotype | Interaction | |
| End-diastolic volume, μl | 65.22 ± 5.70 | 63.53 ± 13.07 | 66.98 ± 7.16 | 73.15 ± 16.76 | NS | NS | NS |
| End-systolic volume, μl | 23.58 ± 8.31 | 23.41 ± 8.30 | 24.72 ± 6.01 | 50.27 ± 14.46 | 0.05 | 0.05 | 0.05 |
| Interventricular septal wall, mm | 1.01 ± 0.22 | 1.26 ± 0.16 | 1.28 ± 0.21 | 1.16 ± 0.37 | NS | NS | NS |
| LV posterior wall, mm | 1.01 ± 0.20 | 1.20 ± .15 | 0.93 ± 0.15 | 0.96 ± 0.25 | NS | NS | NS |
| LV ejection fraction, % | 69.35 ± 11.12 | 70.91 ± 14.19 | 59.93 ± 3.37 | 39.94 ± 8.96 | 0.05 | 0.001 | 0.05 |
| Fractional shortening, % | 39.02 ± 8.43 | 34.35 ± 12.71 | 32.8 ± 4.65 | 19.48 ± 5.11 | 0.05 | 0.001 | 0.05 |
| E, m/s | 638.4 ± 85.4 | 728.5 ± 186 | 675.8 ± 72.2 | 655.3 ± 131.4 | NS | NS | NS |
| A, m/s | 447.3 ± 60.3 | 355.3 ± 55.27 | 423.1 ± 33.5 | 322.7 ± 133.5 | 0.01 | NS | NS |
| E/A | 1.31 ± 0.16 | 1.68 ± .51 | 1.92 ± 0.24 | 1.85 ± 0.27 | NS | 0.05 | NS |
| Deceleration time, ms | 27.3 ± 5.1 | 17.6 ± 3.2 | 19.6 ± 3.7 | 19.4 ± 1.3 | 0.01 | 0.05 | 0.05 |
| Heart rate, beats/min | 493 ± 45 | 520 ± 60 | 549 ± 50 | 481 ± 49 | NS | NS | NS |
Values are means ± SD; n =6–9 mice/group.
NTg mice, nontransgenic mice; MHC-DGAT1 Tg mice, α-myosin heavy chain promoter of diacylglycerol acyltransferase 1 (MHC-DGAT1) transgenic mice; LV, left ventricular; E, early mitral inflow filling velocity; A, late mitral filling velocity.
Significance was assessed by two-way ANOVA. NS, nonsignificant.
Fig. 3.

Cardiac function is impaired in MHC-DGAT1 Tg mice treated with ANG II. A: representative transmitral blood flow Doppler images. Mitral inflow early filling velocity (E), mitral inflow late filling (atrial contraction) velocity (A), and deceleration time (DT) are shown in the first image and quantified as the E-to-A ratio (E/A; C) and DT (D). B: representative M-mode echocardiographs from NTg and MHC-DGAT1 Tg sham and ANG II-treated mice. Intaventricular septal (IVS), LV posterior wall (LVPW), and LV internal diameter (LVID) in systole (s) and diastole (d) are shown in the first image. E and F: fractional shortening (FS; E) and LV ejection fraction (EF; F) were quantified. n = 6 NTg, NTg + ANG II, and MHC-DGAT1 Tg mice and 9 MHC-DGAT1 Tg + ANG II mice.
The effect of ANG II on the lipid profile in the MHC-DGAT1 Tg heart was examined using lipidomic analysis. There was a significant alteration in the lipid profile between NTg and MHC-DGAT1 Tg hearts, with changes identified in >20% of all measured species (P < 0.05, q = 0.05). Genotype was associated with a larger perturbation in cardiac lipids than treatment with ANG II. There were 141 significantly altered lipids identified between the 2 genotypes, and 23 significantly altered lipids were identified as due to treatment with ANG II. Major perturbations included an increase in TG levels and a decrease in ceramide levels in MHC-DGAT1 Tg hearts compared with NTg hearts (Table 2). Interestingly, treatment with ANG II resulted in a reduction in selected TG (TG 48:0 and TG 53:3) in MHC-DGAT1 Tg hearts. This was significantly and inversely correlated with increases in six different ceramide species.
Table 2.
Cer and TG levels in MHC-DGAT1 Tg hearts compared with NTg hearts
| NTg Hearts |
MHC-DGAT1 Tg Hearts |
P Values |
|||||
|---|---|---|---|---|---|---|---|
| Species | Sham | ANG II | Sham | ANG II | ANG II | Genotype | Interaction |
| Cer(d18:1/20:0) | 853 ± 190 | 1,650 ± 620 | 817 ± 130 | 1,150 ± 360 | 0.0015 | 0.09 | 0.14 |
| Cer(d16:1/23:0) | 188 ± 31 | 391 ± 120 | 200 ± 35 | 258 ± 81 | 0.0004 | 0.06 | 0.03 |
| Cer(d18:1/19:0) | 135 ± 26 | 300 ± 120 | 139 ± 310 | 188 ± 65 | 0.0013 | 0.08 | 0.06 |
| Cer(d18:1/22:0)H | 625 ± 130 | 1250 ± 430 | 664 ± 13 | 820 ± 230 | 0.0016 | 0.08 | 0.04 |
| Cer(d18:2/18:1) | 38.7 ± 6.7 | 90 ± 38 | 39.4 ± 9.6 | 54.5 ± 18 | 0.0013 | 0.06 | 0.05 |
| Cer(d18:2/20:1) | 54.8 ± 8.1 | 122 ± 47 | 58.2 ± 12 | 75.5 ± 25 | 0.0013 | 0.07 | 0.04 |
| TG 48:0 | 971 ± 510 | 594 ± 170 | 1460 ± 380 | 721 ± 250 | 0.0010 | 0.04 | 0.22 |
| TG 53:3 | 242 ± 42 | 179 ± 56 | 282 ± 45 | 210 ± 31 | 0.0014 | 0.07 | 0.77 |
Cer, ceramide (where d refers to 1,3 dihydroxy and is followed by the number of carbons in each of the side chains). The number of double bonds present is noted after the colon. Triglycerides (TG) are listed with the total number of carbons in the side chains and number of double bonds of all chains.
Lipotoxicity has been associated with the generation of ROS in a variety of experimental models (8, 47). Similarly, ANG II has been shown to promote the generation of intracellular ROS in target tissues (13). We hypothesized that the synergistic response seen when these two pathological stimuli are combined might be linked to enhanced production of ROS. We assessed ROS levels by nuclear staining with dihydroethidium, biochemical measurement of 4-hydroxynonenal, a measure of lipid peroxidation, and the DNA adduct 8-hydroxydeoxyguanosine. These were not different in NTg hearts in the presence or absence of ANG II or in MHC-DGAT1 Tg hearts treated with vehicle. However, the hearts of ANG II-treated MHC-DGAT1 Tg mice demonstrated a significant increase in ROS levels by all three measures compared with control mice (Fig. 4, A–D).
Fig. 4.

Role of ROS in MHC-DGAT1 Tg hearts treated with ANG II. A: fresh frozen LV sections stained with the ROS-sensitive dye dihydroethidium (DHE). Representative fluorescent images are shown. B: DHE-stained images were quantified and normalized to NTg control fluorescence intensity (n = 4 for each group). C: lipid peroxidation as assessed by the quantification of 4-hydroxynonenal (HNE) content in LV tissue (n = 6 for each group). D: quantification of 8-hydroxydeoxyguanosine (8-OHdG), a marker of DNA oxidative stress, in LV samples (n = 6 for each group). E: gene expression analysis of SOD2. F: SOD2 activity normalized to milligrams of soluble protein (n = 6 for each group). G: Gene expression analysis of NADPH oxidase (Nox)4. Gene expression results were normalized to GADPH expression and compared with the NTg sham group. n = 6 NTg, NTg + ANG II, and MHC-DGAT1 Tg mice and 9 MHC-DGAT1 Tg + ANG II mice.
We next examined the expression of genes known to be involved in ROS generation or degradation in the heart (42). SOD2 is a mitochondrial enzyme responsible for degrading superoxide species in that organelle. It is believed to be important regulator of ROS levels in a variety of tissues including those of the cardiovascular system (12). We found that SOD2 gene expression was decreased in MHC-DGAT1 Tg hearts (Fig. 4E), whereas SOD1 and catalase expression were unaffected (data not shown). There was no further decrease in SOD2 expression in MHC-DGAT1 Tg mice treated with ANG II. SOD2 activity was reduced in NTg mice treated with ANG II, MHC-DGAT1 Tg mice, and MHC-DGAT1 mice treated with ANG II (Fig. 4F).
ANG II has been shown to increase ROS generation through a mechanism involving activation of Nox (16), multisubunit enzymes that catalyze the production of superoxide from oxygen and NADPH. Expression of Nox subunits has been shown to be regulated by ANG II in a variety of other systems (2, 30, 58). We found that Nox4 expression was increased in ANG II-treated MHC-DGAT1 Tg mouse hearts (Fig. 4G). There were no significant differences in expression of Nox2, p22phox, p47phox, and p67phox, as assessed by quantitative PCR (data not shown).
We found that MHC-DGAT1 Tg mice exposed to ANG II demonstrated significant cardiac fibrosis (Fig. 2). Fibrosis has been linked to a variety of signaling molecules in the heart (10, 20), including TGF-β (53). We found that expression of TGF-β2 (Fig. 5A), but not TGF-β1 or TGF-β3 (data not shown), was increased in MHC-DGAT1 Tg LVs exposed to ANG II. TGF-β exerts its biological activity through binding to a cell surface receptor complex of TGFBR1 and TGFBR2 (36). We measured the relative expression of TGFBR1 and TGFBR2 and found a significant increase in TGFBR1 but not TGFBR2 gene expression in MHC-DGAT1 Tg hearts treated with ANG II (Fig. 5, B and C). Similar increases in TGFBR1 protein were demonstrated in myocardial membrane preparations (Fig. 5, D and E). The level of p-Smad2 was used as a marker of TGF-β activity (14). p-Smad2 was measured by Western blot analysis and normalized to total Smad2. Consistent with TGF-β pathway activation (14), p-SMAD2 signals were increased in MHC-DGAT1 Tg ventricular extracts treated with ANG II (Fig. 5, F and G) suggesting that ANG II, in the setting of myocyte steatosis, triggers an increase in TGF-β signaling that is likely to play a key role in the enhanced fibrosis seen in this model.
Fig. 5.

Role of transforming growth factor (TGF)-β in MHC-DGAT1 Tg hearts. A–C: gene expression analysis of TGF-β2 (A), TGF-β receptor (TGFBR)1 (B), and TGFBR2 (C). Results were normalized to GADPH expression and compared with the NTg sham group (n = 6 for each group). D: representative Western blot analysis of TGFBR1 expression (top) from membrane preparations of NTg and MHC-DGAT1 Tg hearts. Expression of β-tubulin is shown as a loading control (bottom). E: results were quantified, and TGFBR1 expression was normalized to β-tubulin (n = 4 for each group). F: representative Western blot analysis of phosphorylated (p-)Smad2 (top) and total Smad2 (bottom). G: results were quantified and normalized to total Smad2 (n = 4 for each group). H: representative Masson's trichrome-stained LV micrographs in sham and ANG II-treated NTg and MHC-DGAT1 Tg mice treated with IgG or TGF-β-neutralizing antibody (NAb), where blue-green stain indicates fibrosis. Bar = 50 μm. I: quantification of the fibrotic area, expressed as a percentage of tissue area (n = 4 for each group). J: Western blot analysis of p-Smad2 (top) and total Smad2 (bottom) in MHC-DGAT1 Tg hearts treated with ANG II and either IgG or TGF-β NAb.
TGF-β NAb has previously been shown to reduce fibrosis in models of cardiac hypertrophy (53), and so we sought to determine if blockade of TGF-β would decrease cardiac fibrosis in MHC-DGAT 1 Tg mice treated with ANG II. NTg and MHC-DGAT1 Tg mice were treated with vehicle or ANG II and TGF-β NAb or IgG control antibodies. Treatment began 1 day before placement of the osmotic pumps and continued over the 14-day course, as previously described (53). ANG II treatment resulted in a similar increase in blood pressure in TGF-β NAb- and IgG-treated mice (data not shown). Interestingly, treatment with TGF-β NAb resulted in significantly less cardiac fibrosis in MHC-DGAT1 Tg mice treated with ANG II (Fig. 5, H and I). This was associated with a reduction in ANG II-dependent p-Smad2 activity in MHC-DGAT1 Tg hearts (Fig. 5J). There was no effect of TGF-β NAb on the low levels of fibrosis in NTg mice. Similarly, there was no change in fibrosis in MHC-DGAT1 Tg mice treated with control IgG antibody (data not shown).
DISCUSSION
We have shown that MHC-DGAT1 Tg mice demonstrate a lower threshold for the development of cardiomyopathy in response to ANG II compared with NTg control mice. The relatively modest dose of ANG II (500 ng·kg−1·min−1 for 14 days) used in our study resulted in a similar increase in blood pressure in both NTg and MHC-DGAT1 Tg mice. However, cardiac size, fibrosis, and systolic dysfunction were much more prominent in MHC-DGAT1 Tg mice exposed to ANG II. We have also shown that ANG II treatment in the setting of the MHC-DGAT1 Tg model results in an increase in ROS levels in the heart and a synergistic activation of the TGF-β pathway, both key mechanisms thought to play a role in the pathogenesis of heart disease (48, 53). Our results suggest that expression of DGAT1 and the resulting alteration in lipid content render the cardiac myocyte more vulnerable to independent pathological stresses, such as those engendered by ANG II.
Echocardiographic analysis of ANG II-treated MHC-DGAT1 Tg mice demonstrated a marked reduction in systolic function compared with sham-treated MHC-DGAT1 mice or NTg mice exposed to ANG II (Fig. 3 and Table 1). The effects of ANG II on the cardiovascular system have been well described, and the use of angiotensin-converting enzyme (ACE) inhibitors as well as angiotensin receptor blockers are cardioprotective (51) in a variety of cardiovascular diseases (4). Interestingly, several studies have shown an increase in expression of the ANG II receptor in models of diabetic cardiomyopathy (15, 25). In addition, Patel et al. (41) have shown that ACE2 is increased in hearts of the diabetic Akita mouse. ACE2 metabolizes ANG II to yield ANG(1–7) and thereby functions as a negative regulator of the renin-angiotensin system. Interestingly, ACE2-null animals demonstrate increased cardiomyopathy in the setting of the diabetic Akita mouse. Our results support these previous studies and suggest that lipid accumulation in the cardiac myocyte potentiates the effect of ANG II, resulting in significant systolic dysfunction.
It should be noted that Liu et al. (31, 32) reported an independently generated, cardiac-selective transgenic mouse expressing DGAT1 that failed to show significant cardiac dysfunction. In their model, DGAT1 expression was shown to improve cardiac function in the setting of enhanced Tg expression of acyl-CoA synthetase (ACS) (MHC-DGAT1; MHC-ACS) or peroxisome proliferator-activated receptor (PPAR)-γ (MHC-DGAT1; MHC-PPAR-γ) (31, 32). Both cardiac Tg expression of ACS and PPAR-γ were shown to result in increased free fatty acid levels in the heart, which were reduced in double-DGAT1 Tg mice, suggesting that increased DGAT1 activity and the resulting sequestration of fatty acids into TG was cardioprotective. The differences between this model and that reported here are not fully understood but may be partially explained by the differences in background mouse strains used in the respective studies. The background strain in our previous report (17) and the experiments presented here were carried out using DBA2/J, whereas Liu et al. used FVB (29) or C57B6 (31, 32).
In an effort to understand the alterations in myocardial lipid content due to both MHC-DGAT1 Tg and treatment with ANG II, we performed lipidomic analysis. Interestingly, >20% of lipid species were significantly altered when NTg and MHC-DGAT1 Tg hearts were compared, whereas only 3% of lipid species were responsive to ANG II administration. There was a modest increase in select TG species in MHC-DGAT1 Tg hearts compared with NTg control hearts. However, treatment with ANG II resulted in a reduction in TG in both NTg and MHC-DGAT1 Tg mice. The role of TG in the pathogenesis of heart disease has been difficult to establish, and several studies have suggested that accumulation of TG can occur without significant cardiac dysfunction (32). In addition, sequestration of potentially toxic lipid species into a neutral TG may improve heart function in selected models of lipotoxicity (31, 32). Our results suggest that accumulation of TG may also serve as a source of toxic lipid metabolites when these hearts are exposed to a pathological stress, such as ANG II infusion. Additional support for this hypothesis is provided by studies involving Tg expression of ATGL in the heart. The ATGL Tg mouse demonstrates decreased cardiac TG and apparent protection from pressure overload resulting from thoracic aortic constriction (27). Ceramides, which have been linked to cardiac dysfunction (40), were increased in response to ANG II in both NTg and MHC-DGAT1 Tg mouse hearts. Differences in ceramide levels between the two mouse lines were not significant; if anything, the trend was toward lower levels in MHC-DGAT1 Tg versus NTg animals. This suggests that increased ceramide levels, per se, cannot fully explain the cardiovascular phenotype in MHC-DGAT1 Tg mice treated with ANG II.
Oxidative stress has been shown to be intimately involved in the genesis of cardiovascular disease, and it is thought to play a major role in ANG II-induced cardiac dysfunction. We found that measures of oxidative damage were increased in MHC-DGAT1 Tg hearts treated with ANG II. Noteworthy, at the doses used in this study, ANG II in NTg mice did not result in a significant increase in ROS. This result led us to hypothesize that the MHC-DGAT1 heart might have a reduced capacity to handle oxidative stress at baseline. Interestingly, we found that expression of the antioxidant enzyme SOD2 was reduced in MHC-DGAT1 Tg hearts. SOD2 is localized to the mitochondria, and SOD2-null mice have been shown to develop lethal cardiomyopathy (63). Cardiomyocytes isolated from heterozygous SOD2 gene knockout mice display increased oxidative damage and decreased mitochondrial function (60). It is worth noting that PPAR-γ coactivator-1α is thought to be required for SOD2 expression (49, 59), and PPAR-γ coactivator-1α expression has been shown to be decreased in the MHC-DGAT1 Tg heart (17).
ANG II increases ROS generation through a mechanism involving the activation of Nox (16), a multisubunit enzyme that catalyzes the production of superoxide from oxygen and NADPH. Nox2 and Nox4 have been shown to be expressed in the heart and are regulated by ANG II (2, 30, 58). While we saw no change in the expression of Nox2 in the MHC-DGAT1 heart, Nox4 was increased in the presence of ANG II. A previous study (1) has reported an increase in Nox4 expression in models of hypertrophy. However, the role of Nox4 in the pathogenesis of heart dysfunction is not entirely clear. Zhang et al. (67) provided evidence for a cardioprotective role for Nox4 and reported that Nox4 gene deletion resulted in increased hypertrophy and fibrosis. However, an independently derived cardiac-specific Nox4-null mouse demonstrated a reduction in cardiac hypertrophy and fibrosis in response to pressure overload (29). The reasons behind these seemingly discrepant findings are unclear, and additional studies will be required to clarify the role of Nox4 in MHC-DGAT1 Tg mice treated with ANG II. It is noteworthy that both SOD2 and Nox4 are expressed in the mitochondria. There is a well-known link between mitochondrial dysfunction and cardiomyopathy as well as recent evidence suggesting that the mitochondrion is a major target of ANG II-dependent oxidative stress (11, 12).
Expression of DGAT1 leads to cardiac fibrosis (17), which is increased in the setting of ANG II treatment, similar to what has been demonstrated in other animal models (9, 56, 65, 66). Fibrosis is also seen in the inducible, cardiac myocyte-selective ATGL-null mouse, implying that myocyte lipid accumulation results in a pathological signal that promotes a profibrotic response in the cardiac fibroblast (26). The TGF-β signaling system has been shown to be a key mediator of fibrosis in the heart (53). ANG II has been shown to enhance expression of TGF-β in the myocyte, and Schultz et al. (44), using the TGF-β−/− Rag1−/− mouse, have shown that TGF-β is necessary for ANG II-induced hypertrophy and cardiac dysfunction. We demonstrated a reduction in TGF-β activity, as assessed by reduced p-Smad levels, and myocardial fibrosis in the presence of TGF-β NAb, confirming the importance of this pathway in mediating fibrosis. At the ANG II concentrations used in our study, we did not see increased expression or enhanced activity of the TGF-β pathway in the absence of cardiac steatosis. However, in MHC-DGAT1 Tg mice, we saw an increase in cardiac expression of TGF-β2 and TGFBR1 as well as activation of TGF-β signaling after treatment with ANG II. The fact that such changes were only seen in the presence of steatosis suggests that lipid accumulation modulates ANG signaling pathways in a manner that leads to an amplification in the profibrotic activity of the TGF-β system.
In summary, the presence of cardiac steatosis in the MHC-DGAT 1 Tg mouse potentiates the deleterious effects of ANG II in the heart. This synergistic interaction appears to promote activation of ROS and TGFβ-dependent signaling pathways. Cardiac lipid accumulation, as seen in diabetes mellitus and morbid obesity, often coexists with other pathological factors that promote cardiovascular disease (e.g., hypertension and ischemic heart disease). The latter are often accompanied by activation of the renin-angiotensin system. Collectively, our studies would suggest that this latter activation may be a more potent stimulus for cardiac dysfunction and ultimately cardiac disease when coupled with steatosis of the cardiac myocyte.
GRANTS
This work was supported by National Institutes of Health Grants HL-45637 (to D. G. Gardner), HL-086158 (to D. J. Glenn), and 1-U24-DK-097154 (to O. Fiehn and D. Grapov) and by funding from the University of California-San Francisco Academic Senate (to D. G. Gardner and D. J. Glenn).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.J.G. and D.G.G. conception and design of research; D.J.G., M.C.C., W.N., Y.Z., D.G., and O.F. performed experiments; D.J.G., M.C.C., W.N., D.G., and O.F. analyzed data; D.J.G., M.C.C., and D.G.G. interpreted results of experiments; D.J.G. prepared figures; D.J.G. drafted manuscript; D.J.G. and D.G.G. edited and revised manuscript; D.J.G., M.C.C., and W.N. approved final version of manuscript.
REFERENCES
- 1.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res 106: 1253–1264, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-Induced cardiac hypertrophy in mice. Circulation 105: 293–296, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J Royal Stat Soc Series B Methodol 57: 289–300, 1995. [Google Scholar]
- 4.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation 115: 3213–3223, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Brecher P. Angiotensin II and cardiac fibrosis. Trends Cardiovasc Med 6: 193–198, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol 296: H1660–H1665, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res 96: 225–233, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107: 813–822, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christoffersen C, Bollano E, Lindegaard MLS, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144: 3483–3490, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res 89: 265–272, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal 16: 1492–1526, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108: 837–846, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Cavanagh EMV, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res 89: 31–40, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol 51: 600–606, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, Kajstura J. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin II- dependent. Lab Invest 80: 513–527, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302: 148–158, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glenn DJ, Wang F, Nishimoto M, Cruz MC, Uchida Y, Holleran WM, Zhang Y, Yeghiazarians Y, Gardner DG. A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension 57: 216–222, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab 15: 805–812, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grapov D, Newman JW. imDEV: a graphical user interface to R multivariate analysis tools in Microsoft Excel. Bioinformatics 28: 2288–2290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 122: 23–35, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312: 734–737, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 17: 1076–1085, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichihara S, Senbonmatsu T, Price E, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation 104: 346–351, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med 347: 305–313, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Khatter JC, Sadri P, Zhang M, Hoeschen RJ. Myocardial angiotensin II (Ang II) receptors in diabetic rats. Ann NY Acad Sci 793: 466–472, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Kienesberger PC, Pulinilkunnil T, Nagendran J, Young ME, Bogner-Strauss JG, Hackl H, Khadour R, Heydari E, Haemmerle G, Zechner R, Kershaw EE, Dyck JR. Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc Res 99: 442–451, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kienesberger PC, Pulinilkunnil T, Sung MMY, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 32: 740–750, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koopman R, Schaart G, Hesselink MK. Optimisation of oil red O staining permits combination with immunofluorescence and automated quantification of lipids. Histochem Cell Biol 116: 63–68, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA 107: 15565–15570, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. J Biol Chem 278: 12094–12100, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284: 36312–36323, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Yu S, Khan RS, Homma S, Schulze PC, Blaner WS, Yin Y, Goldberg IJ. Diacylglycerol acyl transferase 1 overexpression detoxifies cardiac lipids in PPARγ transgenic mice. J Lipid Res 53: 1482–1492, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis. Circ Res 91: 1103–1113, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F, D'Amico M, Paolisso G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J Lipid Res 50: 2314–2323, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massagué J. TGFβ in cancer. Cell 134: 215–230, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 116: 1170–1175, 2007. [DOI] [PubMed] [Google Scholar]
- 38.McGavock JM, Victor RG, Unger RH, Szczepaniak LS. Adiposity of the heart, revisited. Ann Intern Med 144: 517–524, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 48: 1229–1237, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49: 2101–2112, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel VB, Bodiga S, Basu R, Das SK, Wang W, Wang Z, Lo J, Grant MB, Zhong J, Kassiri Z, Oudit GY. Loss of angiotensin-converting enzyme-2 exacerbates diabetic cardiovascular complications and leads to systolic and vascular dysfunction: a critical role of the angiotensin II/AT1 receptor axis. Circ Res 110: 1322–1335, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41a.R Core Team. R: a Language and Environment for Statistical Computing (online). http://web.mit.edu/r_v3.0.1/fullrefman.pdf [10 December 2014].
- 42.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med 50: 777–793, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet 369: 1208–1219, 2007. [DOI] [PubMed] [Google Scholar]
- 44.Schultz JE, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest 109: 787–796, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004. [DOI] [PubMed] [Google Scholar]
- 47.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 55: 798–805, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 8: 132–140, 2002. [DOI] [PubMed] [Google Scholar]
- 49.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127: 397–408, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Sugino H, Ozono R, Kurisu S, Matsuura H, Ishida M, Oshima T, Kambe M, Teranishi Y, Masaki H, Matsubara H. Apoptosis is not increased in myocardium overexpressing type 2 angiotensin II receptor in transgenic mice. Hypertension 37: 1394–1398, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Symeonides P, Koulouris S, Vratsista E, Triantafyllou K, Ioannidis G, Thalassinos N, Katritsis D. Both ramipril and telmisartan reverse indices of early diabetic cardiomyopathy: a comparative study. Eur J Echocardiogr 8: 480–486, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires TGF-β. J Clin Invest 120: 3520–3529, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thakker GD, Frangogiannis NG, Bujak M, Zymek P, Gaubatz JW, Reddy AK, Taffet G, Michael LH, Entman ML, Ballantyne CM. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am J Physiol Heart Circ Physiol 291: H2504–H2514, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Trygg J, Wold S. Orthogonal projections to latent structures (O-PLS). J Chemometrics 16: 119–128, 2002. [Google Scholar]
- 56.Ueno M, Suzuki J, Zenimaru Y, Takahashi S, Koizumi T, Noriki S, Yamaguchi O, Otsu K, Shen WJ, Kraemer FB, Miyamori I. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am J Physiol Endocrinol Metab 294: E1109–E1118, 2008. [DOI] [PubMed] [Google Scholar]
- 57.Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology 144: 5159–5165, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II induced hypertrophy in vascular smooth muscle cells. J Biol Chem 271: 23317–23321, 1996. [DOI] [PubMed] [Google Scholar]
- 59.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res 66: 562–573, 2005. [DOI] [PubMed] [Google Scholar]
- 60.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol 281: H1422–H1432, 2001. [DOI] [PubMed] [Google Scholar]
- 61.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta 1801: 311–319, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep 14: 517–531, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273: 28510–28515, 1998. [DOI] [PubMed] [Google Scholar]
- 64.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 111: 419–426, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaman AK, Fujii S, Goto D, Furumoto T, Mishima T, Nakai Y, Dong J, Imagawa S, Sobel BE, Kitabatake A. Salutary effects of attenuation of angiotensin II on coronary perivascular fibrosis associated with insulin resistance and obesity. J Mol Cell Cardiol 37: 525–535, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Zaman AK, Fujii S, Sawa H, Goto D, Ishimori N, Watano K, Kaneko T, Furumoto T, Sugawara T, Sakuma I, Kitabatake A, Sobel BE. Angiotensin-converting enzyme inhibition attenuates hypofibrinolysis and reduces cardiac perivascular fibrosis in genetically obese diabetic mice. Circulation 103: 3123–3128, 2001. [DOI] [PubMed] [Google Scholar]
- 67.Zhang M, Brewer AC, Schröder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA 107: 18121–18126, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Takagawa J, Sievers RE, Khan MF, Viswanathan MN, Springer ML, Foster E, Yeghiazarians Y. Validation of the wall motion score and myocardial performance indexes as novel techniques to assess cardiac function in mice after myocardial infarction. Am J Physiol Heart Circ Physiol 292: H1187–H1192, 2007. [DOI] [PubMed] [Google Scholar]
- 69.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc Natl Acad Sci USA 97: 1784–1789, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]