

Abstract
Reduced fetal glucose supply, induced experimentally or as a result of placental insufficiency, produces an early activation of fetal glucose production. The mechanisms and substrates used to fuel this increased glucose production rate remain unknown. We hypothesized that in response to hypoglycemia, induced experimentally with maternal insulin infusion, the fetal liver would increase uptake of lactate and amino acids (AA), which would combine with hormonal signals to support hepatic glucose production. To test this hypothesis, metabolic studies were done in six late gestation fetal sheep to measure hepatic glucose and substrate flux before (basal) and after [days (d)1 and 4] the start of hypoglycemia. Maternal and fetal glucose concentrations decreased by 50% on d1 and d4 (P < 0.05). The liver transitioned from net glucose uptake (basal, 5.1 ± 1.5 μmol/min) to output by d4 (2.8 ± 1.4 μmol/min; P < 0.05 vs. basal). The [U-13C]glucose tracer molar percent excess ratio across the liver decreased over the same period (basal: 0.98 ± 0.01, vs. d4: 0.89 ± 0.01, P < 0.05). Total hepatic AA uptake, but not lactate or pyruvate uptake, increased by threefold on d1 (P < 0.05) and remained elevated throughout the study. This AA uptake was driven largely by decreased glutamate output and increased glycine uptake. Fetal plasma concentrations of insulin were 50% lower, while cortisol and glucagon concentrations increased 56 and 86% during hypoglycemia (P < 0.05 for basal vs. d4). Thus increased hepatic AA uptake, rather than pyruvate or lactate uptake, and decreased fetal plasma insulin and increased cortisol and glucagon concentrations occur simultaneously with increased fetal hepatic glucose output in response to fetal hypoglycemia.
Keywords: fetal, glucose, glucose production, liver, hypoglycemia
glucose is the primary fuel for fetal oxidative metabolism (14, 18). It is delivered to the fetus by facilitated diffusion across the placenta according to its maternal-fetal concentration gradient (17), eliminating the need for endogenous glucose production by the fetus (20). Indeed, studies using catheters across the liver in normal fetal sheep have shown that there is net hepatic glucose uptake, rather than hepatic glucose output (42, 47). The absence of fetal hepatic glucose output is advantageous for the fetus as it helps maintain the maternal-fetal glucose concentration gradient and transfer of maternally derived glucose to the fetus. Data in fetal sheep, however, demonstrate an early activation of glucose production when fetal glucose supply is reduced experimentally and in cases of intrauterine growth restriction (IUGR) produced by placental insufficiency (20, 38, 44, 45). The early activation of hepatic glucose production may be a beneficial adaptation to maintain glucose supply to vital organs as placental glucose supply is diminished. Importantly, our recent data in the IUGR sheep fetus demonstrate that increased glucose production is not suppressed by insulin, suggesting the early development of hepatic insulin resistance (44). If persistent after birth, however, these adaptations in glucose metabolism and insulin sensitivity could have adverse consequences by promoting glucose production in excess of glucose utilization capacity, contributing to persistent postnatal hyperglycemia (15, 21). Furthermore, this early activation of glucose production in fetal life may underlie increased susceptibility to hepatic insulin resistance, inappropriate glucose production, and diabetes later in life (23, 31, 33). Therefore, understanding the mechanisms underlying the premature activation of fetal hepatic glucose production is important to develop strategies to prevent these adverse outcomes.
Reduced glucose supply to the fetus is a pregnancy complication associated with placental insufficiency and IUGR. Experimental models in pregnant sheep of reduced fetal glucose supply, producing physiological hypoglycemia, include acute hypoglycemia induced by maternal fasting for several days (10, 20), prolonged maternal insulin infusions (5, 6, 17, 38, 46), or placental insufficiency resulting in IUGR (28, 44, 45, 50). In these studies, increased fetal glucose production rates were identified when fetal glucose utilization rates remained greater than the net rate of glucose uptake from the placenta (19). Evidence for hepatic glucose production in these models is supported by increased hepatic gluconeogenic gene expression and enzyme activity (10, 38, 46). Hepatic glucose output, however, has not been directly measured in these models. Fetal hepatic metabolism has been measured directly using hepatic catheterization in late gestation fetal sheep under normal conditions. The normal fetal liver has a net uptake of glucose, lactate, and most amino acids (AAs) that exceeds its rate of oxidative metabolism and anabolic activity and thus simultaneously releases glutamate, pyruvate, and, to a lesser amount, serine, aspartate, and ornithine (3, 42, 47). Little is known about the mechanisms regulating uptake of carbon substrates by the fetal liver under conditions when glucose production occurs.
We hypothesized that following 1 and 4 days (d1 and d4, respectively) of maternal hypoglycemia, the fetus would progressively adapt to reduced glucose supply by activating hepatic glucose production and increasing net hepatic glucose output. We also hypothesized that the increase in hepatic glucose output would be supported by increased net hepatic uptake of gluconeogenic substrates, including lactate and AAs, along with coordinated hormonal signals that would support gluconeogenesis. To test these hypotheses, we used a sheep model of reduced fetal glucose supply produced by maternal insulin-induced hypoglycemia and catheterization of the fetal liver to directly measure hepatic glucose and substrate metabolism. Our results demonstrate that fetal hypoglycemia for 4 days results in activation of glucose production and increased net glucose output by the fetal liver. These fetal hepatic glucose fluxes occur in parallel with coordinated changes in hepatic AA metabolism and increased endocrine signals that contribute to gluconeogenesis.
MATERIALS AND METHODS
Sheep model of fetal hypoglycemia.
Experiments were conducted in Columbia-Rambouillet adult ewes with singleton pregnancies. Surgery was performed to place fetal and maternal catheters (125 ± 2 days gestational age) according to methods previously reported from our laboratory (22, 42, 49). Catheterized vessels for blood sampling included the maternal femoral artery (sample name = A), umbilical vein (g), fetal abdominal aorta (a), and fetal left hepatic vein (h) and infusion catheters were placed in maternal femoral vein and fetal brachial vein. All catheters were tunneled subcutaneously through a flank incision on the ewe and kept within a plastic pouch attached to the ewe's skin. The catheters were flushed every other day with heparinized saline. All ewes were allowed to recover postoperatively for 3 to 6 days and were kept in individual carts and given ad libitum diet of alfalfa pellets, water, and mineral supplements. All animal procedures were in compliance with guidelines of the United States Department of Agriculture, the National Institutes of Health, and the American Association for the Accreditation of Laboratory Animal Care. The animal care and use protocols were approved by the University of Colorado Denver Institutional Animal Care and Use Committee.
Fetal metabolic studies.
Three consecutive metabolic studies were performed in each maternal-fetal pair under normal (basal) conditions and following 1 (d1) and 3–5 d (d4) of fetal hypoglycemia. For each metabolic study, 3H20 and [U-13C]glucose tracers were infused as a 3-ml bolus (44 uCi 3H20, 50 or 100 mg [13C]glucose) followed by continuous infusion at 3 ml/h (0.63 uCi/min 3H20, 0.75, or 1.5 mg/min [13C]glucose) to measure hepatic blood flow inputs and glucose metabolism. Indocyanine green (ICG) dye was infused to measure hepatic blood flow (bolus: 2.1–2.9 mg; rate: 0.03–0.04 mg/min) as previously described (22). After 90–120 min, once steady state was reached, four consecutive blood draws were obtained every 20–30 min from the maternal artery (A), umbilical vein (g), left hepatic vein (h), and fetal artery (a). Before the start of the tracer infusions, baseline blood draws were obtained from each vessel for tracer background corrections. During the draw period, fetal blood was replaced isovolumetrically with 50 ml of heparinized maternal blood.
One to two days after the basal period metabolic study was complete, the hypoglycemic treatment was started. To induce hypoglycemia, a continuous, variable rate infusion of insulin was initiated into the maternal vein (infusate concentration = 1.7 U/ml, Humulin R; Eli Lilly, Indianapolis, IN) in 0.5% BSA (Sigma, St. Louis, MO; in 0.9% NaCl). Maternal arterial plasma glucose was measured at least twice daily and the maternal insulin infusion was adjusted to produce and maintain a 50% reduction relative to the basal period (2, 5, 6). Fetal metabolic studies described above were repeated on d1 and d4 of hypoglycemia. All pregnant ewes displayed normal behavior and maintained normal feed intake during the hypoglycemic treatment.
Blood sample analysis.
Blood samples were immediately analyzed for hematocrit, pH, Po2, Pco2, oxygen-hemoglobin saturation, and oxygen content using a blood gas analyzer (ABL 520 Radiometer) and for plasma glucose and lactate (YSI; Yellow Springs Instrument 2700) (28, 44, 45). Pyruvate concentrations were determined in deproteinized whole blood samples (42). Plasma AAs were measured by HPLC using a Dionex 300 model 4500 analyzer (Dionex, Sunnyvale, CA) (37).
Hepatic blood flow and substrate uptake rates.
Left hepatic blood flow was estimated using ICG concentrations, which were measured in plasma samples from umbilical vein, fetal artery, and left hepatic vein as previously described (22). Blood flow to the left hepatic lobe was calculated by application of the Fick principle. ICG uptake by the left lobe, (Rup)L, was calculated as
where Rinf equals rate of infusion into the fetus. Left hepatic blood flow was then calculated as
where (ho) is the average concentration of ICG in the umbilical vein (g) and fetal artery (a) representing the blood entering the liver, (h) equals the ICG concentration in the left hepatic vein leaving the liver, and (ho-h) represents the ICG concentration difference across the left hepatic lobe.
3H2O concentrations, adjusted for baseline, were used to estimate the contribution of umbilical venous blood and fetal arterial blood to the left hepatic lobe (22, 49). The fraction of left hepatic blood flow derived from the umbilical vein was calculated as:
where Fg is the fraction of flow contributed by the umbilical vein, (a) represents the arterial concentration of 3H20, (h) is the left hepatic vein concentration, and (g) is the umbilical vein concentration. The fraction of left hepatic flow derived from the artery is as follows:
These fractional inputs were used to calculate the hepatic input (hi) concentrations for each substrate as follows:
where (substrate)g is the substrate concentration in the umbilical vein and (substrate)a is the concentration in the left hepatic artery. Net hepatic uptake of oxygen, glucose, pyruvate, lactate, and individual AAs by the left lobe of the fetal liver was calculated by application of the Fick principle as follows:
where F is hepatic blood or plasma flow (ml/min), (substrate)hi is the substrate concentration of the blood or plasma entering the liver, and (substrate)h is the substrate concentration of the blood or plasma leaving the liver as measured in the left hepatic vein (22). To calculate the rate of carbon uptake for a substrate, the net uptake rate for each substrate was multiplied by the number of carbon atoms in that substrate.
Glucose tracer metabolism.
Glucose tracer enrichments [molar percent excess (MPE)] were measured in fetal artery, umbilical vein, and hepatic vein plasma samples. Plasma (0.01 ml) was mixed and with 0.2 ml of water and 0.15 ml of 0.3 N ZnSO4, followed by 0.15 ml of 0.3 N Ba(OH)2. After centrifugation at 16,000 g for 4 min, the supernatant was transferred to a glass test tube and dried under vacuum. Glucose was converted to the aldononitrile peracetate derivative for GC/MS analysis. To the dried residue, 0.1 ml of hydroxylamine hydrochloride (20 mg/ml in anhydrous pyridine) was added. The pyridine solution was incubated at 90°C for 30 min. After cooling, 0.1 ml of acetic anhydride was added and the solution was incubated for another 30 min at 90°C. After cooling, 1 ml of 1 M HCl was added, followed by 2 ml of chloroform. After vortexing for 0.5 min, the aqueous phase was removed and discarded. The chloroform solution was washed two more times with 1 ml of 1 M HCl and finally with 1 ml of water. After being washed, chloroform was removed under reduce pressure. The residue was dissolved in 0.05 ml acetonitrile and analyzed on GC/MS using chemical ionization. Glucose [U-13C]enrichment was monitored at m/z of 334/328 ratio. Glucose MPE was calculated using the difference in peak area ratios between unenriched (baseline) and enriched samples.
Whole blood glucose concentrations were calculated based on plasma measurements as described previously (19) and used for calculations. Total hepatic glucose utilization was calculated as the product of blood flow and ratio of hepatic [U-13C]glucose difference (hi-h) to hi MPE as previously described in studies measuring hepatic metabolism in the canine (32). Total hepatic glucose production represents the sum of net hepatic glucose output and total hepatic glucose utilization.
Fetal hormones.
Fetal arterial plasma samples were stored at −70°C and analyzed for insulin, glucagon, norepinephrine, and cortisol (28, 29).
Fetal and fetal liver weights.
At the end of the d4 study period, the mother and fetus were euthanized. The fetus and fetal right and left hepatic lobes were weighed. The location of the hepatic catheter in the left lobe was confirmed. In two fetuses, the hypoglycemic period was continued for 4–5 days before necropsy was performed for additional studies not described here.
Statistical analysis.
Data were analyzed from a total of six fetuses, all of which underwent complete basal, d1, and d4 studies. For each study, the average value for the variable of interest across the draw period (draws 1–4) was used in statistical analysis. Data were analyzed by one-way mixed model ANOVA with main effect of study (basal, d1, and d4) and random effect of fetus to account for repeated measures using SAS software (PROC MIXED). When the overall ANOVA was significant (P < 0.05), individual posttest comparisons were made using least square means (PDIFF option). In the basal study, the glucose production rate was tested against a theoretical mean of zero by one-sample t-test and Wilcoxon signed rank test. Mixed model linear regression (PROC MIXED), accounting for repeated measures on each fetus, was used to determine the relationship between hepatic glucose output and AA uptake or hormone concentrations. Statistical significance was declared at P < 0.05.
RESULTS
Maternal insulin infusion produces maternal and fetal hypoglycemia.
Metabolic studies were performed in normal late gestation fetal sheep during basal conditions and were repeated in the same fetuses after d1 and d4 of hypoglycemia. Maternal plasma glucose concentrations were 48% lower on d1 and 63% lower on d4 relative to basal conditions before the start of the maternal insulin infusion (Fig. 1A). Similarly, fetal plasma glucose concentrations were 46 and 58% lower on d1 and d4 (Fig. 1B). Fetal plasma glucose concentrations were ∼30% of maternal concentrations during all study periods, indicative of normal maternal to fetal net placental glucose transport (Fig. 1C). Maternal hematocrit, oxygen content, and lactate concentrations were similar during all study periods (Table 1). Fetal hematocrit, pH, oxygen content, and lactate concentrations were similar during all study periods, but fetal whole blood pyruvate concentrations increased by 28% (Table 1). Fetal weight was 2.77 ± 0.26 kg, total liver weight was 97 ± 16 g, and left hepatic lobe weight was 32 ± 6 g at the end of study.
Fig. 1.
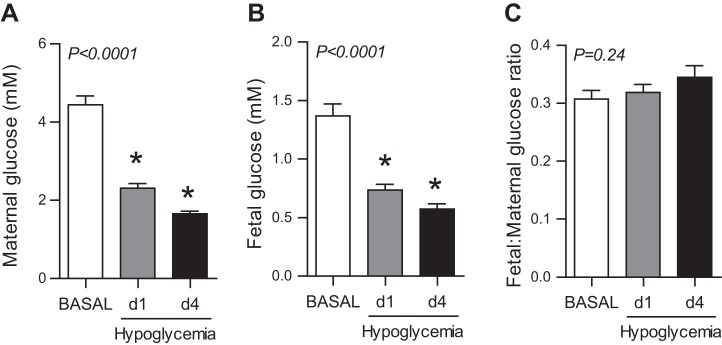
Maternal insulin infusion produces maternal and fetal hypoglycemia. Plasma glucose concentrations in maternal (A) and fetal (B) artery measured during the basal period and on day (d)1 and d4 following hypoglycemia induced by maternal insulin infusion. C: ratio of fetal to maternal plasma arterial glucose concentration during the basal, d1, and d4 study periods. Means ± SE are shown. The overall ANOVA P value is shown, and when significant (P < 0.05), individual comparisons were made. *P < 0.05 vs. basal period.
Table 1.
Maternal and fetal characteristics during basal and hypoglycemic studies
| Hypoglycemia |
||||
|---|---|---|---|---|
| Basal | d1 | d4 | ANOVA† | |
| Maternal artery | ||||
| Hematocrit, % | 31.4 ± 1.6 | 30.7 ± 1.4 | 30.9 ± 1.4 | 0.62 |
| Oxygen content (wb), mM | 5.73 ± 0.29 | 5.64 ± 0.24 | 5.65 ± 0.26 | 0.77 |
| Lactate (pl), mM | 0.62 ± 0.08 | 0.82 ± 0.04 | 0.72 ± 0.08 | 0.13 |
| Fetal artery | ||||
| Fetal age, days | 128.2 ± 0.4 | 130.7 ± 0.4* | 134.0 ± 0.3* | |
| Hypoglycemic period, days | 1 ± 0.0 | 4.3 ± 0.2 | ||
| pH | 7.34 ± 0.004 | 7.35 ± 0.003 | 7.33 ± 0.018 | 0.64 |
| Hematocrit, % | 33.0 ± 0.8 | 32.1 ± 1.2 | 30.2 ± 1.5* | <0.01 |
| Oxygen content, mM | 2.89 ± 0.18 | 2.63 ± 0.25 | 2.77 ± 0.30 | 0.29 |
| Lactate (pl), mM | 1.92 ± 0.12 | 1.89 ± 0.21 | 1.59 ± 0.20 | 0.40 |
| Pyruvate (wb), mM | 0.13 ± 0.01 | 0.16 ± 0.01* | 0.17 ± 0.01* | <0.05 |
| Insulin, ng/ml | 0.36 ± 0.05 | 0.21 ± 0.04* | 0.18 ± 0.02* | <0.005 |
| Cortisol, ng/ml | 8.5 ± 1.5 | 9.0 ± 0.9 | 13.3 ± 2.4* | <0.05 |
| Glucagon, pg/ml | 41.5 ± 7.7 | 58.7 ± 10.4 | 77.2 ± 15.7* | <0.05 |
| Norepinephrine, pg/ml | 736 ± 75 | 885 ± 201 | 695 ± 119 | 0.50 |
| Glucagon:insulin, molar ratio | 2.1 ± 0.4 | 5.3 ± 1.2* | 7.4 ± 1.6* | <0.005 |
| Hepatic flow and oxygen uptake | ||||
| Left hepatic wb flow, ml/min | 134 ± 24 | 131 ± 21 | 134 ± 34 | 0.99 |
| Left hepatic pl flow, ml/min | 90 ± 16 | 88 ± 13 | 93 ± 24 | 0.96 |
| Left hepatic input, umbilical vein, % | 89 ± 4 | 93 ± 6 | 93 ± 5 | 0.42 |
| Left lobe oxygen uptake, μmol/min) | 71.6 ± 11.4 | 94.6 ± 16.5 | 76.1 ± 15.7 | 0.21 |
| Total hepatic oxygen uptake, % | 217 ± 31 | 282 ± 38 | 230 ± 41 | 0.23 |
| Hepatic:total fetal oxygen uptake, % | 20.6 ± 2.4 | 26.4 ± 2.7 | 26.0 ± 4.2 | 0.33 |
Values are expressed as means ± SE. Measurements obtained during basal period and following d1 and d4 of hypoglycemia; pl, plasma values; wb, whole blood values; d, day.
P < 0.05 vs. basal period.
Overall ANOVA P values are presented.
Hypoglycemia increases fetal hepatic glucose output.
Plasma concentrations of glucose and other substrates were measured in samples obtained from the left hepatic vein to directly determine hepatic output and from the umbilical vein and fetal artery to determine hepatic glucose input (hi). Substrate uptake across the fetal liver represents the difference between input and output (hi-ho). The percentage of left lobe hepatic blood supplied by the umbilical vein was 89–93% during the study periods (Table 1), with the remainder of left hepatic input supplied by fetal arterial blood. Left hepatic plasma flow and blood flow rates were not different from basal study values in subsequent study periods (Table 1). Fetal hepatic oxygen uptake was similar between basal and d4 periods, with a trend toward an increase on d1 (P = 0.10, d1 vs. basal). Total hepatic oxygen uptake represented 20–26% of fetal oxygen uptake during each study period (Table 1). Overall, hepatic oxygen consumption remained constant in response to hypoglycemia with a potential increase acutely on d1.
During the basal period, there was net hepatic glucose uptake (Fig. 2A). In response to hypoglycemia, the fetal liver transitioned from net glucose uptake in the basal period to neutral balance between uptake and output on d1 to net glucose output on d4 (Fig. 2A). Fetal hepatic glucose utilization, measured with [U-13C]glucose tracer, decreased on d1 and d4 (Fig. 2B). As a result, by d4 the rate of hepatic glucose production (representing sum of net balance and utilization) was nearly twofold higher compared with the basal period (Fig. 2C). Fetal hepatic glucose production is further demonstrated by a lower plasma [U-13C]glucose MPE ratio (h/hi) across the fetal liver on d4 (Fig. 2D).
Fig. 2.
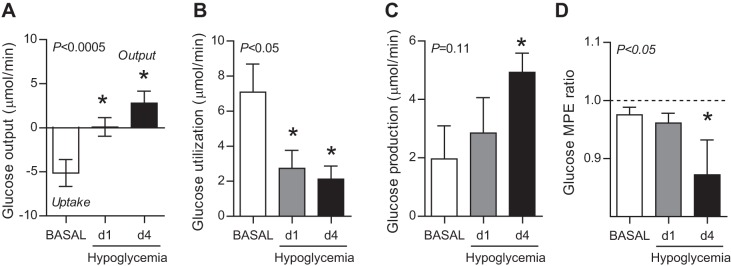
Fetal hypoglycemia increases hepatic glucose production. Rates of net hepatic glucose output (A), hepatic glucose utilization (B), and hepatic glucose production (C; sum of output and utilization) measured during the basal period and on d1 and d4 of hypoglycemia in whole blood. D: ratio of [U-13C]glucose tracer MPE measured across the fetal liver (h/hi). Means ± SE are shown. The overall ANOVA P value is shown. *P < 0.05 vs. basal period.
Increased hepatic AA uptake and decreased hepatic glutamate output during fetal hypoglycemia.
The net hepatic uptake rates of substrates that could be used for glucose production were measured. Fetal hypoglycemia decreased net hepatic lactate uptake by 63% (Fig. 3A) and increased net hepatic pyruvate output by twofold (Fig. 3B). Individual AA net uptake rates were measured across the fetal liver. Under basal conditions, the fetal liver had a net output of serine and glutamate and a net uptake of the other AAs (Table 2). During hypoglycemia, net hepatic glutamate output decreased on d1 and d4 (Table 2). Net hepatic uptake of threonine, glycine, alanine, and lysine increased on d1, while uptake of only glycine remained increased on d4. As a result of these individual AA changes, total hepatic AA net uptake increased threefold on d1 yet was similar between basal and d4 study periods (Fig. 3C). Among all three study periods, net hepatic glucose output correlated inversely with net hepatic glutamate output and directly with net glycine uptake (Fig. 3D) but not with net total AA uptake (P = 0.17).
Fig. 3.

Fetal hepatic substrate uptake during hypoglycemia. Hepatic uptake rates of lactate (A), pyruvate (B), and total amino acids (C) during the basal period and on d1 and d4 following maternal insulin infusion. Means ± SE are shown. Overall, ANOVA P value is shown and when significant (P < 0.05), individual comparisons were made. *P < 0.05 vs. basal period. D: correlation of hepatic glucose output with hepatic total amino acid, glutamate, and glycine uptake rates. Linear regression equation and P value are shown.
Table 2.
Hepatic plasma amino acid uptake
| Hypoglycemia |
||||
|---|---|---|---|---|
| Uptake, μmol/min) | Basal | d1 | d4 | ANOVA† |
| Taurine | −0.18 ± 0.20 | −0.17 ± 0.42 | −0.38 ± 0.55 | 0.91 |
| Aspartate | −0.27 ± 0.08 | −0.26 ± 0.09 | −0.13 ± 0.14 | 0.59 |
| Threonine | 1.27 ± 0.62 | 3.31 ± 0.73* | 1.05 ± 0.42 | <0.05 |
| Serine | −4.92 ± 1.32 | −2.42 ± 0.80 | −6.81 ± 2.72 | 0.22 |
| Asparagine | 0.52 ± 0.31 | 1.38 ± 0.32 | −1.00 ± 1.89 | 0.36 |
| Glutamate | −10.03 ± 2.13 | −5.52 ± 1.62* | −3.66 ± 1.21* | <0.01 |
| Glutamine | 6.41 ± 0.64 | 7.56 ± 1.26 | 5.97 ± 1.02 | 0.44 |
| Proline | 0.85 ± 0.28 | 1.65 ± 0.46 | 1.61 ± 0.55 | 0.26 |
| Glycine | 1.94 ± 0.67 | 3.99 ± 0.75* | 3.73 ± 0.33* | <0.01 |
| Alanine | 3.68 ± 0.73 | 6.21 ± 1.13* | 5.64 ± 1.13 | 0.08 |
| Citruline | 0.02 ± 0.16 | 0.09 ± 0.20 | −0.13 ± 0.19 | 0.65 |
| Valine | 0.15 ± 0.63 | 1.03 ± 0.62 | 0.63 ± 0.34 | 0.53 |
| Cysteine | −0.02 ± 0.04 | 0.10 ± 0.06 | −0.27 ± 0.35 | 0.46 |
| Methionine | 1.29 ± 0.38 | 1.13 ± 0.29 | 1.17 ± 0.29 | 0.90 |
| Isoleucine | 0.45 ± 0.15 | 0.46 ± 0.20 | 0.35 ± 0.10 | 0.87 |
| Leucine | 0.98 ± 0.24 | 1.09 ± 0.25 | 0.77 ± 0.24 | 0.45 |
| Tyrosine | 1.73 ± 0.15 | 1.79 ± 0.26 | 1.73 ± 0.57 | 0.99 |
| Phenylalanine | 1.28 ± 0.21 | 1.60 ± 0.33 | 1.56 ± 0.49 | 0.40 |
| Tryptophan | 0.23 ± 0.08 | 0.47 ± 0.24 | 0.47 ± 0.41 | 0.62 |
| Ornithine | −0.71 ± 0.19 | −0.70 ± 0.30 | −0.81 ± 0.35 | 0.90 |
| Lysine | 1.59 ± 0.36 | 2.49 ± 0.50* | 1.96 ± 0.20 | 0.08 |
| Histidine | 0.74 ± 0.17 | 0.80 ± 0.19 | 0.72 ± 0.06 | 0.92 |
| Arginine | 2.87 ± 0.49 | 3.52 ± 0.60 | 3.23 ± 0.47 | 0.44 |
| Glucogenic amino acid‡ | 19.71 ± 3.58 | 30.59 ± 5.09* | 22.76 ± 4.54 | 0.09 |
Values are expressed as means ± SE. Measurements obtained during basal period and following d1 and d4 of hypoglycemia.
P < 0.05 vs. basal period.
Overall ANOVA P values are presented.
Sum of theonine, asparigine, glutamine, proline, glycine, alanine, and valine.
Given these changes in hepatic substrate uptake, we next evaluated these substrate flux rates relative to carbon number to estimate carbon balance across the fetal liver (Table 3). On d1 and d4, increased glucose carbon output was paralleled with decreased lactate carbon uptake and increased pyruvate carbon output. Total carbon uptake derived from AAs increased only on d1. The sum of carbon substrate uptakes (lactate, pyruvate, and AAs) tended to increase on d1 (P = 0.12, d1 vs. basal) but was similar between basal and d4 periods. Thus increased net AA uptake, rather than lactate or pyruvate uptake, may provide carbon substrates for initially increased net hepatic glucose output but not with sustained net hepatic glucose output.
Table 3.
Hepatic carbon balance
| Hypoglycemia |
||||
|---|---|---|---|---|
| Uptake, g atom carbon/min | Basal | d1 | d4 | ANOVA† |
| Glucose | 30.8 ± 9.1 | −0.6 ± 6.4* | −16.9 ± 8.2* | <0.0005 |
| Lactate | 39.6 ± 3.5 | 19.3 ± 6.5* | 10.5 ± 6.7* | <0.005 |
| Pyruvate | −10.0 ± 1.9 | −21.9 ± 3.7* | −19.8 ± 3.7* | <0.05 |
| Amino acid | 65 ± 22 | 148 ± 34* | 101 ± 38 | <0.05 |
| Substrate‡ | 97 ± 24 | 146 ± 37 | 94 ± 43 | 0.18 |
| Total§ | 134 ± 30 | 144 ± 36 | 79 ± 40 | 0.11 |
| CO2 | −45 ± 11 | −80 ± 31 | −40 ± 14 | 0.27 |
Values are expressed as means ± SE. Measurements obtained during basal period and following d1 and d4 of hypoglycemia.
P < 0.05 vs. basal period.
Overall ANOVA P values are presented.
Sum of lactate, pyruvate, and amino acid carbon uptake rates.
Sum of glucose, lactate, pyruvate, and amino acid carbon uptake rates.
Hepatic carbon balance.
As a result of these substrate fluxes, total carbon balance across the fetal liver was maintained on d1 relative to the basal period, and tended to decrease on d4 (P = 0.09, d4 vs. basal; Table 3). Furthermore, net total carbon uptake by the fetal liver exceeded oxygen uptake during all study periods, supporting an overall sustained activation of anabolic pathways in the liver. Interestingly, hepatic CO2 output tended to increase only on d1 (P = 0.19, d1 vs. basal; Table 3), consistent with the trend for increased O2 uptake on d1, suggesting a potentially increased overall rate of hepatic oxidative metabolism on d1 but similar rates between basal and d4 study periods.
Endocrine responses to fetal hypoglycemia support hepatic glucose production.
Fetal hypoglycemia resulted in a 40% reduction in fetal insulin concentrations on d1 and d4 (Table 1). Fetal cortisol and glucagon levels increased twofold by d4 (Table 1). There was no change in norepinephrine concentration among the study periods (Table 1). The glucagon:insulin ratio, as well as the concentrations of cortisol and glucagon, had a positive correlation with hepatic glucose output, and insulin concentrations were inversely related to hepatic glucose output (Fig. 4). Thus lower plasma insulin concentrations combined with increased plasma cortisol and glucagon concentrations support increased hepatic glucose production.
Fig. 4.
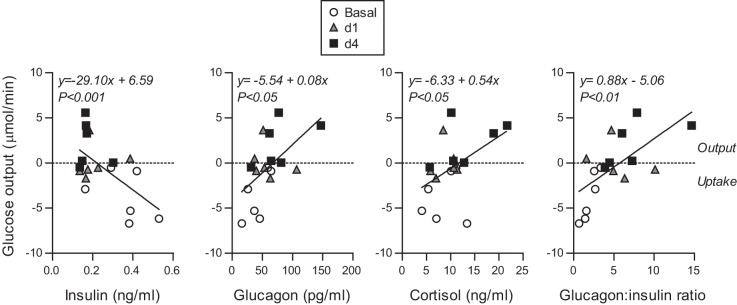
Fetal hormonal changes correlate with hepatic glucose output. Correlation between hepatic glucose output and insulin, glucagon, cortisol concentrations and glucagon:insulin molar ratio. Linear regression equation and P value is shown.
DISCUSSION
These data demonstrate that fetal hypoglycemia resulting from reduced maternal glucose supply produced net hepatic glucose output when measured directly across the liver in late gestation fetal sheep. The fetal liver had increased net AA uptake, which likely provided carbon substrates for glucose production, at least during the early stages of hepatic glucose production in response to reduced glucose supply and plasma concentrations. There also was reduced hepatic glutamate output, supporting a redirection of hepatic AA carbon into glucose production. These coordinated changes in substrate metabolism were paralleled by decreased plasma insulin and increased cortisol and glucagon concentrations, supporting the activation of fetal hepatic glucose production.
Increased hepatic uptake of AA and coordinated changes in the shuttling of intrahepatic AA likely provide carbon substrates for hepatic glucose production in response to fetal hypoglycemia. On d1, total AA uptake by the liver increased, driven by increased uptake of threonine, glycine, alanine, and lysine. By d4, hepatic total AA uptake tended to remain increased, with a significant increase only in glycine uptake. Furthermore, on d1 and d4, hepatic glutamate output decreased, demonstrating retained carbon from AA. Interestingly, hepatic lactate and pyruvate uptakes were not increased on either d1 or d4. Under normal conditions, the fetal liver has a net uptake of most AAs, with total AA carbon uptake (g atoms) exceeding oxygen uptake (30, 42), similar to our findings during the basal period. Furthermore, under basal conditions, the fetal liver releases glutamate, rather than glucose. During fetal life, it is speculated that glutamate release by the fetal liver under normal conditions may serve to limit hepatic oxygen requirements for AA oxidation by shuttling glutamate to the placenta where the oxidation can be completed (1, 42). In addition, glutamate output substitutes for glucose output to maintain low fetal glucose concentrations, which enable the transfer of maternally derived glucose across the placenta (1, 42). This reciprocal relationship between glutamate and glucose output by the fetal sheep liver has also been demonstrated in response to an acute (20 h) supraphysiological fetal glucagon infusion (42). Our results support this relationship between fetal hepatic glutamate and glucose output in response to a physiological reduction in fetal glucose supply, as the hepatic glutamate output decreases as hepatic glucose output increases.
Increased cortisol and glucagon concentrations in the hypoglycemic fetus likely prime the liver for glucose production. Cortisol and glucagon are important activators of fetal glucose production during late gestation (12, 36). Indeed, premature elevations in fetal cortisol and a lower insulin:glucagon ratio have been found after chronic (2–8 wk) fetal hypoglycemia and in placental insufficiency models, both of which result in increased fetal glucose production (38, 44). Furthermore, adrenalectomized fetal sheep demonstrate a blunted response to the activation of glucose production in response to maternal fasting (9). Fetal dexamethasone infusion also increases hepatic PEPCK and G6Pase activity in rodents and sheep but does not induce hepatic glucose output when measured in sheep (13, 34, 47). In addition to directly activated glucose production and gluconeogenic gene expression, elevated cortisol concentrations have been proposed to precede increases in catecholamine concentrations (12), which could further potentiate glucose production. The absence of an increase in norepinephrine in our model suggests a lesser role for catecholamines in mediating the activation of glucose production in response to hypoglycemia. Experimental fetal glucagon administration in fetal sheep induces endogenous hepatic glucose production and decreased glutamate output (4, 35, 42). Thus our results support a role for increased cortisol and glucagon in activating fetal hepatic glucose production.
Decreased fetal insulin concentrations in response to hypoglycemia also may contribute to increased fetal glucose production. Insulin is the primary hormone responsible for suppressing gluconeogenic gene expression and glucose production (7, 36). Since fetal hypoglycemia results in decreased fetal insulin secretion (5, 39, 40), hypoinsulinemia is found in hypoglycemic models with increased glucose production (5, 46). Whether hypoinsulinemia alone is sufficient for the induction of glucose production remains unclear, as pancreatectomized fetuses fail to induce glucose production (10, 11), yet streptozotocin treated fetuses have increased glucose production (16). Differences between these models may reflect differences in counter-regulatory hormone responses, as glucagon production is prevented by pancreatectomy but is not affected by streptozotocin (10, 16). In our study, since hypoglycemic fetuses had both hypoinsulinemia and increased counter-regulatory hormones, we are unable to determine the independent or synergistic effects of these endocrine signals to regulate fetal glucose production and output in response to reduced glucose supply and plasma concentrations.
Our study shows that hepatic blood flow and oxygen uptake are maintained, or even increased acutely, during hypoglycemia. We found that the umbilical vein supplies 89–93% of the blood supply to the left lobe, consistent with other studies (22, 30, 41, 42). Fetal hepatic oxygen uptake was ∼21% of fetal oxygen consumption during basal period, also similar to prior studies (22, 41). Hepatic oxygen consumption tended to increase on d1, up to 28% of fetal consumption, and this could be due to increased AA oxidation and increased overall hepatic metabolism, since hepatic AA uptake and hepatic CO2 production increased on d1. Overall, the absence of a decrease in hepatic oxygen uptake on d1 or d4 also supports that hepatic oxidative metabolism is maintained during acute hypoglycemia. Preservation of carbon uptake during hypoglycemia suggests that the fetal liver may be a privileged organ, as it continues to be perfused by nutrient rich umbilical vein blood, even when the fetus experiences hypoglycemia.
Studies are underway to determine the effect of hypoglycemia in this model on coordinated changes in AA uptake and metabolism among the uterus, placenta, fetus, fetal muscle, and fetal liver. We speculate that there is increased AA release by fetal skeletal muscle tissues and increased fetal AA uptake from the placenta to supply the liver with AA carbon for glucose production, although previous studies with maternal fasting for 5 days demonstrated conflicting results, showing either increased fetal AA uptake (48) or no change (26, 27).
We speculate that glycogenolysis also might contribute to net hepatic glucose output, although we were unable to measure this directly, one of the limitations of our study. Other studies in fetal sheep after maternal fasting for 2 days demonstrate increased activity of G6Pase and decreased hepatic glycogen content in the presence of increased fetal plasma cortisol and norepinephrine concentrations (12). However, after 5 days of maternal fasting, fetal hepatic glycogen content was normalized (24). Furthermore, studies after chronic hypoglycemia (2–8 wk) or chronic placental insufficiency demonstrate preserved hepatic glycogen content, despite evidence of fetal glucose production (38, 46). Therefore, glycogenolysis cannot be the sole source of glucose production during prolonged hypoglycemia due to glucose and general nutrient insufficiency (8, 25, 28, 38). Other substrates, including glycerol and free fatty acids (FFA) fuel hepatic glucose production postnatally; however, in the fetus, lipids provide little fuel for oxidative metabolism and net glycerol uptake by the fetus under normal conditions is quantitatively small (0.6 μmol·min−1·kg−1) (43). Furthermore, heparin was used during blood sampling which compromises our ability to measure FFA. Thus, in our study, total carbon uptake by the liver decreases on d4 and AA uptake alone is not sufficient to provide all the carbon for net hepatic glucose output. Additional studies are needed to test role of glycogenolysis or glycerol and FFA.
In conclusion, our study is the first to demonstrate a transition in fetal hepatic metabolism that develops to support hepatic glucose production in response to hypoglycemia. Initially after 1 day of hypoglycemia, there is increased hepatic AA uptake to compensate for reduced glucose and lactate carbon uptake. After 4 days of hypoglycemia, there was a sustained reduction in plasma insulin concentrations and sustained increases in plasma cortisol and glucagon concentrations, which we speculate induced fetal hepatic gluconeogenic gene expression leading to gluconeogenic activity. As a result of increased AA utilization and specifically decreased hepatic glutamate output, we speculate that AA carbons fuel hepatic glucose output on d1 and d4. Overall, our results provide new insights into the mechanisms underlying the premature activation of fetal hepatic glucose production, which is important for developing strategies to prevent adverse outcomes due to persistently increased hepatic glucose production in neonatal and postnatal periods.
GRANTS
Research support was from National Institutes of Health Grants K01-DK-090199 (to S. R. Thorn), K12-HD057022 (Building Interdisciplinary Research Careers in Women's Health; to L. D. Brown), R01-DK-088139 and K08-HD-060688 (to P. J. Rozance), and T32-HD07186 (to W. W. Hay, Jr., with S. S. Houin as a trainee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.S.H., P.J.R., W.W.H.J, and S.R.T. conception and design of research; S.S.H., R.B.W., and S.R.T. performed experiments; S.S.H. and S.R.T. analyzed data; S.S.H., P.J.R., L.D.B., W.W.H.J., R.B.W., and S.R.T. interpreted results of experiments; S.S.H. and S.R.T. prepared figures; S.S.H. drafted manuscript; S.S.H., P.J.R., L.D.B., W.W.H.J., R.B.W., and S.R.T. edited and revised manuscript; S.R.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Alex Cheung for assistance with isotope enrichments and David Caprio, Karen Trembler, Gates Roe, Larry Toft, and Nicole Isenberg for assistance with studies, all at the University of Colorado.
REFERENCES
- 1.Battaglia FC, Regnault TR, Teng C, Wilkening RB, Meschia G. The role of glutamate in fetal hepatic glucogenesis. Forum Nutr 56: 82–84, 2003. [PubMed] [Google Scholar]
- 2.Carver TD, Anderson SM, Aldoretta PA, Esler AL, Hay WW Jr. Glucose suppression of insulin secretion in chronically hyperglycemic fetal sheep. Pediatr Res 38: 754–762, 1995. [DOI] [PubMed] [Google Scholar]
- 3.Cetin I, Fennessey PV, Sparks JW, Meschia G, Battaglia FC. Fetal serine fluxes across fetal liver, hindlimb, and placenta in late gestation. Am J Physiol Endocrinol Metab 263: E786–E793, 1992. [DOI] [PubMed] [Google Scholar]
- 4.Devaskar SU, Ganguli S, Styer D, Devaskar UP, Sperling MA. Glucagon and glucose dynamics in sheep: evidence for glucagon resistance in the fetus. Am J Physiol Endocrinol Metab 246: E256–E265, 1984. [DOI] [PubMed] [Google Scholar]
- 5.DiGiacomo JE, Hay WW Jr. Fetal glucose metabolism and oxygen consumption during sustained hypoglycemia. Metabolism 39: 193–202, 1990. [DOI] [PubMed] [Google Scholar]
- 6.DiGiacomo JE, Hay WW Jr. Regulation of placental glucose transfer and consumption by fetal glucose production. Pediatr Res 25: 429–434, 1989. [DOI] [PubMed] [Google Scholar]
- 7.Edgerton DS, Lautz M, Scott M, Everett CA, Stettler KM, Neal DW, Chu CA, Cherrington AD. Insulin's direct effects on the liver dominate the control of hepatic glucose production. J Clin Invest 116: 521–527, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ezekwe MO. Effects of maternal starvation on some blood metabolites, liver glycogen, birth weight and survival of piglets. J Anim Sci 53: 1504–1510, 1981. [DOI] [PubMed] [Google Scholar]
- 9.Fowden AL, Forhead AJ. Adrenal glands are essential for activation of glucogenesis during undernutrition in fetal sheep near term. Am J Physiol Endocrinol Metab 300: E94–E102, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fowden AL, Forhead AJ. Insulin deficiency alters the metabolic and endocrine responses to undernutrition in fetal sheep near term. Endocrinology 153: 4008–4018, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Fowden AL, Hay WW Jr. The effects of pancreatectomy on the rates of glucose utilization, oxidation and production in the sheep fetus. Q J Exp Physiol 73: 973–984, 1988. [DOI] [PubMed] [Google Scholar]
- 12.Fowden AL, Mundy L, Silver M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J Physiol 508: 937–947, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franko KL, Giussani DA, Forhead AJ, Fowden AL. Effects of dexamethasone on the glucogenic capacity of fetal, pregnant, and nonpregnant adult sheep. J Endocrinol 192: 67–73, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Hay WW., Jr Recent observations on the regulation of fetal metabolism by glucose. J Physiol 572: 17–24, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hay WW., Jr Strategies for feeding the preterm infant. Neonatology 94: 245–254, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hay WW Jr, Meznarich HK, and Fowden AL. The effects of streptozotocin on rates of glucose utilization, oxidation, and production in the sheep fetus. Metabolism 38: 30–37, 1989. [DOI] [PubMed] [Google Scholar]
- 17.Hay WW Jr, Molina RA, DiGiacomo JE, Meschia G. Model of placental glucose consumption and glucose transfer. Am J Physiol Regul Integr Comp Physiol 258: R569–R577, 1990. [DOI] [PubMed] [Google Scholar]
- 18.Hay WW Jr, Myers SA, Sparks JW, Wilkening RB, Meschia G, Battaglia FC. Glucose and lactate oxidation rates in the fetal lamb. Proc Soc Exp Biol Med 173: 553–563, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Hay WW Jr, Sparks JW, Quissell BJ, Battaglia FC, Meschia G. Simultaneous measurements of umbilical uptake, fetal utilization rate, and fetal turnover rate of glucose. Am J Physiol Endocrinol Metab 240: E662–E668, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Hay WW Jr, Sparks JW, Wilkening RB, Battaglia FC, Meschia G. Fetal glucose uptake and utilization as functions of maternal glucose concentration. Am J Physiol Endocrinol Metab 246: E237–E242, 1984. [DOI] [PubMed] [Google Scholar]
- 21.Hays SP, Smith EO, Sunehag AL. Hyperglycemia is a risk factor for early death and morbidity in extremely low birth-weight infants. Pediatrics 118: 1811–1818, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Holcomb RG, Wilkening RB. Fetal hepatic oxygen consumption under normal conditions in the fetal lamb. Biol Neonate 75: 310–318, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Jaquet D, Deghmoun S, Chevenne D, Collin D, Czernichow P, Levy-Marchal C. Dynamic change in adiposity from fetal to postnatal life is involved in the metabolic syndrome associated with reduced fetal growth. Diabetologia 48: 849–855, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Kaneta M, Liechty EA, Moorehead HC, Lemons JA. Ovine fetal and maternal glycogen during fasting. Biol Neonate 60: 215–220, 1991. [DOI] [PubMed] [Google Scholar]
- 25.Lafeber HN, Rolph TP, Jones CT. Studies on the growth of the fetal guinea pig. The effects of ligation of the uterine artery on organ growth and development. J Dev Physiol 6: 441–459, 1984. [PubMed] [Google Scholar]
- 26.Lemons JA, Schreiner RL. Amino acid metabolism in the ovine fetus. Am J Physiol Endocrinol Metab 244: E459–E466, 1983. [DOI] [PubMed] [Google Scholar]
- 27.Lemons JA, Schreiner RL. Metabolic balance of the ovine fetus during the fed and fasted states. Ann Nutr Metab 28: 268–280, 1984. [DOI] [PubMed] [Google Scholar]
- 28.Limesand SW, Rozance PJ, Smith D, Hay WW Jr. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 293: E1716–E1725, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Limesand SW, Rozance PJ, Zerbe GO, Hutton JC, Hay WW Jr. Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology 147: 1488–1497, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Marconi AM, Battaglia FC, Meschia G, Sparks JW. A comparison of amino acid arteriovenous differences across the liver and placenta of the fetal lamb. Am J Physiol Endocrinol Metab 257: E909–E915, 1989. [DOI] [PubMed] [Google Scholar]
- 31.Martin-Gronert MS, Ozanne SE. Experimental IUGR and later diabetes. J Intern Med 261: 437–452, 2007. [DOI] [PubMed] [Google Scholar]
- 32.McGuinness OP, Fugiwara T, Murrell S, Bracy D, Neal D, O′Connor D, Cherrington AD. Impact of chronic stress hormone infusion on hepatic carbohydrate metabolism in the conscious dog. Am J Physiol Endocrinol Metab 265: E314–E322, 1993. [DOI] [PubMed] [Google Scholar]
- 33.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85: 571–633, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest 101: 2174–2181, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philipps AF, Dubin JW, Matty PJ, Raye JR. Influence of exogenous glucagon on fetal glucose metabolism and ketone production. Pediatr Res 17: 51–56, 1983. [DOI] [PubMed] [Google Scholar]
- 36.Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol 54: 885–909, 1992. [DOI] [PubMed] [Google Scholar]
- 37.Rozance PJ, Crispo MM, Barry JS, O'Meara MC, Frost MS, Hansen KC, Hay WW Jr, Brown LD. Prolonged maternal amino acid infusion in late-gestation pregnant sheep increases fetal amino acid oxidation. Am J Physiol Endocrinol Metab 297: E638–E646, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rozance PJ, Limesand SW, Barry JS, Brown LD, Thorn SR, LoTurco D, Regnault TR, Friedman JE, Hay WW Jr. Chronic late-gestation hypoglycemia upregulates hepatic PEPCK associated with increased PGC1alpha mRNA and phosphorylated CREB in fetal sheep. Am J Physiol Endocrinol Metab 294: E365–E370, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rozance PJ, Limesand SW, Hay WW Jr. Decreased nutrient-stimulated insulin secretion in chronically hypoglycemic late-gestation fetal sheep is due to an intrinsic islet defect. Am J Physiol Endocrinol Metab 291: E404–E411, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Rozance PJ, Limesand SW, Zerbe GO, Hay WW Jr. Chronic fetal hypoglycemia inhibits the later steps of stimulus-secretion coupling in pancreatic beta-cells. Am J Physiol Endocrinol Metab 292: E1256–E1264, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Rudolph AM. Hepatic and ductus venosus blood flows during fetal life. Hepatology 3: 254–258, 1983. [DOI] [PubMed] [Google Scholar]
- 42.Teng C, Battaglia FC, Meschia G, Narkewicz MR, Wilkening RB. Fetal hepatic and umbilical uptakes of glucogenic substrates during a glucagon-somatostatin infusion. Am J Physiol Endocrinol Metab 282: E542–E550, 2002. [DOI] [PubMed] [Google Scholar]
- 43.Teng CC, Tjoa S, Fennessey PV, Wilkening RB, Battaglia FC. Transplacental carbohydrate and sugar alcohol concentrations and their uptakes in ovine pregnancy. Exp Biol Med (Maywood) 227: 189–195, 2002. [DOI] [PubMed] [Google Scholar]
- 44.Thorn SR, Brown LD, Rozance PJ, Hay WW Jr, Friedman JE. Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 62: 65–73, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thorn SR, Regnault TR, Brown LD, Rozance PJ, Keng J, Roper M, Wilkening RB, Hay WW Jr, Friedman JE. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces messenger ribonucleic acid translation initiation and nutrient sensing in fetal liver and skeletal muscle. Endocrinology 150: 3021–3030, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thorn SR, Sekar SM, Lavezzi JR, O'Meara MC, Brown LD, Hay WW Jr, Rozance PJ. A physiological increase in insulin suppresses gluconeogenic gene activation in fetal sheep with sustained hypoglycemia. Am J Physiol Regul Integr Comp Physiol 303: R861–R869, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Timmerman M, Teng C, Wilkening RB, Fennessey P, Battaglia FC, Meschia G. Effect of dexamethasone on fetal hepatic glutamine-glutamate exchange. Am J Physiol Endocrinol Metab 278: E839–E845, 2000. [DOI] [PubMed] [Google Scholar]
- 48.van Veen LC, Teng C, Hay WW Jr, Meschia G, and Battaglia FC. Leucine disposal and oxidation rates in the fetal lamb. Metabolism 36: 48–53, 1987. [DOI] [PubMed] [Google Scholar]
- 49.Vaughn PR, Lobo C, Battaglia FC, Fennessey PV, Wilkening RB, Meschia G. Glutamine-glutamate exchange between placenta and fetal liver. Am J Physiol Endocrinol Metab 268: E705–E711, 1995. [DOI] [PubMed] [Google Scholar]
- 50.Vuguin P, Raab E, Liu B, Barzilai N, Simmons R. Hepatic insulin resistance precedes the development of diabetes in a model of intrauterine growth retardation. Diabetes 53: 2617–2622, 2004. [DOI] [PubMed] [Google Scholar]