

Abstract
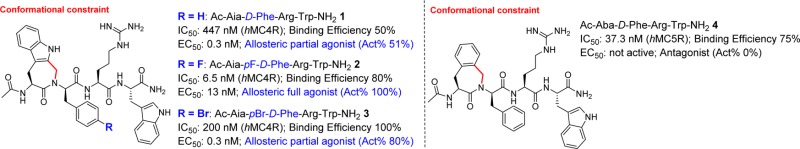
To address the need for highly potent, metabolically stable, and selective agonists, antagonists, and inverse agonists at the melanocortin receptor subtypes, conformationally constrained indolo- and benzazepinone residues were inserted into the α-MSH pharmacophore, His6-Phe7-Arg8-Trp9-domain. Replacement of His6 by an aminoindoloazepinone (Aia) or aminobenzazepinone (Aba) moiety led to hMC4R and hMC5R selective agonist and antagonist ligands, respectively (tetrapeptides 1 to 3 and 4, respectively). In peptides 1 to 3 and depending on the para-substituent of the d-Phe residue in position 2, the activity goes from allosteric partial agonism (1, R = H) to allosteric full agonism (2, R = F) and finally allosteric partial agonism (3, R = Br).
Keywords: Melanocortin ligands, constrained aminoazepinones, hMC4R and hMC5R molecular modeling and ligand docking, fluorine peptidomimetics, allosteric hMC4R ligands
The endogenous melanocortin peptides include the α-, β-, and γ-melanocyte stimulating hormones (MSHs). These peptide hormones derive from posttranslational processing of the pro-opiomelanocortin (POMC) prehormone.1−4 They play a role in a wide range of biological and physiological responses such as feeding and learning behavior, sexual function, and energy homeostasis. The melanocortin peptides operate through interactions with the G protein-coupled melanocortin receptors in both the peripheral and central nervous system and activate the adenylate cyclase second messenger signal transduction cascade.1−4
To date five melanocortin receptor subtypes have been identified.1−4 It has been observed that the primary native ligand for MC1R, MC3R, MC4R, and MC5R is α-melanocyte stimulating hormone (α-MSH, Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2). The native ligands possess neither a high selectivity for any of the above-mentioned MC receptors5 nor a long half-life time as they are rapidly hydrolyzed by proteases. In order to establish the biological functions of the individual MC receptors, the need for highly potent, metabolically stable, and selective agonists, antagonists, and inverse agonists for the four receptors MC1R, MC3R, MC4R, and MC5R became apparent.5 Since all melanocortin peptides possess a central and conserved “His-Phe-Arg-Trp” pharmacophore tetrapeptide sequence, most studies have focused on modifications at the level of this “message” domain.6,7
Initially, α-MSH was modified through the replacement of Met4 by the pseudoisosteric amino acid norleucine (Nle) to prevent oxidation of its side chain, and the configuration of Phe7 was inverted to d-Phe7 to enhance stability against proteases. The resulting peptide [Nle4,d-Phe7]-α-MSH (MT-I) revealed not only to be more potent than α-MSH but also to have a higher in vivo stability and improved pharmacokinetics.8 In addition, molecular modeling and NMR studies showed that the bioactive conformation of several cyclic ligands involved a β-turn structure at the level of the tetrapeptide pharmacophore. One of these potent cyclic analogues was a lactam (Ac-Nle-c[Asp-His-d-Phe-Arg-Trp-Lys]-NH2, MT-II), which proved to be an extremely potent, but nonselective, agonist of human MC1R, MC3R, MC4R, and MC5R.9,10
In order to design potent and more selective peptide melanocortin ligands, several interesting structural modifications of MT-II were reported.11 The replacement of His6 by Pro in MT-II by Grieco et al. resulted in retention of agonist potency for most MC receptors.12 This was the first indication that His6 was not crucial for agonism. Additionally, MT-II-based MCR antagonists, such as SHU-9119 (Ac-Nle4-c[Asp5-His6-d-Nal(2′)7-Arg8-Trp9-Lys10]-NH2) were developed.11,13 Upon substitution of His6 in SHU-9119 by conformationally restricted amino acids, selective antagonists for the hMC3R and hMC4R were discovered.14,15 Hence, the importance of position 6 with regard to activity and MCR selectivity was demonstrated.14,15
Previously, we replaced the His6-d-Phe7 dipeptide segment in MT-II by an Aba-Xxx motif (with Aba = 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one, Figure 1).16 Such azepinones are frequently used in our group and by others for the improvement of peptide stability, selectivity, and potency.16−21 Aba can be viewed as a topographically constrained Phe building block. Molecular modeling indicated a good backbone overlap of all Aba-containing analogues with the proposed conformation of MT-II.16 On the basis of the analysis of Cα(i)–Cα(i + 3) distances and the distances between the CO group of Asp5 and the NH group of Arg8, the β-turn conformation was, however, not adopted by these analogues. The local constraint in the Aba-containing linear analogue of MT-II (Aba-1, Figure 1) was not sufficient to induce efficient binding affinity. Gratifyingly, the cyclic lactam analogue Aba-2 with the “Aba6-d-Phe7” motif proved to be a selective hMC3R antagonist (IC50 of 50 nM at hMC3R). We concluded that the Aba building block needed to be used in conjunction with a global conformational constraint from these results since the linear analogue Aba-1 did not bind at concentrations up to 10 μM.16
Figure 1.

[Aba]MT-II analogues Aba-2–4 and the linear equivalent Aba-1.16
Haskell-Luevano and co-workers prepared a large series of analogues of the linear tetrapeptide Ac-His-d-Phe-Arg-Trp-NH2 in which the Phe7 residue was modified via the replacement of other aromatic residues and various substituents at the para-position of the benzyl side chain of d-Phe7. They obtained an improved selectivity for the mouse (m)MC3R versus the mMC4R as well as a differentiation of agonist versus antagonist activity.22,23
This information could pave the way to the development of low molecular weight peptidomimetics as the tetrapeptide ligands are of intermediate molecular weight.22−24 Proneth and co-workers modified the melanocortin tetrapeptide at the para-position of the d-Phe7 side chain. Halogenation at this position was used to unravel important side chain determinants for differentiation of the agonist versus antagonist activity of the mMC3R.23 By insertion of a pF-d-Phe or pCl-d-Phe residue, they achieved good to moderate activation of mMC1R and mMC3R and excellent activation of mMC4R and mMC5R.23
The study presented herein employs ligand–receptor structure–activity relationship (SAR) studies of locally constrained pharmacophore tetrapeptides 1–5 (Figure 2) to investigate the requirements for receptor selectivity and for differentiation of agonist versus antagonist activity at the human melanocortin receptors MC1R, MC3R, MC4R, and MC5R. Since the MC2R is only activated by ACTH, it has been excluded from this study. Aiming at compact and constrained tetrapeptide analogues, we decided to replace the His6 residue in the tetrapeptide pharmacophore with Aia, Aba, and Ata scaffolds (Figure 2) and to evaluate their influence on receptor selectivity and agonist versus antagonist activity (Table 1).
Figure 2.

Locally constrained melanocortin tetrapeptides 1–5, containing the Aia (1–3), Aba (4), and Ata (5) scaffolds.
Table 1. Agonist versus Antagonist Functional Bioactivities of Modified Tetrapeptides 1–5 at the Human Melanocortin Receptors in Comparison with MT-IIa as a Cyclic Lactam Analogue of α-MSHb.
|
hMC1R |
hMC3R |
hMC4R |
hMC5R |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| peptide | sequence | IC50 (nM) | %BE | EC50 (nM) | Act% | IC50 (nM) | %BE | EC50 (nM) | Act% | IC50 (nM) | %BE | EC50 (nM) | Act% | IC50 (nM) | %BE | EC50 (nM) | Act% |
| 1 | Ac-Aia-d-Phe-Arg-Trp-NH2 | 1352 | 50 | NA | 0 | NB | 52 | 20 | 447 | 50 | 0.3 | 51 | 636 | 80 | NA | 0 | |
| 2 | Ac-Aia-p F-d-Phe-Arg-Trp-NH2 | 258 | 40 | 226 | 50 | 5000 | 50 | NA | 0 | 6.5 | 80 | 13 | 100 | 167 | 80 | 4112 | 50 |
| 3 | Ac-Aia-p Br-d-Phe-Arg-Trp-NH2 | 300 | 80 | 700 | 100 | 100 | 100 | 2435 | 50 | 200 | 100 | 0.3 | 80 | 70.3 | 100 | NA | 0 |
| 4 | Ac-Aba-d-Phe-Arg-Trp-NH2 | 594 | 80 | 1000 | 70 | >2000 | 59 | 2000 | 40 | 220 | 80 | 227 | 80 | 37.3 | 75 | NA | 0 |
| 5 | Ac-Ata-d-Phe-Arg-Trp-NH2 | 4540 | 60 | 750 | 100 | 1000 | 55 | 55 | 20 | 200 | 80 | 203 | 80 | 137 | 70 | NA | 0 |
| MT-II | Ac-N le-c [Asp-His-d-Phe-Arg-Trp-Lys]-NH2 | 1 ± 0.1 | 100 | 1.02 ± 0.4 | 100 | 2.0 ± 0.1 | 100 | 5.1 ± 0.3 | 100 | 2.3 ± 0.85 | 100 | 2.1 ± 0.6 | 100 | 4.2 ± 1.3 | 100 | 5.7 ± 2.2 | 100 |
MT-II = Ac-Nle-c[Asp-His-d-Phe-Arg-Trp-Lys]-NH2.
IC50 = concentration of peptide at 50% specific binding (N = 4). NB = 0% of 125I-NDP-α-MSH displacement observed at 10 μM. %BE (binding efficiency) = maximal % of 125I-NDP-α-MSH displacement observed at 10 μM. EC50 = Effective concentration of peptide that was able to generate 50% maximal intracellular cAMP accumulation (N = 4). Act% = % of cAMP produced at 10 μM ligand concentration, in relation to MT-II. NA = 0% cAMP accumulation observed at 10 μM. The peptides were tested at a range of concentration from 10–10 to 10–5 M.
The affinity for tetrapeptide ligands 1–5 was evaluated by competition binding experiments that were carried out using HEK293 cells, which stably expressed the human MC1, MC3, MC4, and MC5 receptors.15 The activity at these receptors was evaluated in the same cells using a cAMP assay.15 The binding affinities, expressed as IC50 values, and the cAMP accumulation data are compared to MT-II and presented in Table 1. Introduction of the Aia-d-Phe dipeptidomimetic in the linear pharmacophoric sequence (tetrapeptide 1) resulted in weak micromolar antagonist affinity for the hMC1R, nanomolar partial agonism at hMC3R (EC50 = 52 nM), excellent partial agonism at hMC4R (EC50 = 0.3 nM), and moderate antagonism at the hMC5R. The fact that 1 was unable to displace the radioligand at the hMC3R (and with low potency at the hMC4R) is an indication of allosteric binding at these receptors. Fluorination of the para-position of d-Phe7 led to enhanced activity at the hMCRs,23 as low nanomolar selective agonist activity (IC50 = 6.5 nM; EC50 = 13 nM) was indeed observed at hMC4R for tetrapeptide 2. In addition, this substitution gave moderate nanomolar binding as well as weak partial agonism at hMC1R and hMC5R. In contrast to the pF-d-Phe7 tetrapeptide 2, which resulted in a selective hMC4R agonist, the pBr-d-Phe7 tetrapeptide 3 showed significant loss in binding affinity to hMC4R (IC50 = 200 nM), but resulted in a potent allosteric partial agonist (EC50 = 0.3 nM and 80 activity %) at this receptor. In addition, tetrapeptide 3 is a weak agonist at hMC1R, a partial agonist at hMC3R, and a moderate antagonist at hMC5R (IC50 = 70 nM). These affinities reflect the importance of the presence and nature (i.e., van der Waals radius) of the halogen substituent, next to that of the conformationally constrained Aia residue. Whereas the full agonist activity observed in Aia tetrapeptide 2 against hMC4R renders it a good candidate for further modifications, tetrapeptide 1 might also serve as a good template for the development of an allosteric hMC4R partial agonist. The aminobenzazepinone Aba-d-Phe analogue 4 displayed very weak agonist activity on hMC4R (IC50 = 220 nM). In contrast, it resulted in a selective antagonist for hMC5R (IC50 = 37 nM), which renders it an ideal template for the investigation of hMC5R antagonism. In contrast to the Aia and Aba analogues, the Ata-d-Phe analogue 5 showed no specific subtype receptor selectivity. We did not, therefore, focus on this analogue in subsequent docking studies (see below).
The preferential conformations of the most promising peptidic ligands 2 and 4 were analyzed by 2D ROESY NMR spectroscopy in DMSO solution at room temperature. The resulting distance constraints and homonuclear 3JHN,HA couplings (typically 7–9 Hz) were employed for restraining main chain conformations in both cases. Unfortunately, conformational analysis by NMR was unable to determine a preferred topology for tetrapeptides 2 and 4 since the topologies of the ten lowest energy conformers were all substantially different. We concluded, therefore, that preorganization in solution was absent or very limited. In contrast to earlier studies,25 this result could be explained by the fact that the conformational constraint was each time introduced as the first amino acid of the tetrapeptide sequence (Figure 2).
Peptides 2 and 4 were docked into the active state model of hMC4R proposed by Mosberg et al.,26 which was also used in the docking study of MT-II and SHU-9119 by Grieco and co-workers.27 Upon docking of the peptide ligands, multiple binding poses were generated and clustered, yielding two clusters for peptide 2 and one larger cluster for analogue 4. The binding modes selected are high-ranking poses, which occurred multiple times among the best scoring models. The binding mode for both peptides positioned the conformationally restricted Aia or Aba residues in a hydrophobic pocket at the bottom of the extracellular binding site as shown in Figures 3 and 4b,c. The obtained docking poses of the peptides were compared with the earlier proposed binding mode of the pharmacophore of [Nle4,d-Phe7]-α-MSH, His6-d-Phe7-Arg8-Trp9, also shown in Figures 3 and 4a.26 The residues of [Nle4,d-Phe7]-α-MSH (dark brown) are buried slightly deeper into the binding site than peptides 2 (magenta) and 4 (purple). This could reflect the fact that [Nle4,d-Phe7]-α-MSH is more potent than the presented peptides. The 2D ligand interaction maps of [Nle4,d-Phe7]-α-MSH, peptides 2 and 4, together with a detailed comparison of the interactions found in the binding poses, are presented in the Supporting Information. Peptide 2 has a slightly higher number of hydrophobic and H-bonding interactions. In particular, the Aia residue is in contact with six hydrophobic residues (two of which are the conserved residues Phe261 and His264), while the Aba residue in 4 is only in contact with two hydrophobic residues. Furthermore, the fluorinated d-Phe in peptide 2 is in contact with three residues: hydrophobic interactions to Met130 and Ile129 and one polar interaction to the CO of Asp126, with Phe284, Phe261, and His264 also in the vicinity. The nonfluorinated d-Phe in peptide 4, however, features only three intermolecular contacts. Contacts to the fluorine could further stabilize the receptor interaction with peptide 2, and the larger van der Waals radius of the fluorinated benzyl in pF-d-Phe might contribute to favorable conformational changes for the induction of agonism.
Figure 3.
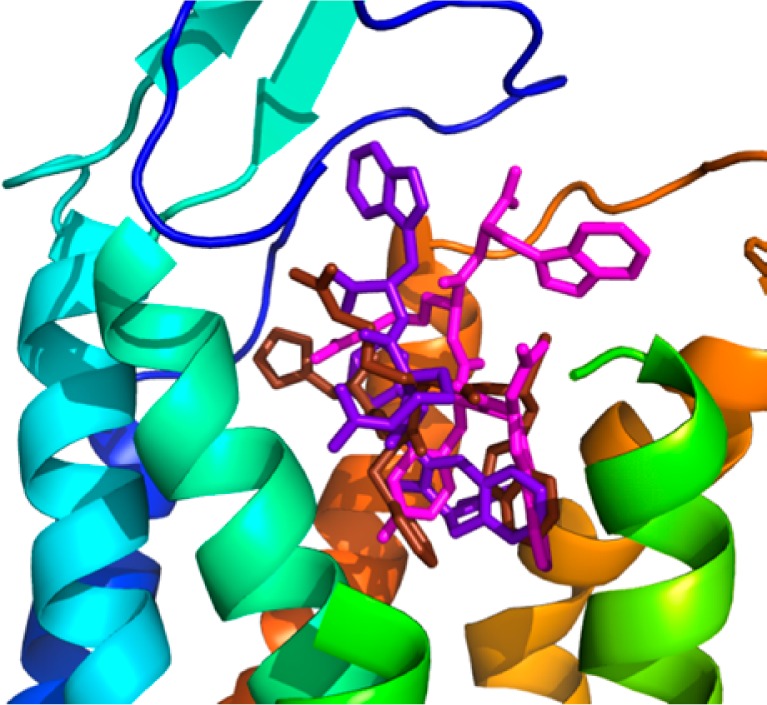
Combined docking poses for Aia peptide 2 (magenta, calculated affinity: −10.8 kcal/mol), Aba peptide 4 (purple, calculated affinity: −10.0 kcal/mol), and [Nle4,d-Phe7]-α-MSH (dark brown) in hMC4R. Extracellular part of TMH4 (green helix) and -5 (yellow helix) are hidden for better visibility of the poses.
Figure 4.

Best docking pose for (A) [Nle4,d-Phe7]-α-MSH (dark brown), (B) Aia peptide 2 (magenta), and (C) Aba peptide 4 (purple) in hMC4R. Extracellular part of TMH4 (green helix) and -5 (yellow helix) are hidden for better visibility of the poses.
In order to understand the selective hMC5R antagonist activity of peptidomimetic 4, we compared the binding of peptides 4 and 2 to hMC5R as shown in Figure 5. The best docking pose of Aba-containing peptide 4 in hMC5R is very similar to the one in hMC4R: the Aba and d-Phe residues position themselves at the bottom of the hydrophobic part of the binding pocket and interact with conserved residues, such as Phe254. The Aia peptide 2, however, either fails to dock into the binding site or assumes a docking pose in which the Aia residue is located outside the binding pocket, while Trp and d-Phe residues occupy the hydrophobic interior. This suggests that binding of peptide 2 to hMC5R is sterically hindered by the bulkiness of the Aia residue, while the Aba residue in peptide 4 fits into the binding pocket but is unable to activate the receptor.
Figure 5.
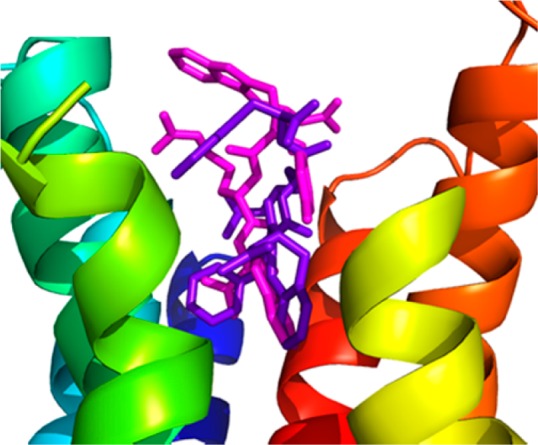
Combined docking poses for Aia peptide 2 (magenta, calculated affinity: −8.0 kcal/mol) and Aba peptide 4 (purple, calculated affinity: −9.4 kcal/mol) in hMC5R. Extracellular part of TMH4 (green helix) and -5 (yellow helix) are hidden for better visibility of the poses.
In conclusion, the insertion of constrained aminobenzo- and indoloazepinone-based residues into the core melanocortin tetrapeptides has resulted in compact and selective human melanocortin receptor ligands. The nature (steric bulk) and substitution pattern (para-halogenation) are important structural features for the modulation of receptor affinity, selectivity, and activity in these constrained peptides. Peptide 2 is a very potent and selective agonist for hMC4R, rendering it a good candidate for studies related to obesity and diabetes. Moreover, the newly discovered antagonist activity of peptide 4 at hMC5R can prove very useful to studies investigating anxiety and other neurological disorders. As further modification of the azepinone cores can improve and fine-tune the pharmacodynamics of these ligands, they represent valuable tools toward the unravelling of the individual role of each receptor subtype, but they may also be used for the development of specific receptor ligands with a distinct activity profile (i.e., allosteric/orthosteric full/partial agonist, neutral antagonist, or inverse agonist).
Glossary
Abbreviations
- Aba
4-amino-1,2,4,5-tetrahydro-3H-2-benzazepin-3-one
- ACTH
adrenocorticotropic hormone
- Aia
4-amino-1,4,5,10-tetrahydroazepino[3,4-b]indol-3(2H)-one
- Ata
7-amino-7,8-dihydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepin-6(5H)-one
- cAMP
cyclic(′-adenosine monophosphate)
- GPCR
G-protein coupled receptor
- MC1R
melanocortin-1 receptor
- MC2R
melanocortin-2 receptor
- MC3R
melanocortin-3 receptor
- MC4R
melanocortin-4 receptor
- MC5R
melanocortin-5 receptor
- MCR
melanocortin receptor
- MSH
melanocyte-stimulating hormone
- POMC
pro-opiomelanocortin
- SAR
structure–activity relationship
- α-MSH
α-melanocyte stimulating hormone
- β-MSH
β-melanocyte stimulating hormone
- γ-MSH
γ-melanocyte stimulating hormone
Supporting Information Available
Complete experimental details along with the characterization of the synthesized tetrapeptides 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ O.V.D.P and K.F contributed equally to this work. The manuscript was written through contributions of all authors. O.V.D.P., S.B., D.F., K.B., and D.T. were in charge of the peptide synthesis; I.Z., M.C., and V.J.H. for the functional bioactivities of tetrapeptides 1–5 at the human melanocortin receptors; O.V.D.P., K.F., and J.C.M. for the conformational NMR analysis of tetrapeptides 2 and 4; and K.F., I.Z., M.C., and V.J.H. for the homology modeling of hMC4R and hMC5R and ligand docking. All authors have given approval to the final version of the manuscript.
The work of O.V.D.P., K.F., J.C.M., S.B., D.F., K.B.,and D.T. was supported by the Research Foundation–Flanders (FWO Vlaanderen) and the Agency for Innovation by Science and Technology (IWT). The bioassay studies were supported in part by grants from the U.S. Public Health Service, National Institutes of Health, DK017420, GM 108040, and DA06284.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hruby V. J.; Wilkes B. C.; Cody W. L.; Sawyer T. K.; Hadley M. E. Melanotropins: Structural, Conformational and Biological Considerations in the Development of Superpotent and Superprolonged Analogues. Pept. Protein Rev. 1984, 3, 1–64. [Google Scholar]
- Cone R. D.; Lu D.; Kopula S.; Vage D. I.; Klungland H.; Boston B.; Chen W.; Orth D. N.; Pouton C.; Kesterson R. A. The Melanocortin Receptors: Agonists, Antagonists, and the Hormonal Control of Pigmentation. Recent Prog. Horm. Res. 1996, 51, 287–318. [PubMed] [Google Scholar]
- Fan W.; Boston B. A.; Kesterson R. A.; Hruby V. J.; Cone R. D. Role of Melanocortinergic Neurons in Feeding and the agouti Obesity Syndrome. Nature 1997, 385, 165–168. [DOI] [PubMed] [Google Scholar]
- Huszar D.; Lynch C. A.; Fairchild-Huntress V.; Dunmore J. H.; Smith F. J.; Kesterson R. A.; Boston B. A.; Fang Q.; Berkemeir L. R.; Gu W.; Cone R. D.; Campfield L. A.; Lee F. Targeted Disruption of the Melanocortin-4-Receptor Results in Obesity in Mice. Cell 1997, 88, 131–141. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Cai M.; Cain J.; Nyberg J.; Trivedi D. Design of Novel Melanocortin Receptor Ligands: Multiple Receptors, Complex Pharmacology, the Challenge. Eur. J. Pharmacol. 2011, 660, 88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby V. J.; Wilkes B. C.; Hadley M. E.; Al-Obeidi F.; Sawyer T. K.; Staples D. J.; de Vaux A. E.; Dym O.; de Lauro Castrucci A.-M.; Hintz M. F.; Riehm J. R.; Rao R. R. α-Melanotropin: the Minimum Active Sequence in the Frog Skin Bioassay. J. Med. Chem. 1987, 30, 2126–2130. [DOI] [PubMed] [Google Scholar]
- de Lauro Castrucci A.-M.; Hadley M. E.; Sawyer T. K.; Wilkes B. C.; Al-Obeidi F.; Staples D. J.; de Vaux A. E.; Dym O.; Hintz M. F.; Riehm J. P.; Rao K. R.; Hruby V. J. α-Melanotropin: the Minimal Active Sequence in the Lizard Skin Bioassay. Gen. Comp. Endrocrinol. 1989, 73, 157–163. [DOI] [PubMed] [Google Scholar]
- Sawyer T. K.; Sanfilippo P. J.; Hruby V. J.; Engel M. H.; Heward C. B.; Burnett J. B.; Hadley M. E. [Nle4, D-Phe7]-α-Melanocyte Stimulating Hormone: a Highly Potent α-Melanotropin with Ultralong Biological Activity. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 5754–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Obeidi F. A.; Hadley M. E.; Pettitt B. M.; Hruby V. J. Design of a New Class of Superpotent Cyclic α-Melanotropins based on Quenched Dynamic Simulations. J. Am. Chem. Soc. 1989, 111, 3413–3416. [Google Scholar]
- Al-Obeidi F. A.; de Lauro Castrucci A.-M.; Hadley M. E.; Hruby V. J. Potent and Prolonged Acting Cyclic Lactam Analogues of α-Melanotropin: Design based on Molecular Dynamics. J. Med. Chem. 1989, 32, 2555–2561. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Cai M.; Grieco P.; Han G.; Kavarana M.; Trivedi D. Exploring the Stereostructural Requirements of Peptide Ligands for the Melanocortin Receptors. Ann. N.Y. Acad. Sci. 2003, 994, 12–20. [DOI] [PubMed] [Google Scholar]
- Grieco P.; Cai M.; Mayorov A. V.; Trivedi D.; Hruby V. J. Structure-Activity Studies of New Melanocortin Peptides Containing an Aromatic Amino Acid at the N-Terminal Position. Peptides 2006, 27, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby V. J.; Lu D.; Sharma S. D.; de Lauro Castrucci A.; Kesterson R. A.; Al-Obeidi F. A.; Hadley M. E.; Cone R. D. Cyclic Lactam α-Melanotropin Analogues of Ac-Nle4-c[Asp5, D-Phe7, Lys10]α-MSH(4–10)-NH2 with Bulky Aromatic Amino Acids at Position 7 Show High Antagonist Potency and Selectivity at Specific Melanocortin Receptors. J. Med. Chem. 1995, 38, 3454–3461. [DOI] [PubMed] [Google Scholar]
- Grieco P.; Lavecchia A.; Cai M.; Trivedi D.; Weinberg D.; MacNeil T.; Van der Ploeg L. H. T.; Hruby V. J. Structure-Activity Studies of the Melanocortin Peptides: Discovery of Potent and Selective Affinity Antagonists for the hMC3R and hMC4R Receptors. J. Med. Chem. 2002, 45, 5287–5294. [DOI] [PubMed] [Google Scholar]
- Cai M.; Cai C.; Mayorov A. V.; Xiong C.; Cabello C. M.; Soloshonok V. A.; Swift J. R.; Trivedi D.; Hruby V. J. Biological and Conformational Study of β-Substituted Prolines in MT-II Template: Steric Effects Leading to Human MC5 Receptor Selectivity. J. Pept. Res. 2004, 63, 116–131. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Mayorov A. V.; Cai M.; Tymecka D.; Chandler K. B.; Palmer E. S.; Van Rompaey K.; Misicka A.; Tourwé D.; Hruby V. J. Novel Selective Human Melanocortin-3 Receptor Ligands: Use of the 4-Amino-1,2,4,5-Tetrahydro-2-Benzazepin-3-One (Aba) Scaffold. Bioorg. Med. Chem. Lett. 2007, 17, 2492–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourwé D.; Verschueren K.; Frycia A.; Davis P.; Porreca F.; Hruby V. J.; Toth G.; Jaspers H.; Verheyden P.; Van Binst G. Conformational Restriction of Tyr and Phe Side Chains in Opioid Peptides: Information about Preferred and Bioactive Side-Chain Topology. Biopolymers 1996, 38, 1–12. [DOI] [PubMed] [Google Scholar]
- Ballet S.; Frycia A.; Piron J.; Chung N. N.; Schiller P. W.; Kosson P.; Lipkowksi A. W.; Tourwé D. Synthesis and Biological Evaluation of Constrained Analogs of the Opioid Peptide H-Tyr-D-Ala-Phe-Gly-NH2 using the 4-Amino-2-Benzazepin-3-One Scaffold. J. Pept. Res. 2005, 66, 222–230. [DOI] [PubMed] [Google Scholar]
- Ballet S.; De Wachter R.; Van Rompaey K.; Tömböly C.; Feytens D.; Töth G.; Quartara L.; Cucchi P.; Meini S.; Tourwé D. Bradykinin Analogs Containing the 4-Amino-2-Benzazepin-3-One Scaffold at the C-Terminus. J. Pept. Sci. 2007, 13, 164–170. [DOI] [PubMed] [Google Scholar]
- Le Diguarher T.; Ortuno J.-C.; Shanks D.; Guilbaud N.; Pierré E.; Raimbaud E.; Fauchère N.; Hickman J. A.; Tucker G. C.; Casara P. J. Synthesis of N,N′-Disubstituted 3-Aminobenzo[c] and [d]Azepin-2-Ones as Potent and Specific Farnesyl Transferase Inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 767–771. [DOI] [PubMed] [Google Scholar]
- Evans B. E.; Rittle K. E.; Bock M. G.; DiPardo R. M.; Freidinger R. M.; Whitter W. L.; Lundell G. F.; Veber D. F.; Anderson P. S.; Chang R. S. L.; Lotti V. J.; Cerino D. J.; Chen T. B.; Kling P. J.; Kunkel K. A.; Springer J. P.; Hirshfield J. Methods for Drug Discovery: Development of Potent, Selective, Orally Effective, Cholecystokinin Antagonists. J. Med. Chem. 1988, 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- Holder J. R.; Bauzo R. M.; Xiang Z.; Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors. Part 2. Modifications at the Phe Position. J. Med. Chem. 2002, 45, 3073–3081. [DOI] [PubMed] [Google Scholar]
- Proneth B.; Pogozheva I. D.; Portillo F. P.; Mosberg H. I.; Haskell-Luevano C. Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 Modified at the Para Position of the Benzyl Side Chain (DPhe): Importance for Mouse Melanocortin-3 Receptor Agonist versus Antagonist Activity. J. Med. Chem. 2008, 51, 5585–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder J. R.; Bauzo R. M.; Xiang Z.; Haskell-Luevano C. Structure-Activity Relationships of the Melanocortin Tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the Mouse Melanocortin Receptors. 1. Modifications at the His Position. J. Med. Chem. 2002, 45, 2801–2810. [DOI] [PubMed] [Google Scholar]
- Vandormael B.; De Wachter R.; Martins J. C.; Hendrickx P. M. S.; Keresztes A.; Ballet S.; Mallareddy J. R.; Tóth F.; Tóth G.; Tourwé D. Asymmetric Synthesis and Conformational Analysis by NMR Spectroscopy and MD of Aba- and α-MeAba-Containing Dermorphin Analogues. ChemMedChem 2011, 6, 2035–2047. [DOI] [PubMed] [Google Scholar]
- Pogozheva I. D.; Chai B.-X.; Lomize A. L.; Fong T. M.; Weinberg D. H.; Nargund R. P.; Mulholland M. W.; Gantz I.; Mosberg H. I. Interactions of Human Melanocortin 4 Receptor with Nonpeptide and Peptide Agonists. Biochemistry 2005, 44, 11329–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco P.; Brancaccio D.; Novellino E.; Hruby V. J.; Carotenuto A. Conformational Study on Cyclic Melanocortin Ligands and New Insight into their Binding Mode at the MC4 Receptor. Eur. J. Med. Chem. 2011, 46, 3721–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.