

Abstract
Hsp90 C-terminal inhibitors represent a novel and alternative chemotherapeutic approach for the treatment of cancer. Novobiocin was the first natural product identified as an Hsp90 C-terminal inhibitor; however, it manifests poor antiproliferative activity. In contrast to N-terminal inhibitors, novobiocin does not induce the pro-survival heat shock response. Structural investigations on novobiocin have elucidated some structure–activity relationships and several promising compounds. On the basis of structure–activity relationships and computational studies, a library of ring-constrained novobiocin analogues was designed, synthesized, and evaluated in antiproliferative assays. Results obtained from these studies provide insights into the Hsp90 C-terminal binding site, and new analogues that were developed manifest low micromolar to mid-nanomolar antiproliferative activity resulting from Hsp90 inhibition.
Keywords: Heat shock protein 90, Hsp90 C-terminal inhibitors, ring-constrained novobiocin analogues, structure−activity relationship, breast cancer
The 90 kDa heat shock protein 90 (Hsp90) is a molecular chaperone that plays a crucial role in homeostasis as well as an adaptive response to cell stress.1,2 It is a key component of the cellular protein folding machinery and regulates the conformational maturation, activation, and integrity of a wide array of client proteins, many of which are associated with cell signaling, proliferation, and survival.3 However, in cancer cells, many of these client substrates are mutated and/or overexpressed and contribute to malignant transformation and/or the maintenance of oncogenic phenotypes.4 In fact, Hsp90 client proteins (e.g., Her2, Raf1, Akt, MET, Src, CDK4, etc.) are directly associated with all six hallmarks of cancer and are actively pursued as individual therapeutic targets for the treatment of cancer.5,6 Consequently, Hsp90 inhibition provides an opportunity to simultaneously disrupt multiple oncogenic pathways and hence represents an attractive target for the development of cancer chemotherapeutics.
Hsp90 exists as a homodimer with each monomer consisting of three domains; (1) the N-terminal ATP-binding domain, (2) the middle domain, and (3) the C-terminal dimerization domain. The majority of research has focused on the development of Hsp90 N-terminal inhibitors. In fact, 17 small molecules that target this region have undergone clinical investigation for the treatment of cancer.7 Although there has been great interest in the development of these Hsp90 inhibitors, challenges such as concomitant induction of the pro-survival heat shock response, hepatotoxicity, and multidrug resistance have dampened enthusiasm regarding their clinical promise.8 In contrast, Hsp90 C-terminal inhibitors have emerged as an alternative strategy for Hsp90 modulation as these agents manifest similar inhibitory activity without induction of the pro-survival heat shock response and therefore could potentially circumvent some of the limitations associated with N-terminal inhibition.9−11
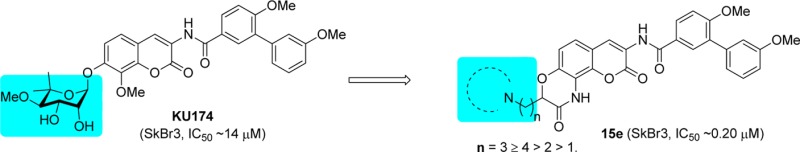
Novobiocin (1), a DNA gyrase inhibitor, was the first natural product identified as a Hsp90 C-terminal inhibitor (Figure 1).12 Unfortunately, it manifests poor antiproliferative activity (IC50 ≈ 700 μM aganist SKBr3 cells) and consequently has limited therapeutic application. Preliminary investigations on novobiocin revealed key structural elements important for Hsp90 activity, which ultimately transformed it from a DNA gyrase inhibitor into a selective Hsp90 inhibitor.13,14 Subsequent studies elucidated additional structure–activity relationships (SARs) and led to the identification of several promising lead molecules (Figure 1) that manifest potent antiproliferative activity against multiple cancer cell lines.15−28 Upon comparison of these lead compounds, the following SARs were observed: (1) the biaryl side chain appears optimal; (2) the amide linker provides hydrogen bonding interactions with the binding pocket; (3) incorporation of a hydrogen-bond acceptor at the 8-position improves inhibitory activity; and (4) modification of the phenolic ether is tolerated and allows for replacement of the complex sugar with simple moieties without compromising inhibitory activity.
Figure 1.

Rationale for ring-constrained novobiocin analogues.
On the basis of these SARs as well as molecular docking studies,28,29 it was hypothesized that lactams containing substituents at the α-position in lieu of the sugar moiety could provide ring-constrained analogues that possess these critical features (Figure 1). Furthermore, as a consequence of ring constraint, the analogues would exhibit less entropic penalty upon binding, compared to their freely rotating counterparts, and therefore could project the side chain into the region normally occupied by the novoise sugar of novobiocin. In addition, replacement of the hydrolytically sensitive ether linkage with a lactam would produce analogues with greater stability.
In support of this hypothesis, compound 9f, which contains the physiologically ionizable pyrrolidine moiety appended to the α-position of the lactam, was designed and docked into the Hsp90 C-terminal model.29 As shown in Figure 2B, compound 9f aligned structurally with the lead compound, KU174.
Figure 2.
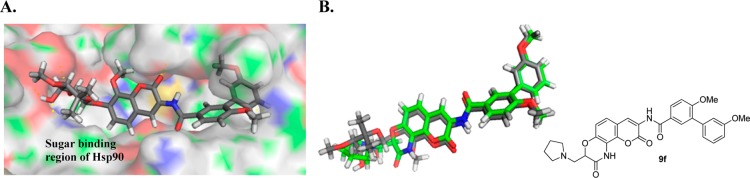
(A) Docked model of KU174 (gray) in the putative Hsp90 C-terminal binding site. (B) Molecular overlay of KU174 (gray) and investigational molecule, 9f (green).
Encouraged by these docking studies, molecules were designed to include a sugar surrogate at the α-position of the lactam and to project into the region typically occupied by the sugar moiety for interactions with the binding pocket. Since no cocrystal structure of Hsp90 bound to C-terminal inhibitors has been solved, there is limited information regarding the size and nature of this binding pocket. Therefore, lactams containing hydrophobic, rigid, flexible, ionizable, and nonionizable substituents were investigated to identify beneficial interactions with the binding site.
Retrosynthetically, we envisioned construction of these lactams from O-alkylation of phenol 6 with α-bromo ester 7, followed by a tandem reduction/ring closing reaction that could be catalyzed upon Pd hydrogenolysis (Scheme 1). Phenol 6 was proposed to result from an Aldol condensation between aldehyde 4 and acid 3, whereas aldehyde 4 could be obtained by the formylation of commercially available nitro resorcinol. Synthesis of acid 3 could be achieved via an amide coupling reaction between the glycine ester and the previously reported biaryl acid 1,1 followed by base-catalyzed hydrolysis.
Scheme 1. Retrosynthetic Analysis of Ring-Constrained Novobiocin Analogues.

As shown in Scheme 2, the synthesis of aromatic analogues began via an amide coupling reaction between biaryl acid 1 and amine 2 to afford amide 3, which underwent an Aldol condensation with benzaldehyde 4 to give acetylated-coumarin 5. Subsequent deacetylation of 5 gave the coumarin intermediate 6, in high yield. Phenol 6 was O-alkylated with the α-bromo esters, 7a–e, to yield the coumarin ethers, 8a–e. The resulting nitro aromatics (8a–e) were then reduced with Pd/C under a hydrogen atmosphere to furnish the corresponding anilines, which underwent an acid-catalyzed cyclization to afford lactams 9a–e. In parallel, a series of amino lactams was synthesized as shown in Scheme 3.
Scheme 2. Synthesis of Aromatic-Containing Lactams.

Reagents and conditions: (a) EDCI, DMAP, Et3N, THF, rt, 12 h, 80%; (b) LiOH·H2O, MeOH/THF/H2O, rt, 12 h, 95%; (c) NaOAc, Ac2O, 110 °C, 3 h, 60%; (d) K2CO3, MeOH, 2 h, 85%; (e) 6, NaH/K2CO3, DMF, 12 h, 20–70%; (f) 10% Pd/C, H2, AcOH, MeOH/EtOAc, rt, 2–3 d, 20–60%.
Scheme 3. Synthesis of Amine-Containing Lactams.

Reagents and conditions: (a) 6, NaH/K2CO3, DMF, 12 h, 20–30%; (b) 10% Pd/C, H2, AcOH, MeOH/EtOAc, rt, 2–3 d, 20–40%; (c) 40% TFA/DCM, DCM, rt, 4 h, 60%.
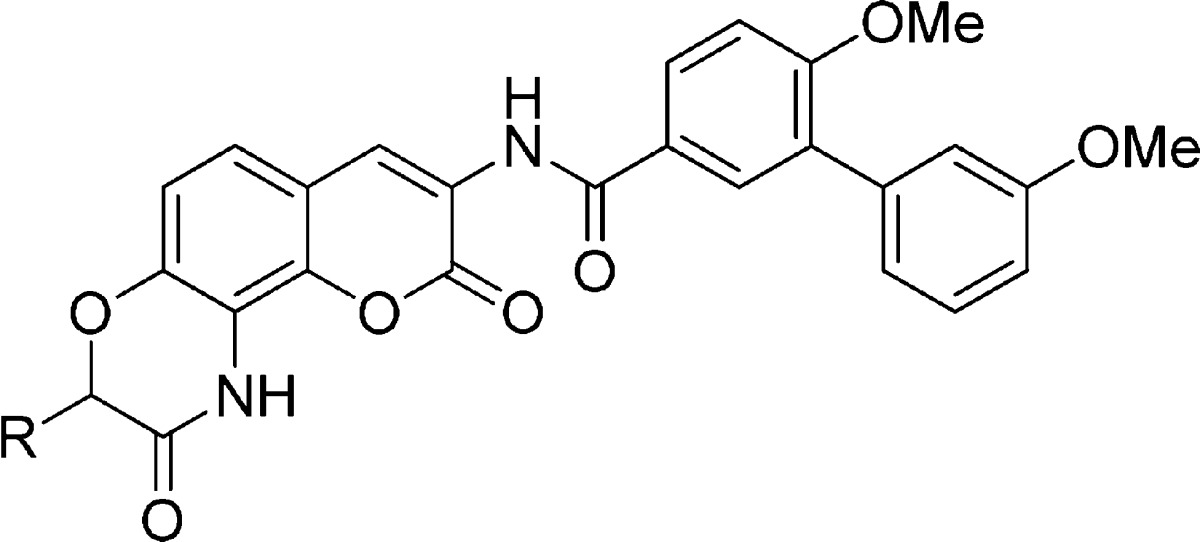
Upon construction, the lactams were evaluated for antiproliferative activity against SKBr3 (estrogen receptor negative, Her2 overexpressing breast cancer cells) and MCF-7 (estrogen receptor positive breast cancer cells) cell lines (Table 1). In general, the aromatic-containing lactams were less effective than their flexible counterparts. Direct attachment of the aromatic moiety to the lactam was detrimental, as compound 9a exhibited an IC50 value of ∼50 μM. Although, the benzyl analogue (9b) manifested improved activity, compounds 9c and 9d were inactive. Interestingly, the inclusion of a polar group onto the aromatic ring resulted in improved activity, as analogue 9e exhibited 3–4-fold greater antiproliferative activity than 9b. These results suggest that a polar moiety attached to a linear tether may be beneficial. Consistent with this hypothesis, compound 9f manifested comparable activity to KU174. Compound 9h, which contains a piperazine moiety, manifested improved antiproliferative activity over compound 9f, while compound 9g was inactive. Interestingly, replacement of the amine with an amide, resulted in inactive analogues (9f vs 9i and 9j), suggesting the physiologically ionizable amine may form key interactions with the protein that are important for antiproliferative activity.
Table 1. Antiproliferative Activity of Aromatic- and Amine-Containing Lactams.
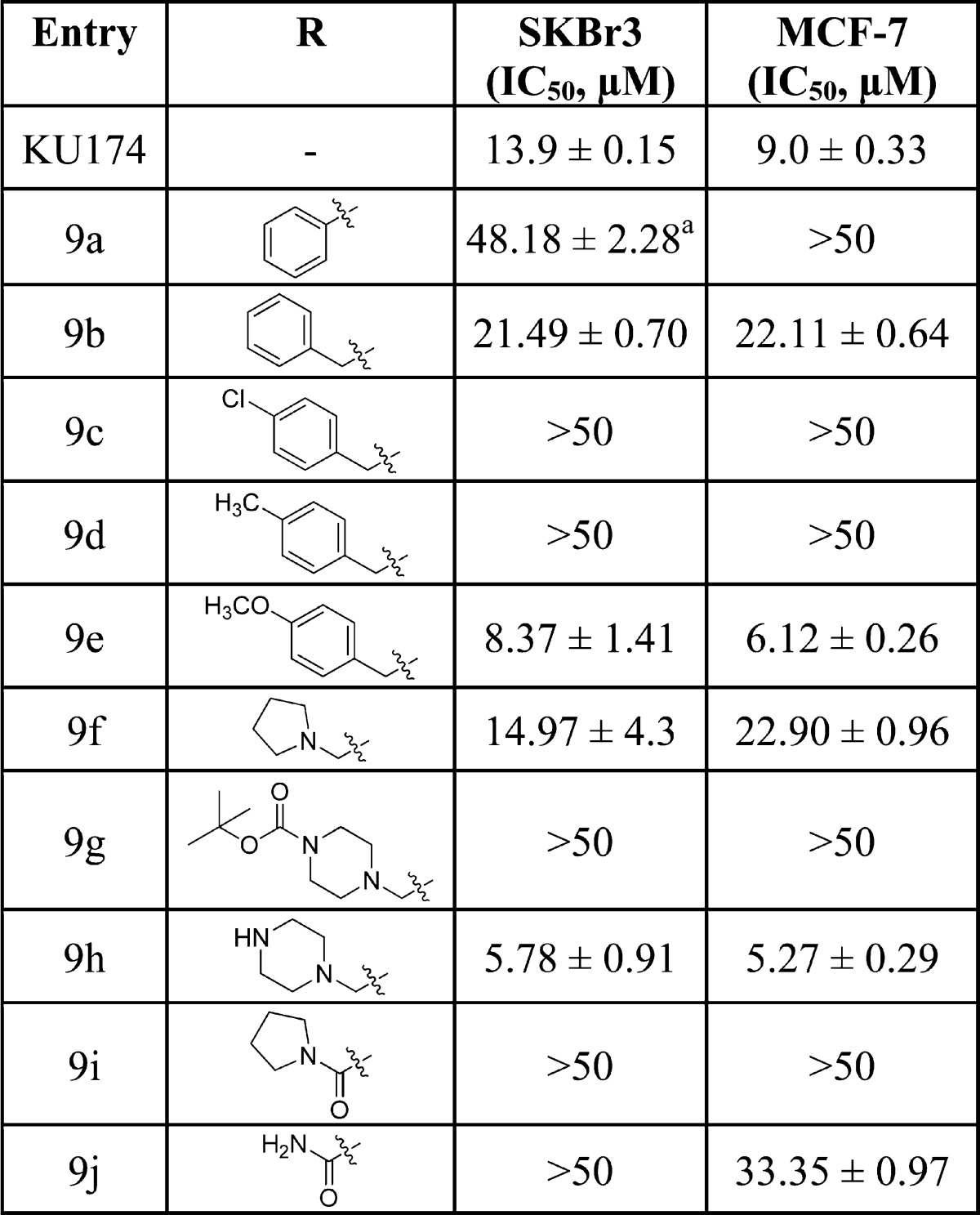
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Overall, preliminary structure–activity relationships suggest an amine and a linear tether are important for antiproliferative activity. To gain further insight into this region of the binding site, amino analogues 12a and 15a, which contain a pyrrolidine moiety tethered to the lactam core, were docked into the Hsp90 C-terminal model (Figure 1S, see Supporting Information). The docking studies suggested the pyrrolidine amine may provide hydrogen bonding interactions with Glu537 within the binding site. Interestingly, this pyrrolidine ring projects into a subpocket that allows for the inclusion of additional substituents. However, homologation of the linker length may diminish hydrogen-bonding interactions and therefore produce less efficacious analogues. Consequently, it was hypothesized that the incorporation of a two- or three-carbon amino tether could project deeper into the hydrophobic subpocket, while maintaining interactions between the ammonium ion and amino acid side chains. The amines were selected based on their potential to provide hydrogen bonding interactions and include various ring sizes and steric bulk.
As shown in Scheme 4, the syntheses of ethylene-containing lactams was initiated by O-alkylation of phenol 6 with 2,4-dibromo ether 10 to give coumarin ether 11 in high yield. The resulting coumarin ether (11) was then treated with primary or secondary amines to give the corresponding products, which were then subjected to tandem reduction/intramolecular ring closing, enlisting a Pd/C catalyst under hydrogen atmosphere to furnish compounds 12a–i.
Scheme 4. Synthesis of Two-Carbon Tethered Lactams.

Reagents and conditions: (a) 6, K2CO3, DMF, rt, 12 h, 78%; (b) R1R2NH, DMF, rt, 12 h; (c) 10% Pd/C, H2, CH3COOH, MeOH/EtOAc, rt, 2–3 d, 28–55%.
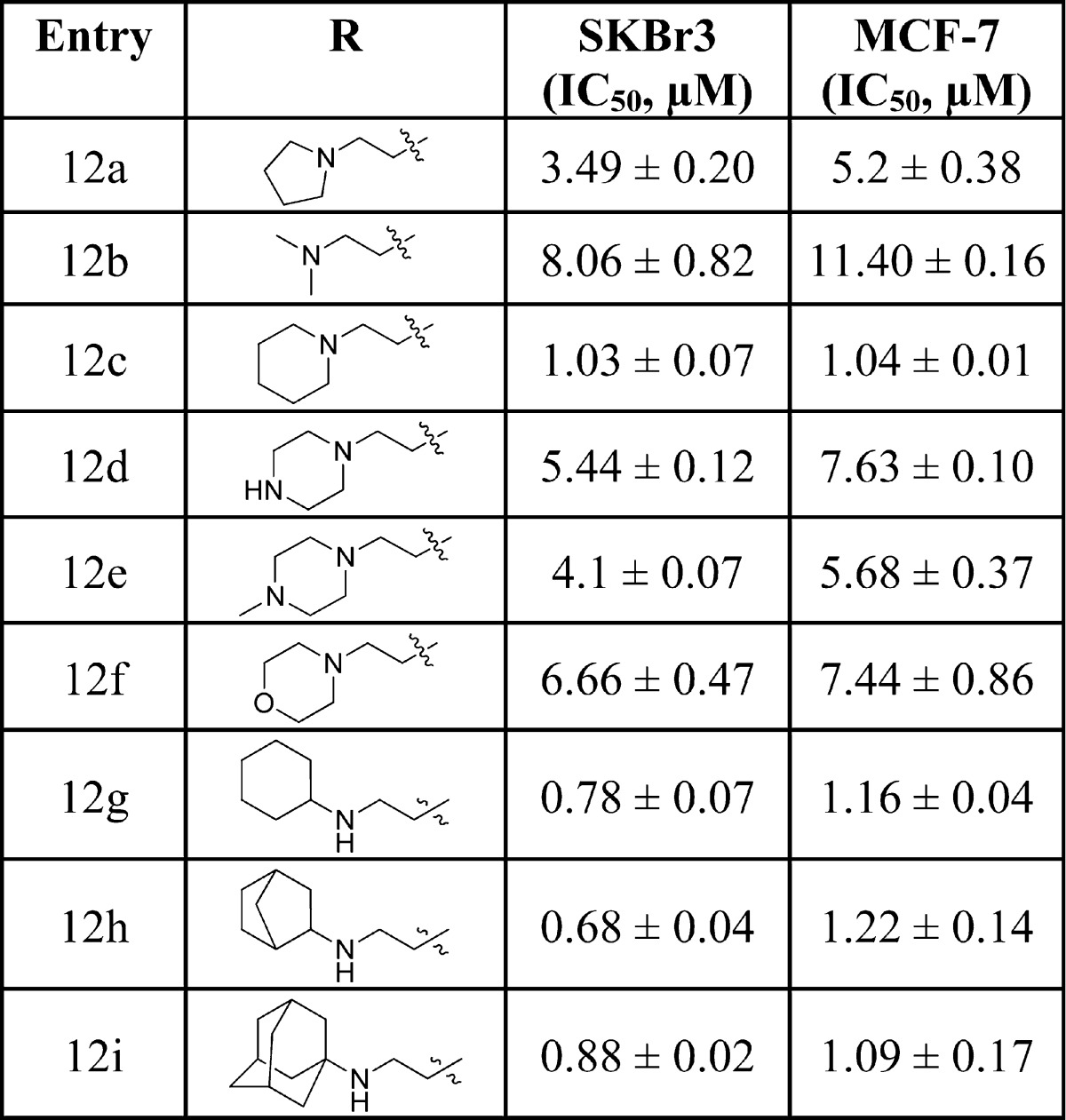
Upon their preparation, these two-carbon tethered lactams were evaluated for antiproliferative activity against SKBr3 and MCF-7 cell lines (Table 2). Consistent with the docking studies, ethylene-containing lactams exhibited improved antiproliferative activity compared to their methylene-containing counterparts (9f vs 12a). Introduction of an acyclic amine (12b) resulted in decreased potency, emphasizing the need for a cyclic ring system. Increasing the ring size to six atoms (12c) maintained activity and suggested that various ring structures could be accommodated. It was found that the inclusion of a second hydrogen bond donor/acceptor (12d, 12e, and 12f) decreased activity, indicating the subpocket may be hydrophobic in nature. Interestingly, the secondary amines manifested better activity than the tertiary amines (12g, 12h, and 12i).
Table 2. Antiproliferative Activity of Two-Carbon Tethered Lactams.
For comparison with 1- and 2-carbon linkers, a series of three-carbon tethers was synthesized as shown in Scheme 5. Confirmation of the optimal linker length was investigated through construction of a 4-carbon tethered lactam 18 (see Supporting Information).
Scheme 5. Synthesis of Three-Carbon Tethered Lactams.

Reagents and conditions: (a) 6, K2CO3, DMF, rt, 12 h, 75%; (b) R1R2NH, DMF, rt, 12 h; (c) 10% Pd/C, H2, CH3COOH, MeOH/EtOAc, rt, 2–3 d, 20–40%.
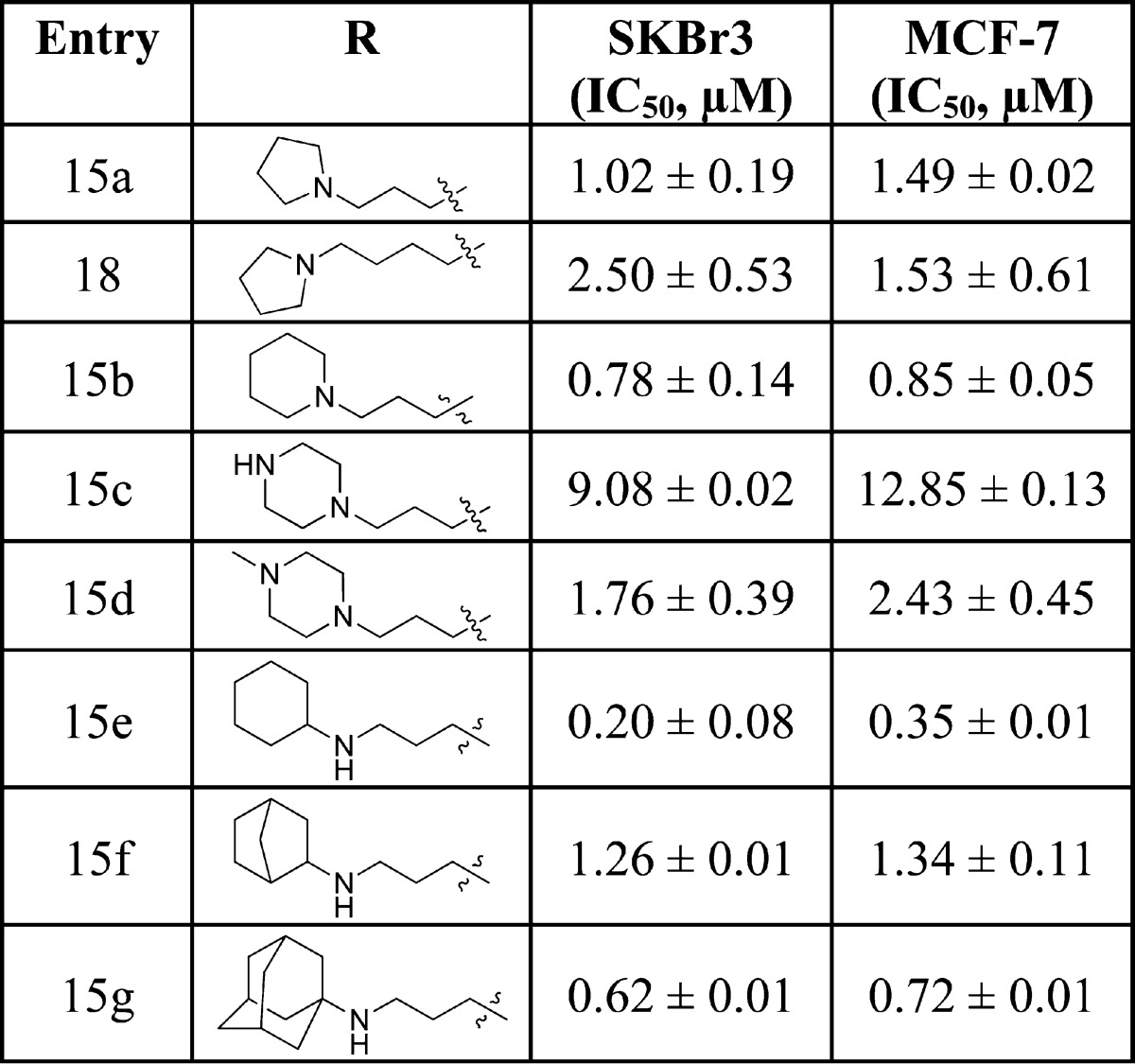
Upon completion, the lactams were evaluated for antiproliferative activity against both cancer cell lines. As shown in Table 3, the analogue containing a three-carbon linker exhibited improved antiproliferative activity over the corresponding 2-carbon tethered lactams (15a vs 12a). In addition, compound 18 showed comparable activity to 15a, suggesting further homologation of the linker length does not provide better interactions with the binding pocket. Similar to the trend observed with ethylene-containing analogues, the piperidine (15b) analogue manifested improved antiproliferative activity, while the piperazine (15c) and N-methylpiperazine (15d) resulted in decreased inhibitory activity. Interestingly, cyclohexylamino analogue 15e manifested better antiproliferative activity compared to the bicycloalkyl and tricyclic amino analogues (15f and 15g), indicating limitations within the hydrophobic subpocket.
Table 3. Antiproliferative Activity Manifested by Three-Carbon Tethered Lactams.

To confirm the antiproliferative activity exhibited by these lactams resulted from Hsp90 inhibition, Western blot analysis was performed on lysates treated with 15b, 15e, and 15g. Actin, an Hsp90-independent protein, was used as a negative control. As shown in Figure 3A, incubation with these compounds induced the degradation of Hsp90-dependent client proteins Her2, Raf-1, and Akt, confirming these analogues manifest their cellular activity through Hsp90 inhibition. In addition, treatment of MCF-7 cells with 15b induced the degradation of the Hsp90-dependent client proteins Her2, Raf-1, and AKt in a concentration-dependent manner (Figure 3B). Actin levels remained unaffected, indicating selective degradation of Hsp90-dependent client proteins. Additionally, Hsp90 levels were unaltered, which is a hallmark exhibited by Hsp90 C-terminal inhibitors.
Figure 3.
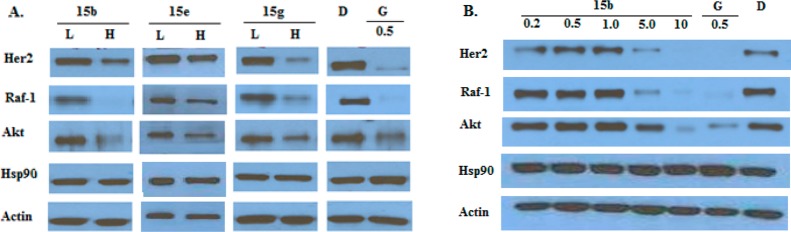
Western blot analyses of lactams after incubation with MCF-7 breast cancer cells for 24 h. (A) Western blot analyses of the Hsp90 client protein degradation after treatment with lactams 15b, 15e, and 15g. (B) Western blot analyses of the Hsp90 client protein degradation after treatment with lactam 15b. Concentrations (in μM) were indicated above each lane. L represents a concentration 1/2 of the antiproliferative activity. H represents a concentration equal to 5-fold of the antiproliferative activity. Geldanamycin (G, 0.5 μM) and dimethyl sulfoxide (D, 100%) serve as positive and negative controls, respectively.
In summary, a series of ring-constrained novobiocin analogues was designed, synthesized, and evaluated against MCF-7 and SKBr3 breast cancer cell lines. Incorporation of ring-constrained analogues led to the development of molecules that manifest submicromolar to mid-nanomolar inhibitory activity. SAR studies revealed that amines attached to a three-carbon linker produced the most efficacious analogues. Furthermore, these studies suggest the presence of a hydrophobic subpocket that can be exploited for the development of more efficacious Hsp90 inhibitors, which will be reported in due course.
Supporting Information Available
Experimental procedures for the synthesis and characterization of new compounds (1H and 13C NMR, HRMS). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors gratefully acknowledge support of this project by the NIH/NCI (CA120458). The NMR support for this work was provided by NSF (9512331).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Taipale M.; Jarosz D. F.; Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [DOI] [PubMed] [Google Scholar]
- Chaudhury S.; Welch T. R.; Blagg B. S. J. Hsp90 as a target for drug development. ChemMedChem 2006, 1, 1331–1340. [DOI] [PubMed] [Google Scholar]
- Donnelly A.; Blagg B. S. J. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop S. C.; Burlison J. A.; Blagg B. S. Hsp90: a novel target for the disruption of multiple signaling cascades. Curr. Cancer Drug Targets 2007, 7, 369–88. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. The hallmarks of cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Blagg B. S. J.; Kerr T. D. Hsp90 inhibitors: Small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med. Res. Rev. 2006, 26, 310–338. [DOI] [PubMed] [Google Scholar]
- Neckers L.; Workman P. Hsp90 molecular chaperone inhibitors: are we there yet?. Clin. Cancer Res. 2012, 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L.; Lindquist S. L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [DOI] [PubMed] [Google Scholar]
- Peterson L. B. B.; Brian S. J. To fold or not to fold: modulation and consequences of Hsp90 inhibition. Future Med. Chem. 2009, 1, 267–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Blagg B. S. J.. Inhibitors of the Hsp90 C-terminus. In Inhibitors of Molecular Chaperones as Therapeutic Agents; RSC Drug Discovery Series; RSC: London, U.K., 2013; Vol. 37 [Google Scholar]
- Davenport J.; Balch M.; Galam L.; Girgis A.; Hall J.; Blagg B.; Matts R. High-throughput screen of natural product libraries for Hsp90 inhibitors. Biology 2014, 3, 101–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu M. G.; Schulte T. W.; Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [DOI] [PubMed] [Google Scholar]
- Burlison J. A.; Neckers L.; Smith A. B.; Maxwell A.; Blagg B. S. J. Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. J. Am. Chem. Soc. 2006, 128, 15529–15536. [DOI] [PubMed] [Google Scholar]
- Yu X. M.; Shen G.; Neckers L.; Blake H.; Holzbeierlein J.; Cronk B.; Blagg B. S. J. Hsp90 inhibitors identified from a library of novobiocin analogues. J. Am. Chem. Soc. 2005, 127, 12778–12779. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Donnelly A. C.; Kusuma B. R.; Brandt G. E. L.; Brown D.; Rajewski R. A.; Vielhauer G.; Holzbeierlein J.; Cohen M. S.; Blagg B. S. J. Engineering an antibiotic to fight cancer: optimization of the novobiocin scaffold to produce anti-proliferative agents. J. Med. Chem. 2011, 54, 3839–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskew J.; Sadikot T.; Morales P.; Duren A.; Dunwiddie I.; Swink M.; Zhang X.; Hembruff S.; Donnelly A.; Rajewski R.; Blagg B.; Manjarrez J.; Matts R.; Holzbeierlein J.; Vielhauer G. Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 2011, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton S. N.; Shawgo M. E.; Matthews S. B.; Lu Y.; Donnelly A. C.; Szabla K.; Tanol M.; Vielhauer G. A.; Rajewski R. A.; Matts R. L.; Blagg B. S. J.; Robertson J. D. KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heat shock protein, exerts potent antiproliferative effects in human leukemic cells. Mol. Pharmacol. 2009, 76, 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly A. C.; Mays J. R.; Burlison J. A.; Nelson J. T.; Vielhauer G.; Holzbeierlein J.; Blagg B. S. J. The design, synthesis, and evaluation of coumarin ring derivatives of the novobiocin scaffold that exhibit antiproliferative activity. J. Org. Chem. 2008, 73, 8901–8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlison J. A.; Avila C.; Vielhauer G.; Lubbers D. J.; Holzbeierlein J.; Blagg B. S. J. Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem. 2008, 73, 2130–2137. [DOI] [PubMed] [Google Scholar]
- Burlison J. A.; Blagg B. S. J. Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org. Lett. 2006, 8, 4855–4858. [DOI] [PubMed] [Google Scholar]
- Huang Y.-T.; Blagg B. S. J. A library of noviosylated coumarin analogues. J. Org. Chem. 2007, 72, 3609–3613. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Reddy Kusuma B.; Blagg B. S. J. Synthesis and evaluation of noviose replacements on novobiocin that manifest antiproliferative activity. ACS Med. Chem. Lett. 2010, 1, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Yan B.; Peterson L. B.; Blagg B. S. J. 3-Arylcoumarin derivatives manifest anti-proliferative activity through Hsp90 inhibition. ACS Med. Chem. Lett. 2012, 3, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Blagg B. S. J. Novobiocin analogues with second-generation noviose surrogates. Biol. Med. Chem. Lett. 2013, 23, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuma B. R.; Khandelwal A.; Gu W.; Brown D.; Liu W.; Vielhauer G.; Holzbeierlein J.; Blagg B. S. J. Synthesis and biological evaluation of coumarin replacements of novobiocin as Hsp90 inhibitors. Biol. Med. Chem. 2014, 22, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly A. C.; Zhao H.; Reddy Kusuma B.; Blagg B. S. J. Cytotoxic sugar analogues of an optimized novobiocin scaffold. MedChemComm 2010, 1, 165–170. [Google Scholar]
- Kyle Hadden M.; Hill S. A.; Davenport J.; Matts R. L.; Blagg B. S. J. Synthesis and evaluation of Hsp90 inhibitors that contain the 1,4-naphthoquinone scaffold. Biol. Med. Chem. 2009, 17, 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. Y.; Shan Z. J.; Zhai H. L.; Li L. N.; Zhang X. Y. Molecular design of anticancer drug leads based on three-dimensional quantitative structure–activity relationship. J. Chem. Inf. Model. 2011, 51, 1999–2006. [DOI] [PubMed] [Google Scholar]
- Matts R. L.; Dixit A.; Peterson L. B.; Sun L.; Voruganti S.; Kalyanaraman P.; Hartson S. D.; Verkhivker G. M.; Blagg B. S. J. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem. Biol. 2011, 6, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.