

Abstract
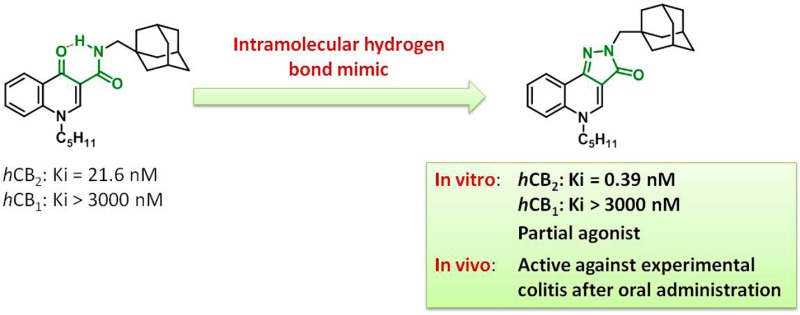
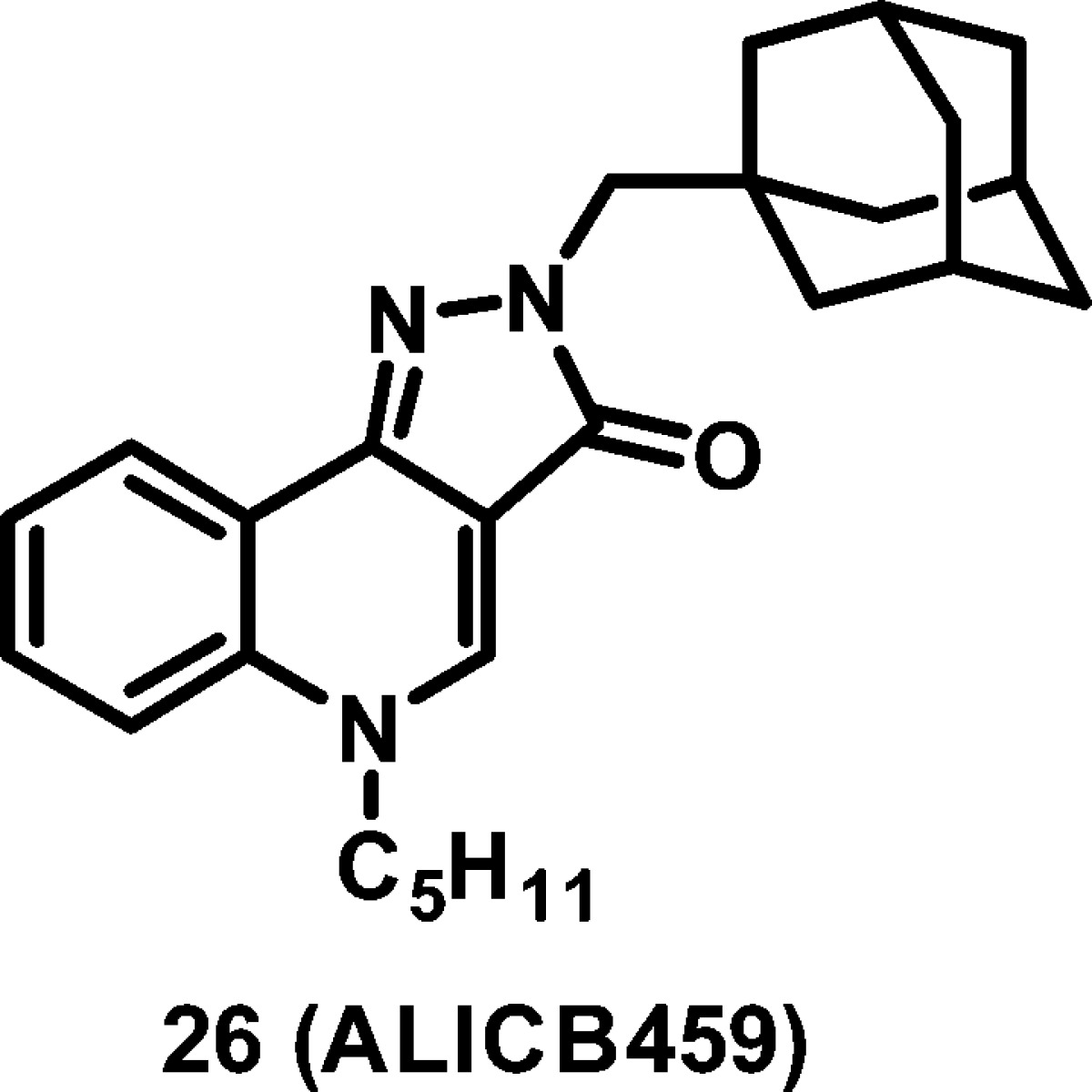
The CB2 cannabinoid receptor has been implicated in the regulation of intestinal inflammation. Following on from the promising activity of a series of 4-oxo-1,4-dihydroquinoline-3-carboxamide, we developed constrained analogues based on a 2H-pyrazolo[4,3-c]quinolin-3(5H)-one scaffold, with improved affinity for the hCB2 receptor and had very high selectivity over the hCB1 receptor. Importantly, the lead of this series (26, hCB2: Ki = 0.39 nM, hCB1: Ki > 3000 nM) was found to protect mice against experimental colitis after oral administration.
Keywords: Cannabinoid receptor, endocannabinoid, colitis, inflammatory bowel disease, quinolone, conformational restriction
Inflammatory bowel disease (IBD) represents a group of chronic inflammatory lesions of unknown etiology that affect the digestive tract.1 The CB2 cannabinoid receptor emerged as a promising therapeutic target in the treatment of these pathologies. Indeed, this GPCR has been identified within the gastrointestinal tract and plays a key role in the regulation of intestinal inflammation.2 Additionally, the CB2 receptor agonists have been shown to exert an anti-inflammatory effect both in intestinal epithelial cells and in experimental models of colitis in mice.
In vitro experiments highlighted that CB2 receptor activation results in suppression of the release of pro-inflammatory cytokines.3 Data from in vivo studies emphasized the importance of this receptor in mediating protection against experimental colitis. Indeed, the CB2 receptor selective agonist JWH133 proved to be efficient in reducing colonic inflammation in models of colitis induced by oil of mustard, dextran sulfate sodium (DSS), or 2,4,6-trinitrobenzenesulfonic acid (TNBS) and in the interleukin 10-deficient mouse model of colitis.4−6 Similarly, the CB2 receptor selective agonist AM1241 was shown to significantly reduce inflammation in the TNBS-induced colitis model.5 The involvement of the CB2 receptor was demonstrated by counteracting the effect with the CB2 antagonist/inverse agonist AM630 or by evaluating the compounds in CB2 receptor deficient mice. Moreover, AM630 was shown to exacerbate colitis in wild-type mice.5 Consistent with these data, we described a series of 4-oxo-1,4-dihydropyridines (CB2 selective agonists) in which the lead compound, ALICB573 (48 in ref (7)), was shown to protect mice against experimental colitis when administered at 10 mg/kg (i.p. injection).7 More recently, two isoxazole-based CB2 agonists were found to be active in the DSS-induced colitis model in mice.8 Taken together, these different studies provide the proof of concept that CB2 agonists are effective in reducing colitis and can therefore be considered as promising agents for the treatment of IBD.
In light of these findings, we aimed to develop new CB2 receptor agonists effective in the treatment of IBD. In terms of ligand profile, the new ligands should (i) display nanomolar affinity for the CB2 receptor, (ii) behave as agonists, (iii) be selective for the CB2 receptor over the CB1 receptor, to limit CNS side effects, and (iv) be active in an experimental model of colitis in rodents.
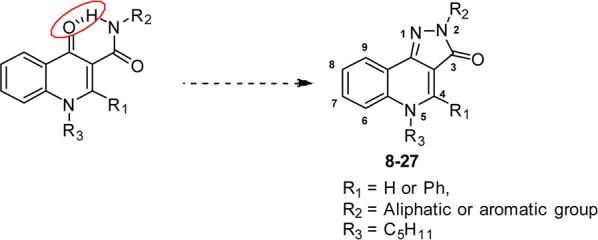
Our groups previously described a series of 4-oxo-1,4-dihydroquinoline-3-carboxamides as selective ligands of the CB2 receptor.9,10 One of the commonly used strategies in drug design to increase affinity and selectivity of a given “flexible” lead for its pharmacological target is to conformationally constrain it to mimic the so-called bioactive conformation. Therefore, we designed constrained analogues of the 4-oxo-1,4-dihydroquinoline-3-carboxamide series based on a 2H-pyrazolo[4,3-c]quinolin-3(5H)-one scaffold (Figure 1).
Figure 1.
Design of conformationally constrained quinolone analogues.
Our strategy relies on the fact that the amide at the C-3 position of the 4-oxo-1,4-dihydroquinoline ring may adopt different conformations, of which one is the result of a hydrogen bond between the amide (C-3) and the heterocyclic carbonyl (Figure 1).11 We hypothesized that this hydrogen-bonded conformation may also be the biologically active conformation of this series of ligands. Accordingly, to accurately orientate the aliphatic or aromatic moiety borne by the amide, we built an additional ring that acts as a permanent conformational lock in place of the intramolecular hydrogen bond. A similar strategy was successfully applied to generate potent topoisomerase II inhibitors.12,13
Thus, we designed, synthesized, and evaluated against both CB1 and CB2 receptors a series of 2H-pyrazolo[4,3-c]quinolin-3(5H)-one based on the already established pharmacophore in the series of 4-oxo-1,4-dihydroquinoline-3-carboxamide.9,10 Therefore, the n-pentyl chain was retained at the N-5 position of the rigid heterocycle and we only varied the moieties at the N-2 position and to a lesser extent the C-4 position.
Key 2H-pyrazolo[4,3-c]quinolin-3(5H)-one compounds 8–27 were prepared by a linear step-by-step synthesis that include the construction of the 4-oxo-1,4-dihydroquinoline scaffold (Scheme 1).14 The latter was obtained using two different synthetic routes depending on the nature of the R1 substituent (at C-4). The first one consisted of a condensation of aniline and diethyl ethoxymethylenemalonate followed by cyclization into 4-oxo-1,4-dihydroquinoline 2.9 Subsequent N-alkylation with 1-bromopentane yielded compound 4. The alternative route, allowing the introduction of a phenyl moiety at C-4, starts with the N-alkylation of isatoic anhydride. The resulting product was then reacted with ethyl benzoylacetate in the presence of sodium hydride to afford the desired 4-oxo-1,4-dihydroquinoline 5.15 The latter as well as its analogue 4 were converted into the more reactive thioxo-derivatives 6 and 7. Finally, the 2H-pyrazolo[4,3-c]quinolin-3(5H)-ones (10–27) were obtained either by reacting 6 and 7 with the corresponding substituted hydrazine or via initial cyclization with monohydrate hydrazine (leading to 8 and 9) and subsequent N-2 alkylation.14
Scheme 1.

The affinities of target compounds 8–27 (Table 1) for the human cannabinoid receptors (hCB1 and hCB2) were determined by a competitive radioligand displacement assay using [3H]-CP-55,940 as the radioligand for both the hCB2 and hCB1 receptors.7 We also investigated the functional activity on the CB2 receptor of four representative compounds of this series using a [35S]-GTPγS binding assay (Table 2).16
Table 1. Binding Affinity of Compounds 8–27 and Reference Compounds for hCB2 Cannabinoid Receptora.
Unless specified otherwise, compounds in this table display Ki values for hCB1 receptor greater than 3000 nM.
Ki (hCB1) > 1000 nM.
Ki (hCB1) = 52.4 ± 1.2 nM.
Table 2. Functional Activity of Selected 2H-Pyrazolo[4,3-c]quinolin-3(5H)-ones and Reference Compounds for hCB2 Cannabinoid Receptor.
| [35S]-GTPγS (hCB2) |
||
|---|---|---|
| Cpd | EC50 (nM) | Emax (%) |
| 16 | 204 ± 41 | 160 ± 3 |
| 24 | 14.3 ± 4.0 | 149 ± 4 |
| 25 | 5.4 ± 1.1 | 141 ± 3 |
| 26 | 64 ± 10 | 163 ± 4 |
| (R)-(+) WIN 55,212-2 | 24.5 ± 1.6 | 207 ± 10 |
| SR144528 | 1.8 ± 0.9 | 21.6 ± 2.7 |
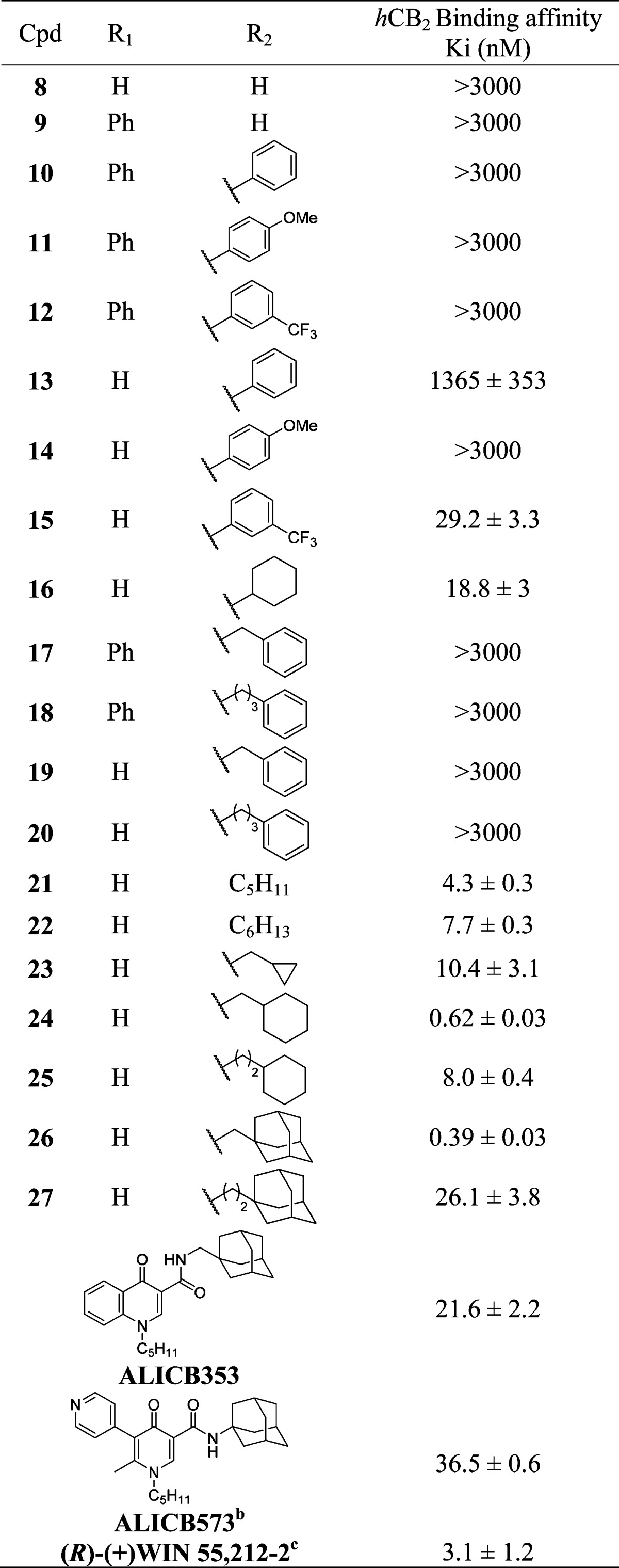
The first step of this study was to evaluate the impact of the phenyl substituent at the C-4 position. While in the 4-oxo-1,4-dihydroquinoline series this modification induced a switch in functional activity (from agonist to inverse agonist),10 in the present case, the substitution was found to be deleterious for the affinity, since none of the C-4 substituted analogues (9–12 and 17–18) displayed activity against CB2 receptor. As a consequence, no functional activity was measured. Conversely, the absence of a substituent at C-4 provides compounds with very high affinity toward the CB2 receptor. Therefore, for the rest of our investigations, we decided to leave this position unsubstituted and vary only the moieties at the N-2 position, which were selected on the basis of the previously established pharmacophore (aromatic or aliphatic groups).
The importance of a substituent at the N-2 position was illustrated by the poor affinity of compound 8 (R2 = H) for both receptor subtypes. Similarly, aromatic groups are generally not well tolerated at this position. Indeed, with the notable exception of 15 (hCB2: Ki = 29.2 nM), none of the compounds presenting an aromatic group at this position (spaced by an alkyl linker or not) displayed significant affinity for the CB2 receptor.
In contrast, the introduction of aliphatic substituents at N-2 is crucial for the affinity, since all evaluated compounds presenting an alkyl or cycloalkyl group at this position were found to bind the CB2 receptor with affinities in the nanomolar range (16 and 21–27). When comparing 16, 24, and 25, it is evident that the distance of the cycloalkyl group from the heterocycle is important, with the methyl linker being optimal. This observation was confirmed when the cycloalkyl was an adamantyl, with 26 (methyl linker) displaying a 67-fold higher affinity than 27 (ethyl linker). In addition, the size of the cycloalkyl group also impacts on the affinity. For instance, when comparing 23, 24, and 26, the compound affinity improves as the cycloalkyl size increases (adamantyl > cyclohexyl > cyclopropyl), with 26 displaying a Ki for hCB2 of 0.39 nM. The drastic loss of affinity when the cycloalkyl is replaced by a phenyl group is noteworthy (24 or 26 vs 19). Lastly, linear alkyl chains are also well tolerated, as evidenced by the high affinity of compounds 21 and 22.
Interestingly, none of the evaluated compounds displayed affinity for the hCB1 receptor (hCB1: Ki > 3000 nM), pointing at this series as highly selective for the hCB2 receptor.
From a functional activity perspective, compounds 16 and 24–26 showed a profile of CB2 partial agonist with EC50 values ranging from 5.4 nM to 204 nM and Emax values on the order of 150% (Table 2). In this assay CB2 receptor full agonist (R)-(+) WIN 55,212-2 displayed an EC50 value of 24.5 nM and an Emax value of 207%, while SR144528 (inverse agonist) had an EC50 of 1.8 nM and an Emax value of 21.6%. The rigidification approach has no effect on the functional activity as the parent 4-oxo-1,4-dihydroquinoline derivatives were also described as agonists.7
It is instructive to compare the CB2 affinity of our lead compound 26 with its analogue in the 4-oxo-1,4-dihydroquinoline series (ALICB353). As can be seen from Table 1, the latter compound displayed a Ki for the hCB2 receptor of 21.6 nM (reported in ref (10) to have a Ki of 50.6 nM). Therefore, the conformational constriction of this 4-oxo-1,4-dihydroquinoline leading to 26 (hCB2: Ki = 0.39 nM) allowed a 55-fold increase in binding affinity.
Our results suggest that the rigidification strategy allows an increase in affinity for the CB2 receptor while not impacting the functional activity. In addition, very high selectivity for the CB2 receptor over the CB1 receptor was achieved. This contrasts with a previously reported study on this series that suggested that the 2H-pyrazolo[4,3-c]quinolin-3(5H)-one was not a good template for designing CB2 selective ligands, since all of the described ligands behaved as low micromolar/high nanomolar dual CB1/CB2 ligands.17 We demonstrate herein that high affinity for the CB2 receptor and selectivity over the CB1 receptor could be achieved using this template, when both the N-2 and N-5 positions are substituted with an alkyl or cycloalkyl group.
To investigate potential binding modes, compound 26 and its 4-quinolone analogue (ALICB353) was docked into an agonist-biased state model of the CB2 receptor.18 In contrast with ALICB353, which produced diverse docking solutions, 26 is not only well tolerated but selects exclusively docking poses that fit in a unique binding mode, satisfying interactions with critical amino acids known to bind reference CB2 agonists (Figure S1 in the Supporting Information). The most representative docking pose for each molecule shows both the adamantyl group in a hydrophobic cavity formed by helices 2 (Phe87, Phe91, Phe94) and 3 (Phe106, Ile110, Val 113), as well as a common hydrogen bond with the hydroxyl group of Ser285 (Figure 2). The docking poses of the central heterocycle and n-pentyl, however, differ slightly for the two compounds. The tricyclic ring of 26 orients orthogonally to the 4-quinolone scaffold of ALICB353, and the n-pentyl chain of 26 extends toward a hydrophobic cavity formed by helices 5 (Phe200, Leu201) and 6 (Val261, Met265) whereas it folds behind the 4-quinolone ring, toward extracellular loop 2 (Leu182, Leu192) in the case of ALICB353.
Figure 2.
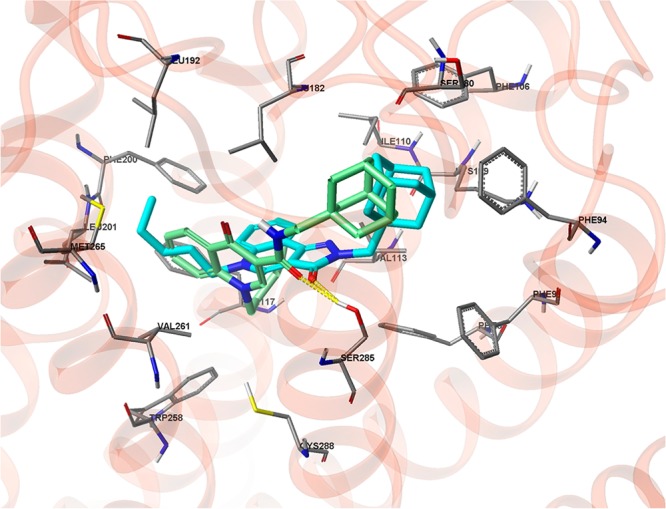
Preferred docking pose for 26 (cyan) and ALICB353 (green) in an agonist-biased state model of the CB2 receptor.
Our lead compound in this series (26)19 was evaluated in a mouse model of acute colitis induced by TNBS using a previously described protocol.20 Compound 26 was orally administered (in carboxymethyl cellulose, CMC) once daily at dosages of 0.1, 1, and 10 mg/kg over 7 days, starting 2 days before colitis induction. Control mice were treated with vehicle only (CMC).
We examined survival rates during the full length of the study and evaluated the body weight loss before euthanasia of the animals (5 days after TNBS administration). We noted that mice treated with 26 show reduced mortality compared to their untreated counterparts (Figure 3A). Moreover, daily treatment with compound 26 attenuated, in a dose-dependent manner, body weight loss generally observed during the development of TNBS colitis (Figure 3B).
Figure 3.
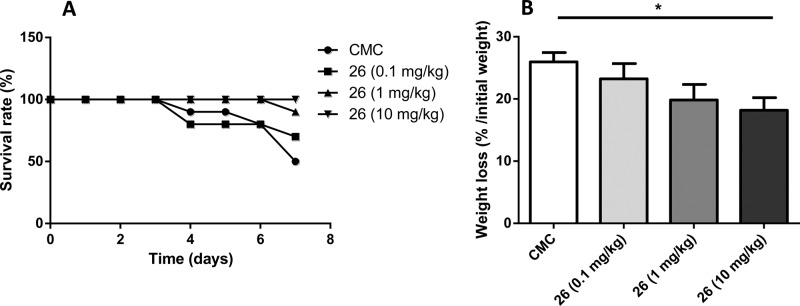
Survival rates and body weight loss evaluation: compound 26 attenuated both mortality and body weight loss (data are the mean ± SEM of 10 mice per group; *p < 0.05; vs vehicle).
After euthanasia of the different groups, the colon of each mouse was examined and damages were assessed using a semiquantitative scoring system. Mice that received the vehicle only showed macroscopic colitis reflected by thickening of the bowel and areas of ulceration (macroscopic score of 4.9, Figure 4A). Compound 26 exerts a dose-dependent decrease in macroscopic score (Figure 4A), with a very strong effect when administered orally at 10 mg/kg (1.7 vs 4.9). This protective effect was confirmed by the results of the histological evaluation. Indeed, 26 was able to attenuate the histological score by 65% (Figure 4B). We also quantified colon levels of TNFα and Il-1β, two cytokines involved in the inflammatory response leading to epithelial injury. As can be seen from Figure 4C and D, compound 26 completely abolished Il-1β mRNA expression but failed to significantly decrease TNFα mRNA expression.
Figure 4.

Macroscopic (A) and histological (B) score. TNFα and IL-1β mRNA levels (C, D) were measured in the colon after TNBS-induced colitis and treatment with compound 26 (data are the mean ± SEM of 10 mice per group; *p < 0.05; ***p < 0.001 vs vehicle).
All together, the above data clearly support that compound 26 protects mice against experimental colitis after oral administration in a dose-dependent manner, an effect attributed in part to the strong anti-inflammatory property of this compound. Although different studies have already emphasized the efficacy of CB2 agonists in colitis,4−8 all were active after i.p. injection, which from a drug development perspective is not desirable. Favorably, compound 26 is active in a mouse model of colitis after oral administration.
Compound 26 has also been profiled for in vitro metabolic stability, plasma protein binding, intestinal absorption, and hERG toxicity (Table 3).21
Table 3. Determination and Evaluation of Selected Physicochemical and in Vitro DMPK-Tox Parameters for Compound 26.
| Parametersa | |
|---|---|
| MW (g/mol) | 403.6 |
| cLogP | 5.6 |
| tPSA | 35.9 |
| In vitro ADME-tox | |
|---|---|
| Human PPB (% free)b | <1 |
| A-B Caco-2 permeability (×10–6 cm·s–1)c | 0.8 |
| P-gp inhibition (% at 10 μM)d | 30.6 |
| Metabolic stability (human liver microsomes, % parent remaining after 1 h)e | 64 |
| hERG (% inhibition of tail current at 1 μM)f | 35.5 |
Determined with ChemDraw Ultra 10.0.
Assessed by equilibrium dialysis (18 h) at 37 °C.
Compound 26 (10 μM) was incubated (0 and 60 min) at 37 °C with Caco-2 cell line (pH 6.5/7.4).
Performed on MDR1-MDCKII cell line, using cellular uptake of calcein AM (30 min incubation/37 °C).
Compound 26 (1 μM) was incubated (0 and 60 min) at 37 °C with human liver microsomes (0.3 mg/mL).
Compound 26 (1 μM) was incubated (5–10 min, cumulatively) at room temperature with hERG HEK 293 cells (conventional whole-cell patch clamp).
It was found that 26 binds strongly to plasma proteins, resulting in less than 1% being in the free form. In the in vitro CACO-2 assay, 26 showed very poor permeability (Papp = 0.8 × 10–6 cm·s–1). The CACO-2 assay, however, might not be reflective of the intestinal permeability in the case of colitis, where there will be substantial intestinal lesions. It is noteworthy that little inhibition of P-gp was observed in the calcein-AM functional assay (30.6% inhibition at 10 μM). Unfortunately compound 26 does show some hERG affinity (35.5% inhibition of tail current at 1 μM) and so may have some toxicity at a high dose. Compound 26 did however show good in vitro metabolic stability, with 64% of compound remaining after 60 min of incubation with human liver microsomes. The DMPK-tox profile of compound 26 needs to be improved, and future optimization regarding this series will have to focus on this particular point.
In summary, the present study shows that 2H-pyrazolo[4,3-c]quinolin-3(5H)-ones, constrained analogues of 4-oxo-1,4-dihydroquinolines, are potent and highly selective CB2 receptor agonists. The rigidification approach applied in this paper resulted in increased affinity for the CB2 receptor while not altering the functional activity. Despite displaying a nonideal in vitro DMPK-Tox profile, compound 26 (ALICB459), the lead of this series, exerts a strong protective effect in the experimental model of TNBS-induced colitis. This effect was achieved after oral administration and was shown to be dose-dependent.
Glossary
Abbreviations
- hCB1&2
human cannabinoid receptor 1 and 2
- DSS
dextran sulfate sodium
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- i.p.
intraperitoneal
- Il-1β
interleukin-1β
- CMC
carboxymethyl cellulose
Supporting Information Available
Experimental procedures and spectroscopic data for all synthesized compounds, detailed pharmacology and all docking poses of 26 and ALICB353 in an agonist-biased state model of the CB2 receptor. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ (J.E.B.) University of Cambridge, Department of Chemistry, Lensfield Road, Cambridge CB2 1EW, United Kingdom.
Author Present Address
⊥ (G.G.M.) Bioanalysis and Pharmacology of Bioactive Lipids Lab, Louvain Drug Research Institute, Université Catholique de Louvain, 72 Avenue E. Mounier, B-1200 Bruxelles, Belgium.
We are grateful for financial support from the Conseil régional du Nord-Pas de Calais and University of Lille 2.
The authors declare no competing financial interest.
Supplementary Material
References
- Podolsky D. K. Inflammatory Bowel Disease. N. Engl. J. Med. 2002, 347, 417–429. [DOI] [PubMed] [Google Scholar]
- Alhouayek M.; Muccioli G. G. The Endocannabinoid System in Inflammatory Bowel Diseases: From Pathophysiology to Therapeutic Opportunity. Trends Mol. Med. 2012, 18, 615–25. [DOI] [PubMed] [Google Scholar]
- Ihenetu K.; Molleman A.; Parsons M. E.; Whelan C. J. Inhibition of Interleukin-8 Release in the Human Colonic Epithelial Cell Line HT-29 by Cannabinoids. Eur. J. Pharmacol. 2003, 458, 207–215. [DOI] [PubMed] [Google Scholar]
- Kimball E. S.; Schneider C. R.; Wallace N. H.; Hornby P. J. Agonists of Cannabinoid Receptor 1 and 2 Inhibit Experimental Colitis Induced by Oil of Mustard and by Dextran Sulfate Sodium. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, 364–371. [DOI] [PubMed] [Google Scholar]
- Storr M. A.; Keenan C. M.; Zhang H.; Patel K. D.; Makriyannis A.; Sharkey K. A. Activation of the Cannabinoid 2 Receptor (CB2) Protects Against Experimental Colitis. Inflamm. Bowel Dis. 2009, 15, 1678–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh U. P.; Singh N. P.; Singh B.; Price R. L.; Nagarkatti M.; Nagarkatti P. S. Cannabinoid Receptor-2 (CB2) Agonist Ameliorates Colitis in IL-10(−/−) Mice by Attenuating the Activation of T Cells and Promoting their Apoptosis. Toxicol. Appl. Pharmacol. 2012, 258, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bakali J.; Gilleron P.; Body-Malapel M.; Mansouri R.; Muccioli G. G.; Djouina M.; Barczyk A.; Klupsch F.; Andrzejak V.; Lipka E.; Furman C.; Lambert D. M.; Chavatte P.; Desreumaux P.; Millet R. 4-Oxo-1,4-dihydropyridines as selective CB2 cannabinoid receptor ligands. Part 2: discovery of new agonists endowed with protective effect against experimental colitis. J. Med. Chem. 2012, 55, 8948–8952. [DOI] [PubMed] [Google Scholar]
- Tourteau A.; Body-Malapel M.; Andrzejak V.; Lemaire L.; Lemoine A.; Mansouri R.; El Bakali J.; Desreumaux P.; Muccioli G. G.; Lambert D. M.; Renault N.; Chavatte P.; Rigo B.; Leleu-Chavain N.; Millet R. 3-Carboxamido-5-aryl-isoxazoles as new CB2 agonists for the treatment of colitis. Bioorg. Med. Chem. 2013, 21, 5383–5394. [DOI] [PubMed] [Google Scholar]
- Stern E.; Muccioli G. G.; Millet R.; Goossens J. F.; Farce A.; Chavatte P.; Poupaert J. H.; Lambert D. M.; Depreux P.; Hénichart J. P. Novel 4-Oxo-1,4-dihydroquinoline-3-carboxamide Derivatives as New CB2 Cannabinoid Receptors Agonists: Synthesis, Pharmacological Properties and Molecular Modeling. J. Med. Chem. 2006, 49, 70–79. [DOI] [PubMed] [Google Scholar]
- Stern E.; Muccioli G. G.; Bosier B.; Hamtiaux L.; Millet R.; Poupaert J. H.; Hénichart J. P.; Depreux P.; Goossens J. F.; Lambert D. M. Pharmacomodulations Around the 4-Oxo-1,4-dihydroquinoline-3-carboxamides, a Class of Potent CB2-Selective Cannabinoid Receptor Ligands: Consequences in Receptor Affinity and Functionality. J. Med. Chem. 2007, 50, 5471–5484. [DOI] [PubMed] [Google Scholar]
- Graybill T. L.; Dolle R. E.; Osifo I. K.; Schmidt S. J.; Gregory J. S.; Harris A. L.; Miller M. S. Inhibition of Human Erythrocyte Calpain I by Novel Quinolinecarboxamides. Bioorg. Med. Chem. Lett. 1995, 5, 387–392. [Google Scholar]
- Wentland M. P.; Perni R. B.; Dorff P. H.; Brundage P.; Castaldi M. J.; Bailey T. R.; Carabateas P. M.; Bacon E. R.; Young D. C.; Woods M. G.; Rosi D.; Drozd M. L.; Kullnig R. K.; Dutko F. J. 3-Quinolinecarboxamides. A Series of Novel Orally-Active Antiherpetic Agents. J. Med. Chem. 1993, 36, 1580–1596. [DOI] [PubMed] [Google Scholar]
- Wentland M. P.; Aldous S. C.; Gruett M. D.; Perni R. B.; Powles R. G.; Danz D. W.; Klingbeil K. M.; Peverly A. D.; Robinson R. G.; Corbett T. H.; Rake J. B.; Coughlin S. A. The Antitumor Activity of Novel Pyrazoloquinoline Derivatives. Bioorg. Med. Chem. Lett. 1995, 5, 405–410. [Google Scholar]
- Crespo M. I.; Gràcia J.; Puig C.; Vega A.; Bou J.; Beleta J.; Doménech T.; Ryder H.; Segarra V.; Palacios J. M. Synthesis and Biological Evaluation of 2,5-Dihydropyrazolo[4,3-c]quinolin-3-ones, a Novel Series of PDE 4 Inhibitors with Low Emetic Potential and Antiasthmatic Properties. Bioorg. Med. Chem. Lett. 2000, 10, 2661–2664. [DOI] [PubMed] [Google Scholar]
- Hayashi H.; Miwa Y.; Ichikawa S.; Yoda N.; Miki I.; Ishii A.; Kono M.; Yasuzawa T.; Suzuki F. 5-HT3 receptor antagonists. 2. 4-hydroxy-3-quinolinecarboxylic acid derivatives. J. Med. Chem. 1993, 36, 617–626. [DOI] [PubMed] [Google Scholar]
- El Bakali J.; Muccioli G. G.; Renault N.; Pradal D.; Body-Malapel M.; Djouina M.; Hamtiaux L.; Andrzejak V.; Desreumaux P.; Chavatte P.; Lambert D. M.; Millet R. 4-Oxo-1,4-Dihydropyridines as Selective CB2 Cannabinoid Receptor Ligands: Structural Insights into the Design of a Novel Inverse Agonist Series. J. Med. Chem. 2010, 53, 7918–7931. [DOI] [PubMed] [Google Scholar]
- Manera C.; Cascio M. G.; Benetti V.; Allarà M.; Tuccinardi T.; Martinelli A.; Saccomanni G.; Vivoli E.; Ghelardini C.; Di Marzo V.; Ferrarini P. L. New 1,8-Naphthyridine and Quinoline Derivatives as CB2 Selective Agonists. Bioorg. Med. Chem. Lett. 2007, 17, 6505–6510. [DOI] [PubMed] [Google Scholar]
- Renault N.; Laurent X.; Farce A.; El Bakali J.; Mansouri R.; Gervois P.; Millet R.; Desreumaux P.; Furman C.; Chavatte P. Virtual Screening of CB2 Receptor Agonists from Bayesian Network and High-Throughput Docking: Structural Insights into Agonist-Modulated GPCR Features. Chem. Biol. Drug Des. 2013, 81, 442–454. [DOI] [PubMed] [Google Scholar]
- Compounds 24 and 25 failed to significantly reduce the colitis macroscopic scores when evaluated at 10 mg/kg (i.p.).
- Desreumaux P.; Dubuquoy L.; Nutten S.; Peuchmaur M.; Englaro W.; Schoonjans K.; Derijard B.; Desvergne B.; Wahli W.; Chambon P.; Leibowitz M. D.; Colombel J. F.; Auwerx J. Attenuation of Colon Inflammation Through Activators of the Retinoid X Receptor (RXR)/Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Heterodimer. A Basis for New Therapeutic Strategies. J. Exp. Med. 2001, 193, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studies performed at Cerep according to their internal procedures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.