

Abstract
The antileukemic agent asparaginase triggers the amino acid response (AAR) in the liver by activating the eukaryotic initiation factor 2 (eIF2) kinase general control nonderepressible 2 (GCN2). To explore the mechanism by which AAR induction is necessary to mitigate hepatic lipid accumulation and prevent liver dysfunction during continued asparaginase treatment, wild-type and Gcn2 null mice were injected once daily with asparaginase or phosphate buffered saline for up to 14 days. Asparaginase induced mRNA expression of multiple AAR genes and greatly increased circulating concentrations of the metabolic hormone fibroblast growth factor 21 (FGF21) independent of food intake. Loss of Gcn2 precluded mRNA expression and circulating levels of FGF21 and blocked mRNA expression of multiple genes regulating lipid synthesis and metabolism including Fas, Ppara, Pparg, Acadm, and Scd1 in both liver and white adipose tissue. Furthermore, rates of triglyceride export and protein expression of apolipoproteinB-100 were significantly reduced in the livers of Gcn2 null mice treated with asparaginase, providing a mechanistic basis for the increase in hepatic lipid content. Loss of AAR-regulated antioxidant defenses in Gcn2 null livers was signified by reduced Gpx1 gene expression alongside increased lipid peroxidation. Substantial reductions in antithrombin III hepatic expression and activity in the blood of asparaginase-treated Gcn2 null mice indicated liver dysfunction. These results suggest that the ability of the liver to adapt to prolonged asparaginase treatment is influenced by GCN2-directed regulation of FGF21 and oxidative defenses, which, when lost, corresponds with maladaptive effects on lipid metabolism and hemostasis.
Keywords: general control nonderepressible 2, amino acid response, antithrombin III, apolipoprotein B-100, eukaryotic initiation factor 2, fibroblast growth factor 21, liver, steatosis, adipose, mice, oxidative stress
nutrient control of mammalian protein synthesis involves a complex interplay of regulators including the eukaryotic initiation factor 2 (eIF2) kinase general control nonderepressible 2 (GCN2). GCN2 is a direct sensor of amino acid supply, activated via binding deacylated tRNAs proximal to the ribosome (41). Activation of GCN2 during dietary amino acid deprivation or imbalance increases eIF2 phosphorylation (2, 25). Phosphorylation of eIF2 functions to slow or stall the initiation step of mRNA translation, reducing general protein synthesis rates (6). The resultant delay in reinitiation rate also alters gene-specific mRNA translation, so as to favor the synthesis of a subset of basic leucine zipper (bZIP) transcription factors that contain upstream open reading frames in the 5′-untranslated region. Synthesis of these bZIP proteins, such as activating transcription factor 4 (ATF4) (48), collectively alters mRNA expression of other bZIP factors as well as genes that alter amino acid synthesis and metabolism, nutrient uptake, redox status, and cell cycle control (26, 27). This realignment of gene expression is called the amino acid response (AAR), and its homeostatic function helps the organism accommodate and if possible adapt to amino acid-deprived conditions (33).
Previous reports from our laboratory and others demonstrate that mice deleted for Gcn2 fail to phosphorylate eIF2 during both dietary and pharmaceutical amino acid deprivation, resulting in hepatic steatosis and liver injury (2, 12, 24, 43, 50). Depletion of circulating asparagine and glutamine is accomplished by asparaginase (37, 39), an effective tumor killing agent in patients diagnosed with acute lymphoblastic leukemia. Among the metabolic complications that correlate with asparaginase, hepatic steatosis and dysfunction feature prominently (13, 21, 42). A means to predict or prevent metabolic complications by asparaginase remains elusive.
In intact mice, asparaginase promotes hepatic lipid accumulation. The degree of steatosis is maximal during the first week of treatment, resolving steadily thereafter (20). To determine if loss of the AAR precludes resolution of hepatic stress to asparaginase, treatment was extended to 14 days in both wild-type and Gcn2 null mice. To clarify the relationship between hepatotoxicity and lipid metabolism during asparaginase treatment, we endeavored to understand the role of the AAR in preventing and/or resolving liver steatosis during asparaginase. To this end, we examined a series of genes and proteins involved in lipid metabolism, including metabolic hormone fibroblast growth factor 21 (FGF21) by the AAR. FGF21 is a circulating hormone that induces hepatic fat oxidation, ketogenesis, and gluconeogenesis in the liver in response to fasting (51). Recent studies demonstrate that amino acid-deficient diets increase expression of FGF21 via ATF4 (18, 34). Herein, we describe for the first time that asparaginase promotes expression of FGF21 in a GCN2-dependent fashion. Loss of FGF21 expression during asparaginase treatment in the GCN2 null mouse corresponded to deranged lipid homeostasis in liver and white adipose tissue, promoting lipotoxicity and hepatic dysfunction.
MATERIALS AND METHODS
Animals and diets.
Male and female Gcn2−/− mice (backcrossed to C57BL/6J for 10 generations) and C57BL/6J wild-type control mice (Jackson Laboratories, Bar Harbor, ME) aged 8–9 wk were housed in plastic cages with wired bottoms. Mice were pair fed the same amount of commercial rodent chow (5001 Laboratory Rodent Diet; LabDiet) as that consumed by Gcn2−/− mice given asparaginase. Mice were exposed to a 12:12-h light-dark cycle and had free access to water. All animal protocols were approved by the Institutional Animal Care and Use Committees at Rutgers, The State University of New Jersey.
l-Asparaginase activity.
l-Asparaginase is an antileukemic agent that depletes circulating levels of asparagine. The activity of asparaginase derived from Escherichia coli (Elspar; Merck) was determined by the Nesslerization technique, as previously described (11, 43). Briefly, the production of ammonia by asparaginase over time was measured and compared with known ammonia standards. Resulting values represented the activity of the enzyme in international units (IU), where one IU equaled the amount of enzyme that catalyzed the formation of 1 μmol of ammonia/min.
Experimental procedure.
Mice received once-daily intraperitoneal injections of either l-asparaginase (3 IU per g body wt) or an equivalent volume of PBS for up to 14 days. The experimental design consisted of four treatment groups: WP, C57BL/6J wild-type mice injected with PBS; WA, wild-type mice injected with asparaginase; GP, Gcn2−/− mice injected with PBS; and GA, Gcn2−/− mice injected with asparaginase. At the start of the experiment, average body weight of each treatment group was ∼20 g (individual mice in each group ranged 17–25 g). Mice that lost greater than ∼20% body wt from the start of treatment and displayed severe morbidity were euthanized. Seven hours after the final injection, mice were euthanized by decapitation and trunk blood was collected to obtain serum. Whole organs or tissues were rapidly dissected, rinsed in ice-cold PBS, blotted, and weighed.
Histology.
Tissues fixed in 4% paraformaldehyde were frozen and then sectioned (10 μm) using a cryostat. Frozen sections were stained with Oil Red O to visualize lipid as described previously (50). Paraffin-embedded liver specimens were sectioned (6 μm) and stained with hematoxylin and eosin to visualize general histology and identify histopathological features under light microscopy.
Hepatic triglyceride secretion.
Very low density lipoprotein-triglyceride (VLDL-TG) secretion rates were calculated from the slope of the regression line describing the rate of rise in blood triglycerides during blockage of intravascular lipolysis after tyloxapol injection (Triton WR1339; Sigma-Aldrich, St. Louis, MO). Tyloxapol is an inhibitor of blood triglyceride clearance, as previously described (28). Briefly, mice were food deprived overnight, and after morning treatment with asparaginase or PBS, mice were intraperitoneally injected with tyloxapol at a dose of 500 mg/kg. A drop of blood was drawn at 20-min intervals, and blood triglyceride concentrations were measured immediately with a handheld meter and test strips (Polymer Technology Systems; CardioChek, Indianapolis, IN). Tyloxapol blocks circulating triglyceride degradation, such that blood triglyceride levels rise linearly, and the linear regression slope (change in concentration/time) indicates secretion rate (28). Rates in units of mg·dl−1·min−1 were converted to absolute rates (mg/min) based on the assumption of 110 ml blood/kg body wt. We consistently found high linearity (R2 ∼0.99) with sufficient data points for precise calculation of slope. Following assessment of the triglyceride secretion rate, mice were euthanized by carbon dioxide inhalation.
Hepatic triglyceride concentration.
Triglycerides were measured from frozen liver tissue (∼40 mg) using the Biovision Colorimetric Triglyceride Quantification Kit (Mountain View, CA), following the manufacturer's instructions.
Protein carbonylation.
Carbonylated proteins are a biomarker of oxidative stress and were measured in livers using the Biovision Protein Carbonyl Content Assay Kit, following the manufacturer's instructions. Briefly, protein carbonyls were tagged by 2,4-dinitrophenyl hydrazones, and the reactant product was measured by spectrophometry analysis at ∼375 nm using spectramax plate reader (Molecular Devices, Sunnyvale, CA).
Lipid peroxidation.
Malondialdehyde (MDA) is a product of lipid peroxidation. The thiobarbituric acid-reactive substance (TBARS) assay is a well-established assay for monitoring and screening lipid peroxidation. MDA forms a 1:2 adduct with thiobarituric acid (TBA) and can be measured colorimetrically using an MDA equivalent standard. We detected the abundance of MDA in liver samples using the OxiSelect TBARS assay kit (Cell Biolabs, San Diego, CA), following the manufacturer's instructions.
Body composition analysis.
Body composition of the mice was determined by magnetic resonance using an EchoMRI instrument (Echo Medical Systems, Houston, TX). Conscious mice were briefly (<6 min) placed in a plastic restraining tube for the period of measurement.
Serum measurements.
The anti-FXa activity of anti-thrombin III (ATIII) in plasma was determined by BIOPHEN Antithrombin 2.5 Kit (ANIARA, West Chester, OH) following to the manufacturer's instructions as previously reported (7a). Protein concentrations of FGF21 were analyzed by solid phase sandwich enzyme-linked immunosorbent assay using the Quantikine Mouse/Rat FGF-21 immunoassay (R&D Systems).
Immunoblot analysis.
Weighed, frozen tissues were transferred to microfuge tubes on ice, and protein lysates were prepared. The homogenates were immediately isolated by centrifugation at 10,000 g for 10 min at 4°C, and measurements of indicated protein levels and phosphorylation states were carried out as previously described (12). Phosphorylation of eIF2 was assessed using an antibody that recognizes the alpha subunit only when it is phosphorylated at serine 51 (Cell Signaling Technology, Beverly, MA); results were normalized for total eIF2α levels with an antibody that recognizes the protein irrespective of phosphorylation state (Santa Cruz Biotechnology, Santa Cruz, CA). Phosphorylation of AMP-activated protein kinase-α (AMPKα) was assessed using an antibody that recognizes the alpha subunit only when it is phosphorylated at threonine 172 (Cell Signaling Technology); results were normalized for total AMPKα levels with an antibody that recognizes the protein irrespective of phosphorylation state (Cell Signaling Technology). Phosphorylation of acetyl-CoA carboxylase (ACC) was assessed using an antibody that recognizes phosphorylation at serine 79 (Cell Signaling Technology); results were normalized for total ACC levels with an antibody that recognizes the protein independent of phosphorylation state (Cell Signaling Technology). ATIII was assessed using an antibody that detects alpha and beta forms of ATIII (Epitomics, Burlingame, CA). Microsomal triglyceride transfer protein (MTP) was purchased from BD Biosciences (San Jose, CA). Apolipoprotein B-100 (ApoB-100) was measured after electrophoresis of samples on 3 to 20% SDS-acrylamide gradient gels. Samples were loaded onto the gradient gel using a 3% SDS-acrylamide stacking gel. After blotting, ApoB was measured using an antibody that detects both ApoB-48 and -100 (Abcam, Cambridge, MA). ApoB-48 was also detected and reacted similarly to ApoB-100. Fatty acid synthase (FAS) was purchased from Cell Signaling Technology, and CHOP/GADD153 antibody was purchased from Santa Cruz Biotechnology (sc-7351). Antibodies to ATF4 (1:500 dilution) and ATF5 (1:300 dilution) were prepared and affinity purified as previously described (53). ATIII, MTP, ApoB-100, and FAS were normalized by glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Blots were developed with enhanced chemiluminesence (Amersham Biosciences). Protein expression was analyzed using a Carestream Gel Logic 6000 imager, and band density was quantitated using carestream molecular imaging software (version 5.0).
Quantitative real-time PCR.
Total RNA was extracted from frozen tissue using TriReagent (Molecular Research Center, Cincinnati, OH) followed by DNase treatment (VersaGene DNase Kit; Gentra Systems) (12). The A260/280 ratio was between 1.8 and 2.0 following RNA purification using the RNeasy mini kit (Qiagen). Levels of mRNAs were determined by quantitative PCR using TaqMan reagents. In this PCR assay, 1 μg of the purified RNA solutions was used for reverse transcription, which was carried out utilizing the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). TaqMan Gene Expression Master Mix and TaqMan Gene Expression Assays (Applied Biosystems) were then used to conduct quantitative PCR.
Amplification and detection were performed using the StepOnePlus Real-Time PCR System (Applied Biosystems) (12). Each mRNA from a single biological sample was measured in triplicate and normalized to 18S ribosomal RNA. Results were obtained by the comparative Ct method and are expressed as fold change with respect to the experimental control (WP).
Data analysis and statistics.
Experimental results were analyzed using SPSS Statistics software (IBM). Data were analyzed using two-way ANOVA to assess main vs. interaction effects, with drug treatment and mouse strain as independent variables. When interaction effects were significant, differences between individual treatment groups were assessed using the least significant difference post hoc test. Means not sharing a common superscripted letter were considered statistically different. The data presented are expressed as means ± SE. The level of significance was set at P < 0.05 for all statistical tests.
RESULTS
GCN2 deletion renders mice unable to survive continued asparaginase treatment.
Asparagine is essential for the growth of lymphoblastic tumors but dispensable for normal tissues (30). Tumor cells that have lost the ability to make asparagine cannot adapt to asparaginase and die (7). On the other hand, somatic tissues possess the capacity to adapt to asparaginase in part via AAR induction of cellular processes (50). The role of GCN2 in the mechanism by which the liver adapts to asparaginase was assessed by injecting asparaginase into wild-type (WA) and Gcn2 null (GA) mice once daily for up to 14 days and comparing outcomes to excipient-treated wild-type (WP) and Gcn2 null (GP) mice. To avoid studying complications secondary to severe morbidity, mice were euthanized when they lost >20% body wt and appeared unwell. Accordingly, all GA mice were euthanized between 8 and 12 days of treatment due to their deterioration in general appearance and behavior concomitant with significant weight loss (Fig. 1, A and B). In contrast, WA mice experienced little to no reduction in body weight and appeared healthy throughout the entire 14-day experiment. Asparaginase significantly reduced food intake in GA mice (Fig. 1C); therefore, subsequent experiments were performed by pair feeding all animals the same amount as the GA mice, which consumed the least amount of food. In these controlled feeding trials, asparaginase treatment still reduced body weight below saline-injected mice (two-way ANOVA P < 0.05 main effect for treatment) with losses in GA mice greatest (Fig. 1D). The pair-feeding trial was conducted for only 8 days to avoid studying secondary morbidities in GA mice.
Fig. 1.

General control nonderepressible 2 (Gcn2) null mice fail to adapt to prolonged asparaginase treatment. A: percentage of each experimental cohort (n = 8–12 per group at day 0) retained in the experiment over the 14-day treatment schedule. None of the GA mice completed the study due to significant loss in body weight coupled with behavioral morbidity. B: body weight change of mice injected with saline or asparaginase between days 0 and 14. C: average daily food intake of mice fed ad libitum for up to 14 days (black bars) or pair fed the same as amount as the GA group for 8 days (white bars). D: body weight change of pair-fed mice injected with saline or asparaginase between days 0 and 8. Values are means ± SE. #Main effect of asparaginase treatment to reduce body weight. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect). WP, wild-type mice given PBS; WA, wild-type mice injected with asparaginase; GP, Gcn2 null mice injected with PBS; GA, Gcn2 null mice injected with asparaginase.
Deletion of GCN2 promotes hepatic lipid accumulation during prolonged asparaginase treatment.
To compare the histopathology resulting from asparaginase treatment in wild-type vs. Gcn2 null mice, frozen liver samples were sectioned and stained to visualize general cellular structure and neutral lipid content. Liver sections taken from WA mice showed minimal changes in histological appearance and low levels of Oil Red O (Fig. 2, A and B). By comparison, GA liver sections showed strong Oil Red O staining with well-defined enlarged fat droplets in the cytosol relative to GP controls, reflecting lipid accumulation. Furthermore, a small but significant increase in liver weight relative to body weight was noted in GA but not WA mice (Fig. 2C). Biochemical analysis confirmed an approximately fourfold increase in triglyceride content in GA livers relative to GP controls, whereas triglyceride content in WA livers was similar to WP controls (Fig. 2D). In a previous experiment, WA mice showed increased hepatic triglyceride after 6 days (50). Thus WA liver had adapted to the amino acid stress, whereas livers from Gcn2 null mice continued to decline metabolically.
Fig. 2.
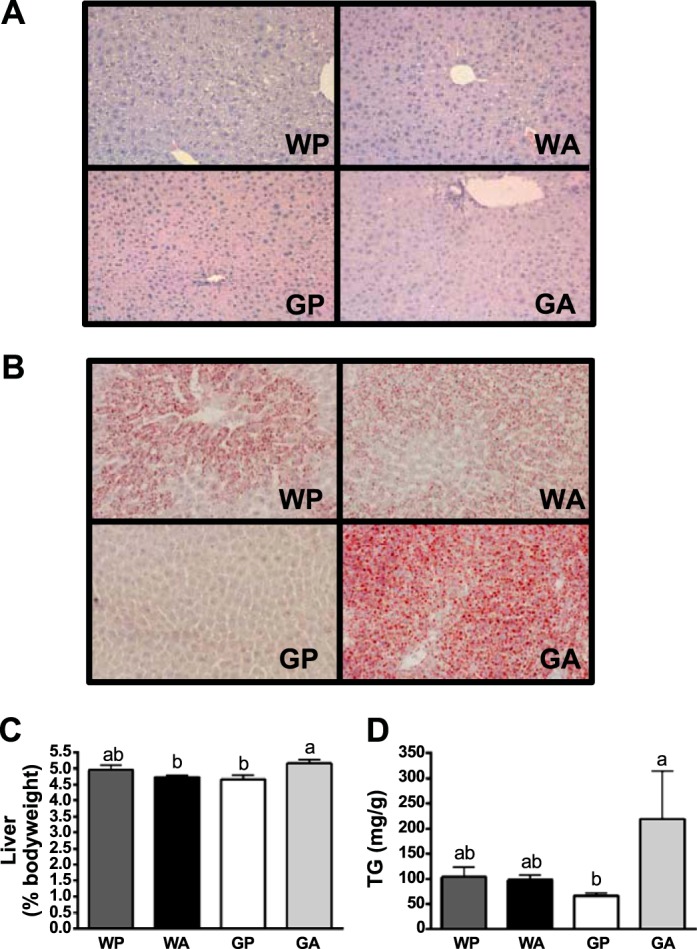
Asparaginase treatment promotes lipid accumulation in liver of Gcn2 null mice. A: paraffin-embedded liver sections (6 μm) were stained with hematoxylin and eosin and images taken using a ×20 objective to visualize general cellular structure. B: neutral lipid content of liver was examined by Oil Red O stain and images captured using a ×20 objective. C: liver mass, expressed relative to body weight. D: liver triglyceride concentrations. Values are means ± SE; n = 5–8 per group. All mice were pair fed the same amount as the GA group. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
GCN2 is the primary regulator of the AAR in liver during prolonged asparaginase treatment.
Consistent with our previous work reporting induction of the AAR in liver by asparaginase following both 1 and 6 days of treatment (12, 50), eIF2 phosphorylation was greatly elevated in WA livers compared with WP controls (Fig. 3A). Treatment of control mice with asparaginase increased hepatic mRNA levels of the transcription factors Atf4 and Atf5, as well as their target genes, eukaryotic translation initiation factor 4E-binding protein 1 (4ebp1), asparagine synthetase (Asns), and CAAT enhancer binding protein homologous protein (Chop) (Fig. 3B). In stark contrast, induction of the hepatic AAR as a result of asparaginase treatment was completely blocked in Gcn2 null mice, a result again consistent with previous reports (12, 50). Examination of protein expression revealed very low or no ATF4 protein expression across treatment groups (data not shown). ATF5 protein increased slightly (P = 0.08) in wild-type mice treated with asparaginase (Fig. 3C), whereas CHOP protein expression significantly increased in the livers of WA but not GA mice (Fig. 3D).
Fig. 3.

Activation of the amino acid response (AAR) pathway in liver by asparaginase requires GCN2. A: asparaginase increases eukaryotic initiation factor 2 (eIF2) phosphorylation in wild-type but not Gcn2 null mice. Data are expressed as band density of serine 51 phosphorylated eIF2 relative to total expression levels of eIF2 alpha subunit by immunoblot analysis. B: quantitative real-time PCR was employed to examine mRNA expression of Atf4, Atf5, Asns, 4ebp1, and Chop. C and D: asparaginase increases protein expression of activating transcription factor 5 (ATF5) and CAAT enhancer binding protein homologous protein (CHOP) in the liver of wild-type but not Gcn2 null mice by immunoblot analysis. All mice were pair fed the same amount as the GA group. Values are means ± SE; n = 5–8 per group except in C and D, which was n = 3–5 per group. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
GCN2 regulates liver FGF21 expression and lipid metabolism during asparaginase treatment.
We next addressed possible mechanisms for the accumulation of lipids in the liver of GA mice. One powerful regulator of hepatic metabolism is FGF21, a peptide hormone expressed in numerous tissues including the liver. Circulating concentrations of FGF21 are reportedly derived from the liver (35). FGF21 causes pleiotropic effects on metabolism, including regulation of fat oxidation, ketogenesis, and gluconeogenesis in the liver (51). Recent reports collectively show hepatic expression and circulating levels of Fgf21 increased by amino acid-deficient diets in a manner requiring GCN2 (18, 34). Similar to protein deficiency, we found an ∼17-fold increase in Fgf21 mRNA levels in liver and an ∼20-fold increase in serum FGF21 protein concentration in WA mice, relative to WP controls (Fig. 4, A and B). Conversely, GP and GA mice possessed low levels of hepatic Fgf21 mRNA and serum Fgf21 levels. An ∼13-fold increase in Fgf21 mRNA expression occurred after a single injection of asparaginase, which was also GCN2 dependent (WP: 0.74 ± 0.35; WA: 9.53 ± 4.58; GP: 1.19 ± 0.28; and GA: 0.92 ± 0.46). Thus hepatic expression and circulating concentrations of FGF21 by asparaginase required GCN2.
Fig. 4.

Induction of fibroblast growth factor 21 (FGF21) by asparaginase requires GCN2. A: hepatic Fgf21 mRNA expression was determined by quantitative real-time PCR. B: FGF21 protein concentration in serum analyzed by enzyme-linked immunoassay. All mice were pair fed the same amount as the GA group. Values are means ± SE; n = 5–8 per group. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
FGF21 is reported to regulate energy metabolism and mitochondrial oxidative function through activation of AMPK and sirtuin (SIRT1) (15). In the current study, large increases in circulating FGF21 in wild-type mice treated with asparaginase were not accompanied by increases in phosphorylation of AMPK or expression of downstream target genes such as peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) (Table 1). Rather, in GA mice, as FGF21 levels were depressed, so was the expression of many hepatic genes involved in mitochondrial function, at least directionally if not significantly. For example, phosphorylation of ACC, a marker of AMPK activity, decreased 25% in GA relative to GP controls (P = 0.07), and expression levels of Pgc1α and Tfam mRNAs were reduced 45% (P = 0.09) and 54% (P = 0.06), respectively. Similar directional trends were also noted in Sirt and Nrf1 mRNA, in total suggesting compromised mitochondrial function in the liver of GA mice.
Table 1.
Liver and adipose responses following 8 days asparaginase treatment in wild-type mice and mice deleted for GCN2
| WP | WA | GP | GA | Strain | Treatment | S × T | |
|---|---|---|---|---|---|---|---|
| Liver | |||||||
| p-AMPKα-Thr172/T-AMPKα | 8.54 ± 1.59 | 9.35 ± 1.60 | 8.33 ± 1.28 | 8.98 ± 1.05 | No | No | No |
| p-ACC-Ser79/T-ACC | 9.73 ± 1.58 | 10.63 ± 1.80 | 8.74 ± 1.01 | 6.53 ± 0.70 | No | No | 0.07 |
| Pgc1α mRNA | 0.83 ± 0.18 | 0.85 ± 0.12 | 1.04 ± 0.21 | 0.57 ± 0.09 | No | No | 0.09 |
| Sirt1 mRNA | 0.94 ± 0.17 | 1.18 ± 0.24 | 1.11 ± 0.21 | 0.83 ± 0.12 | No | No | No |
| Tfam mRNA | 1.00 ± 0.18 | 1.15 ± 0.18 | 1.16 ± 0.22 | 0.53 ± 0.09 | No | No | 0.06 |
| Nrf1 mRNA | 0.91 ± 0.16 | 1.18 ± 0.24 | 1.26 ± 0.27 | 0.82 ± 0.13 | No | No | No |
| Cd36 mRNA | 0.98 ± 0.19 | 0.98 ± 0.18 | 1.05 ± 0.15 | 0.92 ± 0.07 | No | No | No |
| Srebp1 mRNA | 1.13 ± 0.16 | 0.98 ± 0.23 | 1.13 ± 0.18 | 0.84 ± 0.05 | No | No | No |
| Srebp2 mRNA | 1.31 ± 0.22 | 1.14 ± 0.09 | 1.08 ± 0.14 | 0.81 ± 0.27 | No | No | No |
| G6pc | 1.12 ± 0.22 | 1.41 ± 0.39 | 1.12 ± 0.11 | 1.09 ± 0.18 | No | No | No |
| Fsp27 mRNA | 1.05 ± 0.17 | 1.25 ± 0.22 | 1.15 ± 0.25 | 0.85 ± 0.39 | No | No | No |
| Cidea mRNA | 3.65 ± 1.86 | 5.27 ± 2.65 | 7.45 ± 1.80 | 3.02 ± 2.12 | No | No | No |
| Protein carbonylation | 74.27 ± 8.02 | 72.32 ± 7.17 | 78.60 ± 5.08 | 74.70 ± 8.98 | No | No | No |
| White adipose | |||||||
| Asns mRNA | 1.01 ± 0.12 | 0.86 ± 0.06 | 0.83 ± 0.11 | 0.33 ± 0.05 | No | No | 0.05 |
| Pgc1α mRNA | 1.12 ± 0.25 | 1.21 ± 0.17 | 1.23 ± 0.22 | 0.70 ± 0.17 | No | No | 0.1 |
| Sirt1 mRNA | 1.06 ± 0.17 | 1.35 ± 0.14 | 2.04 ± 0.53 | 0.89 ± 0.18 | No | No | No |
| Tfam mRNA | 1.37 ± 0.47 | 0.92 ± 0.08 | 1.18 ± 0.25 | 0.41 ± 0.05 | No | No | 0.1 |
| Nrf1 mRNA | 1.03 ± 0.14 | 1.30 ± 0.16 | 2.03 ± 0.44 | 0.95 ± 0.19 | No | No | No |
| Cd36 mRNA | 1.02 ± 0.10 | 1.73 ± 0.32 | 1.43 ± 0.33 | 0.83 ± 0.15 | No | No | 0.09 |
Values are means ± SE; n = 5–8 per group. WP, wild-type mice injected with PBS; WA, wild-type mice injected with asparaginase; GP, general control nonderepressible 2 null (Gcn2 null) mice injected with PBS; GA, Gcn2 null mice injected with asparaginase. Mice were pair fed the same amount as the GA group. Quantitative real-time PCR was employed to measure relative mRNA levels after 8 days of injections with ASNase or PBS. Target gene transcripts were normalized by 18S rRNA control. Phosphorylation state and protein expression were assessed by immunoblot analyses. Antibodies specific for phosphorylated AMP kinase alpha (p-AMPKα) and acetyl-CoA carboxylase (p-ACC) were quantified relative to total p-AMPKα and total ACC, respectively. Protein carbonylation was measured in mouse livers using the Biovision Protein Carbonyl Content Assay Kit, following the manufacturer's instructions. Protein carbonylation was expressed as nmol carbonyl/mg protein. In all measurements shown, main effects of GCN2 deletion (strain) and asparaginase (treatment) vs. their interaction (S × T) were detected by two-way ANOVA. Statistical trends are noted by the P value; No = not significant, P > 0.1.
Genes related to lipid metabolism were largely unchanged in WA mice compared with WP controls. However, GA mice showed a sharp decline in the levels of gene transcripts related to lipid synthesis, including the transcription factor Pparγ and its target Atp-citrate-lyase and Scd1, as well as Fas mRNA (Fig. 5A) and FAS protein expression (Fig. 5B). In GA mice there was also decreased expression of genes involved in lipid oxidation, including Pparα and its target genes Acox (peroxisomal lipid oxidation) and Acadm (beta-oxidation) and genes facilitating intracellular lipolysis (Atgl) and intravascular lipolysis (Lpl) (Fig. 5A). These results suggest that loss of GCN2 disrupts the expression of hepatic genes involved in lipid synthesis and turnover. No changes in gene expression across treatment groups were found in Cd36, a fatty acid transporter; Srebp1/2, which regulates lipid and cholesterol metabolism; G6pc, encoding the final step in gluconeogenesis; and two lipid droplet protein genes, Fsp27 and Cidea (Table 1).
Fig. 5.

Hepatic triglyceride metabolism and lipid secretion are impaired in Gcn2 null mice administered asparaginase. A: quantitative real-time PCR was employed to examine mRNA expression of Pparα, Pparγ, Fas, Atp citrate lyase, Scd1, Acox, Acadm, Atgl, and Lpl. B: expression of fatty acid synthase (FAS) protein relative to GAPDH was analyzed by immunoblot. C: mice were injected with tyloxapol to inhibit breakdown of triglycerides (TG) in plasma. The rate of appearance of blood TG during this blockade was measured to determine TG secretion from liver. D: protein expression of microsomal triglyceride transfer protein (MTP) relative to GAPDH was analyzed by immunoblot. E: protein expression of apolipoprotein B-100 (ApoB-100) relative to GAPDH was analyzed by immunoblot. Values are means ± SE; n = 5–8 per group. #Main effect of treatment. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
Deletion of GCN2 impairs triglyceride secretion during prolonged asparaginase treatment via reduced ApoB-100 expression.
Both the capacity for lipid synthesis and breakdown appeared reduced; thus neither process could explain the observed hepatic lipid accumulation. Therefore, we next measured triglyceride secretion by the liver. VLDL-TG secretion rates were calculated from the slope of the linear regression describing the rise in blood triglyceride concentration over time after intraperitoneal injection of tyloxapol, an inhibitor of triglyceride clearance (28). Employing this method, we observed a significant reduction in the rate of triglyceride secretion from the liver in GA mice relative to GP controls. In contrast, triglyceride secretion by WA liver tended to increase relative to WP controls (Fig. 5C).
An inability to assemble VLDL-TG could explain the observed impaired triglyceride secretion in GA mice. Therefore, we measured protein expression of MTP and ApoB-100, which are both crucial to the assembly and secretion of triglyceride from liver (40). Contrary to the predicted hypothesis, asparaginase increased MTP protein concentrations in the livers of both WA and GA mice (two-way ANOVA significant main effect, P < 0.05; Fig. 5D). However, asparaginase reduced hepatic ApoB-100 (and ApoB-48, data not shown) protein levels in GA livers (Fig. 5E), consistent with the decreased triglyceride secretion rate in GA mice.
Lipid peroxidation of accumulated lipids leads to liver dysfunction, coagulopathy.
Oxidative stress enhances ApoB-100 degradation (38), and the AAR is important for promoting oxidative stress defenses (27); therefore, we measured the oxidative status in livers following asparaginase treatment. Consistent with this hypothesis, there was a significant increase in MDA concentrations (an indicator of lipid peroxidation) in GA livers (Fig. 6A), along with a decrease in the mRNA expression of the critical endogenous antioxidant GPX1 (Fig. 6B). No significant changes were found in protein carbonylation in liver (Table 1).
Fig. 6.

Loss of AAR promotes oxidative stress and evidence of developing coagulopathy in liver during asparaginase. A: thiobarbituric acid reactive substances assay (TBARS) was performed to test for lipid perodixation in mice livers. Thiobarbituric acid reacts with malondialdehyde (MDA), a by-product of lipid peroxidation, to yield a wavelength at 532 nm that was detected by spectrophotometry analysis. B: quantitative real-time PCR was employed to examine relative mRNA expression of Gpx1. C: quantitative real-time PCR was employed to measure mRNA expression of anti-thrombin III (AtIII). D: levels of ATIII protein at the alpha and beta subunits were measured by immunoblot analysis. Results shown beneath the representative immunoblot are expressed as the density of ATIII relative to GAPDH. E: results show the anti-FXa activity of ATIII in plasma as percentage of WP. All mice were pair fed the same amount as the GA group. Values are means ± SE; n = 5–8 per group. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
Previous studies described asparaginase treatment as causing a “temporary conformational disease,” promoting the retention and/or aggregation of labile secretory proteins made by the liver such as ATIII, a factor regulating hemostasis (29). ATIII activity is reportedly reduced by lipid peroxides and is used as a marker of liver dysfunction and disease (36). To further explore the mechanism by which asparaginase causes reduced ATIII, liver mRNA and protein expression of ATIII, as well as its anti-FXa activity in serum, were measured. In GA mice, significant declines in hepatic mRNA (Fig. 6C) and protein (Fig. 6D) levels of ATIII were noted, as well as significantly reduced ATIII anti-FXa activity in plasma (Fig. 6E) relative to GP controls. While not measured directly, we speculate that other factors involved in regulating blood clotting were also reduced in GA mice, as GA mice treated with asparaginase for longer than 8 days showed evidence of internal bleeding within the peritoneal space, indicating coagulopathy.
Deletion of GCN2 impairs AAR signaling and FGF21 expression in white adipose tissue during prolonged asparaginase.
Asparaginase did not alter relative or total body fat mass, inguinal fat pad weights, or fat-free mass in wild-type mice (Fig. 7, A–D). By comparison, GA mice had reduced total body fat mass (Fig. 7A) and reduced inguinal fat mass (Fig. 7, C and D). To uncover the molecular basis for these differences in body composition, expression levels of AAR targets were investigated in inguinal fat pads. Unlike liver, asparaginase did not activate the AAR in the adipose of wild-type mice (Fig. 7E). Yet, similar to liver, administration of asparaginase to GA mice decreased mRNA expression of Atf5 by approximately threefold in inguinal fat pads relative to GP controls. Also, consistent with decreases in fat mass in GA mice, significant decreases in the expression levels of Fgf21, Pparγ, Fas, Atp-citrate-lyase, and Scd1 (Fig. 7E) were observed, as well as decreased FAS protein expression (Fig. 7F) and a trend for decreased CD36 levels (P = 0.09; Table 1), suggesting decreased lipid transport in GA mice. Finally, a trend for reduced expression of FGF21-regulated genes in the adipose tissue of GA mice was evident, specifically a 43% decline in Pgc1α (P = 0.15), a 57% decrease in Sirt1 (P = 0.12), and a 53% decrease in Nrf1 mRNA (P = 0.10) (Table 1).
Fig. 7.

Gcn2 null mice given asparaginase have reduced body fat. Percent body fat (A) and percent fat-free mass (B) were determined by EchoMRI. C: inguinal fat pads of mice (grams) were weighed after necropsy. D: inguinal fat pad weight expressed relative to body weight. E: quantitative real-time PCR was employed to examine relative mRNA expression of Atf4, Atf5, Fgf21, Pparα, Pparγ, Fas, Atp citrate lyase, Sdcd1, and Acadm. F: levels of FAS were measured by immunoblot analysis. Results shown beneath the representative immunoblot are expressed as the density of FAS relative to total levels of GAPDH control. All mice were pair fed the same amount as the GA group. Values are means ± SE; n = 5–8 per group. #Main effect of treatment. *Main effect of strain. Labeled means without a common letter differ (P < 0.05, drug × strain interaction effect).
DISCUSSION
In the liver, asparaginase inhibits protein synthesis and promotes steatosis and hepatic dysfunction (42, 43, 45). Hepatic steatosis and dysfunction leading to liver failure and secondarily to coagulopathy and thromboembolism are among the most serious side effects associated with asparaginase treatment (10, 23). The data presented in this effort describe how the GCN2-directed AAR controls the ability of the liver to adapt to asparaginase. Loss of GCN2 corresponds with reduced hepatic triglyceride secretion and lipid turnover and resultant hepatic lipid accumulation. Peroxidation of these accumulated lipids alongside other oxidative and inflammatory stress signals (50) collectively promotes hepatic dysfunction, culminating in coagulopathy and morbidity.
Fatty liver could be the result of deranged lipid turnover in liver, impaired triglyceride secretion by the liver, or impaired interorgan fat metabolism. The current work shows all three pathways were affected by asparaginase treatment and influenced by loss of Gcn2. Concerning hepatic lipid turnover, there was a substantial decline in the expression of genes related to lipid synthesis (Fas, Atp-citrate lyase), oxidation (Acox, Acadm), and lipolysis (Lpl, Atgl) in Gcn2 null mice treated with asparaginase. One interpretation of these data is that one or more factors regulated by GCN2 play a permissive role in maintaining lipid metabolism during amino acid stress. These factors maintain expression of relevant genes by counteracting other inhibitory signals or molecular repressors during asparaginase to allow for metabolic adaptation.
An outcome identified in Gcn2 null mice treated with asparaginase was impaired hepatic VLDL-TG secretion in concert with decreased ApoB-100 protein concentrations. We hypothesize the decline in ApoB-100 is a result of increased oxidative stress, a known stimulus for ApoB-100 degradation (22, 38). Previous studies point to the GCN2-eIF2 AAR pathway as critical for regulating oxidative defense systems (5, 14), and Gcn2 null mice fed a leucine-devoid diet demonstrate increased oxidative damage in the liver (3, 16). It may be that sustained loss of the AAR negatively affects production of antioxidants such as GPX1, compromising redox balance. An alternative or additional influence could relate to sortilin 1, a multiligand transmembrane sorting receptor located in the trans-Golgi network and early endosomes (1, 9). Overexpression of sortilin 1 in mice promotes ApoB degradation and reduced VLDL secretion (46). On the other hand, sortilin 1 is decreased in obesity and diabetic models and appears to be repressed by mammalian target of rapamycin (mTOR), extracellular signal-regulated kinase (ERK), and endoplasmic reticulum stress (1, 9). Future studies examining sortilin-1 expression and localization, its binding activity to lipoproteins, and the factors that control its expression during asparaginase in the presence vs. absence of GCN2 are necessary to adequately clarify the relationship between the AAR and sortilin 1 function.
A recent report by Xu et al. (52) describes how Gcn2 null mice exhibit elevated serum triglycerides but reduced hepatic triglyceride storage alongside lower expression levels of the lipid droplet proteins Fsp27 and Cidea. These outcomes were detectible in mice provided a medium-fat (22 kcal%) chow diet but not mice maintained on a low-fat (13 kcal%) chow diet. In the current study, mice were maintained on a low-fat chow diet and so it was not unexpected to find gene expression of Fsp27 and Cidea similar among treatment groups (Table 1). However, we note that the report by Xu et al. (52) also shows Gcn2 null mice to possess increased serum triglyceride levels despite reduced hepatic triglyceride concentrations. Our liver triglyceride data in Fig. 2, B and D, agree with this report, and our triglyceride secretion data extend these findings by suggesting that the livers of Gcn2 null mice have enhanced triglyceride secretion, providing additional explanation as to why Gcn2 null mice have reduced liver triglycerides yet elevated circulating triglycerides.
A third mechanism for metabolic dysfunction relates to impaired interorgan fat metabolism. In other experimental models of fasting and protein restriction, increased circulating FGF21 facilitates mitochondrial oxidative function in adipose, resulting in increased energy expenditure (15, 18, 34). These responses were not identified in mice treated with asparaginase. Instead, mice deleted for Gcn2 demonstrated reduced circulating FGF21 when challenged with asparaginase, which corresponded with dampened mitochondrial function in liver and adipose. We propose that FGF21 is one of the factors that serves to maintain homeostasis and allow for metabolic adaptation during asparaginase treatment. The endocrine actions of FGF21 in this experimental model and differential outcomes in peripheral tissues have yet to be fully explored. Additional exploration of the AAR in the presence vs. absence of GCN2 and/or FGF21 (whole body compared with tissue specific) is warranted to fully understand the metabolic roles of FGF21 in response to asparaginase.
We were also interested in the role of GCN2 in regulation of hemostasis during asparaginase treatment. Upon dissection of mice, we observed that organs of the wild-type mice appeared normal. However, in GA mice, there was pronounced internal bleeding, and some livers appeared necrotic. Consistent with this observation, we found a significant decline in the mRNA and protein levels of ATIII in liver, as well as low ATIII anti-FXa activity in serum. Acquired type I ATIII deficiency is often a side effect of liver failure (36). These data demonstrate that GCN2 has a critical role for regulating the health of the liver and liver dysfunction in GA mice promotes ATIII deficiency, leading to deranged hemostasis.
To what extent ATF4 modulates the transcriptome and directs homeostatic outcomes during asparaginase is unclear. Translational induction of ATF4 by cellular stress is described in detail (31, 48) and, in the case of endoplasmic reticulum stress, demonstrated in both cell lines and tissues from experimental animal models (32, 47). However, this same level of corroboration across model systems cannot be claimed for amino acid deprivation. While in vitro studies report increased ATF4 protein during amino acid withdrawal (4, 26), studies in animals are inconsistent (17, 18, 34, 50). This disparity can be due to a number of reasons aside from previous concerns related to antibody specificity and sensitivity. First, induction of the AAR is dynamic and is tailored to the amplitude and extent of the stress. As such, attempts to visualize both protein and gene expression may not always be successful in a single point in time. We suggest that incongruence between ATF4 protein and gene expression in our model may be related to the time-bound nature of this study. Future time course studies may be necessary to resolve this possibility. Second, it is feasible to consider that in mouse liver other transcription factors such as ATF5 and CHOP may be important contributors to the regulation of the AAR at the level of translational control. Both ATF4 and CHOP are reportedly essential for FGF21 expression during endoplasmic reticulum stress (49), and ATF5 can be regulated in the liver similar to ATF4 (53). At least one report suggests that ATF5 may contribute significantly to the regulation of Asns during amino acid stress in mouse liver (17). Ribosomal profiling and other methods to explore the translational control of gene expression may be necessary to fully understand the contribution of ATF4 to the overall AAR to asparaginase.
In summary, GCN2 is essential for adaptations from asparaginase treatment. Mice deficient in GCN2 are unable to induce the AAR and suffer a greater degree of hepatotoxicity from asparaginase. The present findings are in agreement with two reports correlating polymorphisms in ASNS and ATF5 with adverse outcomes to asparaginase treatment humans undergoing treatment for acute lymphoblastic leukemia (8, 44). The combination of this type of preclinical exploration alongside investigation of leukemic patient profiles may guide the development of tests that successfully predict who is at risk for developing complications arising from asparaginase treatment and facilitate the development of a more individualized and thus effective treatment regimen.
GRANTS
This work was supported by National Institutes of Health Grants HD-070487 (to T. G. Anthony) and GM-49164 (to R. C. Wek).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.J.W., B.A.L., P.S., E.T.M., R.J.A.B., M.E.F., J.D., and T.G.A. performed experiments; G.J.W., B.A.L., P.S., M.E.F., J.D., and T.G.A. analyzed data; G.J.W., J.D., G.C.H., and T.G.A. interpreted results of experiments; G.J.W., P.S., and T.G.A. prepared figures; G.J.W. and T.G.A. drafted manuscript; G.J.W., B.A.L., P.S., E.T.M., R.J.A.B., M.E.F., J.D., G.C.H., R.C.W., and T.G.A. approved final version of manuscript; J.D., G.C.H., R.C.W., and T.G.A. edited and revised manuscript; G.C.H., R.C.W., and T.G.A. conception and design of research.
ACKNOWLEDGMENTS
Present address of G. C. Henderson: Newomics, Emeryville, CA.
REFERENCES
- 1.Ai D, Baez JM, Jiang H, Conlon DM, Hernandez-Ono A, Frank-Kamenetsky M, Milstein S, Fitzgerald K, Murphy AJ, Woo CW, Strong A, Ginsberg HN, Tabas I, Rader DJ, Tall AR. Activation of ER stress and mTORC1 suppresses hepatic sortilin-1 levels in obese mice. J Clin Invest 122: 1677–1687, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anthony TG, McDaniel BJ, Byerley RL, McGrath BC, Cavener DR, McNurlan MA, Wek RC. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem 279: 36553–36561, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Arriazu E, Perez de Obanos MP, Lopez-Zabalza MJ, Herraiz MT, Iraburu MJ. Amino acid deprivation decreases intracellular levels of reactive oxygen species in hepatic stellate cells. Cell Physiol Biochem 26: 281–290, 2010. [DOI] [PubMed] [Google Scholar]
- 4.B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41: 7683–7699, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab 10: 13–26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baird TD, Wek RC. Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv Nutr 3: 307–321, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balasubramanian MN, Butterworth EA, Kilberg MS. Asparagine synthetase: regulation by cell stress and involvement in tumor biology. Am J Physiol Endocrinol Metab 304: E789–E799, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Beeck H, Nagel D, Pindur G, Scharrer I, Preiss A, Seiler D, Hellstern P. Measurement of antithrombin activity by thrombin-based and by factor Xa-based chromogenic substrate assays. Blood Coagul Fibrinolysis 11: 127–135, 2000. [PubMed] [Google Scholar]
- 8.Ben Tanfous M, Sharif-Askari B, Ceppi F, Laaribi H, Gagne V, Rousseau J, Labuda M, Silverman LB, Sallan SE, Neuberg D, Kutok JL, Sinnett D, Laverdiere C, Krajinovic M. Polymorphisms of asparaginase pathway and asparaginase-related complications in children with acute lymphoblastic leukemia. Clin Cancer Res 2014June6[Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi L, Chiang JY, Ding WX, Dunn W, Roberts B, Li T. Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. J Lipid Res 54: 2754–2762, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodmer M, Sulz M, Stadlmann S, Droll A, Terracciano L, Krahenbuhl S. Fatal liver failure in an adult patient with acute lymphoblastic leukemia following treatment with L-asparaginase. Digestion 74: 28–32, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Bunpo P, Cundiff JK, Reinert RB, Wek RC, Aldrich CJ, Anthony TG. The eIF2 kinase GCN2 is essential for the murine immune system to adapt to amino acid deprivation by asparaginase. J Nutr 140: 2020–2027, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bunpo P, Dudley A, Cundiff JK, Cavener DR, Wek RC, Anthony TG. GCN2 protein kinase is required to activate amino acid deprivation responses in mice treated with the anti-cancer agent L-asparaginase. J Biol Chem 284: 32742–32749, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cairo MS. Adverse reactions of L-asparaginase. Am J Pediatr Hematol Oncol 4: 335–339, 1982. [PubMed] [Google Scholar]
- 14.Castilho BA, Shanmugam R, Silva RC, Ramesh R, Himme BM, Sattlegger E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim Biophys Acta 1843: 1948–1968, 2014. [DOI] [PubMed] [Google Scholar]
- 15.Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc Natl Acad Sci USA 107: 12553–12558, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaveroux C, Lambert-Langlais S, Parry L, Carraro V, Jousse C, Maurin AC, Bruhat A, Marceau G, Sapin V, Averous J, Fafournoux P. Identification of GCN2 as new redox regulator for oxidative stress prevention in vivo. Biochem Biophys Res Commun 415: 120–124, 2011. [DOI] [PubMed] [Google Scholar]
- 17.Dang Do AN, Kimball SR, Cavener DR, Jefferson LS. eIF2alpha kinases GCN2 and PERK modulate transcription and translation of distinct sets of mRNAs in mouse liver. Physiol Genomics 38: 328–341, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J 443: 165–171, 2012. [DOI] [PubMed] [Google Scholar]
- 20.Durden DL, Salazar AM, Distasio JA. Kinetic analysis of hepatotoxicity associated with antineoplastic asparaginases. Cancer Res 43: 1602–1605, 1983. [PubMed] [Google Scholar]
- 21.Earl M. Incidence and management of asparaginase-associated adverse events in patients with acute lymphoblastic leukemia. Clin Adv Hematol Oncol 7: 600–606, 2009. [PubMed] [Google Scholar]
- 22.Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J Biol Chem 276: 27855–27863, 2001. [DOI] [PubMed] [Google Scholar]
- 23.Gross MA, Speer RJ, Hill JM. Hepatic lipidosis associated with L-asparaginase treatment. Proc Soc Exp Biol Med 130: 733–736, 1969. [DOI] [PubMed] [Google Scholar]
- 24.Guo F, Cavener DR. The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metab 5: 1–12, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Hao S, Sharp JW, Ross-Inta CM, McDaniel BJ, Anthony TG, Wek RC, Cavener DR, McGrath BC, Rudell JB, Koehnle TJ, Gietzen DW. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 307: 1776–1778, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Henderson GC. Kinetic measurement techniques in the evaluation of lipid metabolism. Curr Drug Discov Technol 10: 209–223, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Hernandez-Espinosa D, Minano A, Martinez C, Perez-Ceballos E, Heras I, Fuster JL, Vicente V, Corral J. L-asparaginase-induced antithrombin type I deficiency: implications for conformational diseases. Am J Pathol 169: 142–153, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill JM, Roberts J, Loeb E, Khan A, MacLellan A, Hill RW. L-asparaginase therapy for leukemia and other malignant neoplasms. Remission in human leukemia. JAMA 202: 882–888, 1967. [PubMed] [Google Scholar]
- 31.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol 23: 5651–5663, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang S, Yan C, Fang QC, Shao ML, Zhang YL, Liu Y, Deng YP, Shan B, Liu JQ, Li HT, Yang L, Zhou J, Dai Z, Jia WP. Fibroblast growth factor 21 is regulated by the IRE1alpha-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem 289: 29751–29765, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilberg MS, Balasubramanian M, Fu L, Shan J. The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr 3: 295–306, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Munzberg H, Hutson SM, Gettys TW, Schwartz MW, Morrison CD. FGF21 is an endocrine signal of protein restriction. J Clin Invest 124: 3913–3922, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, Mohammadi M, Potthoff MJ. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes 63: 4057–4063, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller G. [Acquired antithrombin III deficiency]. Z Gesamte Inn Med 47: 74–77, 1992. [PubMed] [Google Scholar]
- 37.Ollenschlager G, Roth E, Linkesch W, Jansen S, Simmel A, Modder B. Asparaginase-induced derangements of glutamine metabolism: the pathogenetic basis for some drug-related side-effects. Eur J Clin Invest 18: 512–516, 1988. [DOI] [PubMed] [Google Scholar]
- 38.Pan M, Cederbaum AI, Zhang YL, Ginsberg HN, Williams KJ, Fisher EA. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J Clin Invest 113: 1277–1287, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panosyan EH, Grigoryan RS, Avramis IA, Seibel NL, Gaynon PS, Siegel SE, Fingert HJ, Avramis VI. Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer Res 24: 1121–1125, 2004. [PubMed] [Google Scholar]
- 40.Pereira IV, Stefano JT, Oliveira CP. Microsomal triglyceride transfer protein and nonalcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol 5: 245–251, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Qiu H, Dong J, Hu C, Francklyn CS, Hinnebusch AG. The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J 20: 1425–1438, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raetz EA, Salzer WL. Tolerability and efficacy of L-asparaginase therapy in pediatric patients with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 32: 554–563, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Reinert RB, Oberle LM, Wek SA, Bunpo P, Wang XP, Mileva I, Goodwin LO, Aldrich CJ, Durden DL, McNurlan MA, Wek RC, Anthony TG. Role of glutamine depletion in directing tissue-specific nutrient stress responses to L-asparaginase. J Biol Chem 281: 31222–31233, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Rousseau J, Gagne V, Labuda M, Beaubois C, Sinnett D, Laverdiere C, Moghrabi A, Sallan SE, Silverman LB, Neuberg D, Kutok JL, Krajinovic M. ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood 118: 5883–5890, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahoo S, Hart J. Histopathological features of L-asparaginase-induced liver disease. Semin Liver Dis 23: 295–299, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Strong A, Ding Q, Edmondson AC, Millar JS, Sachs KV, Li X, Kumaravel A, Wang MY, Ai D, Guo L, Alexander ET, Nguyen D, Lund-Katz S, Phillips MC, Morales CR, Tall AR, Kathiresan S, Fisher EA, Musunuru K, Rader DJ. Hepatic sortilin regulates both apolipoprotein B secretion and LDL catabolism. J Clin Invest 122: 2807–2816, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 22: 4390–4405, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101: 11269–11274, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wan XS, Lu XH, Xiao YC, Lin Y, Zhu H, Ding T, Yang Y, Huang Y, Zhang Y, Liu YL, Xu ZM, Xiao J, Li XK. ATF4- and CHOP-dependent induction of FGF21 through endoplasmic reticulum stress. Biomed Res Intl 2014: 807874, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson GJ, Bunpo P, Cundiff JK, Wek RC, Anthony TG. The eukaryotic initiation factor 2 kinase GCN2 protects against hepatotoxicity during asparaginase treatment. Am J Physiol Endocrinol Metab 305: E1124–E1133, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woo YC, Xu A, Wang Y, Lam KS. Fibroblast growth factor 21 as an emerging metabolic regulator: clinical perspectives. Clin Endocrinol 78: 489–496, 2013. [DOI] [PubMed] [Google Scholar]
- 52.Xu X, Hu J, McGrath BC, Cavener DR. GCN2 in the brain programs PPARgamma2 and triglyceride storage in the liver during perinatal development in response to maternal dietary fat. PLoS One 8: e75917, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou D, Palam LR, Jiang L, Narasimhan J, Staschke KA, Wek RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem 283: 7064–7073, 2008. [DOI] [PubMed] [Google Scholar]