

Abstract
Patient-derived pluripotent stem cells (PSC) directed to various cell fates holds promise as source material for treating numerous disorders. The availability of precisely differentiated PSC-derived cells will dramatically impact blood component and hematopoietic stem cell therapies, and should facilitate treatment of diabetes, some forms of liver disease and neurologic disorders, retinal diseases, and possibly heart disease. Although an unlimited supply of specific cell types are needed, other barriers must be overcome. This review of the state of cell therapies highlights important challenges. Successful cell transplantation will require optimizing the best cell type and site for engraftment, overcoming limitations to cell migration and tissue integration, and occasionally needing to control immunologic reactivity. Collaboration among scientists, clinicians, and industry is critical for generating new stem cell-based therapies.
Induced pluripotent stem cells (PSC) are generated by reprogramming somatic cells to a pluripotent state by transient expression of pluripotency factors. These cells can self-renew indefinitely and are able to differentiate into any cell lineage (1, 2). The ability to generate PSC from individual patients and differentiate them into an unlimited supply of tissue and organ-specific cells capable of circumventing immunologic rejection following transplantation, could facilitate development of cell-based therapies for the treatment of a variety of debilitating disorders and dramatically change the practice of medicine.
Before these cells can be used in the clinic, a variety of barriers must be overcome. For many diseases it is not yet possible to differentiate PSCs to cells with characteristics identical to those in the organs that need replacement. There are also challenges like scaling up production, eliminating cells with tumor-forming potential, and decreasing the time needed for expansion, differentiation, selection, and testing. Furthermore, treatment of a genetic mutation using autologous cells will often require genetic manipulation, which might result in changes that could increase cancer risk.
Some form of immune suppression may also be required to control cell loss after transplantation, whether due to rejection, an immune response to a genetically corrected protein, or recurrence of autoimmunity, with destruction of the transplant, as might be the case for diabetes. The standard signs of rejection used in solid organ transplantation are not likely to be useful since the sensitivity of functional changes has been shown, following islet transplantation, to be inadequate to diagnose rejection before damage to the engrafted cells is irreversible (3). Of course it might be possible to engineer PSC-derived grafts, with the usual caveats concerning activating oncogenes, so that they would be immunologically inert, and identifiable by an array of imaging strategies.
Although decades of laboratory and clinical investigation have led to successful therapies using hematopoietic cells, few other cell therapies have transitioned from experimental to standard clinical care. Here we discuss the present state of cell therapy in the context of having available differentiated PSC-derived cells. The “gold standard”, blood and hematopoietic stem cell (HSC) transplantation, is highlighted first, followed by an examination of cell therapy for diabetes, liver disease, neurologic and retinal disorders, muscular dystrophies, and heart disease.
Hematopoietic cell based therapies
Many of the principles of cell transplantation derive from our long experience with transfusion of blood products. Infused red blood cells (RBCs), platelets, and HSC are the most widely employed cellular therapies in use today. The relative ease of transfusion and HSC transplantation (HSCT) derives in large part from the intrinsic potential of blood cells to home to and integrate into native niches, give rise to differentiated progeny, and to thereafter egress into the circulation. Thus, HSCT avoids the challenges of restoring integrity and function of more anatomically complex organs like the lung, heart, liver, and brain.
Despite the successes of blood transfusions, isolated hematopoietic stem cells cannot be expanded to the degree needed, and there is limited success with cord blood. In order to eliminate the costly and sometimes unreliable system of volunteer blood supply as well as the risk of transmission of infectious agents, a reliable method for generating an inexhaustible, uniform supply of pathogen-free blood products has tremendous appeal. Furthermore, allogeneic HSCT is associated with considerable treatment-related morbidity and mortality. Thus, transplantation with autologous HSC for the same indications would eliminate the major morbidities of immune mismatch and could potentially expand the range of conditions, including cancers, amenable to HSCT. One of the most promising applications of somatic cell reprogramming is the production of customized pluripotent stem cells followed by gene correction (4), differentiation into HSCs, and auto-transplant with intention to cure any one of dozens of inherited genetic disorders of the blood forming system. Such a proof-of-principle has been achieved for treating murine models of severe combined immune deficiency and sickle cell anemia (4, 5). HSC have been derived from murine embryonic stem cells that manifest the cardinal features of clonal self-renewal and multi-lineage lymphoid-myeloid engraftment in primary and secondary irradiated hosts (6, 7). The derivation of HSC from human PSC has proven elusive, although several examples of low level engraftment have been reported(8–10). Although true HSCs are not yet available, methods exist to produce RBCs (11, 12) and platelets (13) in vitro that are suitable for transfusion. Ultimately, advances in in vitro cell manufacture should soon be able to reduce costs and enable an off-the-shelf supply.
Diabetes
One of several approaches to resolving the long-term complications associated with diabetes has been beta cell replacement by allogeneic whole pancreas or isolated pancreatic islet transplantation, using immune suppression in an attempt to control rejection and recurrent autoimmune destruction of the transplanted tissue. Cadaver pancreas transplantation has been shown to reduce the complications associated with type 1 diabetes (T1D). However, it is a complex surgical procedure associated with significant morbidity and measureable mortality(14). An important lesson from this experience, however, is that conventional immune suppression is able to inhibit recurrent destruction of insulin-producing cells while controlling allograft rejection.
After years of laboratory and clinical investigation, islet transplantation has become a realistic, alternative therapy, which, over the last several decades has become increasingly more successful(15–17). Experience with autologous islet transplantation following total or near-total pancreatic resection for severe chronic pancreatitis and allogeneic islet transplantation for T1D have both been instructive(18). The preferred site for implantation is the liver, and engraftment is accomplished by minimally invasive transcutaneous catheter infusion through the liver into the portal vein. This approach is associated with a small but measureable risk of hemorrhage and partial portal vein thrombosis. More importantly, an Immediate Blood Mediated Inflammatory Response (IBMIR) and pathologic activation of the coagulation system results in a large loss of the infused islets as soon as they come into direct contact with blood(19). As the yield of islets from a chronically injured pancreas is already reduced, this constitutes a limiting factor in trying to reach an islet mass sufficient for insulin independence following autologous islet transplantation. It is also a serious issue following islet allo-transplantation, where success often requires multiple infusions from different donors and transplantation of a much greater islet mass to achieve insulin independence. Although it is not completely understood why so many more islets are required for initial success in allo- than for auto-transplants, this barrier to successful insulin replacement by cell therapy should be resolvable with an inexhaustible supply of donor beta cells.
Failure to achieve long-term insulin independence is more problematic. Five-year insulin independence rates following islet allo-transplantation in selected centers are approaching 50% (17, 20). These improved outcomes have resulted from the transplantation of a larger islet mass and more effective control of rejection and autoimmunity, with late graft loss being attributed to toxicity to beta cells arising from medications that suppress immunity and an inability to diagnose or treat recurrent disease or rejection. Advances in donor cell imaging by genetic engineering of PSCs and the development of new strategies for controlling the immune response should resolve some of these issues.
However, experience with islet autotransplantation indicates a more serious problem for long-term function of engrafted islet cells, independent of the need to control rejection or recurrent autoimmune destruction(21). Although short-term insulin independence has been accomplished, almost all patients are back on insulin therapy after 5 years. This loss of graft function may be attributable to chronic stimulation of an initially marginal intrahepatic beta cell mass that produces metabolic deterioration and loss of beta cells(18). Transplantation of a larger mass of islets may alleviate this problem and result in indefinite graft function. However, the site of engraftment in the liver may also be responsible for poor long-term survival. Islets ectopically engrafted in the liver are known to produce a number of pathologic histologic abnormalities, including extensive amyloid deposition within the islets(22). Thus, it remains unclear whether whole islets can remain functionally intact in the liver over time.
The use of PSC-derived beta cells could resolve many of the above issues. Beta cells that have been dissociated and isolated from intact islets successfully engraft within the liver lobule. This is in contrast to how intact islets engraft, which is by partially remaining within the portal circulation (Figure 1) and requiring neovascularization. Isolated cells may not be as susceptible to forming amyloid deposits, and transplantation of a large mass of individual beta cells might avoid metabolic exhaustion and apoptosis. Dissociated cells do not, however, engraft when infused as individual beta cells but require re-aggregation(23). In addition, individual beta cells function less well when isolated. It has been reported that 30-fold more insulin is released from beta cells within intact islets as compared to that released from purified single beta cells, and re-aggregated beta cells and beta cells aggregated with alpha cells respond up to 4-fold better to glucose challenge than do single beta cells alone(24). Thus, to treat T1D, it may be necessary to transplant PSC-derived beta cells as aggregates or possibly to transplant them with PSC-derived non-beta cells.
Figure 1.
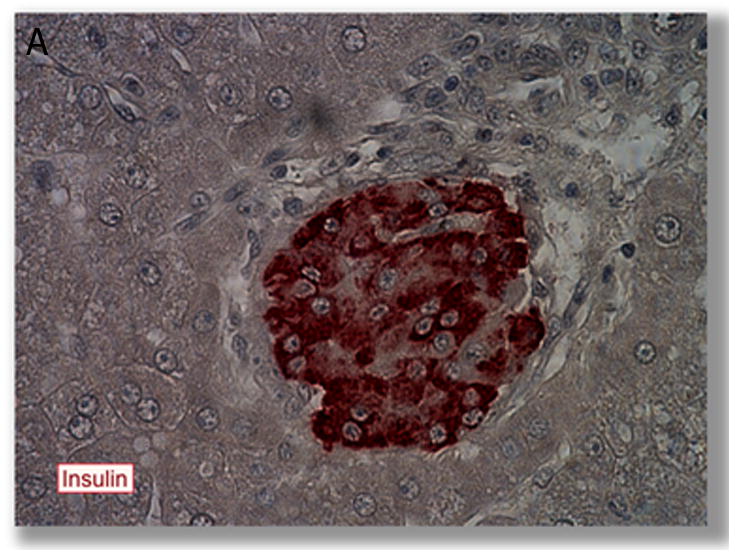
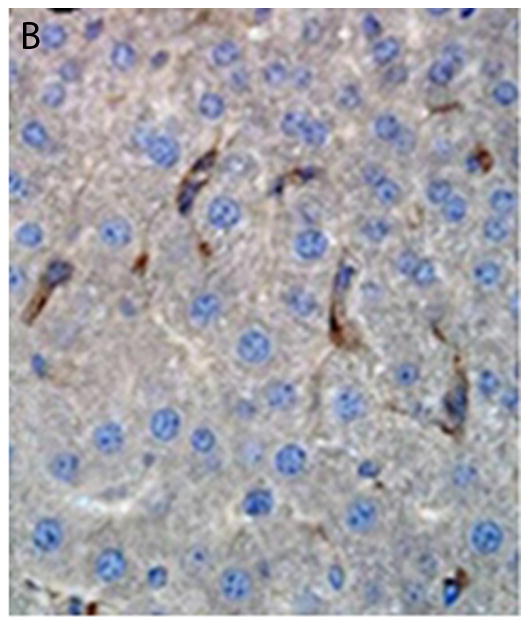
Comparison of intrahepatic engraftment of intact islets versus isolated beta cells following intraportal transplantation. A. Transplanted islet (in brown) within the capillary of the liver following intraportal transplantation. From (28) with permission. B iPS-derived beta cells (in brown) engraft as cells scattered throughout the liver parenchyma, from (150), with permission
Whether PSC–derived surrogate beta cells will have the same capacity to engraft in the liver like primary beta cells is not known. Because they are small, they might pass through the liver into the lungs, as has been described following hepatocyte transplantation. Other sites of transplantation, like the gastric sub-mucosal space, might offer some advantages over the portal vein (Figure 2). Surrogate beta cells could be implanted in the gastic sub-mucosal space endoscopically where they would be accessible for biopsy to monitor the graft status(25).
Figure 2.
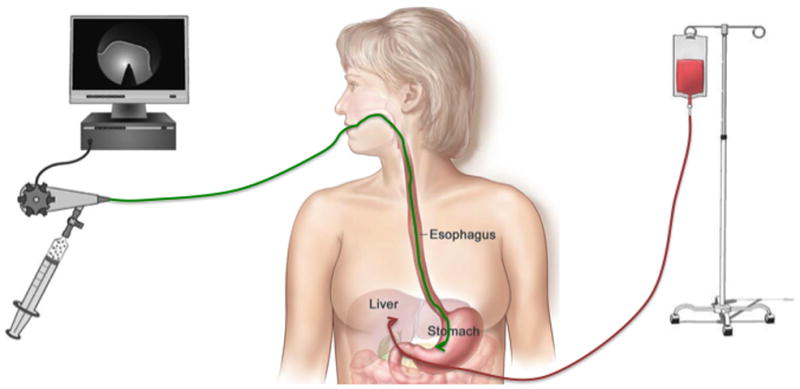
Illustration of traditional portal vein infusion of insulin producing cells for engraftment in the liver versus endoscopic placement of transplanted cells in the gastric sub-mucosal space.
Without concomitant use of immune suppression, autoimmunity will mediate destruction of grafts composed of even autologous cells(26). Whether autoimmunity can be suppressed in a way that is less detrimental to the insulin-producing cells than conventional drug-based allogeneic protection is as yet unknown. Strategies for potentially controlling this process by inducing tolerance are in development(27). In order to modulate immunologically mediated problems, additional transgenes might help confer long-term function and reduce early loss in diabetic recipients. Candidates for this approach are like the ones targeting the tissue factor and the coagulation cascade, or exerting anti-inflammatory activity that have already been tested in cloned pigs(28). An alternative approach is to encapsulate the PSC-derived insulin-producing surrogates, providing a protective barrier from immune rejection and autoimmunity while allowing free exchange of nutrients, waste, and most importantly, insulin and glucose. Unfortunately, thus far these approaches have shown mixed results at least following transplantation of intact islets in diabetic patients, with low levels of circulating C-peptide, and rarely achieving full and lasting insulin independence(29). A variation on this encapsulation and transplant strategy, using PSC-derived beta cells, will soon be tested in clinical trials for the treatment of T1D (30).
Liver Disease
Hepatocyte transplantation holds great promise as a therapy for individuals with life-threatening liver diseases, where organ transplantation is often the only available treatment option. Patients with both acute liver failure and liver-based inborn errors of metabolism, leading to life-threatening extrahepatic complications, are ideal candidates for cell therapy. Numerous studies in rodents have shown that hepatocyte transplantation can reverse acute fulminant hepatic failure (31) and correct liver-based metabolic deficiencies(32–37). Since the native architecture of the liver is intact in these diseases, the transplant procedure involves simple injection of hepatocytes through the portal vein into the liver, where the cells integrate into the host liver and are indistinguishable from the native liver cells (38, 39). Infusion of hepatocytes is a minimally invasive procedure so it can be performed on severely ill patients with relatively low risk. Since the native liver is not removed, the transplanted hepatocytes only need to improve liver function enough to stabilize an acute liver failure patient until their own liver is able to regenerate, or to replace the enzyme deficiency that is missing in liver-based metabolic disorders, a goal similar to that of gene therapy.
Clinical trials of hepatocyte transplantation have only demonstrated the long-term safety of the procedure(40–46). Transplanted hepatocytes have not restored liver function enough to circumvent the need for organ replacement in patients with liver failure, and transplantation has only resulted in partial correction of metabolic disorders (47). Efficacy has been limited by relatively poor initial and long-term engraftment, and an inability to monitor graft function in real time, which makes diagnosing and treating rejection nearly impossible. In acute liver failure, the severity of liver dysfunction requires that the transplanted hepatocytes function immediately, and the lack of a clinically relevant disease model means that the number of cells that need to engraft to reverse hepatic failure is essentially unknown(48, 49). Although animal models of metabolic liver disease recapitulate the human processes better, achieving an adequate level of engraftment is still a problem since the number of donor cells that can be safely transplanted into the liver at any one time is small, usually less than 1% of the liver mass (50). Transplantation of a larger number of cells can lead to severe portal hypertension and translocation of cells into the systemic circulation with embolization to the lungs. Liver-directed radiation has been proposed as a way to facilitate repopulation of the native liver by transplanted hepatocytes, whose viability is relatively short once isolated(51). Preparative radiation inhibits host hepatocyte proliferation and induces post-mitotic hepatocyte death, allowing donor hepatocytes to preferentially proliferate and repopulate the irradiated host liver. This strategy has been employed to completely correct a rodent model of Crigler-Najjar syndrome (52).
An immediately available, inexhaustible supply of functioning donor hepatocytes would allow early intervention in patients with hepatic failure and would allow hepatocytes to be infused over a longer period of time. It is possible that daily large scale portal vein PSC-derived hepatocyte infusions could provide the hepatocyte mass necessary to normalize the encephalopathy, coagulation defects and other life-threatening consequences of hepatic failure, and completely correct liver-based enzyme deficiencies. In addition, unrestricted availability of donor hepatocytes could allow treatment of patients with less severe, but debilitating, liver-based metabolic disorders, which are not now considered candidates for organ transplantation, such as phenylketonuria and partial urea cycle disorders. Whether acute hepatic failure is associated with changes in the local microenvironment that might interfere with engraftment and function of transplanted hepatocytes is not yet known and will need to be addressed with further clinical experience.
Although PSC-derived hepatocytes may help advance cell therapies for acute liver failure and metabolic liver diseases, the vast majority of patients who are in need of lifesaving intervention are patients with end-stage cirrhosis and chronic hepatic failure. Following infusion through the portal vein, hepatocytes have a difficult time entering the hepatic cords through the pathologically expanded extracellular matrix (Figure 3) present in advanced cirrhosis(53). As a result, transplantation by this route generates severe portal hypertension and may produce portal thrombosis. Animal studies suggest that transplantation by direct injection into an extrahepatic site, such as the spleen, can circumvent this engraftment difficulty, improve liver function, and prolong survival in end-stage cirrhosis(43, 54, 55). Even then, however, transplanted hepatocytes provide function for only a period of months. Since it’s not clear that transplanted cells can function for any sustained period of time in the abnormal environment produced by hepatic failure and portal hypertension, the same fate may await hepatocytes transplanted into other extra-anatomic locations, such as the lymph node (56), although diversion of the portal circulation may be able to enhance survival (55, 57). For a variety of reasons, anecdotal reports of hepatocyte transplantation in humans with end-stage cirrhosis has not produced even this level of success (58). Treatment of chronic liver failure might benefit from transplantation of a large mass of hepatocytes into a de-cellularized human or animal liver scaffold (59). When repopulated with donor hepatocytes and non-parenchymal cells, the biohybrid graft might then be vascularized through the portal circulation as an engineered internal auxiliary liver graft. Since this strategy would leave the native cirrhotic liver in place, it would still leave unresolved the management of coexisting portal hypertension and the risk of developing hepatocellular carcinoma in the native liver.
Figure 3.

Normal control liver (behind) and liver with advanced cirrhosis (in front), highlighting the structural differences that make liver failure from cirrhosis difficult to treat by intrahepatic cell therapy. Images courtesey of Dr. Rodney Markin, University of Nebraska Medical Center.
Neurologic and retinal diseases
The brain is arguably the most difficult of organs in which to employ stem cell-based therapeutics; the myriad connections of its neurons and their complex interdependency with macroglia, including astrocytes, oligodendrocytes, and glial progenitor cells, defy precise structural reconstitution. Neurodegenerative disorders, in particular, include diseases of both single and multiple phenotypes, the heterogeneity of which can dictate how amenable each might be to cell-based therapeutics. Some prototypic degenerative dementias, such as Alzheimer’s disease and Lewy body disease, involve a multitude of neuronal phenotypes – and in some cases glial as well, in multisystem atrophy. These multi-phenotypic disorders span anatomic and functional domains, and may exhibit both contiguous and trans-synaptic patterns of spread. For these reasons, they remain poor targets for neuronal replacement strategies at least for the near future.
However, many diseases of the brain involve single cell types, and these conditions lend themselves to phenotype-specific cell replacement, whether by transplantation or by the induction of endogenous neural stem or progenitor cells (60) (Figure 4). Degenerative disorders in which the loss of single phenotypes predominate, especially those in which a single region is differentially affected - such as Parkinson’s disease (PD), in which nigrostriatal neurons are lost before other neurons, and Huntington’s disease (HD), in which medium spiny neuronal loss and striatal atrophy become apparent long before the onset of cortical neuronal loss - have proven more amenable to phenotype-specific cell replacement (61, 62). Memory disorders, which can involve loss of the basal forebrain cholinergic neurons projecting to the hippocampus, have responded to PSC-derived cholinergic neuronal replacement in rodents (63). Yet the memory loss of early Alzheimer’s and frontotemporal dementia typically heralds deterioration across all cognitive modalities; thus, isolated replacement of cholinergic neurons is likely to prove effective in only very selected cases of memory loss – which remain difficult to identify at onset. Nonetheless, for each of these subcortical neuronal phenotypes, animal studies have proven sufficiently promising to justify both the production of cells appropriate for clinical transplantation, and the design of clinical trials by which to evaluate their safety and efficacy.
Figure 4.

Schematic representation of how PSC-derived glial and neural cells populations might be employed to treat a vast array of neurologic and retinal diseases.
Clinical trials of cell transplantation have already been performed in PD and HD (61, 62). These trials used fetal tissues dissected from the regions of interest, so that the specific cell types needed comprised but a fraction of the cells delivered. Perhaps as a result, these early trials yielded largely disappointing results with considerable variability in outcome and frequent untoward side effects, the most prominent being refractory dyskinesias following fetal mesencephalic tissue grafts in PD. Nonetheless, some transplanted PD patients did experience durable benefit, providing at least a proof-of-principal in favor of dopaminergic neuronal transplantation. The generation of midbrain dopaminergic neurons (64, 65) and medium spiny neurons (66, 67) from human PSCs (64–67) thus permit a leap forward in the availability of well-defined engraftable neurons of relative phenotypic homogeneity. This advance should enable a new generation of therapeutic trials less compromised by donor variability and compositional heterogeneity.
Some disorders affect single neuronal types, but are so dispersed throughout the CNS as to present problems of delivery. Several primary epilepsies may derive from deficits in GABAergic interneuronal numbers and function. A number of groups have asked whether transplantation of healthy migration-competent interneurons into epileptic cortex may provide benefit to medication-refractory epileptics. The production of GABAergic interneurons from PSCs (68) has permitted assessment of this promising approach to epileptic therapy. That said, GABAergic neurons comprise a plethora of functionally distinct phenotypes, thus rendering the effects of their transplantation on host neural circuits unpredictable.
Phenotype-specific disorders of the eye may prove more straightforward targets for cell therapy. Loss of the retinal pigment epithelium (RPE) in age-related macular degeneration has been a particular target of interest in that efficient protocols for generating RPE cells from PSCs have been developed (69, 70). PSC-derived RPEs are now in trials for macular degeneration. As more efficient protocols are developed for producing specific retinal phenotypes from PSCs, a broad variety of both intrinsic retinal disorders and optic neuropathies may prove appropriate targets for cell replacement.
Other disorders of single phenotype are not attractive as targets for cell-based therapy due to their multicentric pathology, the non-migratory nature of potential replacement cells, or both. The motor neuronopathies, such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy, are examples. Spinal motor neurons may be generated from PSCs (71–73), and yet their clinical utility has been limited by the multi-segmental nature of motor neuron loss in these diseases, and by our current inability to direct long-distance axonal regrowth and target reinnervation. Cell-based treatment approaches for ALS have thus shifted away from neuronal replacement, towards the delivery of astrocytes, with the goal of correcting underlying glial metabolic deficiencies that may contribute to disease progression in ALS (74). If successful, such studies may herald the use of PSC-derived glia for ALS and related disorders, in which neurons may be the paracrine victims of glial dysfunction.
Diseases of glia may prove especially accessible targets for cell-based therapy. The myelin diseases, which involve the loss or dysfunction of oligodendrocytes, are among the most prevalent and disabling conditions in neurology, and may be particularly appropriate targets for replacement (75). They include multiple sclerosis and white matter stroke, the cerebral palsies, and the hereditary pediatric leukodystrophies. As a result, oligodendrocyte progenitor cells (OPCs), which can give rise to myelinogenic oligodendrocytes, have become of great interest as potential therapeutic agents (76–78). Recently, transplantation of OPCs derived from human PSCs rescued otherwise lethally-hypomyelinated shiverer mice (79). As such, one might expect that efforts underway using tissue-derived cells to treat myelin disorders, such as childhood Pelizaeus-Merzbacher disease (80), and adult progressive multiple sclerosis, may be supplanted by PSC-derived OPCs.
Since OPCs produce astrocytes as well as oligodendrocytes, and are highly migratory, they might prove useful in rectifying the demyelination-associated enzymatic deficiencies of the lysosomal storage disorders, such as Krabbe disease, metachromatic leukodystrophy and Tay-Sachs disease, among others, as well as the astrocytic pathology of vanishing white matter disease(81). In particular, TALEN and CRISPR/Cas gene editing technologies (82) may allow correction of mutations in PSCs, thus allowing autologous and allogeneic therapy to be assessed across the entire range of hereditary leukodystrophies.
Their promise notwithstanding, introducing PSC-derived cells into the postnatal and mature CNS has its own set of risks, that include immune rejection, the induction of neuroepithelial neoplasms and teratomas(65, 80, 83), heterotopic neuronal differentiation and epileptogenesis, and the mass effect that may accompany exuberant cell expansion. Neural stem cells have been reported to have escaped to the ventricular system and subarachnoid space, adventitious growths that may present a risk of hydrocephalus, syringomyelia, or surface venous compression, as well as disruption of CSF flow and waste clearance. The list goes on, and serves to highlight the degree to which we must always be concerned that any cell therapeutic not trigger the law of unintended consequences.
Muscular Dystrophies
Duchenne Muscular Dystrophy (DMD) is a devastating, heritable X-linked muscle disease characterized by progressive muscle weakness due to the lack of dystrophin expression at the sarcolemma of muscle fibers (84–87). Although various approaches have been investigated for delivering dystrophin to dystrophic muscle, there is still no effective treatment that alleviates progression of the disease. Satellite cells play a key role in development of skeletal muscle during embryogenesis and in regeneration of muscle fibers during postnatal life. Upon isolation and culturing, satellite cells re-enter the cell cycle as myoblasts and eventually fuse to form myotubes in vitro (88). Transplantation of normal myoblasts into dystrophin-deficient muscle can create a reservoir of normal myoblasts capable of fusing with one another and with dystrophic muscle fibers, restoring dystrophin to the muscle(89–97).
Myoblast transplantation in animal models and DMD patients can transiently deliver dystrophin and improve the strength of injected dystrophic muscle, but this approach is hindered by poor cell survival, and limited migration of injected cells from the original injection site (89–97). Researchers have used preparative irradiation or injection of myonecrotic agents into the transplantation site to improve efficiency (89–98), and although these approaches have led to an improvement in restoration of dystrophin in mdx mice, success remains limited.
Many scientists consider satellite cells to be the only myogenic cells responsible for muscle growth, regeneration, and repair; however, reports suggest that other cell populations may be able to support muscle regeneration, making them a potential alternative source of cells(99–103). Cells from bone marrow(101, 103), blood vessels(100, 104–106), the neuronal compartment(99, 102), and connective tissues can differentiate toward a myogenic lineage, suggesting that transplantation of these non-satellite cell populations could be effective in improving muscle regeneration (107). Animal studies have shown that many have a very limited capacity to enhance muscle regeneration after transplantation; however, mesoangioblasts, pericytes, and muscle derived stem cells (MDSCs) survive post-implantation (Figure 5) and repair skeletal muscle after injury and disease(96, 100, 104–106, 108). As a result, their human counterparts have been isolated and are undergoing clinical trials for the treatment of stress urinary incontinence (108, 109).
Figure 5.

Importance of cell type for restoration of dystrophin expressing myofibers. Engraftment efficiency for muscle-derived stem cells (left) and myoblasts (right) following transplant.
Most cell therapies for treating DMD employ an intramuscular route of administration; however, systemic delivery would be a far superior method since it is the heart, intercostal muscles, and diaphragm that are involved in the early death of DMD patients and are almost impossible to reach by intramuscular injection. Systemic delivery of stem cells can achieve widespread delivery and leads to dystrophin expression in various muscle groups, although with varying levels of success (110). This technology is still under development and is technically challenging, especially when using small animal models(110, 111).
Although most cell transplant studies for DMD have focused on the use of post-natal stem cells, the progeny of PSCs may eventually be useful for muscle regeneration and repair. A number of stem cell lineages can be easily generated in vitro, but differentiation into skeletal muscle has proven to be difficult(112–116). PSCs transfected with myogenic regulatory factors, in particular Pax3 and Pax7, undergo partial myogenic differentiation and participate in skeletal muscle regeneration(117, 118), whereas MyoD and Myf5 have also been used to produce myoblasts from normal and dystrophic human PSCs (119). Intramuscular transplantation of human skeletal myogenic progenitors derived from PSCs results in durable engraftment, contributing to the satellite cell pool (120). Furthermore, PSCs derived from patients with muscular dystrophies, differentiated into satellite and mesoangioblast-like cells, have been genetically corrected and, following re-introduced into dystrophic mice, ameliorate their dystrophic phenotype (121, 122). Although PSCs have the potential to create an inexhaustible source of therapeutic cells for muscle repair; the availability of large numbers of muscle progenitor cells has not been a major limitation. Poor cell survival and the limited ability of the cells to migrate from their initial site of injection remain the two major hurdles to muscle cell-based therapies, even using PSC-derived cell populations.
Finally, despite the lack of dystrophin at birth, the initiation of acute, severe muscle weakness does not occur in DMD patients until they have reached the age of 4–8 years, which coincides with gradual exhaustion of their muscle progenitor cells (MPCs). Impairment in the myogenic potential of the MPCs isolated from DMD muscle in various animal models has been described (123–125), and recent studies indicate that sparing of the extra-ocular muscles in DMD patients may be related to the existence of a subpopulation of MPCs that that do not become exhausted with age, indicating that muscle weakness in DMD patients is related to MPC exhaustion during disease progression (126). These findings, taken together, suggest that a relationship likely exists between the rapid progression of muscular dystrophy and stem cell exhaustion, and the development of cell therapeutic approaches to delay the exhaustion of MPCs may represent an important alternative avenue to explore for delaying the onset of pathologies associated with muscular dystrophies.
Heart Disease
Heart disease is the most common cause of death worldwide. Because disease can result in the replacement of contractile cardiomyocytes (CMs) with scar tissue, cellular and regenerative therapies hold great promise. A wide variety of cell types have been investigated for their ability to repair the heart. These include skeletal myoblasts, bone marrow-derived cells, cardiac stem cells, and mesenchymal stem cells (127). Most effort has focused on treating ischemic heart disease by infusing cells intravascularly (intravenous, intracoronary, or retrograde coronary sinus), by intramyocardial injection (transendocardial catheter-based injection or epicardial injection), or by scaffold or patch-based epicardial delivery to the myocardium (127).
When skeletal myoblasts are transplanted by direct epicardial injection into the heart, they form stable skeletal muscle grafts that do not couple to the native cardiac muscle (128), and clinical experience in the setting of coronary artery bypass surgery has not demonstrated significant benefit (129). Initial studies in a post-myocardial infarction (MI) mouse model showed that injection of bone marrow-derived c-kit+ and lineage negative (lin−) cells dramatically regenerated myocardium and improved cardiac function (130). Subsequent studies, however, failed to demonstrate myocardial regeneration using this approach (131, 132). Nevertheless, clinical trials evaluating the ability of bone marrow mononuclear cells (BMNCs) to repair the myocardium were initiated, and phase 1 trials of intracoronary BMNC delivery via the re-perfused infarct-related artery hinted at efficacy. Larger phase 2 trials, however, have produced mixed results, ranging from no effect to a modest improvement in ejection fraction (133). In small phase 1 trials, mesenchymal stem cells, autologous cardiac stem cells, and cardiosphere-derived cells have similarly shown possible benefit with respect to left ventricular structure and function in patients post-MI with ischemic cardiomyopathy (133).
Cell survival and engraftment have been major limitations with adult stem cell sources, and the vast majority of transplanted cells are lost within days in most investigations (134). Thus, robust myocardial regeneration has not been observed, and the beneficial effects on cardiac function and structure have been attributed to other mechanisms, largely acting by paracrine signaling to preserve the border-zone around the infarction, reduce apoptosis, blunt adverse remodeling, potentially stimulate endogenous stem cells, modulate inflammation, and promote angiogenesis.
PSCs can differentiate into a range of cell types relevant for cardiac repair including CMs (135), cardiac progenitors (136, 137), endothelial cells (EC) (138), and smooth muscle cells (SMC) (138), which have been tested in small animal models. Protocols for efficient generation of CMs from human PSCs have been developed (139), and purification of CMs has been accomplished using cell surface markers, or glucose-free culture conditions optimized for survival of CMs (140, 141). However, PSC-derived CMs are, in general, a mixture of nodal-like, atrial-like and ventricular-like cells (142) that have limited proliferative capacity, and have a relatively immature phenotype based on electrophysiological characteristics, contractile performance, and metabolic profile. Despite these limitations, mouse PSC-derived CMs are capable of integration into the mouse myocardium, but this is an extremely rare event (113). Survival of transplanted PSC-derived CMs has been a limitation, but by modulating multiple cellular processes a pro-survival cocktail was developed, which enabled the survival of human PSC-derived CMs as islands in athymic rat hearts post-MI, improving cardiac function (143). Transplanted mouse PSC-derived cardiac progenitors improve the function of infarcted mouse hearts (144), and, in guinea pigs, where the heart exhibits an intrinsic heart rate closer to that in man, human PSC-derived CMs engraft and functionally couple to host CMs to improve left ventricular function and reduce ventricular arrhythmias (145).
Preclinical studies are beginning to test PSC cell therapy in large animal models of heart disease. Epicardial delivery of cell sheets composed of PSC-derived CMs in post-MI pig hearts has shown encouraging results (146), and epicardial application of human PSC-derived ECs and SMCs, incorporated into a fibrin based patch, post-MI to the hearts of immune suppressed pigs has reduced infarction size and improved left ventricular function (Figure 6) (147). In addition, direct intramyocardial injection of human PSC-derived CMs in immune suppressed non-human primate hearts post-MI has produced large areas of engraftment, and electrical coupling, but no clear improvement in cardiac function has been observed (148). However, a transient increase in ventricular arrhythmias has occurred, raising a potential safety concern. Transplantation of human PSC-derived cardiac progenitors has been associated with multi-lineage regeneration in the immunosuppressed rhesus monkey heart post-MI (149). Thus, large animal studies are beginning to define which cell preparations and delivery strategies hold promise.
Figure 6.
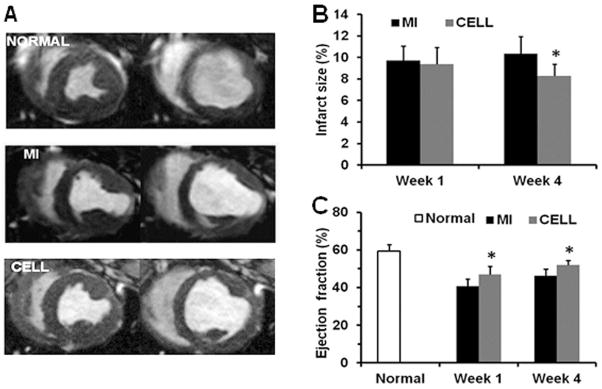
Fibrin patch-based delivery of human PSC-derived ECs and SMCs. A) cardiac MRI from normal, MI, and cell-treatment groups at end systole (left) and end diastole (right); B) after infarction, the cell-treatment produced significantly smaller infarct size (delayed-enhancement MRI); C) and improved ejection fraction compared to MI alone. *P<0.05 vs MI alone. Modified from (147), with permission.
Overall, clinical studies using adult cell sources have not yet demonstrated robust clinical benefits. Animal studies using PSC-derived cardiac cells, however, have shown promising results, with some evidence of improvement in left ventricular function and structure. Strategies that combine cells with bioengineered patches or decellularized cardiac matrices are now also being explored, especially for use in congenital heart disease. At this time, however, the mechanisms by which different cell populations and delivery strategies impact the various cardiac disease states remain poorly understood, and the optimal cell populations to use and the best delivery strategies for clinical translation have not yet been defined. Further research is required.
Conclusions
As discussed above, the generation of an unlimited supply of specific cell types is crucial for cell and regenerative therapies, as they offer great hope for the treatment of a wide spectrum of diseases. However, numerous challenges remain. Recurrent autoimmunity will probably require immune suppression for some diseases, and it is still unclear which specific cell type will be useful for the treatment of disorders such as DMD and heart disease. Optimization will also be needed for the transplant site, as in diabetes, or when dealing with disruption of the extracellular matrix in treating degenerative diseases, as in chronic liver and heart disease, Finally, when the pathologic process is diffuse and migration of transplanted cells is limited, as is the case with Alzheimer’s disease, ALS, and the muscular dystrophies, identifying the best means and location for cell delivery will require further study in many cases. Using autologous cells and genetic engineering should help control rejection and autoimmunity and improve monitoring of short- and long-term engraftment. Despite existing challenges, the availability of an unlimited supply of clinically useful PSC-derived cell populations will facilitate studies into the biology of those cells and will likely assist in the treatment of diabetes, acute hepatic failure, metabolic liver diseases, retinal diseases, PD, HD, and possibly heart disease. Close collaboration between scientists and clinicians, and between academia and industry, will be critical to overcoming remaining challenges to bringing novel therapies to patients in need.
Acknowledgments
We thank E. Tafaleng and J. H. Cummins for providing figures and text editing. This work was supported in part by NIH R01DK48794, P01DK096990, and R01DK099320 (IJF); NINDS, NIMH, the Mathers Charitable Foundation, the National Multiple Sclerosis Society, the Adelson Medical Research Foundation, CHDI, and the New York Stem Cell Research Program (SAG), NIH 1P01AG043376 (JH), U01HL099773 (TJK), and DOD W81XWH-10-1-1055 (MT). IJF is a member of the Scientific Advisory Board of Regenerative Medicine Solutions, Inc, GQD is a member of the Senior Advisory Board of MPM Capital, Inc, SAG is a consultant for Biogen Idec, TJK is a founding shareholder in Cellular Dynamics International and JH is a consultant for Cook Myosite. SAG is an inventor on patents owned by the University of Rochester and Cornell University concerning the use of tissue- and hiPSC-derived glial progenitor cells.
References
- 1.Takahashi K, et al. Cell. 2007 Nov 30;131:861. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JA, et al. Science. 1998 Nov 6;282:1145. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 3.Lacotte S, Berney T, Shapiro AJ, Toso C. Expert Opin Biol Ther. 2011 Jan;11:55. doi: 10.1517/14712598.2011.536530. [DOI] [PubMed] [Google Scholar]
- 4.Hanna J, et al. Science. 2007 Dec 21;318:1920. doi: 10.1126/science.1152092. [DOI] [PubMed] [Google Scholar]
- 5.Rideout WM, 3rd, Hochedlinger K, Kyba M, Daley GQ, Jaenisch R. Cell. 2002 Apr 5;109:17. doi: 10.1016/s0092-8674(02)00681-5. [DOI] [PubMed] [Google Scholar]
- 6.Kyba M, Perlingeiro RC, Daley GQ. Cell. 2002 Apr 5;109:29. doi: 10.1016/s0092-8674(02)00680-3. [DOI] [PubMed] [Google Scholar]
- 7.McKinney-Freeman SL, et al. Blood. 2009 Jul 9;114:268. doi: 10.1182/blood-2008-12-193888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ledran MH, et al. Cell Stem Cell. 2008 Jul 3;3:85. doi: 10.1016/j.stem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Tian X, Woll PS, Morris JK, Linehan JL, Kaufman DS. Stem Cells. 2006 May;24:1370. doi: 10.1634/stemcells.2005-0340. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Menendez P, Cerdan C, Bhatia M. Exp Hematol. 2005 Sep;33:987. doi: 10.1016/j.exphem.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Mazurier C, Douay L, Lapillonne H. Curr Opin Hematol. 2011 Jul;18:249. doi: 10.1097/MOH.0b013e3283476129. [DOI] [PubMed] [Google Scholar]
- 12.Rousseau GF, Giarratana MC, Douay L. Biotechnol J. 2014 Jan;9:28. doi: 10.1002/biot.201200368. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura S, et al. Cell Stem Cell. 2014 Apr 3;14:535. doi: 10.1016/j.stem.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Bottino R, Trucco M, Balamurugan AN, Starzl TE. Best Pract Res Clin Gastroenterol. 2002 Jun;16:457. doi: 10.1053/bega.2002.0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemp CB, Knight MJ, Scharp DW, Lacy PE, Ballinger WF. Nature. 1973 Aug 17;244:447. doi: 10.1038/244447a0. [DOI] [PubMed] [Google Scholar]
- 16.Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Diabetes. 1988 Apr;37:413. doi: 10.2337/diab.37.4.413. [DOI] [PubMed] [Google Scholar]
- 17.Ryan EA, et al. Diabetes. 2005 Jul;54:2060. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 18.Alejandro R, et al. J Clin Invest. 1986 Nov;78:1339. doi: 10.1172/JCI112720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson B, Ekdahl KN, Korsgren O. Curr Opin Organ Transplant. 2011 Dec;16:620. doi: 10.1097/MOT.0b013e32834c2393. [DOI] [PubMed] [Google Scholar]
- 20.Barton FB, et al. Diabetes Care. 2012 Jul;35:1436. doi: 10.2337/dc12-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherland DE, et al. J Am Coll Surg. 2012 Apr;214:409. doi: 10.1016/j.jamcollsurg.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westermark GT, Westermark P, Berne C, Korsgren O. N Engl J Med. 2008 Aug 28;359:977. doi: 10.1056/NEJMc0802893. [DOI] [PubMed] [Google Scholar]
- 23.De Paepe ME, Keymeulen B, Pipeleers D, Kloppel G. Hepatology. 1995 Apr;21:1144. [PubMed] [Google Scholar]
- 24.Pipeleers DG, et al. Diabetes. 1991 Jul;40:908. doi: 10.2337/diab.40.7.908. [DOI] [PubMed] [Google Scholar]
- 25.Echeverri GJ, et al. Am J Transplant. 2009 Nov;9:2485. doi: 10.1111/j.1600-6143.2009.02815.x. [DOI] [PubMed] [Google Scholar]
- 26.Worchester Human Islet Transplantation Group. N Engl J Med. 2006 Sep 28;355:1397. doi: 10.1056/NEJMc061530. [DOI] [PubMed] [Google Scholar]
- 27.Creusot RJ, Giannoukakis N, Trucco M, Clare-Salzler MJ, Fathman CG. Diabetes. 2014 Jan;63:20. doi: 10.2337/db13-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Windt DJ, et al. Am J Transplant. 2009 Dec;9:2716. doi: 10.1111/j.1600-6143.2009.02850.x. [DOI] [PubMed] [Google Scholar]
- 29.Liao SW, et al. Biomaterials. 2013 May;34:3984. doi: 10.1016/j.biomaterials.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulz TC, et al. PLoS One. 7:e37004. doi: 10.1371/journal.pone.0037004. Epub 2012 May 18, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demetriou AA, et al. Hepatology. 1988 Sep-Oct;8:1006. doi: 10.1002/hep.1840080505. [DOI] [PubMed] [Google Scholar]
- 32.De Vree JM, et al. Gastroenterology. 2000 Dec;119:1720. doi: 10.1053/gast.2000.20222. [DOI] [PubMed] [Google Scholar]
- 33.Groth CG, Arborgh B, Bjorken C, Sundberg B, Lundgren G. Transplant Proc. 1977 Mar;9:313. [PubMed] [Google Scholar]
- 34.Matas AJ, et al. Science. 1976 May 28;192:892. doi: 10.1126/science.818706. [DOI] [PubMed] [Google Scholar]
- 35.Rugstad HE, Robinson SH, Yannoni C, Tashjian AH., Jr Science. 1970 Oct 30;170:553. doi: 10.1126/science.170.3957.553. [DOI] [PubMed] [Google Scholar]
- 36.Wiederkehr JC, Kondos GT, Pollak R. Transplantation. 1990 Sep;50:466. doi: 10.1097/00007890-199009000-00021. [DOI] [PubMed] [Google Scholar]
- 37.Yoshida Y, Tokusashi Y, Lee GH, Ogawa K. Gastroenterology. 1996 Dec;111:1654. doi: 10.1016/s0016-5085(96)70029-x. [DOI] [PubMed] [Google Scholar]
- 38.Gupta S, et al. Hepatology. 1999 Feb;29:509. doi: 10.1002/hep.510290213. [DOI] [PubMed] [Google Scholar]
- 39.Ponder KP, et al. Proc Natl Acad Sci U S A. 1991 Feb 15;88:1217. doi: 10.1073/pnas.88.4.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhawan A, Puppi J, Hughes RD, Mitry RR. Nat Rev Gastroenterol Hepatol. 2010 May;7:288. doi: 10.1038/nrgastro.2010.44. [DOI] [PubMed] [Google Scholar]
- 41.Habibullah CM, Syed IH, Qamar A, Taher-Uz Z. Transplantation. 1994 Oct 27;58:951. doi: 10.1097/00007890-199410270-00016. [DOI] [PubMed] [Google Scholar]
- 42.Horslen SP, et al. Pediatrics. 2003 Jun;111:1262. doi: 10.1542/peds.111.6.1262. [DOI] [PubMed] [Google Scholar]
- 43.Mito M, et al. Transplantation. 1979 Dec;28:499. doi: 10.1097/00007890-197912000-00013. [DOI] [PubMed] [Google Scholar]
- 44.Mito M, Kusano M, Kawaura Y. Transplant Proc. 1992 Dec;24:3052. [PubMed] [Google Scholar]
- 45.Muraca M, et al. Lancet. 2002 Jan 26;359:317. doi: 10.1016/S0140-6736(02)07529-3. [DOI] [PubMed] [Google Scholar]
- 46.Sokal EM, et al. Transplantation. 2003 Aug 27;76:735. doi: 10.1097/01.TP.0000077420.81365.53. [DOI] [PubMed] [Google Scholar]
- 47.Fox IJ, et al. N Engl J Med. 1998 May 14;338:1422. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 48.Newsome PN, Plevris JN, Nelson LJ, Hayes PC. Liver Transpl. 2000 Jan;6:21. doi: 10.1002/lt.500060110. [DOI] [PubMed] [Google Scholar]
- 49.Terblanche J, Hickman R. Dig Dis Sci. 1991 Jun;36:770. doi: 10.1007/BF01311235. [DOI] [PubMed] [Google Scholar]
- 50.Bilir BM, et al. Liver Transpl. 2000 Jan;6:32. doi: 10.1002/lt.500060113. [DOI] [PubMed] [Google Scholar]
- 51.Zhou H, et al. Hepatology. 2009;50:303A. [Google Scholar]
- 52.Guha C, et al. Hepatology. 2002 Aug;36:354. doi: 10.1053/jhep.2002.34516. [DOI] [PubMed] [Google Scholar]
- 53.Gagandeep S, et al. J Pathol. 2000 May;191:78. doi: 10.1002/(SICI)1096-9896(200005)191:1<78::AID-PATH587>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 54.Kobayashi N, et al. Hepatology. 2000 Apr;31:851. doi: 10.1053/he.2000.5636. [DOI] [PubMed] [Google Scholar]
- 55.Ribeiro J, et al. Hepatology. 1992 Jan;15:12. doi: 10.1002/hep.1840150104. [DOI] [PubMed] [Google Scholar]
- 56.Hoppo T, Komori J, Manohar R, Stolz DB, Lagasse E. Gastroenterology. 2011 Feb;140:656. doi: 10.1053/j.gastro.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schumacher IK, et al. Hepatology. 1996 Aug;24:337. doi: 10.1002/hep.510240209. [DOI] [PubMed] [Google Scholar]
- 58.Soltys KA, et al. Journal of Hepatology. 2010 Oct;53:769. doi: 10.1016/j.jhep.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uygun BE, et al. Nat Med. 2010 Jul;16:814. doi: 10.1038/nm.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benraiss A, et al. Cell Stem Cell. 2013 Jun 6;12:787. doi: 10.1016/j.stem.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benraiss A, Goldman SA. Neurotherapeutics. 2011 Oct;8:577. doi: 10.1007/s13311-011-0075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindvall O. Nature Biotechnology. 2012 Jan;30:56. doi: 10.1038/nbt.2077. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, et al. Nature Biotechnology. 2013 May;31:440. doi: 10.1038/nbt.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kriks S, et al. Nature. 2011 Dec 22;480:547. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roy NS, et al. Nat Med. 2006;12:1259. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- 66.Carri AD, et al. Development. 2013 Jan 15;140:301. [Google Scholar]
- 67.HDConsortium, . Cell Stem Cell. 2012 Aug 3;11:264. [Google Scholar]
- 68.Nicholas CR, et al. Cell Stem Cell. 2013 May 2;12:573. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buchholz DE, et al. Stem Cells Translational Medicine. 2013 May;2:384. doi: 10.5966/sctm.2012-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stern JH, Temple S. Neurotherapeutics. 2011 Oct;8:736. doi: 10.1007/s13311-011-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roy N, et al. Exp Neurology. 2005;196:224. [Google Scholar]
- 72.Karumbayaram S, et al. Stem Cells. 2009 Apr;27:806. doi: 10.1002/stem.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li XJ, et al. Nature Biotechnology. 2005 Feb;23:215. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- 74.Lepore AC, et al. PLoS One. 2011;6:e25968. doi: 10.1371/journal.pone.0025968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldman SA, Nedergaard M, Windrem MS. Science. 2012 Oct 26;338:491. doi: 10.1126/science.1218071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Archer D, Cuddon P, Lipsitz D, Duncan I. Nat Med. 1997;3:54. doi: 10.1038/nm0197-54. [DOI] [PubMed] [Google Scholar]
- 77.Windrem MS, et al. Nat Med. 2004 Jan;10:93. doi: 10.1038/nm974. [DOI] [PubMed] [Google Scholar]
- 78.Windrem MS, et al. Cell Stem Cell. 2008 Jun 5;2:553. doi: 10.1016/j.stem.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang S, et al. Cell Stem Cell. 2013 Feb 7;12:252. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupta N, et al. Science Translational Medicine. 2012 Oct 10;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goldman SA. Archives of Neurology. 2011 Jul;68:848. doi: 10.1001/archneurol.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li M, Suzuki K, Kim NY, Liu GH, Izpisua Belmonte JC. The Journal of Biological Chemistry. 2014 Feb 21;289:4594. doi: 10.1074/jbc.R113.488247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tuszynski MH, et al. Cell. 2014 Jan 30;156:388. [Google Scholar]
- 84.Hoffman EP, Brown RH, Jr, Kunkel LM. Cell. 1987 Dec 24;51:919. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 85.Watkins SC, Hoffman EP, Slayter HS, Kunkel LM. Nature. 1988 Jun 30;333:863. doi: 10.1038/333863a0. [DOI] [PubMed] [Google Scholar]
- 86.Arahata K, et al. Nature. 1988 Jun 30;333:861. doi: 10.1038/333861a0. [DOI] [PubMed] [Google Scholar]
- 87.Zubrzycka-Gaarn EE, et al. Nature. 1988 Jun 2;333:466. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]
- 88.Bischoff R. Dev Biol. 1986 May;115:129. doi: 10.1016/0012-1606(86)90234-4. [DOI] [PubMed] [Google Scholar]
- 89.Beauchamp JR, Morgan JE, Pagel CN, Partridge TA. J Cell Biol. 1999 Mar 22;144:1113. doi: 10.1083/jcb.144.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fan Y, Maley M, Beilharz M, Grounds M. Muscle Nerve. 1996 Jul;19:853. doi: 10.1002/(SICI)1097-4598(199607)19:7<853::AID-MUS7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 91.Gussoni E, et al. Nature. 1992 Apr 2;356:435. doi: 10.1038/356435a0. [DOI] [PubMed] [Google Scholar]
- 92.Huard J, et al. Muscle Nerve. 1992 May;15:550. doi: 10.1002/mus.880150504. [DOI] [PubMed] [Google Scholar]
- 93.Karpati G, et al. Am J Pathol. 1989 Jul;135:27. [PMC free article] [PubMed] [Google Scholar]
- 94.Mendell JR, et al. N Engl J Med. 1995 Sep 28;333:832. doi: 10.1056/NEJM199509283331303. [DOI] [PubMed] [Google Scholar]
- 95.Morgan JE, Hoffman EP, Partridge TA. J Cell Biol. 1990 Dec;111:2437. doi: 10.1083/jcb.111.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qu Z, et al. J Cell Biol. 1998 Sep 7;142:1257. doi: 10.1083/jcb.142.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vilquin JT, Wagner E, Kinoshita I, Roy R, Tremblay JP. J Cell Biol. 1995 Nov;131:975. doi: 10.1083/jcb.131.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Torrente Y, El Fahime E, Caron NJ, Bresolin N, Tremblay JP. Cell Transplant. 2000 Jul-Aug;9:539. [PubMed] [Google Scholar]
- 99.Clarke DL, et al. Science. 2000 Jun 2;288:1660. doi: 10.1126/science.288.5471.1660. [DOI] [PubMed] [Google Scholar]
- 100.De Angelis L, et al. J Cell Biol. 1999 Nov 15;147:869. doi: 10.1083/jcb.147.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferrari G, et al. Science. 1998 Mar 6;279:1528. doi: 10.1126/science.279.5356.1528. [DOI] [PubMed] [Google Scholar]
- 102.Galli R, et al. Nature Neuroscience. 2000 Oct;3:986. doi: 10.1038/79924. [DOI] [PubMed] [Google Scholar]
- 103.Pittenger MF, et al. Science. 1999 Apr 2;284:143. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 104.Crisan M, et al. Cell Stem Cell. 2008 Sep 11;3:301. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 105.Dellavalle A, et al. Nat Cell Biol. 2007 Mar;9:255. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- 106.Zheng B, et al. Nature Biotechnology. 2007 Sep;25:1025. doi: 10.1038/nbt1334. [DOI] [PubMed] [Google Scholar]
- 107.Blau HM, Brazelton TR, Weimann JM. Cell. 2001 Jun 29;105:829. doi: 10.1016/s0092-8674(01)00409-3. [DOI] [PubMed] [Google Scholar]
- 108.Peault B, et al. Mol Ther. 2007 May;15:867. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- 109.Carr LK, et al. Int Urogynecol J Pelvic Floor Dysfunct. 2008 Jun;19:881. doi: 10.1007/s00192-007-0553-z. [DOI] [PubMed] [Google Scholar]
- 110.Sampaolesi M, et al. Nature. 2006 Nov 30;444:574. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- 111.Bachrach E, et al. Proc Natl Acad Sci U S A. 2004 Mar 9;101:3581. doi: 10.1073/pnas.0400373101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kennedy M, et al. Nature. 1997 Apr 3;386:488. doi: 10.1038/386488a0. [DOI] [PubMed] [Google Scholar]
- 113.Klug MG, Soonpaa MH, Koh GY, Field LJ. J Clin Invest. 1996 Jul 1;98:216. doi: 10.1172/JCI118769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Muller M, et al. FASEB J. 2000 Dec;14:2540. doi: 10.1096/fj.00-0002com. [DOI] [PubMed] [Google Scholar]
- 115.Rinaldi F, Perlingeiro RC. Transl Res. 2013 Nov 14; doi: 10.1016/j.trsl.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vittet D, et al. Blood. 1996 Nov 1;88:3424. [PubMed] [Google Scholar]
- 117.Barberi T, et al. Nat Med. 2007 May;13:642. doi: 10.1038/nm1533. [DOI] [PubMed] [Google Scholar]
- 118.Rohwedel J, et al. Dev Biol. 1994 Jul;164:87. doi: 10.1006/dbio.1994.1182. [DOI] [PubMed] [Google Scholar]
- 119.Goudenege S, et al. Mol Ther. 2012 Nov;20:2153. doi: 10.1038/mt.2012.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Darabi R, et al. Cell Stem Cell. 2012 May 4;10:610. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Filareto A, et al. Nat Commun. 2013;4:1549. doi: 10.1038/ncomms2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tedesco FS, et al. Science Translational Medicine. 2012 Jun 27;4:140ra89. doi: 10.1126/scitranslmed.3003541. [DOI] [PubMed] [Google Scholar]
- 123.Jasmin G, Tautu C, Vanasse M, Brochu P, Simoneau R. Lab Invest. 1984 Feb;50:197. [PubMed] [Google Scholar]
- 124.Liechti-Gallati S, Moser H, Siegrist HP, Wiesmann U, Herschkowitz NN. Pediatr Res. 1981 Nov;15:1411. doi: 10.1203/00006450-198111000-00004. [DOI] [PubMed] [Google Scholar]
- 125.Sacco A, et al. Cell. 2010 Dec 23;143:1059. doi: 10.1016/j.cell.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kallestad KM, et al. Exp Cell Res. 2011 Apr 1;317:873. doi: 10.1016/j.yexcr.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sanganalmath SK, Bolli R. Circ Res. 2013 Aug 30;113:810. doi: 10.1161/CIRCRESAHA.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Leobon B, et al. Proc Natl Acad Sci USA. 2003;100:7808. doi: 10.1073/pnas.1232447100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Menasche P, et al. Circulation. 2008 Mar 4;117:1189. doi: 10.1161/CIRCULATIONAHA.107.734103. [DOI] [PubMed] [Google Scholar]
- 130.Orlic D, et al. Nature. 2001;410:701. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 131.Balsam LB, et al. Nature. 2004;428:668. doi: 10.1038/nature02460. [DOI] [PubMed] [Google Scholar]
- 132.Murry CE, et al. Nature. 2004;428:664. doi: 10.1038/nature02446. [DOI] [PubMed] [Google Scholar]
- 133.Fisher SA, et al. The Cochrane Database of Systematic Reviews. 2014;4:CD007888. doi: 10.1002/14651858.CD007888.pub2. [DOI] [PubMed] [Google Scholar]
- 134.Nguyen PK, Lan F, Wang Y, Wu JC. Circulation Research. 2011 Sep 30;109:962. doi: 10.1161/CIRCRESAHA.111.242909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Caspi O, et al. J Am Coll Cardiol. 2007 Nov 6;50:1884. doi: 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- 136.Moretti A, et al. Cell. 2006 Dec 15;127:1151. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 137.Yang L, et al. Nature. 2008 May 22;453:524. [Google Scholar]
- 138.Ferreira LS, et al. Circ Res. 2007 Aug 3;101:286. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]
- 139.Mummery CL, et al. Circulation Research. 2012 Jul 20;111:344. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dubois NC, et al. Nature Biotechnology. 2011 Nov;29:1011. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tohyama S, et al. Cell Stem Cell. 2013 Jan 3;12:127. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 142.He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Circ Res. 2003;93:32. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- 143.Laflamme MA, et al. Nature Biotechnology. 2007 Sep;25:1015. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 144.Christoforou N, et al. PloS one. 2010;5:e11536. doi: 10.1371/journal.pone.0011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shiba Y, et al. Nature. 2012 Aug 5; [Google Scholar]
- 146.Kawamura M, et al. Circulation. 2012 Sep 11;126:S29. doi: 10.1161/CIRCULATIONAHA.111.084343. [DOI] [PubMed] [Google Scholar]
- 147.Xiong Q, et al. Circulation. 2013 Mar 5;127:997. doi: 10.1161/CIRCULATIONAHA.112.000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chong JJH, Yang X, Don CW, Laflamme MA, Murry CE. Nature. 2014;510:273. [Google Scholar]
- 149.Blin G, et al. J Clin Invest. 2010 Apr 1;120:1125. doi: 10.1172/JCI40120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Alipio Z, et al. Proc Natl Acad Sci U S A. 2010 Jul 27;107:13426. doi: 10.1073/pnas.1007884107. [DOI] [PMC free article] [PubMed] [Google Scholar]