

Abstract
The plastids of ecologically and economically important algae from phyla such as stramenopiles, dinoflagellates and cryptophytes were acquired via a secondary endosymbiosis and are surrounded by three or four membranes. Nuclear-encoded plastid-localized proteins contain N-terminal bipartite targeting peptides with the conserved amino acid sequence motif ‘ASAFAP’. Here we identify the plastid proteomes of two diatoms, Thalassiosira pseudonana and Phaeodactylum tricornutum, using a customized prediction tool (ASAFind) that identifies nuclear-encoded plastid proteins in algae with secondary plastids of the red lineage based on the output of SignalP and the identification of conserved ‘ASAFAP’ motifs and transit peptides. We tested ASAFind against a large reference dataset of diatom proteins with experimentally confirmed subcellular localization and found that the tool accurately identified plastid-localized proteins with both high sensitivity and high specificity. To identify nucleus-encoded plastid proteins of T. pseudonana and P. tricornutum we generated optimized sets of gene models for both whole genomes, to increase the percentage of full-length proteins compared with previous assembly model sets. ASAFind applied to these optimized sets revealed that about 8% of the proteins encoded in their nuclear genomes were predicted to be plastid localized and therefore represent the putative plastid proteomes of these algae.
Keywords: Thalassiosira pseudonana, Phaeodactylum tricornutum, chloroplast, proteome, prediction, technical advance
Introduction
Plastids arose through endosymbiotic processes – a primary endosymbiosis of a cyanobacterium gave rise to red and green algae and the subsequent evolution of plants, and multiple secondary endosymbioses of either a red or a green alga gave rise to a broad diversity of eukaryotic microbes. Marine microalgae with secondary plastids from the red lineage contribute significantly to global biogeochemical cycles and support productive marine food webs (Cavalier-Smith, 1999). Major groups include diatoms, coccolithophores, cryptophytes, dinoflagellates, and apicomplexans. Plastids in these organisms have a complex structure with either three or four membranes, most with the endoplasmic reticulum (ER) as the outermost membrane (Kroth, 2002). The majority of genes from the original endosymbionts were either lost, replaced by genes of the host or transferred to the nucleus of the host; only a minority of the genes was retained on the original endosymbiont genome (Timmis et al., 2004). Of those organisms with a secondary plastid of the red lineage, only cryptophytes possess a remnant nucleus from the endosymbiont – the nucleomorph, which is located in the periplastidic space between the second and third envelope membrane (Curtis et al., 2012).
Consequently, the majority of proteins required for plastid function is encoded in the nucleus and subsequently transported to the plastid. Delivery of nuclear-encoded plastid proteins across multiple membranes requires an efficient protein import system (Gruber et al., 2007), which includes protein transport via the ER. All known nuclear-encoded plastid-localized proteins in cells with secondary plastids of the red lineage possess bipartite N-terminal pre-sequences that consist of an ER-type signal peptide followed immediately by a transit peptide (Kroth, 2002; Patron and Waller, 2007). The transit peptide is cleaved off the mature protein upon completion of the import reaction, likely by a specific stromal processing peptidase (Huesgen et al., 2013).
The transit peptide domains of bipartite plastid targeting pre-sequences commonly begin with a phenylalanine residue at the +1 position after the signal peptide cleavage site (Kroth, 2002; Armbrust et al., 2004; Patron and Waller, 2007), which is crucial for successful plastid protein import (Apt et al., 2002; Kilian and Kroth, 2005; Gruber et al., 2007). The transit peptide contains a high proportion of hydroxylated residues, few negatively charged residues, and a net positive charge (Patron and Waller, 2007), which is also necessary for plastid protein import (Felsner et al., 2010). Other features of the transit peptide, including its length, are less critical for plastid import (Apt et al., 2002; Kilian and Kroth, 2005; Gruber et al., 2007). The phenylalanine at the +1 position of the transit peptide is part of a conserved sequence motif (‘ASAFAP’ motif) surrounding the signal peptide cleavage site in diatoms (Kilian and Kroth, 2005; Gruber et al., 2007), cryptophytes (Gould et al., 2006a; Patron and Waller, 2007), and dinoflagellates (Patron et al., 2005; Patron and Waller, 2007). This distinctive motif is a good marker for identifying nuclear-encoded plastid proteins based on DNA sequence data (Gruber et al., 2007; Gruber and Kroth, 2014).
Here we present the results of a genomewide prediction of nucleus-encoded plastid proteins for the diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum, based on recognition of ASFAP motifs, combined with a composition-based evaluation of the transit peptide downstream of the cleavage site.
Results and discussion
Characterization of the ‘ASAFAP’ motif
The plastid protein prediction was initiated with a set of putative plastid-targeted proteins from the diatoms Thalassiosira pseudonana and Phaeodactylum tricornutum, compiled based on the lists of nucleus-encoded and plastid-targeted proteins published by Armbrust et al. (2004) (for T. pseudonana), Gruber et al. (2007) (for P. tricornutum), and Kroth et al. (2008) (P. tricornutum and T. pseudonana). These proteins were supplemented with additional proteins that are most likely plastid-targeted based on functional annotation. Furthermore, homologues of proteins from the T. pseudonana lists were searched in the P. tricornutum genome and vice versa.
For maximum consistency between the sequence sets for the two species, all sequences found in only one of the organisms were removed from the set. To avoid potential overfitting of the data, we reduced the level of homology within the protein set using an ‘all against all’ BLAST search of the candidate sequences from T. pseudonana and P. tricornutum. Only sequences that paired with one homologue from the other diatom species (instead of a sequence from the same species) were retained to minimize inclusion of gene duplications present in only one organism. The highest level of homology present in the sequence set therefore corresponds to the time of independent evolution since the split between the pennate and centric diatom lineages, which took place approximately 90 million years ago (Bowler et al., 2008). The final set consists of 83 orthologous pairs of putative plastid-targeted protein sequences from T. pseudonana and P. tricornutum (Table S1).
Via proteomic amino-termini profiling, Huesgen et al. (2013) recently identified 1295 unique N-terminus peptides from 939 nuclear-encoded T. pseudonana proteins. These N-terminal peptides in many cases represent N-termini of native functional proteins, after cleavage of N-terminal targeting signals. The peptide list also contains N-termini of proteins that are not processed in vivo, as well as the products of internally cleaved proteins. Searched against our T. pseudonana dataset, 44 of the N-terminal peptides identified by Huesgen et al. (2013) match 36 of the 83 T. pseudonana sequences (Table S1). For 31 of the matches the position of our manually identified signal peptide cleavage site lies between 14 and 95 amino acid residues upstream of the N-terminal peptide, and therefore supports the presence of a transit peptide-like domain that is actually cleaved off. Three of the peptides match the transit peptide domain itself with the peptide starting with the +1 position of the predicted cleavage site or one position further downstream, which supports that the signal peptides as well as transit peptides are cleaved off independently from each other. It should be noted that in one case (T. pseudonana protein ID 270231) there is N-terminal peptide support for both the N-terminus of the transit peptide-like domain with the signal peptide cleaved off as well as for the N-terminus of the putative mature protein after cleavage of a 12-residue transit peptide. Ten of the peptides correspond to N-termini derived from internal cleavage of the protein after 133–1242 residues (Table S1).
Despite the overall divergence between T. pseudonana and P. tricornutum (Bowler et al., 2008), the plastid targeting motifs are similar in both organisms, and also similar to the N-terminal signal found in the list of 63 T. pseudonana transit peptides published by Huesgen et al. (2013) (Figure S1). We therefore combined the sequence sets from T. pseudonana and P. tricornutum to generate a scoring matrix (Table S2) based on the frequency of occurrence of each amino acid weighted by the amount of information at each position in the sequence logo (Figure1). This was used to develop a single plastid protein prediction method.
Figure 1.

Conserved cleavage site motif.Conserved sequence motif surrounding the signal peptide cleavage site for 166 putative plastid-targeted protein sequences from T. pseudonana and P. tricornutum (see Table S1). Sequence logos represent the scoring matrix used to calculate cleavage site and transit peptide scores for SignalP positive sequences. Sequence logos and frequency plots (Schneider and Stephens, 1990) were created with WebLogo (Crooks et al., 2004; http://weblogo.berkeley.edu/).
Prediction of signal peptides and cleavage sites
The signal peptide of plastid-targeted proteins in diatoms and other organisms with secondary plastids can be identified via the prediction program SignalP (Nielsen et al., 1997; Emanuelsson et al., 2007) that has been developed through a number of versions. The most current versions are SignalP 3.0 (Bendtsen et al., 2004) and SignalP 4.1 (Petersen et al., 2011). SignalP 3.0 employs either a neuronal network (NN) or a hidden Markov (HMM) model to identify the signal peptide (Nielsen and Krogh, 1998; Bendtsen et al., 2004), SignalP 4.1 exclusively uses a NN, but can be adjusted to two levels of sensitivity (Petersen et al., 2011). SignalP 3.0 NN recognized a signal peptide in 163 of the 166 test proteins (83 sequences from T. pseudonana and P. tricornutum each, see Table S1) whereas SignalP 3.0 HMM recognized 165 signal peptides (Table S1). SignalP 4.1 identified signal peptides in 141 or 161 of the 166 sequences, depending on the choice of sensitivity (Table S1).
In diatoms, the signal peptide cleavage site is spanned by the ‘ASAFAP’ motif (Kilian and Kroth, 2005; Gruber et al., 2007). In a comparison between manually identified cleavage site motifs with the predictions of the different SignalP variants, we found that the SignalP 3.0 NN prediction identified the ‘ASAFAP’ motif in 150 out of the 166 proteins, whereas the SignalP 3.0 HMM prediction identified the motif in 139 of the 166 proteins. SignalP 4.1, identified 148 of the cleavage site motifs (Table S1), the cleavage site predictions are identical for both sensitivity settings. For most of the tested sequences, cleavage site predictions are identical between the different SignalP versions; deviant predictions are found for all versions, with no particular overlap that would allow conclusions on the presence of non-canonical sequences in our set (Figure S2). Based on the highest level of congruency with the manual motif identifications, we decided to use the NN prediction of SignalP 3.0 for all subsequent analyses, and to additionally evaluate methods to increase the accuracy of the cleavage site predictions via the direct detection of ‘ASAFAP’ motifs.
For this, we used the information in the sequence logo to evaluate potential alternate cleavage sites. Because the highest bit scores within the sequence logo, and thus the greatest discriminating potential, were found on either side of the signal peptide cleavage site (Figure1), proteins were first scored over a 25 amino acid sequence window from −5 to +20 around the SignalP 3.0 NN-predicted signal peptide cleavage site, using the scoring matrix (Table S2) generated from the 166 putative plastid-targeted proteins (Table S1). Next, proteins were scored over a sliding window of five residues, including two positions upstream and downstream of the SignalP predicted cleavage site (Figure2). The ASAFind predicted cleavage site corresponds to the cleavage site position with the highest score.
Figure 2.

Sliding window and cleavage site score calculation.Conserved cleavage site motifs that differ from the SignalP predicted cleavage site are identified by calculating cleavage site scores for 25-position sequence windows surrounding the SignalP predicted cleavage site as shown on the example of the Phaeodactylum tricornutum oxygen evolving enhancer 1 (PtOEE1, GenBank AY191862, Protein ID 20331). See also Gruber et al. (2007) and Kilian and Kroth (2005) for detailed mutational analyses of this sequence.
A signal peptide cleavage site is common to all ER-targeted proteins, including those that are targeted to the ER but not to the plastid. To discriminate between plastid proteins and other secretory proteins, we calculated a transit peptide score, again via the weighted scoring matrix, based on the 20 residues downstream of the signal peptide cleavage site. Thus, this transit peptide score does not evaluate the ER cleavage site itself.
Plastid protein prediction
Because of the general trade-off between sensitivity (the ability to recognize true positives) and specificity (the ability to recognize true negatives), we opted to develop a plastid protein prediction protocol at two confidence levels, tuned for either high sensitivity or high specificity. For the statistical evaluation of our prediction method we compiled a set of reference proteins based on available experimental protein location data (e.g. fusion of reporter genes, proteomic studies or immuno-electron microscopy) for P. tricornutum proteins (Table S3). This dataset included plastid-targeted protein sequences, as well as proteins targeted to other compartments such as the ER and mitochondria. It consisted of 132 proteins, 19 of which were, by coincidence, also included in the sequence set used to calculate the initial sequence logos. The use of largely separate sequence sets for generating the scoring matrix and evaluating the prediction ensures that the reference data are not overfit. Sequences in the reference set were classified as positive if they were experimentally shown to be plastid targeted or as negative if they were experimentally shown to be targeted to another compartment (Table S3).
Plastid-targeted reference proteins were best distinguished from non-plastid-targeted reference proteins by the following protocol (Figure3). If the SignalP 3.0 NN prediction was negative, the sequence was defined as ‘not plastid, SignalP negative.’ If the SignalP 3.0 NN prediction was positive, the window spanning two positions each upstream and downstream of the SignalP NN-identified cleavage site was further evaluated to identify the position with the highest cleavage site score. This position was deemed the ASAFind predicted cleavage site. If the first amino acid of the ASAFind predicted transit peptide was an amino acid other than ‘F’, ‘W’, ‘Y’ or ‘L’, the sequence was defined as ‘not plastid, SignalP positive’. These proteins are candidates for retention in the ER or for other targeting via the secretory system. If an ‘F’, ‘W’, ‘Y’ or ‘L’ residue was present at the first position of the ASAFind predicted transit peptide, the sequence was classified as potentially plastid targeted and evaluated further. If the ASAFind predicted cleavage site coincided with the SignalP prediction and the transit peptide score was higher than 2, the sequence was defined as ‘plastid, high confidence’, otherwise the sequence was defined as ‘plastid, low confidence’ (Figure3).
Figure 3.

Plastid protein prediction.Decision making steps for the prediction of plastid proteins, the ASAFind prediction is based on the results of the signal peptide prediction via SignalP 3.0 neuronal networks (Nielsen and Krogh, 1998; Bendtsen et al., 2004; Emanuelsson et al., 2007).
This protocol was optimized with our reference set as the gold standard. We empirically tested the performance of different prediction approaches and parameters (such as sliding window ranges or score cut-offs) by calculating sensitivity, specificity and Matthews correlation coefficients (MCC) or by receiver operating characteristics (ROC) plot analyses (Baldi et al., 2000; Brown and Davis, 2006; Fawcett, 2006).
Based on these analyses, the ‘plastid, low confidence’ prediction is highly sensitive, while the ‘plastid, high confidence’ prediction is extremely specific (Figure4). The MCC for our method is higher (Table1) than for the specialized prediction server HECTAR (Gschloessl et al., 2008) which combines a number of publically available subcellular localization methods using a Support Vector Machine to produce a prediction. The increase in prediction performance of ASAFind is mainly driven by the enhanced sensitivity of our approach; as a consequence, HECTAR should be used with care when a high sensitivity is desired.
Figure 4.
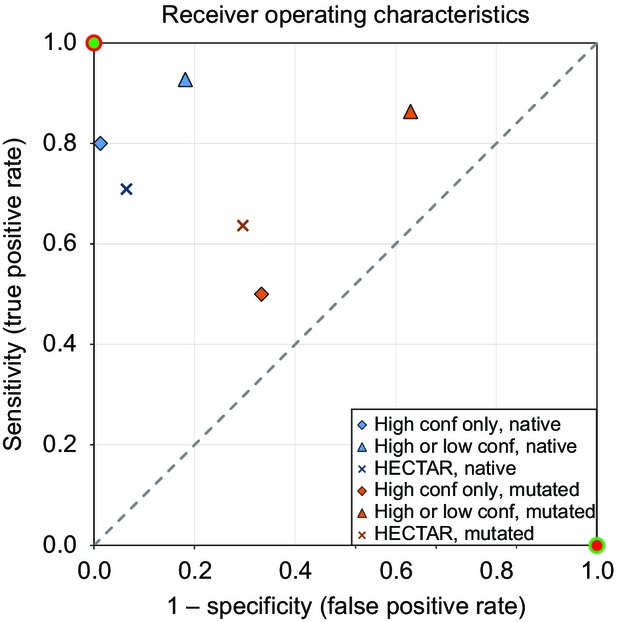
Receiver operating characteristics.Receiver operating characteristics (ROC) plot of plastid protein prediction methods evaluated with sets of native (Tables S3 and S4) or mutated (Table S5) reference sequences, see Table S6 for additional counts/scores. ‘High conf only, native’ are high confidence only predictions tested with the native experimental reference set; ‘high or low conf, native’ are high or low confidence predictions tested with the native experimental reference set; ‘HECTAR, native’ are predictions by HECTAR (Gschloessl et al., 2008) tested with the native experimental reference set (Tables S3 and S4); ‘high conf only, mutated’ are high confidence only predictions tested with the mutated sequences reference set (Table S5); ‘high or low conf, mutated’ are high or low confidence predictions tested with the mutated sequences reference set; ‘HECTAR, mutated’ are predictions by HECTAR (Gschloessl et al., 2008) tested with the mutated sequences reference set. Hypothetical ‘perfect’ (green dot with red ring) and ‘perfect inverse’ (red dot with green ring) predictions are included on the diagram; the dashed line corresponds to random guesses.
Table 1.
Plastid protein prediction statistics. Performance of the plastid protein prediction methods evaluated with Phaeodactylum tricornutum sequences of proteins with experimentally determined intracellular location (see Table S3). See Table S6 for formulas and additional counts/scores, MCC: Matthews correlation coefficient
| High confidence only | High or low confidence | HECTARa | |
|---|---|---|---|
| Sensitivity | 0.80 | 0.93 | 0.71 |
| Specificity | 0.99 | 0.82 | 0.94 |
| MCC | 0.82 | 0.74 | 0.67 |
See Gschloessl et al. (2008).
In addition to the reference protein set, we also collected 49 sequences that were mutated in previous studies to pinpoint the crucial components of the targeting signal (Kilian and Kroth, 2005; Gruber et al., 2007; Felsner et al., 2010) (Table S5). As expected, the prediction methods performed considerably worse with this mutated protein test set (Figure4 and Tables S4–S6), emphasizing that native targeting pre-sequences are under strong selection pressures to maintain their functionality. This result shows that experimentally engineered pre-sequences are useful for the characterization of the exact requirements for the targeting signal as performed by Apt et al. (2002), Felsner et al. (2010), Gruber et al. (2007) or Kilian and Kroth (2005), however, due to the artificial nature of these sequences, they are of limited use as templates for the development of prediction algorithms.
The efficiency of our method depends on the presence of the conserved ‘ASAFAP’ motif. Mernberger et al. (2014) recently developed a motif-independent method for subcellular localization of proteins, with the stated goal of predicting plastid proteins in organisms with limited information on potential protein localization signals. Although their methods were tested with the diatom P. tricornutum, we were unable to compare the performance of their methods with ours because neither the sequences used in their reference set nor the performance metrics are specified in the manuscript and the method is not available for public use. Mernberger et al. (2014) do not compare the performance of their prediction methods to the performance of the dedicated prediction tool HECTAR (Gschloessl et al., 2008), but do compare to other established methods including TargetP (Emanuelsson et al., 2000), WoLF PSORT (Horton et al., 2007) and MultiLoc2 (Blum et al., 2009), none of which was developed to predict protein localization in secondary plastids. Therefore it remains unclear whether the apparent advantage of Mernberger et al.'s (2014) approach over the other tested prediction tools comes from a methodological improvement or from use of specific training sets for the tested organisms.
The performance of our method was also evaluated in the cryptophyte Guillardia theta with 54 homologues of proteins from the P. tricornutum reference set (Curtis et al., 2012). For G. theta, the ‘plastid, low confidence’ prediction has a sensitivity of 0.85 and a specificity of 0.88 (MCC: 0,73), while the ‘plastid, high confidence’ prediction has a sensitivity of 0.70 and a specificity of 0.97 (MCC: 0.72) (Curtis et al., 2012). Use of our diatom-optimized prediction method for other organisms with secondary plastids of the red algal lineage, such as cryptophytes, appears to come with a loss of sensitivity, at similar levels of specificity (compare to Table1).
Gene catalog optimization
The predicted complete proteomes of T. pseudonana and P. tricornutum were optimized to ensure that the datasets used for plastid localization predictions were composed primarily of full-length proteins (Figure5). This was necessary because of the challenges involved in complete gene annotation, especially in non-model organisms, where identification of 5′ exons can be particularly problematic (Yandell and Ence, 2012; Gruber and Kroth, 2014). Indeed, many studies involving diatom protein annotation have resorted to manual extension of original gene models (Kroth et al., 2008; Huesgen et al., 2013). For our optimization, all protein predictions for the two diatoms available through the Joint Genome Institute genome portal (http://www.jgi.doe.gov) were considered (over 50 000 gene models for each genome), including user-defined gene models. The T. pseudonana models were extended in both directions–upstream to the first ‘ATG’ codon and downstream to the first stop codon in the same reading frame. For both diatoms, models over 10 kb in length were assumed to be incorrect and were excluded from the dataset. The resulting protein set for each diatom was further optimized by identifying the longest gene model for a given position on the genome with an N-terminal ATG (initiator methionine codon), with no internal stop codons, with EST support, and with a C-terminal stop codon. The so-called Joint Genome Institute (JGI) ‘filtered’ gene model was selected in cases in which multiple gene models with identical sequences fulfilled these criteria. Identical sequences were removed from the optimized set of gene models. As a final step, gene models derived from unknown chromosome locations were evaluated for inclusion in the optimized set. Proteins that were <95% identical to a protein derived from a known chromosome location were added to the optimized protein datasets for both diatoms (Datasets S1 and S2).
Figure 5.

Gene catalog optimization.Source gene catalogs and procedures applied to obtain the optimized gene catalogs for Thalassiosira pseudonana (Thaps) and Phaeodactylum tricornutum (Phatr), see Datasets S1 and S2.
The resulting optimized datasets were composed of a larger set of predicted proteins than is currently available for download through the JGI genome portal (Table2 and Datasets S1 and S2). The percentage of gene models beginning with an ATG increased substantially in both genomes (from 83 to 96% in T. pseudonana and from 89 to 98% in P. tricornutum, see Table2). The entire dataset was analyzed via SignalP 3.0 NN. The number of predicted proteins with signal peptides also increased, from 12 to 22% of the total proteins in T. pseudonana and from 14 to 24% in P. tricornutum (Table2). Signal peptides, although not conserved directly on the sequence level, nevertheless have to fulfill structural requirements that are a function of their primary sequence (Patron and Waller, 2007). The increased number of predicted signal peptides in our improved gene catalog therefore indicates that the additional sequence regions represent coding sequence under actual selection pressure, as opposed to untranslated regions, that are not under selection pressure to maintain signal peptide features. We also compared the experimentally verified sequences of our reference proteins with either the optimized dataset or the original JGI dataset. In the JGI dataset, gene model translation start sites were identical to those of 77 of the 131 experimentally verified proteins used as our reference sequences. With our optimized dataset, gene model translation start sites were identical to those of 121 of the 132 reference proteins (Table S3). Together, these findings emphasize the enhanced quality of our optimized datasets and reiterate the difficulty of predicted targeting pre-sequences based solely on homology to proteins from closely related organisms.
Table 2.
Gene catalog optimization. Results of the gene catalog optimization procedure in comparison with Thalassiosira pseudonana (Thaps) and Phaeodactylum tricornutum (Phatr) genome assemblies
| Gene models total | Gene models beginning with ‘ATG’ (%) | Gene models with signal peptides (%) | |
|---|---|---|---|
| Thaps v3.0 initial releasea | 11 776 | – | 1384 (12) |
| Thaps v3.0 filtered modelsb | 11 390 | 9477 (83.2) | 2077 (18.24) |
| Thaps optimized catalogc | 13 344d | 12 756 (95.59) | 2915e (21.85) |
| Phatr v2.0 initial releasea | 10 402 | – | 1479 (14) |
| Phatr v2.0 filtered modelsb | 10 025 | 8917 (88.94) | 2070 (20.65) |
| Phatr optimized catalogc | 10 814f | 10 611 (98.12) | 2648g (24.49) |
Plastid proteome prediction
The optimized gene catalogs were used to identify nuclear-encoded plastid proteins in T. pseudonana (Table S7) and P. tricornutum (Table S8). The distribution of transit peptide scores is similar between T. pseudonana and P. tricornutum (Figure6). Both curves are characterized by a sudden decrease in the transit peptide score coinciding with the absence of a phenylalanine residue at the first position of the scored transit peptide. Our transit peptide score cut-off of two was empirically optimized for prediction performance. Analysis of this larger dataset indicates that proteins exceeding this cut-off may lack a phenylalanine residue at the first position of the transit peptide. In these instances, the other amino acids within the scoring matrix window have to contribute much more to the overall score. The scores attained by sequences from the reference set show that a cut-off of two is sufficient to separate plastid-targeted sequences from sequences targeted to other compartments (Figure6).
Figure 6.

Transit peptide score distribution.Distribution of transit peptide scores for the sequence window with the highest cleavage site score of each SignalP positive sequence in the optimized gene catalogs, sequences from the native experimental reference set (Tables S3 and S4) are highlighted in the diagram for Phaeodactylum tricornutum, see Tables S7 and S8 for raw data. BLS, ‘blob’-like structure (Kilian and Kroth, 2005); ER, endoplasmic reticulum.
The T. pseudonana optimized gene catalog contains 2915 proteins with predicted signal peptides and the P. tricornutum optimized gene catalog contains 2648 proteins with predicted signal peptides (Table2). In T. pseudonana 996 proteins are predicted to localize to the plastid with high confidence and 895 proteins are predicted to localize to the plastid in P. tricornutum with high confidence. The higher total number of genes in the T. pseudonana genome (13 344) compared with P. tricornutum (10 814) therefore is largely driven by proteins that are not targeted to the plastid (Table3). Using the ‘plastid, high confidence’ criteria, the G. theta genome contains 755 plastid proteins, at a much higher total number of genes (24 840) (Curtis et al., 2012), so also in this case the genome expansion is mainly driven by genes encoding non-plastid proteins.
Table 3.
Plastid protein prediction results. Predicted plastid proteins in the optimized gene catalogs for Thalassiosira pseudonana and Phaeodactylum tricornutum, numbers in parentheses are percentages of total number of sequences in the optimized gene catalogs, for complete prediction results see Tables S7 and S8
| Thalassiosira pseudonana | Phaeodactylum tricornutum | |
|---|---|---|
| Plastid, high confidence only | 996 (7.46) | 895 (8.28) |
| Plastid, high or low confidence | 1568 (11.75) | 1608 (14.87) |
| Not plastid, SignalP positive | 1347 (10.09) | 1040 (9.62) |
| Not plastid, SignalP negative | 10 429 (78.15) | 8166 (75.51) |
Conclusion
ASAFind combines high sensitivity with high specificity compared to previously published prediction tools (Gschloessl et al., 2008; Mernberger et al., 2014), and provides a powerful method for in silico prediction of plastid proteins in algae with secondary plastids of the red lineage. Furthermore, it allows the user to adjust predictions either in favour of sensitivity or specificity in order to enable the discovery of new plastid proteins (high sensitivity) or to validate sequences (high specificity) predicted by other approaches. We provide here the approximately 8% of proteins encoded in the nuclear genomes of diatoms (T. pseudonana and P. tricorntum) that are predicted to be plastid localized with high specificity and high confidence. This percentage is similar to predictions for the green alga Chlamydomonas reinhardtii (7% nuclear-encoded plastid proteins; Terashima et al., 2011).
Experimental procedures
Annotation of scoring matrix and reference sets
Experimental data were compiled from published studies (Liaud et al., 2000; Apt et al., 2002; Domergue et al., 2003; Kilian and Kroth, 2004, 2005; Kroth et al., 2005; Tanaka et al., 2005; Gould et al., 2006b; Gruber et al., 2007, 2009; Lepetit et al., 2007; Siaut et al., 2007; Sommer et al., 2007; Kitao et al., 2008; Ast et al., 2009; Hempel et al., 2009, 2010; Kitao and Matsuda, 2009; Weber et al., 2009; Bullmann et al., 2010; Felsner et al., 2010; Joshi-Deo et al., 2010; Allen et al., 2011, 2012; Bruckner et al., 2011; Grouneva et al., 2011; Moog et al., 2011; Tachibana et al., 2011; Vugrinec et al., 2011; Sturm et al., 2013). The gene models for the experimentally tested proteins were manually verified (Table S3). The Thalassiosira pseudonana v3.0 (Armbrust et al., 2004; Bowler et al., 2008) and Phaeodactylum tricornutum v2.0 (Bowler et al., 2008) genome databases were accessed online via the United States Department of Energy Joint Genome Institute (JGI) genome portal (http://genome.jgi-psf.org/) (Grigoriev et al., 2012) using TBLASTN and BLASTP (Altschul et al., 1997). If none of the automatically created gene models was complete, gene models were manually edited with the editing function of the JGI genome portal (Grigoriev et al., 2012). Local BLAST (Altschul et al., 1997) searches were performed using the program BioEdit (Hall, 1999).
Software and scripting techniques
For the prediction of signal peptides SignalP v3.0b (Bendtsen et al., 2004) and SignalP4.0 (Petersen et al., 2011) were installed locally on a Linux system running Ubuntu. SignalP was invoked using the ‘euk’ option and proteins were judged SignalP positive based on the NN criterion.
For statistical analyses and formatting, data were processed using Perl scripts (Strawberry Perl for Windows – 5.12.3.0, http://strawberryperl.com/). Statistical figures and the ROC plot were prepared following the methods described in (Baldi et al., 2000; Brown and Davis, 2006; Fawcett, 2006). Sequence logos (Schneider and Stephens, 1990) were prepared using the WebLogo (Crooks et al., 2004) server (http://weblogo.berkeley.edu/).
ASAFind was implemented in Python 2.7 (https://www.python.org/) using Biopython v1.63 (Cock et al. 2009) and is available as a web server at http://rocaplab.ocean.washington.edu/tools/asafind. A standalone version, which offers the option of a custom weight matrix based on sequences of user interest is also available at the same location and as Appendix S1.
Input data
Protein sequences were downloaded from the JGI genome portals for Thalassiosira pseudonana v3.0 (Armbrust et al., 2004; Bowler et al., 2008) and Phaeodactylum tricornutum v2.0 (Bowler et al., 2008) on Aug. 8, 2011 (All Models and User Models) and 5 October 2012 (unmapped models) and processed as described in the results section.
Acknowledgments
We thank Cedar McKay for assistance with the Python and web server implementations of ASAFind, Chris Berthiaume and David Schruth for the extension of T. psuedonana gene models and John M. Archibald for helpful discussions. Partial support for this work was provided by the Dalhousie University Centre for Comparative Genomics and Evolutionary Bioinformatics (AG; CGEB), the German Academic Exchange Service (TM; DAAD) the German Research Foundation (PGK; DFG SFB 969, project A4), the Gordon and Betty Moore Foundation (EVA; GBMF537 and GBMF3776), the National Science Foundation (GR; grant 0629521), the University of East Anglia (TM; UEA) and the University of Konstanz (AG and PGK). The authors declare that they have no competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Conserved transit peptides.
Cleavage site motif identification.
Initial sequence set.
Scoring matrix.
Reference set.
Prediction results for native reference sequences.
Prediction results for mutated reference sequences.
Prediction statistics.
Prediction results for Thalassiosira pseudonana.
Prediction results for Phaeodactylum tricornutum.
Optimized Thalassiosira pseudonana gene catalog.
Optimized Phaeodactylum tricornutum gene catalog.
ASAFind Python script.
References
- Allen AE, Dupont CL, Obornik M, et al. Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature. 2011;473:203–207. doi: 10.1038/nature10074. [DOI] [PubMed] [Google Scholar]
- Allen AE, Moustafa A, Montsant A, Eckert A, Kroth PG, Bowler C. Evolution and functional diversification of fructose bisphosphate aldolase genes in photosynthetic marine diatoms. Mol. Biol. Evol. 2012;29:367–379. doi: 10.1093/molbev/msr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apt KE, Zaslavkaia L, Lippmeier JC, Lang M, Kilian O, Wetherbee R, Grossman AR, Kroth PG. In vivo characterization of diatom multipartite plastid targeting signals. J. Cell Sci. 2002;115:4061–4069. doi: 10.1242/jcs.00092. [DOI] [PubMed] [Google Scholar]
- Armbrust EV, Berges JA, Bowler C, et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science. 2004;306:79–86. doi: 10.1126/science.1101156. [DOI] [PubMed] [Google Scholar]
- Ast M, Gruber A, Schmitz-Esser S, Neuhaus HE, Kroth PG, Horn M, Haferkamp I. Diatom plastids depend on nucleotide import from the cytosol. Proc. Natl Acad. Sci. USA. 2009;106:3621–3626. doi: 10.1073/pnas.0808862106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Brunak S, Chauvin Y, Andersen CA, Nielsen H. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Blum T, Briesemeister S, Kohlbacher O. MultiLoc2: integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinformatics. 2009;10:274. doi: 10.1186/1471-2105-10-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowler C, Allen AE, Badger JH, et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 2008;456:239–244. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
- Brown CD, Davis HT. Receiver operating characteristics curves and related decision measures: a tutorial. Chemometr. Intell. Lab. Syst. 2006;80:24–38. [Google Scholar]
- Bruckner CG, Rehm C, Grossart HP, Kroth PG. Growth and release of extracellular organic compounds by benthic diatoms depend on interactions with bacteria. Environ. Microbiol. 2011;13:1052–1063. doi: 10.1111/j.1462-2920.2010.02411.x. [DOI] [PubMed] [Google Scholar]
- Bullmann L, Haarmann R, Mirus O, Bredemeier R, Hempel F, Maier UG, Schleiff E. Filling the gap, evolutionarily conserved Omp85 in plastids of chromalveolates. J. Biol. Chem. 2010;285:6848–6856. doi: 10.1074/jbc.M109.074807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. Principles of protein and lipid targeting in secondary symbiogenesis: euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J. Eukaryot. Microbiol. 1999;46:347–366. doi: 10.1111/j.1550-7408.1999.tb04614.x. [DOI] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis BA, Tanifuji G, Burki F, et al. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature. 2012;492:59–65. doi: 10.1038/nature11681. [DOI] [PubMed] [Google Scholar]
- Domergue F, Spiekermann P, Lerchl J, Beckmann C, Kilian O, Kroth PG, Boland W, Zahringer U, Heinz E. New insight into Phaeodactylum tricornutum fatty acid metabolism. Cloning and functional characterization of plastidial and microsomal delta12-fatty acid desaturases. Plant Physiol. 2003;131:1648–1660. doi: 10.1104/pp.102.018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Fawcett T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006;27:861–874. [Google Scholar]
- Felsner G, Sommer MS, Maier UG. The physical and functional borders of transit peptide-like sequences in secondary endosymbionts. BMC Plant Biol. 2010;10:223. doi: 10.1186/1471-2229-10-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SB, Sommer MS, Hadfi K, Zauner S, Kroth PG, Maier UG. Protein targeting into the complex plastid of cryptophytes. J. Mol. Evol. 2006a;62:674–681. doi: 10.1007/s00239-005-0099-y. [DOI] [PubMed] [Google Scholar]
- Gould SB, Sommer MS, Kroth PG, Gile GH, Keeling PJ, Maier UG. Nucleus-to-nucleus gene transfer and protein retargeting into a remnant cytoplasm of cryptophytes and diatoms. Mol. Biol. Evol. 2006b;23:2413–2422. doi: 10.1093/molbev/msl113. [DOI] [PubMed] [Google Scholar]
- Grigoriev IV, Nordberg H, Shabalov I, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouneva I, Rokka A, Aro EM. The thylakoid membrane proteome of two marine diatoms outlines both diatom-specific and species-specific features of the photosynthetic machinery. J. Proteome Res. 2011;10:5338–5353. doi: 10.1021/pr200600f. [DOI] [PubMed] [Google Scholar]
- Gruber A, Kroth P. Plant Metabolism. New York, NY: Humana Press; 2014. Deducing intracellular distributions of metabolic pathways from genomic data; pp. 187–211. [DOI] [PubMed] [Google Scholar]
- Gruber A, Vugrinec S, Hempel F, Gould SB, Maier UG, Kroth PG. Protein targeting into complex diatom plastids: functional characterisation of a specific targeting motif. Plant Mol. Biol. 2007;64:519–530. doi: 10.1007/s11103-007-9171-x. [DOI] [PubMed] [Google Scholar]
- Gruber A, Weber T, Bartulos CR, Vugrinec S, Kroth PG. Intracellular distribution of the reductive and oxidative pentose phosphate pathways in two diatoms. J. Basic Microbiol. 2009;49:58–72. doi: 10.1002/jobm.200800339. [DOI] [PubMed] [Google Scholar]
- Gschloessl B, Guermeur Y, Cock JM. HECTAR: a method to predict subcellular targeting in heterokonts. BMC Bioinformatics. 2008;9:393. doi: 10.1186/1471-2105-9-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Hempel F, Bullmann L, Lau J, Zauner S, Maier UG. ERAD-derived preprotein transport across the second outermost plastid membrane of diatoms. Mol. Biol. Evol. 2009;26:1781–1790. doi: 10.1093/molbev/msp079. [DOI] [PubMed] [Google Scholar]
- Hempel F, Felsner G, Maier UG. New mechanistic insights into pre-protein transport across the second outermost plastid membrane of diatoms. Mol. Microbiol. 2010;76:793–801. doi: 10.1111/j.1365-2958.2010.07142.x. [DOI] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesgen PF, Alami M, Lange PF, Foster LJ, Schroder WP, Overall CM, Green BR. Proteomic amino-termini profiling reveals targeting information for protein import into complex plastids. PLoS One. 2013;8:e74483. doi: 10.1371/journal.pone.0074483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Deo J, Schmidt M, Gruber A, Weisheit W, Mittag M, Kroth PG, Buchel C. Characterization of a trimeric light-harvesting complex in the diatom Phaeodactylum tricornutum built of FcpA and FcpE proteins. J. Exp. Bot. 2010;61:3079–3087. doi: 10.1093/jxb/erq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian O, Kroth PG. Presequence acquisition during secondary endocytobiosis and the possible role of introns. J. Mol. Evol. 2004;58:712–721. doi: 10.1007/s00239-004-2593-z. [DOI] [PubMed] [Google Scholar]
- Kilian O, Kroth PG. Identification and characterization of a new conserved motif within the presequence of proteins targeted into complex diatom plastids. Plant J. 2005;41:175–183. doi: 10.1111/j.1365-313X.2004.02294.x. [DOI] [PubMed] [Google Scholar]
- Kitao Y, Matsuda Y. Formation of macromolecular complexes of carbonic anhydrases in the chloroplast of a marine diatom by the action of the C-terminal helix. Biochem. J. 2009;419:681–688. doi: 10.1042/BJ20082315. [DOI] [PubMed] [Google Scholar]
- Kitao Y, Harada H, Matsuda Y. Localization and targeting mechanisms of two chloroplastic beta-carbonic anhydrases in the marine diatom Phaeodactylum tricornutum. Physiol. Plant. 2008;133:68–77. doi: 10.1111/j.1399-3054.2008.01053.x. [DOI] [PubMed] [Google Scholar]
- Kroth PG. Protein transport into secondary plastids and the evolution of primary and secondary plastids. Int. Rev. Cytol. 2002;221:191–255. doi: 10.1016/s0074-7696(02)21013-x. [DOI] [PubMed] [Google Scholar]
- Kroth PG, Schroers Y, Kilian O. The peculiar distribution of class I and class II aldolases in diatoms and in red algae. Curr. Genet. 2005;48:389–400. doi: 10.1007/s00294-005-0033-2. [DOI] [PubMed] [Google Scholar]
- Kroth PG, Chiovitti A, Gruber A, et al. A model for carbohydrate metabolism in the diatom Phaeodactylum tricornutum deduced from comparative whole genome analysis. PLoS One. 2008;3:e1426. doi: 10.1371/journal.pone.0001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepetit B, Volke D, Szabo M, Hoffmann R, Garab G, Wilhelm C, Goss R. Spectroscopic and molecular characterization of the oligomeric antenna of the diatom Phaeodactylum tricornutum. Biochemistry. 2007;46:9813–9822. doi: 10.1021/bi7008344. [DOI] [PubMed] [Google Scholar]
- Liaud MF, Lichtle C, Apt K, Martin W, Cerff R. Compartment-specific isoforms of TPI and GAPDH are imported into diatom mitochondria as a fusion protein: evidence in favor of a mitochondrial origin of the eukaryotic glycolytic pathway. Mol. Biol. Evol. 2000;17:213–223. doi: 10.1093/oxfordjournals.molbev.a026301. [DOI] [PubMed] [Google Scholar]
- Mernberger M, Moog D, Stork S, Zauner S, Maier UG, Hullermeier E. Protein sub-cellular localization prediction for special compartments via optimized time series distances. J. Bioinform. Comput. Biol. 2014;12:1350016. doi: 10.1142/S0219720013500169. [DOI] [PubMed] [Google Scholar]
- Moog D, Stork S, Zauner S, Maier UG. In silico and in vivo investigations of proteins of a minimized eukaryotic cytoplasm. Genome Biol. Evol. 2011;3:375–382. doi: 10.1093/gbe/evr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Krogh A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc. Int. Conf. Intell Syst. Mol. Biol. 1998;6:122–130. [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Patron NJ, Waller RF. Transit peptide diversity and divergence: a global analysis of plastid targeting signals. BioEssays. 2007;29:1048–1058. doi: 10.1002/bies.20638. [DOI] [PubMed] [Google Scholar]
- Patron NJ, Waller RF, Archibald JM, Keeling PJ. Complex protein targeting to dinoflagellate plastids. J. Mol. Biol. 2005;348:1015–1024. doi: 10.1016/j.jmb.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Petersen T, Brunak S, von Heijne G, Nielsen H. Signal1 4.01 discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siaut M, Heijde M, Mangogna M, Montsant A, Coesel S, Allen A, Manfredonia A, Falciatore A, Bowler C. Molecular toolbox for studying diatom biology in Phaeodactylum tricornutum. Gene. 2007;406:23–35. doi: 10.1016/j.gene.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Sommer MS, Gould SB, Lehmann P, Gruber A, Przyborski JM, Maier U-G. Der1-mediated preprotein import into the periplastid compartment of Chromalveolates? Mol. Biol. Evol. 2007;24:918–928. doi: 10.1093/molbev/msm008. [DOI] [PubMed] [Google Scholar]
- Sturm S, Engelken J, Gruber A, Vugrinec S, Kroth PG, Adamska I, Lavaud J. A novel type of light-harvesting antenna protein of red algal origin in algae with secondary plastids. BMC Evol. Biol. 2013;13:159. doi: 10.1186/1471-2148-13-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Allen AE, Kikutani S, Endo Y, Bowler C, Matsuda Y. Localization of putative carbonic anhydrases in two marine diatoms, Phaeodactylum tricornutum and Thalassiosira pseudonana. Photosynth. Res. 2011;109:205–221. doi: 10.1007/s11120-011-9634-4. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Nakatsuma D, Harada H, Ishida M, Matsuda Y. Localization of soluble beta-carbonic anhydrase in the marine diatom Phaeodactylum tricornutum. Sorting to the chloroplast and cluster formation on the girdle lamellae. Plant Physiol. 2005;138:207–217. doi: 10.1104/pp.104.058982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima M, Specht M, Hippler M. The chloroplast proteome: a survey from the Chlamydomonas reinhardtii perspective with a focus on distinctive features. Curr. Genet. 2011;57:151–168. doi: 10.1007/s00294-011-0339-1. [DOI] [PubMed] [Google Scholar]
- Timmis JN, Ayliffe MA, Huang CY, Martin W. Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004;5:123–135. doi: 10.1038/nrg1271. [DOI] [PubMed] [Google Scholar]
- Vugrinec S, Gruber A, Kroth PG. Protein targeting into complex plastids - support for the translocator model. Endocytobiosis Cell Res. 2011;21:59–63. [Google Scholar]
- Weber T, Gruber A, Kroth PG. The presence and localization of thioredoxins in diatoms, unicellular algae of secondary endosymbiotic origin. Mol. Plant. 2009;2:468–477. doi: 10.1093/mp/ssp010. [DOI] [PubMed] [Google Scholar]
- Yandell M, Ence D. A beginner's guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012;13:329–342. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Conserved transit peptides.
Cleavage site motif identification.
Initial sequence set.
Scoring matrix.
Reference set.
Prediction results for native reference sequences.
Prediction results for mutated reference sequences.
Prediction statistics.
Prediction results for Thalassiosira pseudonana.
Prediction results for Phaeodactylum tricornutum.
Optimized Thalassiosira pseudonana gene catalog.
Optimized Phaeodactylum tricornutum gene catalog.
ASAFind Python script.