

Abstract
Although phytoplankton are the major source of marine dissolved organic matter (DOM), their blooms are a global problem that can greatly affect marine ecological systems, especially free-living bacteria, which are the primary DOM degraders. In this study, we analyzed free-living bacterial communities from Xiamen sea during an Akashiwo sanguine bloom using Illumina MiSeq sequencing of 16S rRNA gene amplicons. The bloom was probably stimulated by low salinity and ended after abatement of eutrophication pollution. A total of 658,446 sequence reads and 11,807 OTUs were obtained in both bloom and control samples with Alpha-proteobacteria and Gamma-proteobacteria being the predominant classes detected. The bloom decreased bacterial diversity, increased species evenness, and significantly changed the bacterial community structure. Bacterial communities within the bloom were more homogeneous than those within the control area. The bacteria stimulated by this bloom included the SAR86 and SAR116 clades and the AEGEAN-169 marine group, but a few were suppressed. In addition, many bacteria known to be associated with phytoplankton were detected only in the bloom samples. This study revealed the great influence of an A. sanguinea bloom on free-living bacterial communities, and provided new insights into the relationship between bacteria and A. sanguinea in marine ecosystems.
Phytoplankton blooms are a worldwide ecological problem due to eutrophication pollution1,2,3,4,5,6, which can stimulate phytoplankton growth and proliferation. The majority of organic matter in the marine environment, particularly dissolved organic matter (DOM), is produced by phytoplankton7,8,9. Free-living bacteria in these environments hydrolyze most of the DOM10, preventing its accumulation during periods of high DOM production such as during phytoplankton blooms11. The free-living bacteria account for most of the total bacterial production during a bloom12. The interactions between phytoplankton and bacteria are often intertwined, with extracellular products being produced by both in coastal marine environments9,13. Bacteria can stimulate14 or inhibit15 phytoplankton growth and can even kill phytoplankton4,6,16,17. Therefore, it is necessary to understand how phytoplankton blooms affect marine microbial communities, and determine whether microbial communities are able to control phytoplankton blooms.
Many molecular tools have been utilized to investigate bacterial communities associated with phytoplankton blooms, such as 16S ribosomal DNA (rDNA) clone libraries18, polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE)11,19, flow cytometry20, terminal restriction fragment length polymorphism18, fluorescence in situ hybridization21,22, metatranscriptomics23 and high-throughput sequencing24,25. These studies reveal the close relationship between specific phytoplankton blooms and their associated bacterial communities, and which bacterial species may be crucial for the regulation of bloom dynamics and succession. For example, algal-derived organic matter has been shown to control populations of Bacteroidetes, Gamma-proteobacteria and Alpha-proteobacteria21; and the Alteromonas group can greatly influence the flux of organic matter by proliferation during development of diatom blooms, while Bacteroidetes may contribute to bloom decomposition26. These studies greatly expanded our understanding of bloom processes as well as our ability to forecast and prevent blooms. The high-throughput sequencing technologies have developed quickly in recent years, and Illumina MiSeq sequencing has become the most popular since it can generate a multi-million sequence reads of partial 16S rRNA genes to meet the throughput demands of environmental microbial ecology studies and reduce cost27.
The dinoflagellate Akashiwo sanguine is an alga that causes blooms world-wide. It is eurythermal and euryhaline28 and commonly causes spring or summer red tides, especially when the seawater salinity and temperature are relatively low29. A few studies of this dinoflagellate have been carried out and have shed light on some bloom dynamics. For example, the raphidophyte Chattonella antiqua and A. sanguinea inhibit each other's growth30; the raphidophyte Heterosigma akashiwo inhibits A. sanguinea via allelochemicals and direct cell contact to influence bloom formation31. Xiuning Du et al. studied an A. sanguinea bloom along the central Oregon coast and made speculations that the algal seed came from the Washington coast where a massive bloom of A. sanguinea had been observed32; and the potential impact on the environmental quality of estuarine water has also been investigated2. A. sanguinea is harmful to birds, fish33 and abalone larvae29. Since 2008, A. sanguinea blooms frequently occur off the coast of Xiamen. Little research has been reported on the bacterial communities associated with these blooms except for our previous research based on PCR-DGGE, which suggested that bacteria may play an important role in the negative regulation of A. sanguinea-blooms2.
In order to examine the free-living bacterial community during an A. sanguinea bloom in more detail 16S rRNA genes of the free-living bacteria were sequenced using the Illumina MiSeq. The aims of this study were to find out which free-living bacterial taxa were dominant during the bloom, how the free-living bacterial diversity and community structure were influenced by this bloom, and whether there were any bacterial taxa that could negatively regulate this bloom. The results showed that the A. sanguinea bloom significantly changed the free-living bacterial community structure and stimulated most bacterial taxa; however, the bloom decreased bacterial diversity while increasing species evenness.
Methods
Study sites and sample collection
The A1 bloom site (N 24°35′53.40″, E 118°9'29.67″) and the H1 control site (N 24°36'56.31'', E 118°9'15.92'') were selected in or near the area where an A. sanguinea bloom had occurred along the Xiamen coast (Figure 1). Twenty liters of near-surface (0.5 m) sea water was collected from both the bloom and control sites during (31 July, 1–4 August, 2011) and after (7 August, 2011) the bloom. Pre-autoclaved polypropylene sampling vials were used for sampling.
Figure 1. Location of sampling sites.

This map was created based on PhotoShop (Version CS5) by CYY.
Environmental parameters
Identification of phytoplankton species, phytoplankton cell counts, concentration of chlorophyll a, dissolved inorganic phosphorus (DIP), nitrate, nitrite, and ammonia, the density of A. sanguinea and bacteria, chemical oxygen demand (COD) and bacterial productivity were determined as previously described2,34. Dissolved inorganic nitrogen (DIN) was calculated by summing the molarity of NO3−, NO2− and NH4+. The N:P ratio was calculated by dividing DIN values by DIP values. The silico-molybdenum blue spectrophotometric method was used for the determination of silicate. The eutrophication index (EI) was calculated as follows:
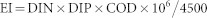 |
where DIN is the dissolved inorganic nitrogen content in mg/L; DIP is the dissolved inorganic phosphorus content in mg/L; and COD is the chemical oxygen demand in mg/L.
DNA Extraction and preparation
Water samples (500 mL) were filtered through 5-μm diameter pore-size filters (Millipore, US) to remove particle attached cells. The filtrate was filtered again through 0.22-μm diameter pore-size filters as soon as the samples were taken to the laboratory. Filters were stored at −70°C until analysis (the 5-μm diameter pore-size filters were taken for a comparison study with DGGE). DNA extraction was performed as previously described2. DNA quality was assessed using 260/280 nm and 260/230 nm ratios with a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, US). Final DNA concentrations were quantified with PicoGreen34 using a FLUOstar Optima microplate reader (BMG Labtech, Germany).
16S rRNA gene amplification and Illumina MiSeq sequencing
Primers 515F (5′-GTGCCAGCMGCCGCGG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) targeting the V4 hyper variable regions of bacterial 16S rRNA genes were selected35. Both forward and reverse primers were tagged with adapter, pad and linker sequences. Each barcode sequence (12 mer) was added to the reverse primer for pooling multiple samples into one run of sequencing. All primers were synthesized by Invitrogen (Life Technologies, USA).
PCR amplification was performed in triplicate using a Gene Amp PCR-System 9700 (Applied Biosystems, USA) in a total volume of 25 μL containing 2.5 μL 10 × PCR bufferII and 0.5 units of AccuPrime™ Taq DNA Polymerase High Fidelity (Life Technologies, USA), 0.4 μM of each primer, and 10 ng template DNA. Thermal cycling conditions were as follows: an initial denaturation at 94°C for 1 min, and 35 cycles at 94°C for 20 s, 53°C for 25 s, and 68°C for 45 s, with a final extension at 68°C for 10 min.
Following amplification, 2 μL of PCR product was used to confirm successful amplification using agarose gel (1%) electrophoresis. The triplicate PCR reactions for each sample preparation were combined and quantified with PicoGreen: 200 ng of PCR product from each sample was pooled for each sequencing run. The pooled mixture was purified with a QIAquick Gel Extraction Kit (QIAGEN Sciences, USA) and analyzed on an Agilent 2100 Bioanalyzer using High Sensitivity DNA Chips (Agilent Technologies, Germany) for size distribution, and then was re-quantified with PicoGreen.
As described in the MiSeq Reagent Kit Preparation Guide (Illumina, USA), the purified mixture was diluted and denatured to obtain an 8 pM sample DNA library, and mixed with an equal volume of 8 pM PhiX (Illumina, San Diego, CA, USA). Finally, 600 μL of the mixture library was loaded with read 1, read 2 and index sequencing primers35 on a 300-cycle (2 × 150 paired ends) kit, and run on a MiSeq at the Institute for Environmental Genomics at the University of Oklahoma.
Assignment of sequence reads to samples
After we obtained the raw sequences, the PhiX sequences were removed. The remaining raw sequences of 16S rRNA gene were sorted and distinguished by unique sample tags. Since each sample had a unique tag, all sequence reads with the same tag were assigned to the same sample. Based on the primer sequences, each sample could be further separated into two (forward and reverse) regions. Finally, the tag, both primers and spacers were trimmed based on Btrim36.
Combination and data preprocessing
Forward and reverse sequences were merged by overlapping paired-end reads using FLASH37 with a required overlap length of 10–100 bp. For all 30 samples, the number of merged reads ranged from 16,632 to 36,368 and the average merged read length was 250 bp. Low quality fragments were removed before Chimera detection and removal using U-Chime38. For all 30 samples, the number of reads ranged from 13,600 to 31,639. Samples were randomly resampled at 13,600 sequences. OTUs were classified using UCLUST at a 97% similarity level39. Taxonomic assignment was performed using the RDP classifier40 (http://rdp.cme.msu.edu). All statistical analyses were performed in R41. Dissimilarity tests were based on the Bray-Curtis dissimilarity index using analysis of similarities (ANOSIM)42, non-parametric multivariate analysis of variance (adonis)43, and multi-response permutation procedures (MRPP)44. Monte Carlo permutation was used to test statistical significances. Statistics were performed running the Vegan package (v.1.15–1)45 in R. The data comparison between the two sites was performed only for samples on 31 July and 2-4August (days 1, 3, 4, and 5 during the bloom) since there was insufficient DNA extracted from the control site on 1 and 7 August.
Results
Overview of sequencing analysis
After processing, 658,446 high quality sequences remained with an average length of 253 bases. A total of 11,807 OTUs were generated after clustering at a 97% similarity level and 5,672 OTUs were singletons. Species richness estimates of control and bloom samples were quite high (14,392.85 and 12,882.17, respectively) using the Chao estimator, while the rarefaction curves of control and bloom samples (Figure 2) were still far from saturation. A relatively large number (5,601) of OTUs failed to be assigned into any genus with a confidence level higher than 50% (Table 1), suggesting the presence of many potentially novel bacteria in the sea area near Xiamen. A higher percentage of unclassified sequences were detected in the control area (33.6%) than in the bloom area (23.3%). Archaea accounted for 0.016% (19 OTUs) of the total population, including the phyla Euryarchaeota and Thaumarchaeota. The bacteria were from 30 phyla, 75 classes, 156 orders, 292 families, and 542 genera. Proteobacteria (91%) was the most abundant phylum with 54.8% contributed by Alpha-proteobacteria and 35.1% by Gamma-proteobacteria. The top 10 most abundant sequences at class, order, family and genus levels accounted for 98.9, 92.7, 84.8 and 76.9% of all sequences, respectively (Figure 3).
Figure 2. Rarefaction curves of bloom and control samples.

Table 1. Ratio of unclassified sequences at different taxonomic levels.
| Relative abundance (%) | |||
|---|---|---|---|
| Taxonomy | Total | Bloom | Control |
| Phylum | 0.03 | 0.04 | 0.02 |
| Class | 0.19 | 0.17 | 0.22 |
| Order | 1.27 | 1.08 | 1.48 |
| Family | 4.07 | 3.89 | 4.43 |
| Genus | 28.87 | 26.30 | 31.91 |
Figure 3. Composition of the top 10 taxa at the (A) class, (B) order, (C) family, and (D) genus level for all samples.

Environmental parameters
A prolonged bloom dominated by A. sanguinea (mixed with a small amount of Skeletonema costatum and Alexandrium tamarense) occurred in site A1 from 31 July to 4 August 2011 in the sea area near Xiamen. A distinctly colored border line in the water caused by this bloom appeared between A1 and the control area (H1). The water properties and chemical analysis results are shown in Table 2. Based on the dynamics of algal density, 31 July, 2 and 3 August were during the bloom (B); 1 and 4 August were the bloom peak (BP; named BP1 and BP2, respectively); and 5 August was after the bloom (AB).
Table 2. Properties of samples during the Akashiwo sanguinea bloom in 2011 (Only the samples for comparison (bloom vs. control) are shown).
| Samples | T | Sal | pH | SP | DO | NO2− | NO3− | NH4+ | DIN | DIP | N/P | SiO4− | COD | CHLa | EI | BACT | A. sa | TALG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1-1 | 31 | 24 | 7.99 | 2.8 | 5.25 | 0.032 | 0.186 | 0.009 | 0.227 | 0.021 | 11 | 0.87 | 4.66 | 67.91 | 4.84 | 4.60 × 1011 | 1.08 × 105 | 1.32 × 105 |
| A1-2 | 31 | 26 | 7.97 | 19.0 | 4.66 | 0.029 | 0.194 | 0.009 | 0.232 | 0.032 | 7.33 | 0.84 | 2.04 | 51.9 | 3.32 | 4.11 × 1011 | 2.05 × 105 | 2.36 × 105 |
| A1-3 | 30.8 | 28 | 7.89 | 15.5 | 4.57 | 0.030 | 0.355 | - - | - - | 0.026 | - - | 0.95 | 2.95 | 23.32 | - - | 9.96 × 1011 | 1.55 × 105 | 1.84 × 105 |
| A1-4 | 30 | 29 | 7.87 | 14.2 | 4.46 | 0.030 | 0.296 | 0.028 | 0.354 | 0.020 | 18.03 | 0.90 | 2.01 | 33.09 | 3.09 | 3.11 × 1011 | 0.81 × 105 | 0.99 × 105 |
| A1-5 | 30.5 | 25 | 7.84 | 13.8 | 3.9 | 0.013 | 0.117 | 0.018 | 0.147 | 0.010 | 15.38 | 0.57 | 1.89 | 52.97 | 0.59 | 2.86 × 1011 | 1.70 × 105 | 1.89 × 105 |
| A1-8 | 31 | 30 | 7.82 | 10.1 | 3.51 | 0.026 | 0.255 | 0.078 | 0.359 | 0.027 | 13.20 | 1.02 | 1.38 | 21.3 | 3.00 | - - | 0.10 × 105 | 0.37 × 105 |
| H1-1 | 31 | 24 | 7.89 | 13.6 | 6.17 | 0.043 | 0.183 | 0.002 | 0.228 | 0.012 | 18.96 | 0.79 | 1.45 | 38.65 | 0.88 | 1.77 × 1011 | 0.18 × 105 | 0.41 × 105 |
| H1-2 | 31 | 24 | 7.87 | 10.6 | - - | 0.027 | 0.213 | 0.026 | 0.266 | 0.013 | 20.87 | 0.56 | 0.97 | 28.13 | 0.74 | 1.00 × 1011 | 0.08 × 105 | 0.18 × 105 |
| H1-3 | 31 | 25 | 7.84 | 10.4 | 3.65 | 0.023 | 0.186 | 0.031 | 0.2393 | 0.024 | 10.06 | 0.68 | 1.26 | 31.31 | 1.59 | 0.75 × 1011 | 0.22 × 105 | 0.35 × 105 |
| H1-4 | 30.5 | 29 | 7.78 | 14.6 | 3.76 | 0.015 | 0.194 | 0.030 | 0.2385 | 0.012 | 20.24 | 0.79 | 1.36 | 21.58 | 0.85 | 1.23 × 1011 | 0.63 × 105 | 0.76 × 105 |
| H1-5 | 30.5 | 29 | 7.75 | 15.0 | 3.73 | 0.023 | 0.230 | 0.040 | 0.2927 | 0.009 | 34.02 | 0.94 | 1.04 | 23.14 | 0.58 | 2.38 × 1011 | 0.08 × 105 | 0.14 × 105 |
| H1-8 | 31.8 | 26 | 7.61 | 13.0 | 6.13 | 0.039 | 0.375 | 0.122 | 0.535 | 0.050 | 10.61 | 1.51 | 1.14 | 20.81 | 6.87 | - - | 0.33 × 105 | 0.36 × 105 |
The sample numbers indicate time (days), 1: 31 July; 2–5: 1–3 August; 8: 7 August. T: temperature, °C; Sal: salinity,%; SP: suspended particles, mg/L; DO: dissolved oxygen, mg/L; NO2−: nitrite nitrogen, mg/L; NO3−: nitrate nitrogen, mg/L; NH4+: ammonia nitrogen, mg/L; DIN: dissolved inorganic nitrogen, mg/L; DIP: dissolved inorganic phosphorus, mg/L; N/P: ratio of dissolved inorganic nitrogen and DIP; SiO4−: silicate mg/L; COD: chemical oxygen demand, mg/L; CHLa: chlorophyll a, ug/L; EI: Eutrophication index; BACT: bacterial density, cells/mL; A. sa: Akashiwo sanguinea density; TALG: total algal density; - -: not available.
All environmental variables showed distinct differences between the bloom and control areas during the sampling periods. Compared to the control area, pH was much higher in A1 although the pH for both A1 and H1 decreased during the bloom. Dissolved oxygen (DO) showed the same pattern except that it was higher in H1 on day 1 and after the bloom. The concentration of suspended particles reached a peak on BP1.
Nitrite and nitrate dynamics were complex but both declined to their lowest levels on BP2 and then recovered by AB. Ammonia nitrogen showed a generally increasing trend in the bloom area except on BP2. DIP concentration was higher in A1, declined to its lowest level on BP2, and then recovered after the bloom. In general, the bloom area had much higher inorganic N and P concentrations and a lower N/P ratio. The silicate concentration was higher in A1 except on BP2 and AB. Silicate concentrations were negatively correlated with A. sanguine density, but recovered dramatically by AB. The much higher COD observed in A1, indicating the bloom outbreak, was associated with the level of water pollution.
In general, A1 had a higher chlorophyll a concentration than H1 during the bloom because of the high algal density. The EI, based on the inorganic nutrient levels, was higher in A1 than in H1 during the bloom (except for BP2) and indicated that the bloom area had a serious eutrophication status (EI > 1). The EI dynamics were in contrast to the algal density dynamics in that EI decreased as the bloom increased. On day 2 the algal density peaked, decreased and then reached a second peak on day 5. During this same time period, the EI declined to its lowest levels. The EI recovered after the bloom, indicating that this bloom was able to reduce the eutrophication pollution of the sea water. Bacterial density was also higher in the bloom area and total algal density coincided well with A. sanguinea cell numbers.
Overall effects of the A. sanguine bloom on free-living bacterial communities
Comparison of the microbial community composition was made between the bloom and control samples for days 1, 3, 4 and 5, these being the only days when the DNA was sufficient in the control area. The 16S rRNA sequences obtained were subjected to random re-sampling at 13,600 sequences for each sample, and sequences appearing in only one of three replicates were removed to minimize errors.
Bacterial species richness and diversity
Rarefaction curves based on a 97% cluster similarity showed a remarkable difference in the free-living bacterial diversity between A1 and H1. The bloom samples showed a lower bacterial diversity than the control samples, except on day 1 (Figure 4); otherwise, the estimated bacterial diversity was more stable during the bloom. However, Shannon diversity showed no significant difference between A1 and H1.
Figure 4. Rarefaction curves (A) and species richness (B) estimates for bloom and control samples using the Chao estimator method.

Bacterial composition and community structure
The bacterial community structure of the bloom and control samples at the genus level is shown in Figure 5. A dissimilarity test (Table 3) based on the adonis function showed that the free-living bacterial community structure in A1 was significantly different from that of H1, indicating that the bacterial community significantly changed during the algal bloom. Detrended correspondence analysis (DCA) results also showed that samples from the bloom area were clustered together in the center of the ordination plot (Figure 6), while control samples were scattered around the bloom samples, indicating that the bacterial community structures were more similar or stable within the bloom. In addition, bacterial community diversity evenness (Figure 7) was much higher in A1 than in H1. Simpson evenness increased alongside the bloom; while it decreased in the control area.
Figure 5. Bacterial community structure at the genus level.

Only the top 10 genera are listed, (B): bloom, (C): control.
Table 3. Dissimilarity tests of bacterial communities between bloom and control areas.
| Euclidean | Manhattan | Jaccard | Bray | |||||
|---|---|---|---|---|---|---|---|---|
| Time | F | p | F | p | F | p | F | p |
| Day 1 | 0.28 | <0.01 | 0.46 | 0.03 | 8.67 | <0.01 | 2.95 | <0.01 |
| Day 3 | 0.10 | 0.07 | 0.59 | <0.01 | 6.95 | <0.01 | 5.63 | <0.01 |
| Day 4 | 0.22 | 0.01 | 0.41 | <0.01 | 8.17 | <0.01 | 2.76 | <0.01 |
| Day 5 | 0.21 | <0.01 | 0.31 | 0.05 | 5.81 | <0.01 | 1.81 | 0.02 |
Figure 6. Detrended correspondence analysis (DCA) of the bacterial community.

Figure 7. Free-living bacterial Simpson evenness of the bloom and control groups.

Unclassified bacteria
Except on day 1, the bloom area had a much lower percentage of unclassified bacteria than the control area, indicating that the bloom environment had more known bacterial species although the percentage increased along with the bloom (Figure 8). At day 3 and later, the unclassified Rhodobacteraceae group and the SAR86 clade were the most dominant groups and, compared to the control area, A1 had fewer Rhodobacteraceae and more SAR86. In addition, A1 had more AEGEAN-169 marine group. In A1, the unclassified Rhodobacteraceae group increased as the bloom increased and decreased again after the bloom. Excluding the top 10 genera, the other genera increased after the bloom.
Figure 8. Unclassified sequence ratios of bloom and control areas at the genus level.

Bacteria closely associated with the A. sanguinea bloom
T-test results for the top 10 genera showed that, on day 1, A1 had a significantly higher relative abundance of Loktanella (P = 0.04) and the NS5 marine group (P = 0.02), but lower Litoricola (P = 0.04). On day 3, A1 had more SAR86 (P < 0.01) and SAR116 (P = 0.03) clades, AEGEAN-169 (P = 0.02) and the NS5 marine group (P < 0.01), and OM60 (NOR5) clade (P < 0.01), but fewer Vibrio (P = 0.02). On day 4, the bloom area also had fewer Vibrio (P = 0.04) and on day 5, A1 had more of the SAR116 clade (P = 0.04).
The response ratio results in Figure 9 showed that a total of 119 genera were significantly different between A1 and H1. The number of taxa that were significantly different between the two sites increased during the bloom. Most of the taxa had much higher relative abundance in A1 except on day 1 (when 12 genera increased and 45 genera decreased). Most known bacteria, such as the predominant genera SAR86, OM60 (NOR5) and the SAR116 clade, and the NS5 marine group, exhibited a higher relative abundance in A1. The Proteobacteria, especially the Alpha-proteobacteria and Gamma-proteobacteria showed the most complex changes.
Figure 9. Heat map illustrating the-fold change of genera identified in comparisons between the bloom vs. control areas.

Red-orange colors indicate an x-fold increase in abundance in the first comparison indicators, blue colors indicate a decrease. Percentages indicate relative abundances (total = 88.56% of all sequences). The color scale indicates the magnitude of the response ratio.
Spearman's correlation analysis between A. sanguinea and bacteria showed that many bacteria had significantly positive correlations with this alga, for example the OCS155 marine group (P = 0.02), the Marinoscillum (P = 0.03), ML602J-37 of Cytophagia (P = 0.4), the NS3a marine group (P = 0.02), AEGEAN-169 marine group of Rhodospirillaceae (P = 0.04), the SAR116 (P < 0.01) and SAR86 clades (P = 0.048) of the Flavobacteriaceae, Piscirickettsia (P = 0.04), and Acholeplasma (P = 0.01). In contrast, Roseibacterium (P = 0.04), Sulfitobacter (P = 0.048), Inhella (P < 0.01) of the Comamon adaceae, Pseudidiomarina (P = 0.04) and Bermanella (P = 0.03) had a negative correlation with A. sanguinea. Figure 10 shows the relative abundance dynamics of some related dominant genera during this bloom and they showed a much higher relative abundance in A1. Besides, Candidatus Aquilunai strain, the NS3 a marine group, the OM43 clade, Piscirickettsia and Salinihabitans showed an increase in the bloom. Salinihabitans of the Rhodobacteraceae significantly decreased in A1 compared to H1 but it recovered after the bloom, suggesting that an A. sanguinea bloom could restrain its growth.
Figure 10. Relative abundance of bacterial genera in the bloom and control areas.

In general, most bacteria, such as the dominant SAR86 and SAR116 clades, and AEGEAN-169 marine group increased during the bloom. These bacteria could be benefitting from the organic matter released from the algae. A total of 37 genera appeared only in the bloom area (Table 4),most from the Proteobacteria.
Table 4. Unique free-living bacteria in the bloom area.
| Phylum | Class | Genus |
|---|---|---|
| Actinobacteria | Actinobacteria | Microbacteriaceae-DS001 |
| ML602J-51 | ||
| Bacteroidetes | Cytophagia | Cyclobacteriaceae uncultured 128 |
| Flavobacteria | Winogradskyella | |
| Sphingobacteria | Aureispira | |
| Chloroflexi | SAR202 clade | SAR202 clade |
| Deferribacteres | Deferribacteres | PAUC34f |
| Firmicutes | Bacilli | Bacillus |
| Verrucomicrobia | Opitutae | Pelagicoccus |
| WCHB1-60 | WCHB1-60 | WCHB1-60 |
| Proteobacteria | Alpha-proteobacteria | 4-Org1-14 |
| E6aD10 | ||
| Methylobacterium | ||
| Hoeflea | ||
| Dinoroseobacter | ||
| Phaeobacter | ||
| Roseovarius | ||
| OM75 clade | ||
| Rhodovibrio | ||
| Thalassobaculum | ||
| Rickettsia | ||
| Caedibacter | ||
| Delta-proteobacteria | GR-WP33-58 | |
| Epsilon-proteobacteria | Sulfurospirillum | |
| Gamma-proteobacteria | BD2-7 | |
| Haliea | ||
| Colwellia | ||
| Psychrosphaera | ||
| Rheinheimera | ||
| Trabulsiella | ||
| Chromohalobacter | ||
| Cobetia | ||
| ZD0405 | ||
| Enhydrobacter | ||
| EV818SWSAP88 | ||
| Piscirickettsia | ||
| Solimonas |
Discussion
Algae performing photosynthesis consume CO2 and release O2. As their numbers increase during a bloom, photosynthesis rates increase resulting in higher pH and DO in the bloom area, which in turn promotes the growth of the phytoplankton46. However, as a bloom event ends, algal death and decomposition uses up much of the oxygen in the water and results in decreasing pH and DO. As well as affecting pH and DO, inorganic nutrients were also consumed by the bloom since NO3−, NO2− and DIP concentrations were negatively related to algal density in A1. While these nutrients were consumed by the bloom, the inorganic nutrient concentration was still higher in the bloom than in the control area. The bloom also had a lower N/P ratio indicating that A. sanguine required more N than P. Unbalanced consumption of N and P also resulted in the N/P ratio of the water changing within the bloom. Low salinity and high nutrient content from eutrophication pollution are probably important requirements for the A. sanguinea bloom formation28,47. In addition, we found that silicon was important for the growth of A. sanguine since the consumption of silicon was concurrent with A. sanguinea density during this bloom. Information on silicon consumption by A. sanguinea is limited, however, one report notes that silicon concentration decreased alongside an A. sanguinea bloom but recovered after the bloom44. A bloom might thus relieve eutrophication pollution in sea water by significant consumption of inorganic nutrients. In general, nutrient consumption continued during the bloom. A. sanguine consumed a large amount of NO3−, DIP and SiO4− but less NH4+. While higher COD and EI were needed for bloom formation, the bloom could reduce COD and relieve eutrophication pollution. The bacterial community increased in density in response to the bloom.
The free-living bacterial communities in the bloom and control areas were studied using Illumina sequencing for the first time. Although, in this study, more than 650,000 bacterial sequences and 11,807 OTUs were obtained in the Xiamen sea area, more than half of the OTUs were singletons and the rarefaction curve was unsaturated, indicating the high free bacterial diversity in this coastal area. In addition, 5601 OTUs could not be assigned to any genera with a confidence level higher than 50%, suggesting the presence of abundant novel bacteria in this area. The Proteobacteria was the predominant bacterial phylum while the Alpha-proteobacteria was the predominant class. Alpha-proteobacteria are noted as abundant free-living bacterioplankton both in coastal and open-ocean habitats48. Bacteria of both high-nutrient and low-chlorophyll ocean regions and an iron fertilized bloom in the Kerguelen ocean are also dominated by Alpha-proteobacteria49. An intriguing finding was the presence of a higher proportion of unclassified sequences in the control versus the bloom area. These unclassified sequences could either have been novel and therefore could not be classified into any of the known lineages or they belonged to less well-studied lineages and so there were few or no sequence representatives. The higher proportion of these unknown sequences in the control area was probably due to the less stable environmental conditions, while the bloom area was more stable. It has been confirmed that certain water stability conditions are necessary for bloom formation and sustainability50,51,52, since hydrology (specifically freshwater discharge, flushing and residence time) greatly influences both nutrient delivery to, and cycling in, affected waters53. Additionally, bloom samples would probably be of more interest and would be given greater attention on marine microbial studies. Similarly, in the terrestrial environment, there are more unclassified bacteria in fell-fields (treeless rock strewn areas) compared to vegetated plots in a range of Antarctic habitats54.
Compared to the control area, free-living bacterial diversity was lower and evenness much higher in the bloom area, and bacterial composition of the bloom samples was more consistent based on the DCA profile. Since bacterial diversity was relatively stable in the bloom area, this bloom did not change very much in terms of bacterial diversity. In contrast, the bacterial community structure was significantly changed, which is consistent with the results of a study in Norwegian coastal waters where bacterial diversity and community composition were stable during a spring phytoplankton bloom20. The bacterial community structure often depends on nutrient composition and phytoplankton exudate composition is species-specific, so that particular phytoplankton results in a specific bacterial community55,56. The bloom area had a higher proportion of the SAR86 group than the control area, and this group is abundant in bloom communities18. SAR86 is a clade of the Gamma-proteobacteria and is one of the most abundant uncultivated microbial groups in ocean surface water57,58,59. SAR86 maybe an aerobic chemoheterotroph based on some complete genomes58 and has the ability to consume the wide range of lipids and polysaccharides found in seawater. The biochemical composition of the organic matter produced during a bloom could greatly influence this group, and the results of our study indicated that SAR86 greatly benefitted during A. sanguine blooms. The AEGEAN-169 marine group was another dominant group with a higher relative abundance in the bloom area, but there is little relevant ecological information available on this group.
Comparison of the bacterial community structures between bloom and control areas showed that the largest difference occurred on day 3 (during the bloom). However, day 1 (during the bloom) was quite different from the other days. Since phytoplankton can release abundant organic matter that would provide bacteria with carbon and energy sources. The bloom would be expected to enhance most bacteria, particularly dominant species which may be adept at using this resource, such as the SAR116 and OM60 (NOR5) clades, and the AEGEAN-169 and NS5 marine groups. Among these, the SAR116 clade is a unique Alpha-proteobacterial group, and its members are widely found in general marine areas60,61, as well as in bloom areas18,24. They could take advantage of the organic matter released from the phytoplankton. The AEGEAN-169 clade is closely related to SAR11, which is extremely abundant in the ocean and feed on dissolved organic carbon and nitrogen. The OM60 (NOR5) clade is a group of Gamma-proteobacteria which is widespread in the euphotic zone of coastal areas62,63. The abundance of this clade increases in phytoplankton blooms64,65 and they are positively correlated with chlorophyll fluorescence in some sea areas66. The reason for this may be that these bacteria can benefit from organic matter, such as dimethylsulfoniopropionate or dimethylsulfide, released from the phytoplankton65.
Among the predominant bacteria which increased in this bloom was the Candidatus Aquiluna strain from the Microbacteriaceae, which was first isolated from a lake67 while the first strain of this genus to be sequenced was isolated from an Arctic fjord68. However, information concerning its relationship to phytoplankton blooms is limited67. The OM43 clade of the Methylophilaceae is widespread in the coastal environment69,70 and is associated with phytoplankton populations and primary productivity23. This clade was found during a diatom bloom off the Oregon coast22. They may also be able to use the methanol and other C1 compounds produced by phytoplankton70,71. Information on ML602J-37 of Cytophagia, Salinihabitans, Piscirickettsia and the NS3a marine group is limited. Roseobacter had a higher abundance in the bloom area on day 3 (5.6 ± 0.49%), when algal density was lowest, but this is not as high as has been observed in other studies11,18,72,73. Most of the unique bacterial genera that appeared only in the bloom area are known to be associated with algal blooms: Winogradskyella is heterotrophic and many isolates are associated with algae74,75 and phytoplankton blooms76; Aureispira was was found during a Microcystis bloom77; SAR202 clade organisms are ubiquitous and they usually occur in mesopelagic and deeper zones78,79, their appearance in the surface water of bloom areas indicate their presence is closely associated with DOM levels78; Rheinheimera shows anti-algal activity80; Roseovarius81 and Bacillus82 are also shown to have algicidal activity; Rickettsia83, Hoeflea84 and Methylobacterium85 can be symbiotic in algae; Dinoroseobacter may have a beneficial relationship with algae86 and can be a symbiont of dinoflagellates87; and Colwellia plays a key role in remineralizing organic matter generated from primary production88.
This study found a high bacterial diversity and abundant novel species in the sea area near Xiamen with Alpha-proteobacteria and Gamma-proteobacteria being the predominant classes detected. The low salinity and high eutrophication levels in this area were ideal for the formation of the A. sanguinea bloom. This bloom stimulated the growth of those bacterial taxa which could take advantage of the organic matter released by the phytoplankton, such as the SAR86 clade, the AEGEAN-169 marine group and some novel bacterial communities from the Rhodobacteraceae. As the A. sanguinea bloom dramatically influenced free-living bacterial communities in many aspects and this study furthered our understanding of its ecological process. Once bacterial communities have changed, so as their ecological functions and which would be studied with functional gene arrays (e.g. GeoChip) in the future.
Author Contributions
Decision to publish: T.L.Z. and J.Z.Z. Conceived and designed the experiments: C.Y.Y., Y.L., Y.Y.Z., W.Z., Y.T., J.Z.Z. and T.L.Z. Performed the experiments: C.Y.Y., Y.L., B.J.Z., J.D.N., L.Y.W. and Z.L.H. Analyzed the data: C.Y.Y., Y.L., J.D.N., L.Y.W. and Z.L.H. (C.Y.Y. and Y.L., prepared figures 1–6 and Table 1–4. J.D.N., L.Y.W. and Z.L.H. prepared figures 7–10.) Contributed reagents/materials/analysis tools: Y.Y.Z., W.Z., Y.T. and T.L.Z. Wrote the paper: C.Y.Y. and Y.L. All authors reviewed the manuscript.
Acknowledgments
The Natural Science Foundation of China (41376119, 40930847), the Public Science and Technology Research Funds for Ocean Projects (201305016, 201305022) and the Program for Changjiang Scholars and Innovative Research Team in University (41121091) are gratefully acknowledged. Professor John Hodgkiss of The University of Hong Kong is thanked for his assistance with English.
References
- Anderson D. M., Glibert P. M. & Burkholder J. M. Harmful algal blooms and eutrophication: nutrient sources, composition, and consequences. Estuaries 25, 704–726 (2002). [Google Scholar]
- Yang C. et al. Bacterial community dynamics during a bloom caused by Akashiwo sanguinea in the Xiamen Sea Area, China. Harmful Algae 20, 132–141 (2012). [Google Scholar]
- Cai W., Wang H., Tian Y., Chen F. & Zheng T. Influence of a bacteriophage on the population dynamics of toxic dinoflagellates by lysis of algicidal bacteria. Appl. Environ. Microb. 77, 7837–7840 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. et al. Toxicity of algicidal extracts from Mangrovimonas yunxiaonensis strain LY01 on a HAB causing Alexandrium tamarense. J. Hazard. Mater. 278, 372–381 (2014). [DOI] [PubMed] [Google Scholar]
- Wang B. et al. An algicidal protein produced by bacterium isolated from the Donghai Sea, China. Harmful Algae 13, 83–88 (2012). [Google Scholar]
- Li Y. et al. Towards molecular, physiological, and biochemical understanding of photosynthetic inhibition and oxidative stress in the toxic Alexandrium tamarense induced by a marine bacterium. Appl. Microbio. Biot. 98, 4637–4652 (2014). [DOI] [PubMed] [Google Scholar]
- Anderson G. & Zeutschel R. Release of dissolved organic matter by marine phytoplankton in coastal and offshore areas of the northeast Pacific Ocean. Limnol. Oceanogr. 15, 402–407 (1970). [Google Scholar]
- Biddanda B. & Benner R. Carbon, nitrogen, and carbohydrate fluxes during the production of particulate and dissolved organic matter by marine phytoplankton. Limnol. Oceanogr 42, 506–518 (1997). [Google Scholar]
- Agustí S. & Duarte C. Phytoplankton lysis predicts dissolved organic carbon release in marine plankton communities. Biogeosciences 10, 1259–1264 (2013). [Google Scholar]
- Karner M. & Herndl G. Extracellular enzymatic activity and secondary production in free-living and marine-snow-associated bacteria. Mar. Biol. 113, 341–347 (1992). [Google Scholar]
- Riemann L., Steward G. F. & Azam F. Dynamics of Bacterial Community Composition and Activity during a Mesocosm Diatom Bloom. Appl. Environ. Microb. 66, 578–587 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelboe M., Søndergaard M., Letarte Y. & Borch N. H. Attached and free-living bacteria: Production and polymer hydrolysis during a diatom bloom. Microbial. Ecol. 29, 231–248 (1995). [DOI] [PubMed] [Google Scholar]
- Baines S. B. & Pace M. L. The production of dissolved organic matter by phytoplankton and its importance to bacteria: patterns across marine and freshwater systems. Limnol. Oceanogr. 36, 1078–1090 (1991). [Google Scholar]
- Cole J. J. Interactions between bacteria and algae in aquatic ecosystems. Annu. Rev. Ecol. 13, 291–314 (1982). [Google Scholar]
- Ferrier M., Martin J. & Rooney-Varga J. Stimulation of Alexandrium fundyense growth by bacterial assemblages from the Bay of Fundy. J. Appl. microbial. 92, 706–716 (2002). [Google Scholar]
- Li Y. et al. Tenacibaculum xiamenense sp. nov., an algicidal species isolated from coastal seawater. Int. J. Syst. Evol. Micr. 63, 3481–3486 (2013). [DOI] [PubMed] [Google Scholar]
- Zheng X. et al. A marine algicidal actinomycete and its active substance against the harmful algal bloom species Phaeocystis globosa. Appl. Microbiol. Biot. 97, 9207–9215 (2013). [DOI] [PubMed] [Google Scholar]
- González J. M. et al. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl. Environ. Microb. 66, 4237–4246 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney-Varga J. N. et al. Links between phytoplankton and bacterial community dynamics in a coastal marine environment. Microb. Ecol., 49, 163–175 (2005). [DOI] [PubMed] [Google Scholar]
- Larsen A. et al. Spring phytoplankton bloom dynamics in Norwegian coastal waters: Microbial community succession and diversity. Limnol. Oceanogr. 49, 180–190 (2004). [Google Scholar]
- Teeling H. et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611 (2012). [DOI] [PubMed] [Google Scholar]
- Morris R., Longnecker K. G. & iovannoni S. Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ. microbial. 8, 1361–1370 (2006). [DOI] [PubMed] [Google Scholar]
- Rinta-Kanto J. M., Sun S., Sharma S., Kiene R. P. & Moran M. A. Bacterial community transcription patterns during a marine phytoplankton bloom. Environ. microbial. 14, 228–239 (2012). [DOI] [PubMed] [Google Scholar]
- Howard E. C. et al. Changes in dimethylsulfoniopropionate demethylase gene assemblages in response to an induced phytoplankton bloom. Appl. Environ. Microb. 77, 524–531 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N. et al. Metagenome of microorganisms associated with the toxic Cyanobacteria Microcystis aeruginosa analyzed using the 454 sequencing platform. Chin. J. Oceanol. Limnol. 29, 505–513 (2011). [Google Scholar]
- Tada Y., Taniguchi A., Sato-Takabe Y. & Hamasaki K. Growth and succession patterns of major phylogenetic groups of marine bacteria during a mesocosm diatom bloom. J. Oceanogr. 68, 509–519 (2012). [Google Scholar]
- Vasileiadis S. et al. Soil bacterial diversity screening using single 16S rRNA gene V regions coupled with multi-million read generating sequencing technologies. PloS one 7, e42671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T. et al. Effects of temperature, salinity, and irradiance on the growth of the dinoflagellate Akashiwo sanguinea. J. Exp. Mar. Biol. Ecol. 342, 226–230 (2007). [Google Scholar]
- Yamasaki Y. et al. Growth inhibition and formation of morphologically abnormal cells of Akashiwo sanguinea (Hirasaka) G. Hansen et Moestrup by cell contact with Cochlodinium polykrikoides Margalef. Mar. Biol. 152, 157–163 (2007). [Google Scholar]
- Qiu X. et al. Growth interactions between the raphidophyte Chattonella antiqua and the dinoflagellate Akashiwo sanguinea. Harmful Algae 11, 81–87 (2011). [Google Scholar]
- Qiu X. et al. Allelopathy of the raphidophyte Heterosigma akashiwo against the dinoflagellate Akashiwo sanguinea is mediated via allelochemicals and cell contact. Mar. Ecol. Prog. Ser. 446, 107–118 (2012). [Google Scholar]
- Du X., Peterson W., McCulloch A. & Liu G. An unusual bloom of the dinoflagellate Akashiwo sanguinea off the central Oregon, USA, coast in autumn 2009. Harmful Algae 10, 784–793 (2011). [Google Scholar]
- Jessup D. A. et al. Mass stranding of marine birds caused by a surfactant-producing red tide. PLoS One 4, e4550 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S. J., Costa J. & Emanuel J. R. PicoGreen Quantitation of DNA: Effective Evaluation of Samples Pre-or Psost-PCR. Nucleic. Acids. Res. 24, 2623–2625 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME. J. 6, 1621–1624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98, 152–153 (2011). [DOI] [PubMed] [Google Scholar]
- Magoč T. & Salzberg S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C., Haas B. J., Clemente J. C., Quince C. & Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- Cole J. R. et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic. Acids. Res. 37, 141–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R. D. C. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. ISBN 3-900051-07-0 (2008) Available at: http://www.R-project.org. (Accessed: 15th January 2013) [Google Scholar]
- Clarke K. R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993). [Google Scholar]
- Anderson M. J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26, 32–46 (2001). [Google Scholar]
- Mielke P. W., Berry K. J., Brockwell P. J. & Williams J. S. A class of nonparametric tests based on multiresponse permutation procedures. Biometrika 68, 720–724 (1981). [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930 (2003). [Google Scholar]
- Hansen P. J. Effect of high pH on the growth and survival of marine phytoplankton: implications for species succession. Aquat. Microb. Ecol. 28, 279–288 (2002). [Google Scholar]
- Qasim S., Bhattathiri P. & Devassy V. The influence of salinity on the rate of photosynthesis and abundance of some tropical phytoplankton. Mar. Biol. 12, 200–206 (1972). [Google Scholar]
- DeLong E. F., Franks D. G. & Alldredge A. L. Phylogenetic diversity of aggregate-attached vs. free-living marine bacterial assemblages. Limnol. Oceanogr. 38, 924–934 (1993). [Google Scholar]
- West N. J., Obernosterer I., Zemb O. & Lebaron P. Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ. Microbiol. 10, 738–756 (2008). [DOI] [PubMed] [Google Scholar]
- Wong K., Lee J. H. & Hodgkiss I. A simple model for forecast of coastal algal blooms. Estuar. Coast. Shelf. S. 74, 175–196 (2007). [Google Scholar]
- Cullen J. J. & MacIntyre J. G. Behavior, physiology and the niche of depth-regulating phytoplankton. Nato. Asi. Series. G. Ecol. Sci. 41, 559–580 (1998). [Google Scholar]
- Watanabe M. et al. Generation of a Chattonella antiqua bloom by imposing a shallow nutricline in a mesocosm. Limnol. Oceanogr. 40, 1447–1480 (1995). [Google Scholar]
- Paerl H. [Cyanobacterial harmful algal blooms: state of the science and research needs]. Nutrient and other environmental controls of harmful cyanobacterial blooms along the freshwater-marine continuum [217–237] (Springer, Berlin, 2008). [DOI] [PubMed] [Google Scholar]
- Yergeau E., Newsham K. K., Pearce D. A. & Kowalchuk G. A. Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ. Microbial. 9, 2670–2682 (2007). [DOI] [PubMed] [Google Scholar]
- Fogg G. The ecological significance of extracellular products of phytoplankton photosynthesis. Bot. Mar. 26, 3–14 (1983). [Google Scholar]
- Sarmento H. & Gasol J. M. Use of phytoplankton-derived dissolved organic carbon by different types of bacterioplankton. Environ. Microbial. 14, 2348–2360 (2012). [DOI] [PubMed] [Google Scholar]
- Molloy S. Marine microbiology: SAR86: streamlined for success. Nat. Rev. Microbiol. 10, 82–83 (2012). [DOI] [PubMed] [Google Scholar]
- Dupont C. L. et al. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME. J. 6, 1186–1199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattenhofer M. et al. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ. Microbial. 11, 2078–2093 (2009). [DOI] [PubMed] [Google Scholar]
- Giovannoni S. J., Britschgi T. B., Moyer C. L. & Field K. G. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345, 60–63 (1990). [DOI] [PubMed] [Google Scholar]
- Giovannoni S. & Rappé M. [Ecological and Applied Microbiology]. Evolution, diversity, and molecular ecology of marine prokaryotes [47–84] (Wiley, New York, 2000). [Google Scholar]
- Spring S. et al. Taxonomy and evolution of bacteriochlorophyll a-containing members of the OM60/NOR5 clade of marine gammaproteobacteria: description of Luminiphilus syltensis gen. nov., sp. nov., reclassification of Haliea rubra as Pseudohaliea rubra gen. nov., comb. nov., and emendation of Chromatocurvus halotolerans. BMC Microbiol. 13, 118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J. C. et al. Polyphyletic photosynthetic reaction centre genes in oligotrophic marine Gammaproteobacteria. Environ. Microbial. 9, 1456–1463 (2007). [DOI] [PubMed] [Google Scholar]
- Beardsley C., Pernthaler J., Wosniok W. & Amann R. Are readily culturable bacteria in coastal North Sea waters suppressed by selective grazing mortality? Appl. Environ. Microb. 69, 2624–2630 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernthaler A. & Pernthaler J. Diurnal variation of cell proliferation in three bacterial taxa from coastal North Sea waters. Appl. Environ. Microb. 71, 4638–4644 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. et al. Biogeography and phylogeny of the NOR5/OM60 clade of Gammaproteobacteria. Syst. Appl. Microbiol. 32, 124–139 (2009). [DOI] [PubMed] [Google Scholar]
- Hahn M. W. Description of seven candidate species affiliated with the phylum Actinobacteria, representing planktonic freshwater bacteria. Int. J. Syst. Evol. Microbiol. 59, 112–117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang I. et al. Genome sequence of “Candidatus Aquiluna” sp. strain IMCC13023, a marine member of the actinobacteria isolated from an Arctic Fjord. J. Bacteriol. 194, 3550–3551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappé M. S., Vergin K. & Giovannoni S. J. Phylogenetic comparisons of a coastal bacterioplankton community with its counterparts in open ocean and freshwater systems. FEMS. Microbiol. Ecol. 33, 219–232 (2000). [DOI] [PubMed] [Google Scholar]
- Giovannoni S. J. et al. The small genome of an abundant coastal ocean methylotroph. Environ. Microbial. 10, 1771–1782 (2008). [DOI] [PubMed] [Google Scholar]
- Heikes B. G. et al. Atmospheric methanol budget and ocean implication. Global. Biogeochem. Cy. 16, 1133, 10.1029/2002GB001895 (2002). [DOI] [Google Scholar]
- Rinta-Kanto J. M. et al. Analysis of sulfur-related transcription by Roseobacter communities using a taxon-specific functional gene microarray. Environ. Microbial. 13, 453–467 (2011). [DOI] [PubMed] [Google Scholar]
- Biers E. J., Sun S. & Howard E. C. Prokaryotic genomes and diversity in surface ocean waters: interrogating the global ocean sampling metagenome. Appl. Environ. Microb. 75, 2221–2229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S. et al. Diversity of Chemotactic Heterotrophic Bacteria Associated with Arctic Cyanobacteria. Curr. Microbial. 66, 64–71 (2013). [DOI] [PubMed] [Google Scholar]
- Nedashkovskaya O. I., Vancanneyt M., Kim S. B. & Zhukova N. V. Winogradskyella echinorum sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from the sea urchin Strongylocentrotus intermedius. Int. J. Syst. Evol. Microbiol. 59, 1465–1468 (2009). [DOI] [PubMed] [Google Scholar]
- Pinhassi J., Nedashkovskaya O. I., Hagström Å. & Vancanneyt M. Winogradskyella rapida sp. nov., isolated from protein-enriched seawater. Int. J. Syst. Evol. Microbiol. 59, 2180–2184 (2009). [DOI] [PubMed] [Google Scholar]
- Cheng C. et al. Pyrosequencing Reveals the Bacterial Community during Microcystis water Bloom in Guanting Reservoir, Beijing. Pro. Eng. 18, 341–346 (2011). [Google Scholar]
- Schattenhofer M. et al. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ. Microbiol. 11, 2078–93 (2009). [DOI] [PubMed] [Google Scholar]
- Bryan M. J. et al. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One 3, e2048 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. M., Lin C. Y. & Sheu S. Y. Investigating antimicrobial activity in Rheinheimera sp. due to hydrogen peroxide generated by l-lysine oxidase activity. Enzyme. Microb. Tech. 46, 487–493 (2010). [DOI] [PubMed] [Google Scholar]
- Fernandes N. et al. Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PloS one 6, e27387 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S. Y., Ishida K., Ito Y., Okada S. & Murakami M. Bacillamide, a novel algicide from the marine bacterium, Bacillus sp. SY-1, against the harmful dinoflagellate, Cochlodinium polykrikoides. Tetrahedron letters 44, 8005–8007 (2003). [Google Scholar]
- Kawafune K., Hongoh Y., Hamaji T. & Nozaki H. Molecular identification of rickettsial endosymbionts in the non-phagotrophic volvocalean green algae. PLoS One 7, e31749 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios L., Arahal D. R., Reguera B. & Marin I. Hoeflea alexandrii sp. nov., isolated from the toxic dinoflagellate Alexandrium minutum AL1V. Int. J. Syst. Evol. Microbiol. 56, 1991–1995 (2006). [DOI] [PubMed] [Google Scholar]
- Zarnowski R., Felske A., Ellis R. J., Geuns J. M. & Pietr S. J. A Methylobacterium-like organism from algal crusts covering silicone rubber electric insulators in Africa. J. Appl. Microbiol. 93, 1012–1019 (2002). [DOI] [PubMed] [Google Scholar]
- Goecke F., Thiel V., Wiese J., Labes A. & Imhoff J. F. Algae as an important environment for bacteria-phylogenetic relationships among new bacterial species isolated from algae. Phycologia 52, 14–24 (2013). [Google Scholar]
- Wagner-Dobler I. et al. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker's guide to life in the sea. ISME. J. 4, 61–77 (2010). [DOI] [PubMed] [Google Scholar]
- Williams T. J. et al. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317 (2013). [DOI] [PubMed] [Google Scholar]