

Abstract
Antagonism of quorum sensing represents a promising new antivirulence approach for the treatment of bacterial infection. The development of a novel series of non-natural irreversible antagonists of P. aeruginosa LasR is described. The lead compounds identified (25 and 28) display potent LasR antagonist activity and inhibit expression of the P. aeruginosa virulence factors pyocyanin and biofilm formation in PAO1 and PA14.
Keywords: antivirulence, Pseudomonas aeruginosa, biofilm inhibition, structure−activity relationship, quorum sensing
The common opportunistic human pathogen P. aeruginosa plays a major role in infections of immunocompromised patients and is of significant medical interest as traditional antibiotic treatments are often ineffective.1−4 A prominent feature in the pathogenesis and drug-resistance of this bacterium is its ability to form a biofilm, a sessile community of cells inhabiting an extracellular polymeric matrix.5 Inhibition of the LasR quorum sensing circuit in P. aeruginosa has been shown to lead to a significant decrease in overall biofilm formation and virulence factor production.6 Further, the importance of quorum sensing in the pathogenicity of P. aeruginosa has been demonstrated in a number of animal models including a Caenorhabditis elegans nematode model,7,8 the neonatal mouse model of pneumonia,9 and a burned mouse model.1 In each of these cases, P. aeruginosa mutants defective in quorum sensing were significantly less virulent than the parent strains. Accordingly, the identification of a potent, drug-like small molecule inhibitor of quorum sensing in P. aeruginosa is anticipated to be of significant medical interest.10−15
The P. aeruginosa LasR quorum sensing receptor is agonized by the natural ligand N-3-oxo-dodecanoyl-l-homoserine lactone (3-oxo-C12 HSL, 1, Figure 1).7,16 A high-throughput screen recently identified a novel non-natural agonist 2.17 In subsequent studies it was determined that this molecule binds in the homoserine lactone binding pocket of the LasR receptor and displays potent agonist activity, comparable to the natural ligand.18,19 While this ligand lacks the metabolically labile homoserine lactone functionality, its agonist activity is therapeutically undesirable. To impart the desired LasR antagonist activity into analogues of 2, we drew inspiration from the recent discovery of the HSL-based antagonist 3.20 In this probe, the placement of electrophilic functionality at the terminus of the fatty-acid tail leads to covalent modification of LasR Cys79 and provides antagonist activity.
Figure 1.

Selected agonists and antagonists of P. aeruginosa LasR quorum sensing.
In this study, we describe our discovery and optimization efforts to identify potent drug-like inhibitors of virulence in P. aeruginosa resulting in a series of irreversible LasR antagonists based on the potent agonist 2.
Informed by the recent crystal structure of 2 complexed to LasR,19 our analogue series focused on the incorporation of electrophilic functionality in place of the 2-chloro benzoate ester in 2 (see Supporting Information). The synthesis of key intermediates 7 and 10 (Scheme 1) proceeded efficiently from commercially available material in three and five steps, respectively. Briefly, 2-methoxyphenylacetic acid (5) was reacted with 2-nitroaniline in the presence of SOCl2 to provide 6, which was subsequently deprotected and brominated to give key intermediate phenol 7. Intermediate 10 was prepared from bromination and benzyl protection of salicylamide (8). The resulting amide 9 was reduced to the amine, coupled with 2-nitrobenzoic acid and deprotected to provide intermediate phenol 10. With these two core scaffolds in hand, we began to investigate the incorporation of pendant electrophilic functionality through reaction of the phenol.
Scheme 1. Synthesis of Intermediates 7 and 10.

Building on precedent for the antagonist activity of isothiocyanate containing ligands (e.g., 3), we evaluated a series of ligands bearing this electrophilic functionality tethered to the free phenol present in 7 and 10 (Scheme 2). EDC coupling of an isothiocyanate-containing carboxylic acid 14 with 7 or 10 provided analogues 11 and 12 containing a three-carbon linker. The synthesis of analogues with shorter linkers was not successful using this route. Isothiocyanate preparation (e.g., 13 to 14) with shorter carbon linkers led a complex mixture of unidentified reaction products. Recognizing the potential for reaction of the carbonyl group with the proximal isothiocyanate in these analogues bearing a shorter linker, we turned our attention to the preparation of a series of ether-linked analogues (Scheme 2b). Therefore, etherification of phenol 6 followed by amine deprotection and isothiocyanate formation yielded ether-linked isothiocyanate analogues 15 and 16.
Scheme 2. Synthesis of Isothiocyanate Analogues.

Evaluation of the LasR antagonist assay of these analogues revealed that incorporation of a pendant isothiocyanate provided a series of LasR antagonists with IC50 values greater than 145 μM (Table 1). Full inhibition of LasR with all of the ligands in this series was observed with no effect on bacterial growth up to 200 μM, as monitored by evaluating OD600. Further, replacement of the ester linkage in analogue 11 with an ether linker (16) was well tolerated with the two analogues showing comparable IC50 values and % inhibition. To evaluate if these analogues were binding in the HSL ligand-binding pocket of LasR and to examine the mechanism of inhibition, we performed a competition-binding assay. As previously shown for HSL analogue 3,20 isothiocyanate-containing analogue 16 is a noncompetitive antagonist of LasR (see Supporting Information).
Table 1. Biological Activity of Isothiocyanate Analogues.
Values determined employing the GFP-based LasR antagonist bioassay (see Supporting Information). All IC50 values are reported as the mean of triplicate analysis with the range defining the 95% confidence interval. Br-HSL (4) was used as a control for antagonism with an IC50 = 16.4 ± 7.4 μM.
Percent inhibition of GFP at 100 μM with respect to Br-HSL (4) as a positive control for LasR antagonism, set at 100% inhibition.
In an effort to enhance the potency (IC50) of our antagonists we evaluated an alternate series of electrophilic analogues with the potential to react with cysteine.21 Preparation of chloroacetamide-containing analogues 17 and 18 proceeded in three steps from intermediate 7 (Scheme 3a). Biological assay of these analogues showed improvement in potency with IC50 values between 74 and 100 μM (Table 2). Additionally, a series of maleimide-containg analogues were prepared (24–28) employing a BOP coupling reaction between intermediate phenol 7 or 10 and the appropriately functionalized carboxylic acid 21–23 (Scheme 3b).
Scheme 3. Synthesis of Chloroacetamide and Maleimide Analogues.

Table 2. Biological Activity of Chloroacetamide and Maleimide Analogues.
| compd | IC50 (μM)a | % inhibition at 100 μMb |
|---|---|---|
| 17 | 73.8 ± 19.4 | 91 |
| 18 | 99.5 ± 21.0 | 79 |
| 24 | 86.8 ± 57.4 | 56 |
| 25 | 3.6 ± 1.9 | 98 |
| 26 | 78.1 ± 69.3 | 30 |
| 27 | 51.9 ± 35.1 | 32 |
| 28 | 1.5 ± 0.5 | 101 |
Values determined employing the GFP-based LasR antagonist bioassay (see Supporting Information). All IC50 values are reported as the mean of triplicate analysis with the range defining the 95% confidence interval. Br-HSL (4) was used as a control for antagonism with an IC50 = 16.4 ± 7.4 μM.
Percent inhibition of GFP at 100 μM with respect to Br-HSL (4) as a positive control for LasR antagonism, set at 100% inhibition.
Evaluation of the biological activity of the maleimide series of analogues revealed several potent LasR antagonists (Table 2). Within this series strict dependence on the length of the linker was observed. In the analogues derived from intermediate 7, the optimal linker length was two-carbon atoms (i.e., 25), with an IC50 value of 3.6 μM. This analogue represents one of the most potent antagonists of LasR yet discovered.20,22−28 Ligands with either a one-carbon linker or a three-carbon linker demonstrated diminished potency, with more than a 22-fold increase in IC50 values. A similar dependence on linker length is found with analogues derived from intermediate 10. In this series, however, the optimal linker length is a three-carbon linker (i.e., 28) with best-in-class antagonist activity of 1.5 μM. The analogue containing a two-carbon linker (27) is 34-fold less potent.
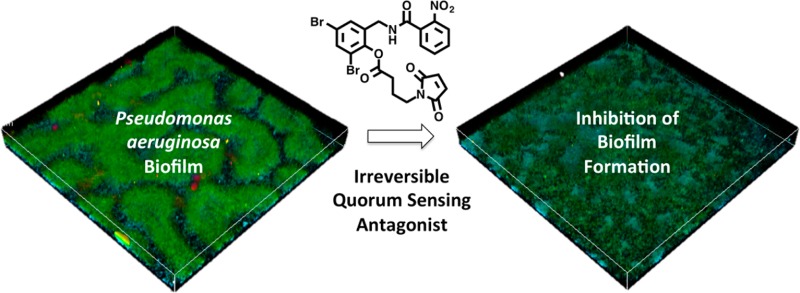
Having identified potent antagonists of LasR (25 and 28), we examined their mechanism of inhibition. We first evaluated if these analogues bind in the HSL binding pocket of LasR and act as noncompetitive antagonists as previously observed with 16. In this competition-binding assay, the concentration of the antagonist is held constant while evaluating the agonist dose–response curve of the native agonist, 3-oxo-C12 HSL (Figure 2a and Supporting Information). We observe consistent EC50 values for the native agonist; however, higher concentrations of 25 or 28 lead to diminished maximal activation of LasR. This feature is consistent with ligands 25 and 28 acting as irreversible antagonists of LasR.
Figure 2.
(a) Competition assay characteristic of irreversible LasR inhibition by 28. (b) Antagonist bioassay for 28 and succinimide 29 with (S)-2-(4-bromophenyl)-N-(2-oxotetrahydrofuran-3-yl) acetamide (Br-HSL) as a positive control for antagonism. (c) The structures and biological activity of succinimide containing ligands 29 and 30.
We then looked to differentiate between two potential mechanisms of irreversible inhibition. In the first, irreversible modification of LasR by an inhibitor renders the antagonist activity of the ligand noncompetitive. In contrast, formation of a covalent bond between ligand and receptor may represent a key feature in both the ligand binding kinetics and the antagonist activity of the molecule. If the first mechanism is operative, covalent attachment to LasR simply serves to render an antagonist ligand irreversible. In contrast, in the second mechanism, the bond formation process is responsible for antagonism.
To differentiate between the two possible mechanisms of inhibition we prepared succinimide-containing ligands 29 and 30 (Figure 2c). These ligands are nearly identical to lead antagonists 25 and 28 in terms of their steric and electronic properties; however, because of the absence of the maleimide, these ligands would be unable to covalently modify LasR. Evaluation of the biological activity of 29 and 30 reveals that these ligands do not display LasR antagonist activity and instead are LasR agonists (Figure 2b,c). The agonist activity observed in the absence of a cysteine-reactive maleimide supports a mechanism in which covalent modification of LasR is critical for the antagonist activity of this class of molecules.
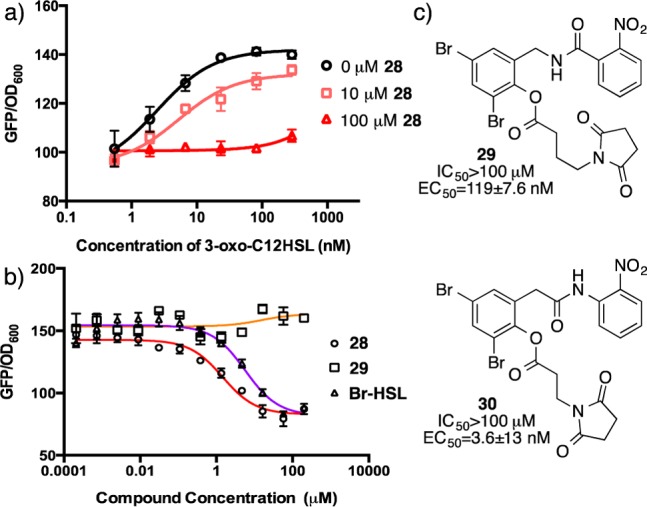
Having established that the potent LasR antagonists 25 and 28 are noncompetitive and that the presence of an electrophilic maleimide is critical for the inhibitory activity of these molecules, we examined computationally the binding of these ligands with LasR.29 Docking of these ligands with LasR reveals that the maleimide is in close proximity to Cys79 within the HSL ligand-binding pocket (Figure 3a). The reactivity of a free cysteine with an electrophilic maleimide is well-known,21 and the reactivity of LasR Cys79 with an HSL-based ligand bearing a pendant isothiocyanate (i.e., 3) has been established.20 Together, our results are consistent with the mechanism of inhibition for 25 and 28 involving covalent modification of Cys79 in the LasR HSL-binding pocket.
Figure 3.
(a) Lead inhibitor 28 docked in the ligand binding pocket of LasR highlighting amino acid residues Y56, W60, D73, and C79. (b) Pharmacophore model showing the low energy docking pose of 28 (black) compared to lower activity ligands 24 (gray) and 26 (yellow). (c) Pharmacophore model showing the low energy docking pose of 28 (black) compared to high activity ligand 25 (gray) and median activity ligand 27 (pink).
Given the requirement of covalent bond formation with Cys79 for antagonism, the observation of a strict dependence of IC50 on linker length is not unexpected. Variation of linker length would be expected to most directly influence placement of the electrophilic maleimide in an orientation suitable for reaction with Cys79; however, the effect of linker length is not independent of the structure of the analogue core. The optimal linker length for a maleimide ligand derived from phenol 6 is two carbon atoms (i.e., 25) whereas the optimal linker length for the maleimide derived from 9 is three carbon atoms (i.e., 28). This observation suggests that linker length to the electrophile is not an independent activity determinant but rather linker length is coupled to an additional critical binding determinant(s).
Analysis of the ligands with potent activity provides guidelines for the development of a pharmacophore model to describe the observed activity (Figure 3b,c). The pharmacophore model was built by identifying the key binding determinants in the most potent analogue (28) in comparison to the remaining analogues in this series (24–27).30 Using a pharmacophore model derived from the low energy docking pose for 28 (Figure3a and see Supporting Information) we evaluated a conformer library of ligands 24–27 to determine the fit of these ligands to our model. As anticipated by the antagonist activity of all ligands in this series, structural deviation from the pharmacophore model defined by 28 is subtle. The analogues displaying the lowest potency (24 and 26) maintain structural overlap for much of the model; however, they deviate in their positioning of the critical maleimide functionality (Figure 3b). In contrast, the potent antagonist 25 (gray, Figure 3c) maintains complete structural overlap with the pharmacophore model generated from 28, supporting the accuracy of this model. We note, however, that ligand 27 (pink, Figure 3c), which has median potency within this series also appears to fully satisfy our pharmacophore model.
Having identified two potent antagonists of LasR quorum sensing, we evaluated the ability of these analogues to inhibit virulence factor expression in wild-type clinical isolates of P. aeruginosa. We focused on quantifying the inhibition of the virulence factor pyocyanin and the inhibition of biofilm formation using P. aeruginosa strains PAO1 (ATCC 10145) and PA14.31
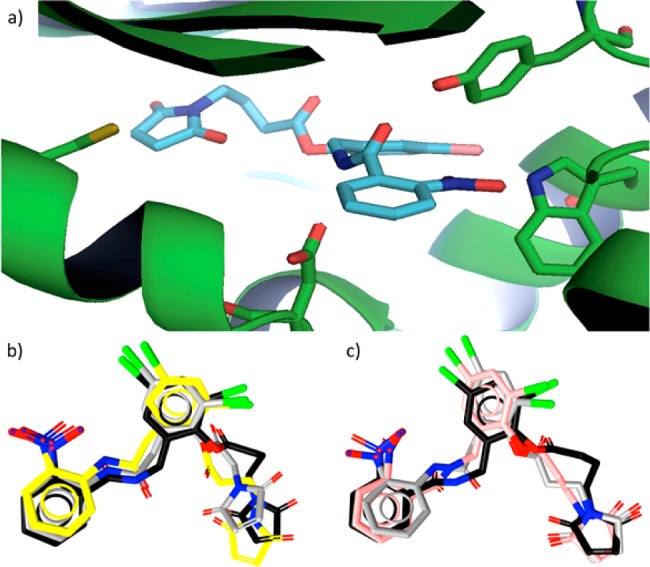
The inhibition of pyocyanin in these strains was first evaluated. Pyocyanin is a blue-green redox-active pigment and a prominent virulence factor in PAO1 and PA14. Lead quorum sensing inhibitor, 28, provided potent dose-dependent inhibition of this virulence factor (Figure 4). We observed a decrease in absorbance (A695) in cell-free supernatants from P.aeruginosa cultures that is characteristic of decreasing levels of pyocyanin. To further support that this change in absorbance is pyocyanin dependent, we performed an extraction and acidification of the cell-free supernatants selective for pyocyanin before measuring absorbance (A520, see Supporting Information). Using both methods, we similarly observe a decrease in absorbance, which is consistent with a decrease in pyocyanin expression in the presence of 28. Importantly, in this assay, no effect on growth is observed in either strain as assessed by measurement of cell density (A600). Higher levels of pyocyanin expression in untreated samples of PA14 are noted when compared to PAO1 and the inhibition of pyocyanin expression in PAO1 is achieved at somewhat lower antagonist concentrations, consistent with the enhanced virulence of the PA14 strain.8,31,32
Figure 4.
Inhibition of pyocyanin expression in P. aeruginosa PAO1 and PA14 by 28. Pyocyanin production was determined in triplicate by direct detection of pyocyanin absorbance (A695). No inhibition of growth was observed.
The ability of 28 to inhibit biofilm formation in static cultures of P. aeruginosa PA14 was evaluated using a crystal violet staining protocol with 48 h biofilms. Similar to the inhibition of the virulence factor pyocyanin, potent, dose-dependent inhibition of biofilm formation is observed in the presence of lead antagonist 28 (Figure 5a).
Figure 5.
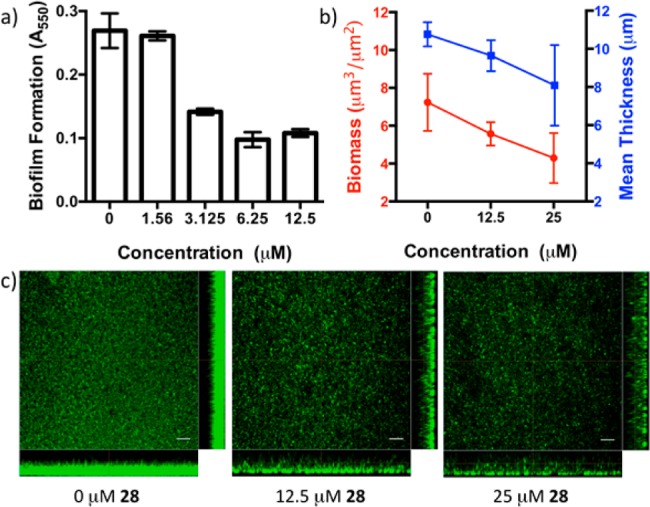
Inhibition of biofilm formation in P. aeruginosa PA14 in the presence of 28. (a) PA14 biofilm formation evaluated by crystal violet staining (A550). (b) Biofilm biomass and thickness determined from confocal fluorescence microscopy images using the program Comstat2.34,34b (c) Confocal fluorescence microscopy images of PA14 biofilms. Confocal images were collected in duplicate and imaging was repeated three times, representative images are shown. Scale bar = 25 μm.
To further evaluate the potent inhibitory effect on biofilm formation observed in the presence of 28, we imaged a series of 36 h P. aeruginosa PA14 biofilms grown in rich media (Luria broth) using confocal fluorescence microscopy after staining with SYTO-9 nucleic acid stain.33 The images collected display a dose-dependent reduction in biofilm thickness and overall biomass (Figure 5b,c). Repeating this assay using defined media similarly shows a striking decrease in biofilm biomass, thickness, and overall morphology, confirming the virulence factor inhibitory activity of 28 (see Supporting Information).
We have identified a series of potent irreversible inhibitors of P. aeruginosa LasR, which display antivirulence activity against PAO1 and the hyper-virulent strain PA14. The mechanism of inhibition for our lead analogues in this series has been characterized enabling the development of a pharmacophore model to describe the potent irreversible antagonist activity observed. Within this series, we find that antagonist activity is dependent on the presence of a pendant electrophile (e.g., maleimide). Our lead inhibitor shows potent, dose-dependent inhibition of the P. aeruginosa virulence factor pyocyanin and biofilm formation while possessing no bactericidal or bacteriostatic activity. The potent biological activity of analogues 25 and 28, together with enhanced metabolic stability in comparison to other reported LasR antagonists (e.g., 3) provides for the expectation that this series will serve as new leads in the development of an antivirulence therapy for the prominent Gram-negative bacterial pathogen P. aeruginosa.
Acknowledgments
We gratefully acknowledge Dr. Bonnie L. Bassler (Princeton University) for providing bacterial strains used in this study, Dr. Venkat Venkataraman and Ms. Hao Wu (Rowan University School of Osteopathic Medicine) for assistance with the confocal fluorescence microscopy, Dr. Martin F. Semmelhack and Dr. Christian Ventocilla (Princeton University) for collection of HRMS data, Mr. Joseph N. Capilato and Ms. Amy L. Warren (Rowan University) for assistance with compound synthesis, and Ms. Lydia M. Hanna (Rowan University) for assistance with the virulence factor assays.
Supporting Information Available
Synthesis, characterization (1H NMR, 13C NMR, and LC–MS spectra and chromatograms along with full tabulated data), bioassay results with dose–response curves for all new compounds, competition assay for 16 and 25, pyocyanin analysis, additional confocal fluorescence microscopy data, additional docking poses, and supplemental discussion. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally to this work.
This work was financially supported by Rowan University CSM and SEED grants and by a Rowan University New Faculty Research Startup Grant.
The authors declare no competing financial interest.
Supplementary Material
References
- Rumbaugh K. P.; Griswold J. A.; Iglewski B. H.; Hamood A. N. Contribution of Quorum Sensing to the Virulence of Pseudomonas aeruginosa in Burn Wound Infections. Infect. Immun. 1999, 67, 5854–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert P. A. Mechanisms of Antibiotic Resistance in Pseudomonas Aeruginosa. J. R. Soc. Med. 2002, 95, 22. [PMC free article] [PubMed] [Google Scholar]
- Driscoll J.; Brody S.; Kollef M. The Epidemiology, Pathogenesis and Treatment of Pseudomonas Aeruginosa Infections. Drugs 2007, 67, 351–368–368. [DOI] [PubMed] [Google Scholar]
- Hirsch E. B.; Tam V. H. Impact of Multidrug-Resistant Pseudomonas Aeruginosainfection on Patient Outcomes. Expert Rev. Pharmacoeconomics Outcomes Res. 2010, 10, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musk J.; Dinty J.; Hergenrother P. J. Chemical Countermeasures for the Control of Bacterial Biofilms: Effective Compounds and Promising Targets. Curr. Med. Chem. 2006, 13, 2163–2177. [DOI] [PubMed] [Google Scholar]
- Smith R. S.; Harris S. G.; Phipps R.; Iglewski B. The Pseudomonas aeruginosa Quorum-Sensing Molecule N-(3-Oxododecanoyl)homoserine Lactone Contributes to Virulence and Induces Inflammation in Vivo. J. Bacteriol. 2002, 184, 1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson J. P.; Gray K. M.; Passador L.; Tucker K. D.; Eberhard A.; Iglewski B. H.; Greenberg E. P. Structure of the Autoinducer Required for Expression of Pseudomonas aeruginosa Virulence Genes. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. W.; Rahme L. G.; Sternberg J. A.; Tompkins R. G.; Ausubel F. M. Pseudomonas aeruginosa Killing of Caenorhabditis elegans Used to Identify P. aeruginosa Virulence Factors. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 2408–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. B.; DiMango E.; Bryan R.; Gambello M.; Iglewski B. H.; Goldberg J. B.; Prince A. Contribution of Specific Pseudomonas aeruginosa Virulence Factors to Pathogenesis of Pneumonia in a Neonatal Mouse Model of Infection. Infect. Immun. 1996, 64, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen T. B.; Givskov M. Quorum-Sensing Inhibitors as Anti-Pathogenic Drugs. Int. J. Med. Microbiol. 2006, 296, 149–161. [DOI] [PubMed] [Google Scholar]
- Rasko D. A.; Sperandio V. Anti-Virulence Strategies to Combat Bacteria-Mediated Disease. Nat. Rev. Drug Discovery 2010, 9, 117–128. [DOI] [PubMed] [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. Targeting Virulence: a New Paradigm for Antimicrobial Therapy. Nat. Chem. Biol. 2007, 3, 541–548. [DOI] [PubMed] [Google Scholar]
- Khmel I. A.; Metlitskaya A. Z. Quorum Sensing Regulation of Gene Expression: a Promising Target for Drugs Against Bacterial Pathogenicity. Mol. Biol. 2006, 40, 169–182. [PubMed] [Google Scholar]
- Rutherford S. T.; Bassler B. L.. Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harbor Perspect. Med. 2012, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaSarre B.; Federle M. J. Exploiting Quorum Sensing to Confuse Bacterial Pathogens. Microbiol. Mol. Biol. Rev. 2013, 77, 73–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez P. N.; Koch G.; Thompson J. A.; Xavier K. B.; Cool R. H.; Quax W. J. The Multiple Signaling Systems Regulating Virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müh U.; Hare B. J.; Duerkop B. A.; Schuster M.; Hanzelka B. L.; Heim R.; Olson E. R.; Greenberg E. P. A Structurally Unrelated Mimic of a Pseudomonas aeruginosa Acyl-Homoserine Lactone Quorum-Sensing Signal. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16948–16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhari J. S.; Kinoyama I.; Struss A. K.; Pullanikat P.; Lowery C. A.; Lardy M.; Janda K. D. Synthesis and Molecular Modeling Provide Insight Into a Pseudomonas aeruginosa Quorum Sensing Conundrum. J. Am. Chem. Soc. 2011, 133, 3840–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.; Nair S. K. Molecular Basis for the Recognition of Structurally Distinct Autoinducer Mimics by the Pseudomonas aeruginosa LasR Quorum-Sensing Signaling Receptor. Chem. Biol. 2009, 16, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara N.; Mashiach R.; Amar D.; Krief P.; Spieser S. A. H.; Bottomley M. J.; Aharoni A.; Meijler M. M. Covalent Inhibition of Bacterial Quorum Sensing. J. Am. Chem. Soc. 2009, 131, 10610–10619. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müh U.; Schuster M.; Heim R.; Singh A.; Olson E. R.; Greenberg E. P. Novel Pseudomonas aeruginosa Quorum-Sensing Inhibitors Identified in an Ultra-High-Throughput Screen. Antimicrob. Agents Chemother. 2006, 50, 3674–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske G. D.; O’Neill J. C.; Miller D. M.; Wezeman R. J.; Mattmann M. E.; Lin Q.; Blackwell H. E. Comparative Analyses of N-Acylated Homoserine Lactones Reveal Unique Structural Features That Dictate Their Ability to Activate or Inhibit Quorum Sensing. ChemBioChem 2008, 9, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske G. D.; O’Neill J. C.; Miller D. M.; Mattmann M. E.; Blackwell H. E. Modulation of Bacterial Quorum Sensing with Synthetic Ligands: Systematic Evaluation of N-Acylated Homoserine Lactones in Multiple Species and New Insights Into Their Mechanisms of Action. J. Am. Chem. Soc. 2007, 129, 13613–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattmann M. E.; Blackwell H. E. Small Molecules That Modulate Quorum Sensing and Control Virulence in Pseudomonas aeruginosa. J. Org. Chem. 2010, 75, 6737–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske G. D.; Mattmann M. E.; Blackwell H. E. Evaluation of a Focused Library of N-Aryl l-Homoserine Lactones Reveals a New Set of Potent Quorum Sensing Modulators. Bioorg. Med. Chem. Lett. 2008, 18, 5978–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni N.; Li M.; Wang J.; Wang B. Inhibitors and Antagonists of Bacterial Quorum Sensing. Med. Res. Rev. 2009, 29, 65–124. [DOI] [PubMed] [Google Scholar]
- O’Loughlin C. T.; Miller L. C.; Siryaporn A.; Drescher K.; Semmelhack M. F.; Bassler B. L. A Quorum-Sensing Inhibitor Blocks Pseudomonas aeruginosa Virulence and Biofilm Formation. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17981–17986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D Pharmacophores Derived From Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [DOI] [PubMed] [Google Scholar]
- Rahme L. G.; Stevens E. J.; Wolfort S. F.; Shao J.; Tompkins R. G.; Ausubel F. M. Common Virulence Factors for Bacterial Pathogenicity in Plants and Animals. Science 1995, 268, 1899–1902. [DOI] [PubMed] [Google Scholar]
- Mahajan-Miklos S.; Tan M. W.; Rahme L. G.; Ausubel F. M. Molecular Mechanisms of Bacterial Virulence Elucidated Using a Pseudomonas aeruginosa-Caenorhabditis elegans Pathogenesis Model. Cell 1999, 96, 47–56. [DOI] [PubMed] [Google Scholar]
- Müsken M.; Di Fiore S.; Römling U.; Häussler S. A 96-Well-Plate–Based Optical Method for the Quantitative and Qualitative Evaluation of Pseudomonas aeruginosa Biofilm Formation and Its Application to Susceptibility Testing. Nat. Protoc. 2010, 5, 1460–1469. [DOI] [PubMed] [Google Scholar]
- Heydorn A.; Nielsen A. T.; Hentzer M.; Sternberg C.; Givskov M.; Ersbøll B. K.; Molin S. Quantification of Biofilm Structures by the Novel Computer Program COMSTAT. Microbiology 2000, 146Pt 102395–2407. [DOI] [PubMed] [Google Scholar]
- Vorregaard M.; Ersbøll B. K.; Yang L.; Haagensen J. A. J.; Heydorn A.; Molin S.; Sternberg C. Personal communication; http://www.comstat.dk.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.