

Abstract
Purpose.
To understand the relationship between rapamycin loading/release and surface chemistries of porous silicon (pSi) to optimize pSi-based intravitreal delivery system.
Methods.
Three types of surface chemical modifications were studied: (1) pSi-COOH, containing 10-carbon aliphatic chains with terminal carboxyl groups grafted via hydrosilylation of undecylenic acid; (2) pSi-C12, containing 12-carbon aliphatic chains grafted via hydrosilylation of 1-dodecene; and (3) pSiO2-C8, prepared by mild oxidation of the pSi particles followed by grafting of 8-hydrocarbon chains to the resulting porous silica surface via a silanization.
Results.
The efficiency of rapamycin loading follows the order (micrograms of drug/milligrams of carrier): pSiO2-C8 (105 ± 18) > pSi-COOH (68 ± 8) > pSi-C12 (36 ± 6). Powder X-ray diffraction data showed that loaded rapamycin was amorphous and dynamic drug-release study showed that the availability of the free drug was increased by 6-fold (compared with crystalline rapamycin) by using pSiO2-C8 formulation (P = 0.0039). Of the three formulations in this study, pSiO2-C8-RAP showed optimal performance in terms of simultaneous release of the active drug and carrier degradation, and drug-loading capacity. Released rapamycin was confirmed with the fingerprints of the mass spectrometry and biologically functional as the control of commercial crystalline rapamycin. Single intravitreal injections of 2.9 ± 0.37 mg pSiO2-C8-RAP into rabbit eyes resulted in more than 8 weeks of residence in the vitreous while maintaining clear optical media and normal histology of the retina in comparison to the controls.
Conclusions.
Porous silicon–based rapamycin delivery system using the pSiO2-C8 formulation demonstrated good ocular compatibility and may provide sustained drug release for retina.
Keywords: intravitreal drug delivery, sirolimus, porous silicon, rabbit eye, surface chemistry, drug loading and release, sustained release, hydrosilylation, oxidation, silanization
Mild oxidation and subsequent silanization of the porous silicon (pSi) rendered the resultant pSi particles optimized for rapamycin loading/release as an intravitreal injectable delivery system. The system slowly released rapamycin and safely resided in rabbit vitreous more than 8 weeks.
Introduction
Age-related macular degeneration (AMD) and diabetic retinopathy are the leading causes of blindness in the world and there are very limited treatment options. Wet AMD and proliferative retinopathy are associated with neovascularization because the stressed RPE cells secret VEGF at the disease site.1,2 Two anti-VEGF agents have been recently approved for these conditions3,4 and require monthly or bimonthly intraocular injections to neutralize VEGF. Intraocular injection frequencies of 6 to 12 times a year remain a major burden on patients and health care providers. In addition, there are two major components of pathologies for wet AMD: excessive VEGF and unwanted cell proliferation and scarring; the current anti-VEGF agents cannot provide the solution to vision-destroying scarring.
In RPE cells, VEGF and other cytokines are induced by the mammalian target of rapamycin (mTOR) or the hypoxia-inducible factor-1 pathway by various growth factors and inflammatory cytokines.5,6 Rapamycin (RAP), also known as sirolimus, prevents mitogen-induced hypoxia-inducible factor-1 and hypoxia-inducible factor-1–dependent transcription and secretion of VEGF.5,7 Therefore, RAP may be of use in therapy of wet AMD and proliferative retinopathy. Indeed, preclinical studies have shown that RAP has a therapeutic effect on AMD-like retinopathy.8,9 Due to low solubility of RAP in water (2.6 μg/mL),10 local ocular use is limited. It is also difficult to enhance the solubility of sirolimus by salt generation because of the lack of an ionizable group in the pH range 1 to 10.10,11 Rapamycin is a potent immunosuppressant administered orally for refractory uveitis; however, side effects are associated with systemic use.12
Age-related macular degeneration and diabetic retinopathy are chronic and lifetime diseases. A localized system that maintains an optimal (therapeutic and nontoxic) drug level at the disease site can mitigate the systemic side effects from oral or intravenous administrations.
Porous silicon (pSi) has been reported in recent years as a carrier for the controlled release of drugs.13–15 In contrast to conventional mesoporous silica obtained by sol-gel or precipitation routes, significant control of the pSi microstructure is possible by tuning the electrochemical etching parameters.16,17 In our previous reports, controlled release was achieved by covalent bonding of drugs to pSi particles.14,18 However, drug-loading efficiency by covalent bonding was low (<10% by weight). Another useful feature of pSi is its readily modified surface chemistry to optimize the drug loading and release according to the properties of the drug payloads.13,19 It has been observed that certain chemistries can slow the degradation of the pSi matrix or enhance the release of poorly soluble active pharmaceutical ingredients.20,21
In the current study, we aimed to develop a novel surface chemistry of pSi for infiltration RAP loading and delivery in the context of ocular use for the treatment of AMD and diabetic retinopathy.
Materials and Methods
Porous Silicon Microparticle Fabrication
Porous silicon microparticles were prepared by anodic electrochemical etch of highly doped, (100)-oriented, p-type silicon wafers (boron-doped, 1.1 mΩ·cm resistivity; Siltronix, Inc., Archamps, France), in an electrolyte consisting of a 3:1 (vol:vol) solution of 48% aqueous hydrofluoric acid (HF) and ethanol (Fisher-Scientific, Pittsburg, PA, USA) as we previously described.22 The current density waveform generates a porosity modulation in the pSi layer that displays a sharp peak in the optical reflectance spectrum at approximately 600 nm. The waveform was etched into the silicon wafer for a total of 400 seconds. The resulting porous layer was then removed from the silicon substrate and ultrasonicated in ethanol for 30 minutes in a FS5 dual action ultrasonic cleaner (Thermo Fisher Scientific, Pittsburg, PA, USA) to generate the microparticles. Then it was rinsed with ethanol three times and preserved in ethanol.
Surface Modification of pSi Microparticles
The freshly made pSi microparticles were modified following three different strategies, as described below.
Hydrosilylation of undecylenic acid. The first type of pSi particle, namely pSi-COOH, resulted from chemical modification of freshly etched pSi by microwave-assisted hydrosilylation in the presence of neat undecylenic acid (10-undecenoic acid). This procedure results in a functional pSi surface grafted with C-10 alkyls containing terminal carboxyl groups (-COOH). A significant quantity of the surface hydrides (Si-H) remaining after the hydrosilylation reaction were eliminated by air oxidation at 150°C for 48 hours in a ceramic boat inside a muffle furnace (Thermo Fisher Scientific), as previously described.23 The functionalized samples were kept under vacuum in a desiccator.
Hydrosilylation of 1-dodecene. The second type of pSi particle, designated pSi-C12, resulted from chemical modification of freshly etched pSi by microwave-assisted hydrosilylation of 1-dodecene, as previously described.24 The functionalized samples were kept under vacuum in a desiccator.
Silanization of partially oxidized pSi with methoxy(dimethyl)octylsilane. The third type of pSi particle, designated pSiO2-C8, was prepared from pSi by air oxidation followed by covalent grafting of hydrocarbon chains to the porous silica surface with a silanization reaction. Initially, the samples were placed in a ceramic boat inside a muffle furnace at room temperature. The temperature was ramped to 600°C at a rate of 10°C per minute, held at 600°C for 1 hour, then slowly cooled to room temperature inside the furnace to minimize the stress in the samples during the transient cooling. This oxidation treatment removes Si-H and partially converts silicon (Si) to silica (SiO2). To generate Si-OH terminated surfaces for further coupling of the organosilane, oxidized pSi particles were treated with 4% (vol/vol) hydrochloric acid aqueous solution (HCl 37 wt%; Sigma-Aldrich Corp., St Louis, MO, USA), shaken for 1 hour at room temperature and washed with deionized water (DI H2O). Approximately 40 mg hydroxyl-terminated pSiO2 microparticles were suspended in ethanol, transferred to a Schlenk flask and dried under vacuum overnight. Then 8.3 mmol of methoxy(dimethyl)octylsilane (98 wt%; Sigma-Aldrich) per gram of pSi microparticles (Sigma-Aldrich) were dissolved in anhydrous toluene and added via syringe under nitrogen flow to yield an 8 wt% final solution. The flask was heated at 120°C in a nitrogen atmosphere and allowed to react overnight with constant shaking. The particles were then washed with toluene (Sigma-Aldrich), dimethylformamide (Sigma-Aldrich), ethanol (Fisher-Scientific), and ether (Sigma-Aldrich), three times each, dried at room temperature, and stored under vacuum in a desiccator.
Loading of RAP Into pSi Microparticles
Drug loading was achieved by the impregnation method using a pressure gradient. Approximately 20 mg functionalized pSi microparticles were transferred to a glass vial, then sealed with a rubber septum and placed under vacuum at less than 0.002 atm for approximately 30 minutes. Thereafter, connection to the vacuum pump was closed and 0.4 mL of an infiltrating solution of 50 g/L RAP (LC Labs, Woburn, MA, USA) in acetone (HPLC grade; Sigma-Aldrich) was injected into the vial. This vacuum-assisted infiltration was used to improve penetration of the drug solution into the pores. Infiltrating liquid flowed rapidly into the pores of pSi microparticles, which were soaked for 10 minutes before breaking the vacuum. The vials were tightly closed with their cap, protected from direct light with aluminum foil wrapping, and placed in a rotator shaker at 20 rpm (position 40, Model 24; Reliable Scientific, Inc., Nesbit, MS, USA) at room temperature. After incubation overnight, the particles were separated from the drug solution by aspirating the supernatant with a pipette, without additional rinsing. The remaining solvent was slowly evaporated under vacuum at room temperature. The dried particles were stored under a protective argon atmosphere until further analysis.
The amount of RAP loaded into pSi microparticles was determined by a solvent extraction method, in which 400 μL acetone (HPLC grade; Sigma-Aldrich) was used to extract RAP from 1 mg pSi particles. The Eppendorf tubes were wrapped with aluminum foil to protect the drug from direct light and were shaken in a rotator at 40 rpm (position 60, Model 24; Reliable Scientific, Inc.) at room temperature overnight. Empty functionalized particles not containing drug were extracted following the same procedure, and were used as a control. For RAP quantitation, both samples and controls were spiked with 1000 ng ascomycin (ASC; LC Laboratories, Woburn, MA, USA) (10 μL of a 100 μg/mL ASC solution in acetone) as an internal standard. Additionally, the empty particle controls were spiked with 1000 ng RAP (LC Laboratories). The pSi microparticles were removed from solution by centrifugation at 10,000 rpm for 10 minutes. The extracts were transferred to an Eppendorf tube, dried, and then suspended in mobile phase A (see below) before injection of 0.001% into a chromatographic column. The internal standard was calibrated by determining, after extraction of the sample, the ratio of the analyte peak height to the internal standard peak height.
The drug was analyzed through positive ion mode electrospray ionization (ESI) liquid chromatography-mass spectrometry (LC-MS/MS) using an Agilent 1260 HPLC chromatographic system coupled with a Thermo LCQdeca-MS spectrometer. Mobile phase A was 2.5% by volume methanol (CH3OH, HPLC grade; Sigma-Aldrich) and 0.1% formic acid (HCOOH; Sigma-Aldrich) in water. Pure methanol with 0.1% by volume formic acid was used as mobile phase B. The LC-ESI-MS/MS was operated under selected reaction monitoring (SRM) scan mode to detect RAP and the spiked internal standard ASC.25 Rapamycin was dissolved into methanol (HPLC grade; Sigma-Aldrich), and then infused to the ESI source to tune the instrument and obtain the fingerprint of RAP under LC-MS and LC-MS/MS analysis. The sodium salt of the molecular ion peak ([M+Na]+) of RAP at m/z 936.5 and the typical MS/MS spectrum of this molecular ion peak were observed. Liquid chromatography with MS/MS detection at m/z 936.5, peaked at a retention time of 12.38 ± 0.10 minutes. The most intense daughter ion peak at m/z 614.2 was chosen for SRM in subsequent RAP detection using LC-MS/MS analysis. Liquid chromatography–ESI-MS/MS on m/z 814.3 ([M+Na]+ molecular ion peak of internal standard ASC), followed by SRM at its most intense fragmental peak (m/z 604.1) confirmed the presence of the internal standard ASC, as expected (not shown). The internal standard ASC showed a retention time of 12.10 ± 0.10 minutes. By using standard controls (release media spiked with known amount of RAP and ASC), the concentration of intact RAP was determined.
In Vitro RAP Release and pSi Matrix Degradation
Dynamic drug release from an intravitreal drug depot was simulated using a syringe pump (NE-1000; New Era Pump Systems, Farmingdale, NY, USA) and a custom chamber with 1.5 mL effective circulation volume to mimic rabbit vitreous.26 Approximately 6-mg particles loaded with RAP corresponding to each of the three formulations of this study, pSi-COOH-RAP, pSi-C12-RAP, and pSiO2-C8-RAP, were suspended in three chambers filled with 1.5 mL Hank's Balanced Salt Solution (HBSS). The syringes were filled with HBSS and the pumps were set to a flow rate of 1 μL/min.27 The entire chamber was maintained at 37°C and pumped continuously over 30 days. At the same time every day, the diffusate was collected and stored at −80°C until the analysis.
On analysis of the samples, in addition to quantitate RAP, elemental Si was quantified as Si dissolved in the collected solution by inductively coupled plasma-optical emission spectroscopy (ICP-OES) as reported previously.28 The purpose is to monitor the relation between drug release and silicon crystal degradation: synchronized drug release and pSi degradation is preferable for intravitreal drug-delivery application.
The optimal formulation of pSiO2-C8 was subjected to further in vitro release using the benchmark crystalline RAP (free drug) as the concurrent control. The drug-release experiment was conducted with a modified dialysis method by using a tubular cellulose dialysis bag (Spectra Por Biotech grade, MWCO 3.5-5 kDa #133192; Spectrum Labs, CA). The mass of pSiO2-C8-RAP microparticles was 5.38 mg (loaded with 1 mg RAP);1 mg commercial RAP was used as a free drug control. The dialysis bags were immersed in 1.5 mL DI H2O in a glass vial, mimicking the volume of rabbit vitreous. The vials were shaken at 150 rpm and 37°C in an orbital shaker (model TSSM1; Chemglass Life Sciences, Vineland, NJ, USA). The dissolution medium submerging the dialysis bag was sampled and replaced with fresh DI H2O every other day for 19 days. Aliquots were stored at +4°C until further analysis.
Physicochemical Characterization of pSi Microparticles
Scanning electron microscopy (SEM). The average particle size and pore size of pSi microparticles were determined from plan-view images of randomly selected particles (n >10) using a Phillips XL30 field emission SEM operating at an accelerating voltage of 5 kV (FEI Phillips, Hillsboro, OR, USA). Samples were sputter-coated with iridium to increase conductivity and improve image quality. Scanning electron microscopy monitored the pSi microparticle surface before functionalization as well as after the drug-loading procedure and the in vitro release studies.
Fourier transform infrared (FTIR) spectroscopy. Surface chemistry modifications and drug loading were characterized by FTIR spectroscopy using attenuated total reflectance (ATR) mode on a Nicolet 6700 Smart-iTR spectrometer (Thermo Fisher Scientific). Sample spectra were collected from 600 to 4000 cm−1 in absorbance mode, with a resolution of 1 cm−1. Each spectrum was the average of 128 scans.
Gas sorption analysis. Both surface chemistry modification and drug loading can reduce pSi specific surface area and total pore volume, which was studied using gas sorption analysis. The textural properties of the particles were analyzed by nitrogen adsorption at −196°C on an ASAP 2020 adsorption analyzer applying a volumetric technique (Micromeritics, Norcross, GA, USA). Approximately 50 mg of the pSi sample was transferred to a preweighed sample tube and degassed at 105°C for a minimum of 12 hours or until the outgas rate was lower than 5 mm Hg per minute. The sample was reweighed to obtain a consistent mass of the degassed pSi. The samples were then manually degassed for at least 2 hours before N2 isotherm. The specific surface area (m2/g) of the particles was calculated from the adsorption branch of the isotherms using the Brunauer-Emmett-Teller (BET) model.29 The pore volume was calculated from a single adsorption point (P/P0 = 0.99).
Elemental analysis. The chemical composition of the samples was analyzed by elemental analysis (CHNS/O system) in a CHNS/O 2400 Series II thermo analyzer (Perkin Elmer, Waltham, MA, USA) to evaluate the mass loading of drug in the pSi carriers.
Powder X-ray diffraction (XRD). To assess the status of loaded drug in the pores, powder XRD was carried out using a D8 Advance diffractometer (Bruker, Billerica, MA, USA). Data were collected on powder samples at room temperature using a LynxEye detector (Bruker AXS, Inc., Madison, WI, USA). The X-ray generator was operated at 40 kV and 25 mA using CuKα (λ = 1.5418 Å), with a scan speed of 0.5 seconds per step, a step size of 0.02°, and a 2θ range of 5 to 75°.
In Vitro Cytotoxicity of Released RAP Toward Endothelial EA.hy926 Cells
Cellular assays were performed on human umbilical vein cells (EA.hy926, ATCC CRL-2922; American Type Culture Collection, Manassas, VA, USA) to assess the functional toxicity of released RAP. A WST-1 (4-[3-(4-Iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) toxicity assay was used to test cell viability. The EA.hy926 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM)/F12 (1:1; Gibco Invitrogen, Paisley, UK) and supplemented with 10% fetal bovine serum (Gibco Invitrogen) and 1% antibiotic-antimycotic (100×; stabilized with 10,000 units penicillin, 10 mg streptomycin, and 25 μg amphotericin B per milliliter [Sigma-Aldrich]) in an incubator (Heraeus Instruments GmbH, Hanau, Germany) for 24 hours to reach 60% to 70% confluence. The cell medium was replaced with 100 μL of mixture of the samples and DMEM/F-12 medium (25% sample and 75% medium) in each well. After 5 days, 10 μL of a solution of WST-1 (Roche Diagnostics, Indianapolis, IN, USA) was added to each well, and the plates returned to the incubator. The optical density at 440 nm (OD440) of each well was measured on a UV-Vis spectrophotometer (Spectra Max Plus 384; Molecular Devices, Sunnyvale, CA, USA) at 0.5-hour intervals.
The data were analyzed for the following samples at various concentrations: (a) control RAP, as received, diluted with DI H2O to 1 ng/mL, 10 ng/mL, and 100 ng/mL (Water for Injection [WFI]-quality, cell culture grade; Cellgro, Manassas, VA, USA). A RAP stock solution (1 mg/mL) in dimethylsulfoxide (DMSO, ATCC 4-X, American Type Culture Collection) was used as the starting solution. Final concentrations of DMSO were 0.001% to 0.01% and 0.1%, respectively. Negative controls were prepared by dilution of a DI H2O and DMSO solution with DI H2O to final DMSO concentrations of 0.001%, 0.01%, and 0.1%; (b) RAP released from drug-loaded pSi particles, diluted with DI H2O to 1, 10, and 100 ng/mL as the highest concentration. Final RAP concentrations were 0.25 nM, 2.5 nM, and 25 nM, respectively; (c) silicic acid released from non–drug-loaded pSi particles (empty), equivalent in weight to the drug-loaded particles used in (b), following the same dilution strategy as in actual samples.
General and Ocular Safety in Rabbits
Six pigmented New Zealand Red rabbits (Western Oregon Rabbit Company, Philomath, OR, USA) were used for this study. Only one eye of each rabbit was used for drug injection and the contralateral eye was injected with PBS and served as normal control. All six rabbits had intravitreal injections of 3 mg (2.9 ± 0.39 mg after mass balance) straight etched pSiO2-C8 formulation in 100 μL. Following the intravitreal injections anterior segments of the eyes were monitored by slit-lamp biomicroscopy and the posterior segments of the eyes by indirect ophthalmoscopy. Intraocular pressure and fundus photos of both eyes were recorded on day 4, day 14, and day 56 using Tonopen (Medtronic, Jacksonville, FL, USA) and fundus camera (Canon F-A; Canon, Inc., Tokyo, Japan). Three rabbits were killed 2 weeks after the injection as a short-term safety study and the other three were killed 8 weeks as a long-term safety study. Before rabbits were killed, ERG was performed on every rabbit eye. After they were killed, the eye globes were fixed in 4% gluteraldehyde for histology processing. 17 The weight of each animal was recorded immediately before the injection and before they were killed to monitor possible weight loss from the toxicity of the delivery system and the drug.
Data Analysis
The data from in vitro release using the microdialysis bag were compared between SiO2-C8-RAP and Free RAP using sampling time points as matched pairs with the Wilcoxon signed rank test. Results from the cell viability tests are expressed as means ± SD from at least three independent experiments. The data were analyzed by one-way ANOVA. Multiple comparisons were performed for all pairs using Tukey-Kramer HSD, alpha = 0.05. For IOP and ERG parameters, a paired t-test was used to compare the injected eyes with their untouched contralateral eyes. These tests were performed using JMP statistical software (version 11; SAS Institute, Inc., Cary, NC, USA). The criterion for significance was P less than 0.05 for all the comparisons.
Results
Characterization of pSi Microparticles
The particle dimensions were 43 ± 8 μm in width, 70 ± 16 μm in length, and 25 ± 2 μm thick, as measured by SEM. The pore size was found to be 13 ± 5 nm (Fig. 1).
Figure 1.

(A) Scanning electron microscope images of a sample of freshly etched pSi microparticles. (B) A close-up SEM view of one of the microparticles, showing the mesoporous structure.
Surface Chemistry and Drug Loading
Hydride terminated freshly etched pSi microparticles were reacted in (1) 1-dodecene to attach hydrophobic, covalently bonded alkyl groups via thermal hydrosilylation (namely Si-C12) (Fig. 2A); (2) reacted in undecylenic acid to attach covalently bonded alkyl groups ending in a carboxyl (-COOH) group via thermal hydrosilylation, followed by thermal oxidation in air at 150°C (namely Si-COOH) (Fig. 2B); and (3) partially oxidized in air at 600°C and modified with hydrophobic covalently bonded alkyl groups via silanization reaction (namely SiO2-C8) (Fig. 2C).
Figure 2.
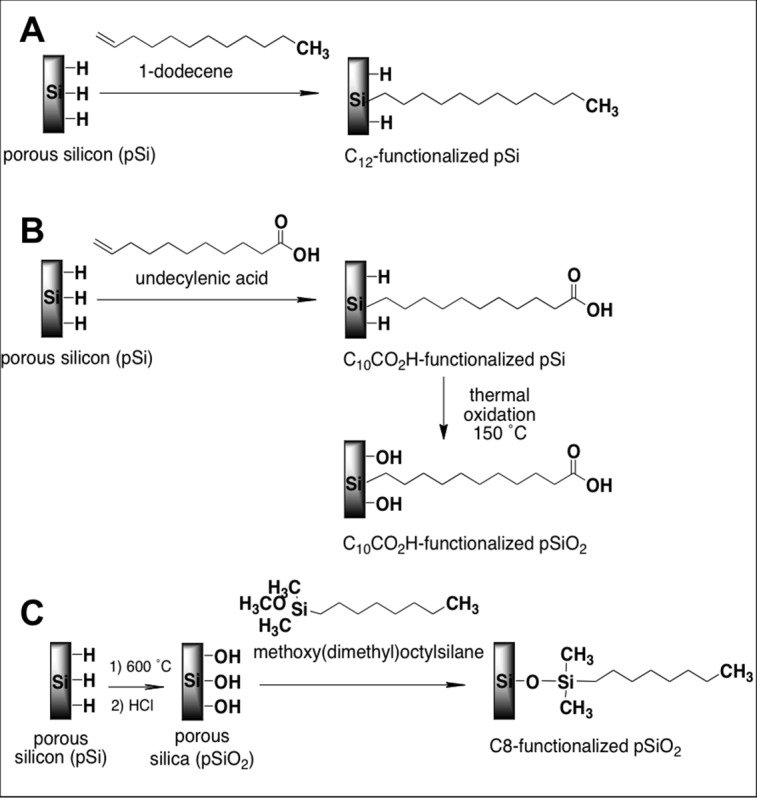
Schematic of pSi surface functionalization reactions. (A) Hydrosilylation of 1-dodecene (pSi-C12). (B) Hydrosilylation of undecylenic acid and further thermal oxidation (pSi-COOH). (C) Silanization of partially oxidized pSi (SiO2) with methoxy(dimethyl)octylsilane (pSiO2-C8).
Rapamycin was loaded into the pSi microparticles following surface modification.
Drug-loading capacity was quantified by LC-MS and expressed as micrograms RAP per milligrams of silicon. The calculated loading capacity was 36 ± 6 μg/mg (n = 4) for pSi-C12-RAP, 68 ± 8 μg/mg (n = 7) for pSi-COOH-RAP, and 105 ± 18 μg/mg (n = 7) for pSiO2-C8-RAP microparticle formulations.
The drug loading was confirmed by measuring the percentage of nitrogen using elemental analysis (CHNS/O). It is assumed that the entire detected content of nitrogen in the sample comes from loaded RAP. In addition, the detected N content for the pure pSi and the functionalized pSi was less than 0.01 wt %. The calculated drug-loading capacity by this method was 17 wt% for pSi-C12-RAP, 22 wt% for pSi-COOH-RAP, and 28 wt% for pSiO2-C8-RAP. These values confirm the successful drug loading and follow a similar trend as compared to those calculated after LC-MS.
Fourier transform infrared spectroscopy also confirmed the surface chemistry treatments of the pSi microparticles as well as the drug loading.
The FTIR spectra of freshly etched pSi as well as functionalized particles are shown in Figure 3A. Freshly etched pSi displays bands that are characteristic of surface hydride species. The band at 2100 cm−1 is attributed to the stretching modes of Si-H bonds and the absorbance observed at 910 cm−1 is attributed to Si-H deformation modes. After surface functionalization, characteristic bands corresponding to C-H vibrational modes are present in all samples. The bands at 2960 cm−1, 2929 cm−1, and 2857 cm−1 are assigned to stretching modes of C-H bonds, whereas the bands at 1450 cm−1 and 1378 cm−1 are attributed to deformation modes of C-H bonds. The characteristic stretching mode of a carbonyl group C = O at 1720 cm−1 confirms successful hydrosilylation of undecylenic acid. The band at 3412 cm−1 is attributed to O-H stretching modes. The hydrogen-terminated surface of the as-prepared pSi particles was converted to silicon dioxide (SiO2) during the thermal oxidation process, as evidenced by the characteristic Si-O-Si absorbance bands at approximately 1069 cm−1. After hydrosilylation, the Si-H stretching bands were present, indicating the microparticles were still hydride-coated.30,31 Remaining surface hydrides Si-H after hydrosilylation of undecylenic acid were eliminated by air oxidation at 150°C, as confirmed by FTIR. Complete conversion of the Si matrix to SiO2 does not occur under these conditions.19,23
Figure 3.
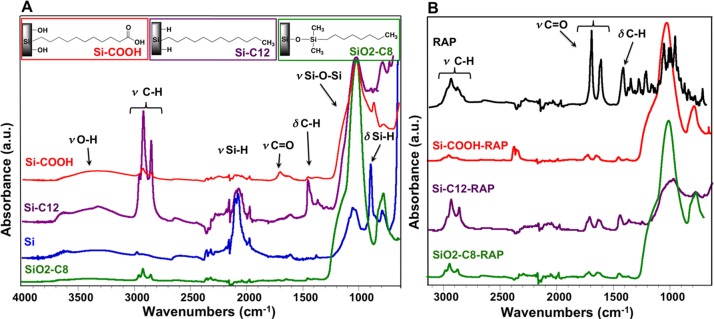
(A) Fourier transform infrared spectra of as-prepared pSi particles before chemical modification, and after functionalization with 1-dodecene (pSi-C12), undecylenic acid (pSi-COOH) and after silanization reaction with alkyl groups (pSiO2-C8). (B) Fourier transform infrared spectra of pSi particles after surface chemical modification and subsequent RAP loading (pSi-C12-RAP, pSi-COOH-RAP, and pSiO2-C8-RAP), as well as spectra of crystalline RAP. Group vibrational assignments are indicated in the text.
The FTIR spectra of drug-loaded pSi microparticles, namely pSi-C12-RAP, pSi-COOH-RAP, and pSiO2-C8-RAP, as well as the spectrum of pure RAP are shown in Figure 3B. The successful loading of RAP is confirmed by the comparison of the infrared spectra of the functionalized pSi microparticles (Fig. 3A) with the infrared spectrum of RAP-loaded pSi microparticles and pure RAP (Fig. 3B). The intensity of the C-H stretching absorption bands, the C-H deformation bands, and the carbonyl C = O stretching bands are larger in the spectra corresponding to drug-loaded pSi microparticles.
The textural properties of the functionalized and drug-loaded samples were studied by nitrogen adsorption/desorption porosimetry. The isotherms corresponding to the functionalized microparticles before and after drug loading are plotted in Figure 4. The isotherms are type IV with H1 hysteresis loops with parallel adsorption and desorption branches, suggesting cylindrical mesopores of approximately constant cross section. The surface area (SBET) and pore volume (VT) of the pSi microparticle formulations as well as the relative polarity of the surface as monitored by the CBET parameter are given in Table 1.
Figure 4.

Nitrogen adsorption/desorption isotherms of pSi microparticle formulations pSi-COOH, pSi-C12, and pSiO2-C8 (from left to right). Each plot shows the isotherms before and after RAP loading.
Table 1.
Textural Parameters of pSi Microparticle Formulations
|
Sample |
SBET, m2/g |
VT, cm3/g Exp |
CBET |
Drug Loaded,* μg/mg |
| pSi† | 458 ± 4 | 1.30 | 31 | NA |
| pSi-COOH | 385 ± 2 | 1.31 | 52 | NA |
| pSi-C12 | 356 ± 2 | 0.82 | 27 | NA |
| pSiO2-C8 | 260 ± 2 | 0.62 | 32 | NA |
| pSiO2‡ | 217 ± 1 | 0.61 | 80 | NA |
| pSi-COOH-RAP | 268 ± 1 | 0.92 | 48 | 68 ± 8 |
| pSi-C12-RAP | 236 ± 2 | 0.56 | 24 | 36 ± 6 |
| pSiO2-C8-RAP | 204 ± 1 | 0.48 | 27 | 105 ± 18 |
CBET, constant related to the affinity between the N2 adsorbate molecules and the pSi surface; SBET, specific surface area calculated at (P/P0) 0.03–0.24; VT, total pore volume at relative pressure (P/P0) equal to 0.99.
Drug-loading capacity calculated by LC-MS analysis.
Freshly etched pSi.
Porous Si converted to fully oxidized to porous silica (SiO2) by thermal oxidation in air at 800°C for 1 hour.
The measured surface area and the total pore volume both decrease after chemical functionalization, and these values are further reduced after drug loading. The observed decreases after functionalization are attributed to the presence of organic groups covalently attached to the matrix surface. The additional decrease in surface area and pore volume after drug loading are consistent with the filling of the inner pore channels with drug.
The CBET parameter is a constant obtained from the BET analysis and is related to the affinity of the solid surface for the adsorbate: the greater the C value, the stronger the interaction. It can be related to the polarity of the samples and it ranges from 80 for the hydrophilic fully oxidized pSiO2 sample to 31 for the hydrophobic freshly etched pSi sample.
Powder XRD spectra are shown in Figure 5. Functionalized pSi microparticles show XRD maxima centered at 2θ = 28°, 48°, 56°, and 69°, corresponding to reflections from the (111), (220), (311), and (400) crystallographic planes of the cubic diamond lattice of Si. During thermal treatment at 600°C, the pSi skeleton is partially oxidized to SiO2, resulting in a diffuse peak at 2θ equals approximately 22° corresponding to amorphous silica.
Figure 5.

The XRD patterns of empty functionalized microparticles pSi-C12, pSi-COOH, and pSiO2-C8; drug-loaded functionalized microparticles pSi-C12-RAP, pSi-COOH-RAP, and pSiO2-C8-RAP; and crystalline RAP. The XRD data were recorded in the 2θ range (5–75°).
The main peaks in the powder XRD pattern of crystalline RAP (Fig. 5) appear at 2θ: 7.2°, 9.9°, 10.2°, 11.1°, 12.5°, 14.5°, 15.3°, 15.5°, 16.2°, 20.0°, 20.4°, and 21.8°. However, no characteristic RAP peaks were observed from the RAP-loaded pSi samples (Fig. 5), even though the quantity of drug analyzed in each experiment was approximately the same. The XRD peaks observed from the pSi samples all could be assigned to either crystalline Si or to amorphous silica. The results are consistent with the formation of an amorphous polymorph of RAP within the nanoscale confines of the pSi carriers.
In Vitro RAP Release and pSi Degradation
Plotted in Figure 6 are RAP release profiles resulting from in vitro dissolution of pSi-COOH-RAP, pSi-C12-RAP, and pSiO2-C8-RAP microparticle formulations that were performed in a flow chamber with HBSS eluent. Drug release and silicon degradation were monitored simultaneously (Fig. 6).
Figure 6.
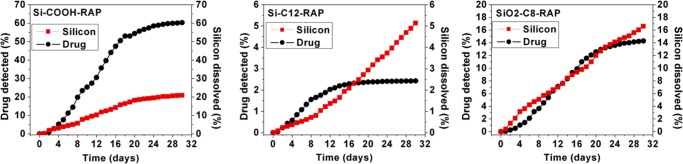
Rapamycin concentration detected by LC-MS/MS from flow chamber in vitro release test in HBSS (black circles, left axis), and cumulative silicon erosion detected as dissolved oxides of silicon by ICP-OES (red squares, right axis), for microparticle formulations pSi-COOH-RAP (left), pSi-C12-RAP (middle), and pSiO2-C8-RAP (right).
The drug-release profiles in Figure 6 show that the pSi-COOH-RAP formulation degrades fastest and releases RAP more rapidly than the other two formulations. During the 30-day test period, we measured 60% of the loaded drug had been released and 20% of the pSi matrix dissolved from pSi-COOH-RAP. Drug release from pSiO2-C8-RAP was less extensive, resulting in 14% of the drug being released and 17% pSi dissolved, whereas the pSi-C12-RAP formulation was the slowest with a 2.5% drug release and 5% pSi dissolved during the 30-day test period. The pSiO2-C8-RAP formulation displayed the closest temporal match between silicon degradation and RAP release. Because of this promising behavior, we performed additional in vitro release experiments using a microdialysis bag (Fig. 7) to directly compare the soluble RAP from the pSiO2-C8-RAP formulation to a solid crystalline RAP control. The appearance of dissolved RAP in the reservoir was then monitored for 19 days using positive ion mode ESI LC-MS/MS. The observed drug half-life was doubled and the drug exposure to retina or drug availability for retina uptake was increased by nearly six times (P = 0.0039, Wilcoxon signed rank test) (Table 2).
Figure 7.

Concentration of RAP transported across a dialysis membrane as a function of time for crystalline RAP (“Free RAP”) compared with the pSiO2-C8-RAP formulation (P = 0.0039, Wilcoxon signed rank test). Solvent was DI H2O and the experiment was maintained at 37°C.
Table 2.
Pharmacokinetic Parameters for pSiO2-C8-RAP Microparticle Formulation
|
Sample |
R2 |
Half-life Lambda_z, d_ |
Tmax, d |
Cmax, μg/mL |
Clast, μg/mL |
AUClast, μg·d/mL |
AUCINF_pred, μg·d/mL |
| Free-RAP | 0.76 | 1.95 | 1.00 | 511.83 | 3.04 | 2605.97 | 2608.89 |
| SiO2-C8-RAP | 0.96 | 4.14 | 3.00 | 2812.69 | 73.95 | 14810.77 | 15178.55 |
AUCINF_pred, area under the concentration-time curve from time 0 to the predicted end time point by extrapolation; AUClast, area under the concentration-time curve from time 0 to the last time point; Clast, drug concentration at the last observation time point; Cmax, maximum concentration detected; Tmax, time at which the concentration was maximum.
To confirm the presence of RAP isomers in the released extracts, we performed LC-MS/MS detection at m/z 936.5 at 12.57 minutes, corresponding to the retention time of the observed shoulder in the chromatograms (Fig. 8). An ion with m/z 936.5 was observed within this peak (Fig. 8C) and the fingerprints of the MS/MS spectrum of m/z 936.5 peak (Fig. 8D) confirmed that this species is an isomer of RAP.
Figure 8.

Electrospray ionization in positive ion mode MS spectra (full scan) (A, C) and the ESI-MS/MS spectra of m/z 936.5 [M+Na]+ peak, where M is RAP (B, D) from the release extracts of crystalline RAP (free drug) collected at day 9 from in vitro release following dialysis method. Liquid chromatography MS/MS detection at m/z 936.5 on extracts was performed at 12.38 minutes (A, B) and 12.57 minutes (C, D), corresponding with the retention time of the peak and the shoulder usually observed in the chromatograms. In both cases, ion with m/z 936.5 was observed within these peaks (A, C) with similar MS/MS fingerprints (B, D), confirming that these species are likely isomers of RAP.
Effect of Released Rapamycin on EA.hy926 Cells
Extracts released from the pSiO2-C8-RAP microparticle formulation from the microdialysis experiment were tested on EA.hy926 endothelial cells. The extracts showed significant inhibitory effect in the proliferation assay, relative to control extracts derived from the dissolution of crystalline commercial RAP (Fig. 9; Table 3). These data, along with the LC-MS data, confirmed that the released RAP was in its intact, biologically active form.
Figure 9.

Viability of EA.hy926 cells determined by WST-1 assay after exposure of cells to the liquid extracts of RAP released from the pSiO2-C8-RAP microparticle formulation or controls. Medium_no cells: 100% culture medium without cells; medium_cells: 100% culture medium and EA.hy926 cells; medium_DIH2O: the cells were treated with a mixture of 75% culture medium and 25% DI H2O; for pctrl_l (positive control, low dose), pctrl_m (positive control, middle dose), and pctrl_h (positive control, high dose), the cells were treated with a mixture of 25% (by volume) of RAP solution and 75% culture medium, at final RAP concentrations of 0.25 ng/mL (low), 2.5 ng/mL (middle), and 25 ng/mL (high). For nctrl_l (negative control, low dose), nctrl_m (negative control, middle dose), and nctrl_h (negative control, high dose), the cells were treated with a mixture of 75% (by volume) culture medium and 25% DI H2O containing the equivalent amount of DMSO contained in the positive controls at concentrations of 0.25 ng/mL (low), 2.5 ng/mL (middle), and 25 ng/mL (high). For Free_0.25ng/mL, Free_2.5ng/mL, and Free_25ng/mL, the cells were treated with a mixture of 25% (by volume) extracts released from free crystalline RAP diluted to final calculated concentrations of 0.25 ng/mL, 2.5 ng/mL and 25 ng/mL, and 75% culture medium. For NoRapSiO2C8_0.25, NoRapSiO2C8_2.5, and NoRapSiO2C8_25, the cells were treated with a mixture of 75% (by volume) culture medium and 25% of extracts released from empty pSiO2-C8 (no drug) formulation following the same dilution protocol as for the positive control samples containing RAP concentrations of 0.25 ng/mL, 2.5 ng/mL, and 25 ng/mL. For SiO2C8_0.25ng/mL, SiO2C8_2.5ng/mL, and SiO2C8_25ng/mL, the cells were treated with a mixture of 75% (by volume) culture medium and 25% of extracts released from the pSiO2-C8 formulation diluted to 0.25 ng/mL, 2.5 ng/mL, and 25 ng/mL.
Table 3.
Connecting Letters Report for Inhibition Assay on EA.hy926 Cells
|
Level |
Mean |
||||||||||
| medium_cells | A | 1.6149833 | |||||||||
| NoRapSiO2C8_2.5 | B | 1.4153250 | |||||||||
| medium_DIH2O | B | C | 1.3688333 | ||||||||
| nctrl_l | B | C | D | 1.3471375 | |||||||
| NoRapSiO2C8_25 | B | C | D | 1.3144125 | |||||||
| pctrl_l | B | C | D | E | 1.2937625 | ||||||
| nctrl_m | B | C | D | E | 1.2763875 | ||||||
| NoRapSiO2C8_0.25 | B | C | D | E | 1.2452875 | ||||||
| nctrl_h | C | D | E | F | 1.2127375 | ||||||
| Free_0.25ng/mL | D | E | F | G | 1.1628125 | ||||||
| SiO2C8_0.25ng/mL | E | F | G | H | 1.1472500 | ||||||
| SiO2C8_2.5ng/mL | F | G | H | I | 1.0363375 | ||||||
| pctrl_m | F | G | H | I | 1.0282000 | ||||||
| Free_25ng/mL | G | H | I | 1.0165375 | |||||||
| SiO2C8_25ng/mL | G | H | I | 1.0058063 | |||||||
| Free_2.5ng/mL | I | 0.9745750 | |||||||||
| pctrl_h | H | I | 0.9730625 | ||||||||
| medium_nocells | J | 0.1434833 |
Levels not connected by same letter are significantly different. Comparisons for all pairs using Tukey-KRAMER HSD, alpha = 0.05. Samples as defined in Figure 9.
Rapamycin derived from the pSiO2-C8-RAP microparticle formulation at a concentration of 2.5 ng/mL or 25 ng/mL showed inhibitory activity equivalent to the medium-dose and high-dose positive controls. The 2.5 ng/mL and 25 ng/mL control samples, which were obtained using the same dialysis setup except using commercial crystalline RAP instead of the microparticle formulation, also showed inhibitory activity equivalent to the medium-dose and high-dose positive controls. The DMSO additive showed no inhibitory effect on cells at concentrations as large as 0.1 % by volume.
In Vivo General and Ocular Safety
Following the intravitreal injections of pSiO2-C8-RAP, no general toxicity or ocular toxicity was observed. The mean body weight for the short-term study animals increased from 2.93 ± 0.15 to 3.23 ± 0.17 kg and from 4.07 ± 0.15 to 4.4 ± 0.25 kg for the long-term study rabbits. The retina was normal and the pSiO2-C8-RAP particles were suspended in clear vitreous humor with clear view of retina and optic nerve (Fig. 10, top). Both IOP and ERGs of the study eyes were comparable with their contralateral eyes (Table 4).
Figure 10.

The top two fundus images were from the same rabbit eye that received RAP-loaded pSiO2-C8 particles. Left image was taken 1 week postinjection and the right image taken 8 weeks postinjection, showing clear view of fundus and RAP-loaded pSiO2-C8 particles in vitreous. The numbers of the particles were fewer in the 8-week image but still visible. The histology images from the 2-week (2wks) study (bottom left) and 8-week study (bottom right) were taken at visual streak, which is indicated by high density of ganglion cells. Compared with the left eye (OS), the study eyes (OD) did not show abnormality in retina and choroid. The separation of RPE from underneath choroid in the frame of 8wks OS was caused during tissue processing.
Table 4.
Drug-Injected Eyes Versus Their Fellow Eyes for IOP and ERG
|
Drug Eyes |
Fellow Eyes |
Difference |
PValue |
|
| ERG b-wave amplitude | 54.14 μV | 53.7 μV | 0.44 | 0.91 |
| ERG a-wave amplitude | −6.62 μV | −5.99 μV | 0.63 | 0.49 |
| IOP | 9.74 mm Hg | 9.89 mm Hg | 0.15 | 0.87 |
Paraffin section and hematoxylin and eosin staining revealed normal retinal structure for both 2-week animals (Fig. 10, bottom left) and 8-week animals (Fig. 10, bottom right), as compared with their fellow eyes.
Discussion
Rapamycin possesses broad biological activity and is being extensively tested in many clinical trials for various diseases worldwide. Due to its low water solubility and short vitreous half-life,32,33 its ocular local use for posterior eye diseases is limited. Most recently, a proprietary RAP depot-forming formulation was reported to be safe for subconjunctival or intravitreal injections and appeared to be effective for uveitis34,35 or macular edema.36 This formulation was reported to provide up to 60 days of sustained release with one administration.37 Intravitreal depot-forming formulation is likely similar to intravitreal triamcinolone acetonide (TA), which provides a sustained drug release; however, vitreous free drug level is excessively high during most of the release course and can cause adverse ocular events, such as high IOP and cataract formation. Even with 4 mg TA intravitreal injection, the vitreous free drug level reaches above 1000 ng/mL.38 Similarly, the recent report from the depot-forming sirolimus demonstrated that even a 220-μg intravitreal injection yielded a free drug concentration near 1000 ng/mL.37 Rapamycin is a very potent drug and high drug concentration could cause adverse effects in a sustained release system. Indeed, a most recent phase I/II clinical trial demonstrated retinal toxicity from intravitreal injection of this proprietary depot-forming sirolimus in eyes with dry AMD.39
In the current study, we were aiming at developing a longer and better sustained delivery system for intravitreal sirolimus. We prepared three types of functionalized pSi microparticles, with varying polarity (hydrophobicity/hydrophilicity), as well as hydrolytic stability against dissolution in aqueous media. The pSi surface functionalization rationale followed in this study involved modifying pSi to make it more hydrophobic to enhance interaction between the hydrophobic RAP drug and the nanocarrier surface for increased drug-loading efficiency. The CBET parameter can be used to rank the order of hydrophilicity: pSiO2 > pSi-COOH > pSiO2-C8 > pSi > pSi-C12 (Table 1). Grafting of 1-dodecene (pSi-C12) created a very hydrophobic surface and in comparison, grafting of undecylenic acid (pSi-COOH) created a mildly hydrophobic surface due to the carboxylic acid group and the further partial oxidation process. Finally, silanization of partially oxidized silicon (pSiO2-C8) created a slightly more hydrophobic surface than that of pSi-COOH. The oxidized silicon surface of both of these sample types is expected to be hydrophilic, but the grafted alkyl chains impart some hydrophobic character that matches hydrophobicity of RAP.
The surface chemistry had an effect on the efficiency of RAP loading, which followed the following order (μg/mg): pSiO2-C8 (105 ± 18) > pSi-COOH (68 ± 8) > pSi-C12 (36 ± 6), as measured by LC-MS. Compared with the covalent drug loading reported previously, the current adsorption RAP loading to pSiO2-C8 was also greater and above 10% by weight. The greater capacity of pSiO2-C8 for the RAP can be ascribed to the hydrophobic surface chemistry combined with the limited steric hindrance offered by the shorter (C8) alkyl chains at the surface. Notably, the XRD data show that the loaded RAP is amorphous in all the pSi formulations studied. This suggests that the nanoscale confines of the pSi carriers restrict the RAP precipitate to an amorphous polymorph. This is an important result, because the crystalline nature of many drugs often results in unfavorable or erratic pharmacokinetics behavior. In particular, the ability of the pSi nanostructure to generate an amorphous polymorph of RAP suggests that it can be used to control the free drug availability of a hydrophobic drug.11,21,40 In the present work, the in vitro dynamic drug release revealed that the availability of the drug for target tissue intake increased by nearly 6-fold (compared with crystalline RAP) when RAP was loaded into the pSiO2-C8 particles.
In our previous work, grafting of alkyl groups has been used to covalently attach drug molecules directly to a pSi matrix, and in that case the drug releases at the same rate as the matrix erodes.22 In the present case, the RAP drug was loaded by infiltration, relying on hydrophobic van der Waals interactions to retain the drug in the porous matrix. The drug-release mechanism from these pSi carriers is thus a combination of diffusion and degradation. Whereas the RAP release profiles are affected by surface chemistry (Fig. 6), they are not as strongly correlated to degradation of the porous matrix as in the systems where the drug is directly bonded to the pSi matrix. We did not observe a burst release of drug in the current systems, probably due to the hydrophobic character of the reservoirs, together with the hydrophobicity of RAP, which does not favor rapid entry of the payload into the aqueous media. The rate of drug release to approximate pSi matrix degradation is an important criterion for the infiltration loaded pSi system; otherwise, empty particles will still be lingering in the vitreous after the therapeutic effect of the payload has disappeared. The data for the pSi-C12-RAP formulation shows that no additional drug is detected after approximately 2 weeks of exposure to the eluent, whereas the dissolved silicon assay indicates that this formulation continuously degrades (by 5%) through the entire 4-week test period. The analysis of the mass spectral data indicated that RAP released from the pSi-C12-RAP formulation also was chemically altered. Thus, it is likely that this formulation also is responsible for the degradation of RAP. It should be pointed out that RAP is itself hydrolytically unstable in aqueous solutions41; therefore, all of the measured drug-release values in this work may be underestimates of the actual. The mass spectral data used to determine drug release in Figure 6 quantify only the intact drug molecule and not the degradation products. Of the three formulations studied, pSiO2-C8-RAP showed the best performance in terms of drug-loading capacity, minimizing chemical degradation of RAP, and simultaneously releasing active drug with simultaneous carrier degradation. Whereas drug release in the pSi-COOH-RAP system seemed to be dominated by drug leaching, release of RAP from the pSiO2-C8-RAP formulation was more tightly correlated with degradation of the pSi matrix (Fig. 6; up to 14% drug released, 17% Si dissolved). We further tested the pSiO2-C8-RAP formulation in a refined in vitro cellular inhibition study using a modified dialysis method. Rapamycin is unstable in PBS and HEPES buffer,41 therefore experiments were carried out in DI H2O. The released drug showed a positive inhibitory effect on endothelial cells, confirming that this formulation releases the drug in a biologically active form. Consistent with the amorphous nature of the drug within the pSi carrier as discussed above, we found that the availability of the free drug was increased by 6-fold compared with crystalline RAP, which has a very low water solubility.
In summary, the current dynamic release study revealed approximately 20% of the total payload leached out within 30 days, which may translate into a sustained release of 150 days. Indeed. the safety study in vivo rabbit eye demonstrated considerable numbers of RAP-loaded pSiO2-C8 particles were still present in rabbit vitreous at 8 weeks after a single intravitreal injection of 2.9 (±0.39) mg of the delivery system (containing 305 μg RAP). In addition, with this system a burst release was not observed, which is a preciously favorable feature in the drug-delivery arena. This system may provide sustained long-term release of RAP and warrants a full in vivo pharmacodynamic investigation.
Acknowledgments
The authors thank Yunxuan Su, PhD, (MS-facility, University of California San Diego) for helpful discussions of LC-MS investigation, and Anna Nilsson for English proofreading of this manuscript.
Supported by National Institutes of Health Grant EY020617 (LC) and National Science Foundation Grant DMR-1210417 (MJS).
Disclosure: A. Nieto, None; H. Hou, None; S.W. Moon, None; M.J. Sailor, Spinnaker Biosciences (I); W.R. Freeman, Spinnaker Biosciences (C, I); L. Cheng, Spinnaker Biosciences (C, I)
References
- 1. Ozaki H, Seo MS, Ozaki K, et al. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol. 2000; 156: 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenthal R, Wohlleben H, Malek G, et al. Insulin-like growth factor-1 contributes to neovascularization in age-related macular degeneration. Biochem Biophys Res Commun. 2004; 323: 1203–1208. [DOI] [PubMed] [Google Scholar]
- 3. Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011; 364: 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heier JS, Brown DM, Chong V, et al. Intravitreal aflibercept (VEGF Trap-Eye) in wet age-related macular degeneration. Ophthalmology. 2012; 119: 2537–2548. [DOI] [PubMed] [Google Scholar]
- 5. Demidenko ZN, Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell Cycle. 2011; 10: 1557–1562. [DOI] [PubMed] [Google Scholar]
- 6. Slomiany MG, Rosenzweig SA. IGF-1-induced VEGF and IGFBP-3 secretion correlates with increased HIF-1 alpha expression and activity in retinal pigment epithelial cell line D407. Invest Ophthalmol Vis Sci. 2004; 45: 2838–2847. [DOI] [PubMed] [Google Scholar]
- 7. Stahl A, Paschek L, Martin G, et al. Rapamycin reduces VEGF expression in retinal pigment epithelium (RPE) and inhibits RPE-induced sprouting angiogenesis in vitro. FEBS Lett. 2008; 582: 3097–3102. [DOI] [PubMed] [Google Scholar]
- 8. Dejneka NS, Kuroki AM, Fosnot J, Tang W, Tolentino MJ, Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol Vis. 2004; 10: 964–972. [PubMed] [Google Scholar]
- 9. Kolosova NG, Muraleva NA, Zhdankina AA, Stefanova NA, Fursova AZ, Blagosklonny MV. Prevention of age-related macular degeneration-like retinopathy by rapamycin in rats. Am J Pathol. 2012; 181: 472–477. [DOI] [PubMed] [Google Scholar]
- 10. Simamora P, Alvarez JM, Yalkowsky SH. Solubilization of rapamycin. Int J Pharm. 2001; 235: 25–29. [DOI] [PubMed] [Google Scholar]
- 11. Kim MS, Kim JS, Park HJ, Cho WK, Cha KH, Hwang SJ. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011; 6: 2997–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shanmuganathan VA, Casely EM, Raj D, et al. The efficacy of sirolimus in the treatment of patients with refractory uveitis. Br J Ophthalmol. 2005; 89: 666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drug delivery applications. J Pharm Sci. 2008; 97: 632–653. [DOI] [PubMed] [Google Scholar]
- 14. Chhablani J, Nieto A, Hou H, et al. Oxidized porous silicon particles covalently grafted with daunorubicin as a sustained intraocular drug delivery system. Invest Ophthalmol Vis Sci. 2013; 54: 1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kovalainen M, Monkare J, Mäkilä E, Salonen J, Lehto VP, Herzing K-H, Järvinen K. Mesoporous silicon (pSi) for sustained peptide delivery: effect of pSi microparticle surface chemistry on peptide YY3-36 release. Pharm Res. 2012; 29: 837–846. [DOI] [PubMed] [Google Scholar]
- 16. Martinez JO, Chiappini C, Ziemys A, et al. Engineering multi-stage nanovectors for controlled degradation and tunable release kinetics. Biomaterials. 2013; 34: 8469–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hou H, Nieto A, Ma F, Freeman WR, Sailor MJ, Cheng L. Tunable sustained intravitreal drug delivery system for daunorubicin using oxidized porous silicon. J Control Release. 2014; 178: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang C, Hou H, Nan K, Sailor MJ, Freeman WR, Cheng L. Intravitreal controlled release of dexamethasone from engineered microparticles of porous silicon dioxide. Exp Eye Res. 2014; 129: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anglin EJ, Cheng L, Freeman WR, Sailor MJ. Porous silicon in drug delivery devices and materials. Adv Drug Deliv Rev. 2008; 60: 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salonen J, Laitinen L, Kaukonen AM, et al. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. J Control Release. 2005; 108: 362–374. [DOI] [PubMed] [Google Scholar]
- 21. Wang F, Hui H, Barnes TJ, Barnett C, Prestidge CA. Oxidized mesoporous silicon microparticles of improved oral delivery of poorly soluble drugs. Mol Pharm. 2010; 7: 227–236. [DOI] [PubMed] [Google Scholar]
- 22. Wu E, Andrew J, Cheng L, Freeman W, Pearson L, Sailor M. Real-time monitoring of sustained drug release using the optical properties of porous silicon photonic crystal particles. Biomaterials. 2011; 32: 1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu EC, Andrew JS, Buyanin A, Kinsella JM, Sailor MJ. Suitability of porous silicon microparticles for the long-term delivery of redox-active therapeutics. Chem Commun (Camb). 2011; 47: 5699–5701. [DOI] [PubMed] [Google Scholar]
- 24. Park JS, Kinsella JM, Jandial DD, Howell SB, Sailor MJ. Cisplatin-loaded porous Si microparticles capped by electroless deposition of platinum. Small. 2011; 7: 2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Streit F, Armstrong VW, Oellerich M. Rapid liquid chromatography–tandem mass spectrometry routine method for simultaneous determination of sirolimus, everolimus, tacrolimus, and cyclosporine A in whole blood. Clin Chem. 2002; 48: 955–958. [PubMed] [Google Scholar]
- 26. Kim JS, Beadle JR, Freeman WR, et al. A novel cytarabine crystalline lipid prodrug: hexadecyloxypropyl cytarabine 3′,5′-cyclic monophosphate for proliferative vitreoretinopathy. Mol Vis. 2012; 18: 1907–1917. [PMC free article] [PubMed] [Google Scholar]
- 27. Davson H, Luck CP. Chemistry and rate of turnover of the ocular fluids of the bush baby (Galago crassicaudatus agisymbanus). J Physiol. 1959; 145: 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nieto A, Hou H, Sailor MJ, Freeman WR, Cheng L. Ocular silicon distribution and clearance following intravitreal injection of porous silicon microparticles. Exp Eye Res. 2013; 116: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gregg SJ, Sing KSW. Adsorption, Surface Area and Porosity. 2nd ed. London: Academic Press Inc.; 1982: 112. [Google Scholar]
- 30. Buriak JM. Organometallic chemistry on silicon and germanium surfaces. Chem Rev. 2002; 102: 1272–1308. [DOI] [PubMed] [Google Scholar]
- 31. Boukherroub R, Petit A, Loupy A, Chazalviel JN, Ozanam F. Microwave-assisted chemical functionalization of hydrogen-terminated porous silicon surfaces. J Phys Chem B. 2003; 107: 13459–13462. [Google Scholar]
- 32. Buech G, Bertelmann E, Pleyer U, Siebenbrodt I, Borchert HH. Formulation of sirolimus eye drops and corneal permeation studies. J Ocul Pharmacol Ther. 2007; 23: 292–303. [DOI] [PubMed] [Google Scholar]
- 33. Douglas LC, Yi NY, Davis JL, Salmon JH, Gilger BC. Ocular toxicity and distribution of subconjunctival and intravitreal rapamycin in horses. J Vet Pharmacol Ther. 2008; 31: 511–516. [DOI] [PubMed] [Google Scholar]
- 34. Sen HN, Larson TA, Meleth AD, Smith WM, Nussenblatt RB. Subconjunctival sirolimus for the treatment of chronic active anterior uveitis: results of a pilot trial. Am J Ophthalmol. 2012; 153: 1038–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nguyen QD, Ibrahim MA, Watters A, et al. Ocular tolerability and efficacy of intravitreal and subconjunctival injections of sirolimus in patients with non-infectious uveitis: primary 6-month results of the SAVE Study. J Ophthalmic Inflamm Infect. 2013; 3: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krishnadev N, Forooghian F, Cukras C, et al. Subconjunctival sirolimus in the treatment of diabetic macular edema. Graefes Arch Clin Exp Ophthalmol. 2011; 249: 1627–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mudumba S, Bezwada P, Takanaga H, et al. Tolerability and pharmacokinetics of intravitreal sirolimus. J Ocul Pharmacol Ther. 2012; 28: 507–514. [DOI] [PubMed] [Google Scholar]
- 38. Inoue M, Takeda K, Morita K, Yamada M, Tanigawara Y, Oguchi Y. Vitreous concentrations of triamcinolone acetonide in human eyes after intravitreal or subtenon injection. Am J Ophthalmol. 2004; 138: 1046–1048. [DOI] [PubMed] [Google Scholar]
- 39. Petrou P, Cunningham D, Shimel K, et al. Intravitreal sirolimus for the treatment of geographic atrophy: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci. 2014; 56: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tang L, Saharay A, Fleischer W, Hartman PS, Loni A, Canham LT, Coffer JL. Sustained antifungal activity from a ketoconazole-loaded nanostructured mesoporous silicon platform. Silicon. 2013; 5: 213–217. [Google Scholar]
- 41. Rouf MA, Bilensoy E, Vural I, Hincal AA. Determination of stability of rapamycin following exposure to different conditions. Eur J Pharm Sci. 2007; 32: S46. [Google Scholar]