

Abstract
Glucagon-like peptide-1 (GLP-1) is an incretin that plays important physiological roles in glucose homeostasis. Produced from intestine upon food intake, it stimulates insulin secretion and keeps pancreatic β-cells healthy and proliferating. Because of these beneficial effects, it has attracted a great deal of attention in the past decade, and an entirely new line of diabetic therapeutics has emerged based on the peptide. In addition to the therapeutic applications, GLP-1 analogs have demonstrated a potential in molecular imaging of pancreatic β-cells; this may be useful in early detection of the disease and evaluation of therapeutic interventions, including islet transplantation. In this Perspective, we focus on GLP-1 analogs for their studies on improvement of biological activities, enhancement of metabolic stability, investigation of receptor interaction, and visualization of the pancreatic islets.
1. Introduction
Glucagon-like peptide-1 (GLP-1) is an endogenous incretin and plays a significant role in glucose homeostasis.1−6 GLP-1 is a 30 amino acid containing peptide secreted from intestinal L-cells in response to food intake. It is produced by post-translational processing of preproglucagon, a precursor of many glucagon related peptides. It exists in two equipotent forms, GLP-1(7–36)-NH2 and GLP-1(7–37), the former being more abundant. It binds to and activates the GLP-1 receptor (GLP-1R) belonging to class B family of G-protein-coupled receptors (GPCRs) in order to exert its regulatory functions. While GLP-1R is found in many organs, including pancreas, brain, heart, kidney, and GI tract,7,8 a recent study revealed that it is exclusively expressed in the pancreatic β-cells and rarely in α- and δ-cells, as shown by colocalization experiments of GLP-1R mRNA and protein with islet cell hormones in mice, rats, and humans.9 Activation of the receptor on the β-cells leads to a rapid increase in the levels of cAMP and intracellular calcium followed by insulin exocytosis in a glucose-dependent manner. Although GLP-1R in the α-cells is <0.2% of that in the β-cells, GLP-1 inhibits glucagon secretion by 50% through modulation of calcium channel activity.10
Since its discovery in the early 1980s,11,12 GLP-1 has attracted significant attention because of its unique antidiabetic functions.13 GLP-1 therapy demonstrated that the peptide potentiates insulin secretion in both healthy and type 2 diabetic patients. Unlike other diabetes drugs, the insulinotropic effect of GLP-1 is self-limiting, as it subsides once the plasma glucose level is lowered to normal range, reducing the risk of hypoglycemia. In addition, GLP-1 regulates postprandial glucose elevation through several other mechanisms, including promoting insulin gene transcription, stimulating pancreatic β-cell proliferation and neogenesis, inhibiting β-cell apoptosis, and blocking glucagon release. It also prevents gastric emptying and induces satiety, leading to body weight decrease. GLP-1 therapy seems to offer cardioprotective effects as well.14
All of these physiological effects underscore GLP-1 as a promising therapeutic agent for treating type 2 diabetes (T2D). However, the endogenous GLP-1 has a very short half-life, owing to rapid metabolic degradation by proteases like dipeptidyl peptidase IV (DPP-IV) and neutral endopeptidase 24.11 (NEP 24.11).15,16 DPP-IV cleaves the peptide bond in Ala8-Glu9, and the resulting metabolite GLP-1(9–36)-NH2 is found to have 100-fold lower binding affinity compared to the intact peptide.17 The metabolite also exhibits negligible agonistic activity (>10000-fold decrease).17 As a result, biologically active GLP-1 constitutes only approximately 10–20% of total plasma GLP-1, as determined by the combination of HPLC, specific radioimmunoassays (RIAs) and sensitive enzyme-linked immunosorbent assays (ELISA).18 Although the metabolite does not induce insulin release or glucose metabolism,19 a potential role was suggested for inhibiting hepatic glucose production in an insulin-resistant state of obesity.20 It also appears to increase glucose disposal independent of insulin effects21 as well as stimulates myocardial glucose uptake and improves systemic hemodynamics.22 On the other hand, NEP 24.11 cleaves GLP-1 at multiple sites, particularly at the peptide bonds in Asp15-Val16, Ser18-Tyr19, Tyr19-Leu20, Glu27-Phe28, Phe28-Ile29, and Trp31-Leu32.15 Although the DPP-IV degradation is a major cause of GLP-1 function loss, an investigation with a specific NEP 24.11 inhibitor candoxatril suggested that endoproteolysis by NEP 24.11 may contribute up to 50% of GLP-1 degradation in vivo.23
To date, a couple of stable GLP-1 agonists have received FDA approval as a new class of diabetic drugs, exenatide (1, GLP-1R agonist) and liraglutide (2, GLP-1 analog). Marketed as Byetta by AstraZeneca, 1 is a synthetic version of exendin-4, a 39 amino acid containing peptide isolated from the saliva of Gila monster.24 Marketed as Victoza by Novo Nordisk, 2 is a GLP-1 analog prepared by substituting Lys34 for Arg and attaching a C16 fatty acid to Lys26 via a γ-glutamyl spacer.25 Marketed as Bydureon (AstraZeneca), exenatide long-acting release (LAR) is a slow releasing subcutaneous depot form of 1.26 In addition, DPP-IV inhibitors have been developed to prolong the activity of the endogenous GLP-1.27 Besides the therapeutic impact, GLP-1 analogs demonstrated high potential as molecular imaging agents for noninvasively assessing pancreatic β-cell mass, as the GLP-1 receptor is highly localized in the β-cells.9
2. Structure–Activity Relationship of GLP-1
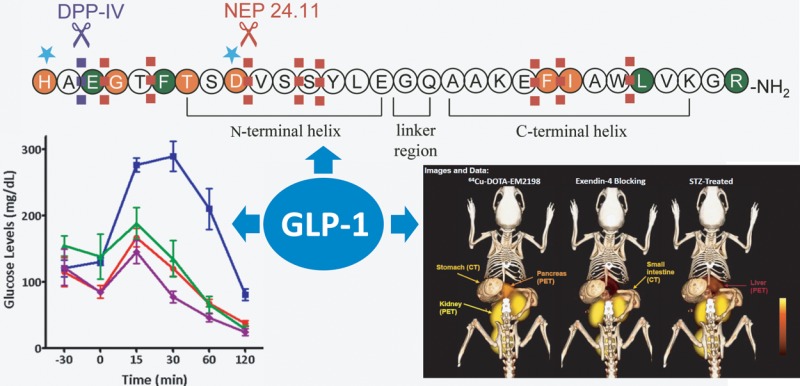
GLP-1, after binding to its receptor, triggers a conformational change of the receptor, leading to activation of an associated Gs protein by exchanging its bound GDP with a GTP. This results in dissociation of Gs protein from the receptor and increases secondary messengers like cAMP and Ca2+. In order to investigate the significance and function of amino acid residues in the sequence of the peptide, alanine scanning studies have been carried out.28,29 Adelhorst and co-workers have systematically replaced each residue with Ala and identified key amino acids strongly contributing to receptor interaction.29 It appears that His7, Gly10, Phe12, Thr13, Asp15, Phe28, and Ile29 play an important role in receptor binding, as marked decrease in binding affinity was caused by each Ala substitution. On the other hand, His7, Gly10, Asp15, and Phe28 are critical for receptor activation, as their Ala substitutions resulted in a substantial decrease in cAMP production. On the basis of these results, it is hypothesized that the N-terminal region of GLP-1 is more critical for activating the receptor, whereas the C-terminal region mainly contributes to receptor binding (Figure 1). However, this concept should be taken cautiously, since many residues in the N-terminal region are still found to be essential for maximal receptor interaction.
Figure 1.
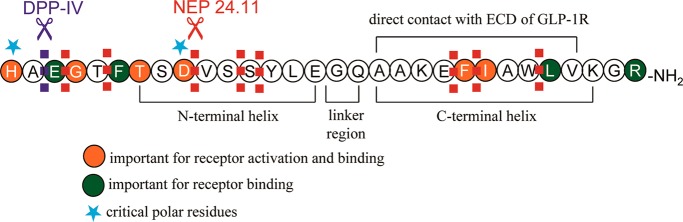
Sequence of GLP-1 and critical residues for receptor interaction.
His7
His7 is found to be a critical residue for both receptor binding and activation, as evidenced by the alanine scanning studies.28,29 A histidine at the first position is, in fact, highly conserved in the glucagon family that interacts with class B GPCRs for their functions.6 Deletion of His7 resulted in a significant loss of affinity (Kd = 33 nM compared to 0.3 nM of GLP-1), and the activity of the truncated peptide became 82% lower than that of GLP-1.30 To investigate the significance of the positively charged side chain, His7 was replaced with Arg and Lys.30 These substitutions substantially reduced receptor binding (270- to 1300-fold) and activation (57–88% lower activity), suggesting a special role of the imidazole ring in receptor interaction. When His7 was substituted for Tyr, it resulted in a significant reduction of binding affinity (Kd = 52 nM) and biological activity (66% of GLP-1 response).31 However, replacement with Phe was found to retain the biological activity close to GLP-1, suggesting aromaticity is an important factor for receptor interaction.32 Taken together, it can be summarized that both the aromatic character and the positive charge of the imidazole ring strongly contribute to receptor recognition and function. However, a bulky aromatic group like the indole ring of Trp reduced potency, indicating that a steric factor should be also considered.32
It was suspected that the α-amino group of His7 plays a role in DPP-IV activity. Gallwitz et al. synthesized several GLP-1 analogs containing alterations at the N-terminus of His7, such as N-methylation, α-methylation, des-amination, and imidazolelactic acid substitution.31 This study revealed that all of the GLP-1 analogs except the α-methylated ones were hardly degraded by DPP-IV in vitro. These modified GLP-1 analogues showed a marginal reduction in receptor affinity (2- to 5-fold), except des-amino-GLP-1 that lost binding by 15-fold. All of these peptides promoted intracellular cAMP production at concentrations comparable to GLP-1. In addition, modification of His7 with N-pyrogluatmylation and N-acetylation decreased both receptor binding (18- to 89-fold) and activation (3- to 6-fold).33 Despite such poor binding, both analogs exerted a potent insulinotropic effect in vitro. Another studies showed that a GLP-1 analog containing a glucitol or mPEG2K attached at the N-terminus of His7 showed outstanding DPP-IV stability.34,35
Ala8
Since Ala8 is the target of DPP-IV, much work has focused on this residue. Substitution of Ala8 for Gly resulted in only a slight decrease in binding affinity (3-fold) with comparable agonist activity to GLP-1.36 [Gly8]-GLP-1 showed a comparable insulinotropic effect in diabetic animal models. The introduction of Gly8 circumvented the DPP-IV degradation and led to a longer half-life, as a single injection of 0.1 nmol of the peptide to diabetic mice normalized fasting hyperglycemia and glucose intolerance for several hours, whereas the activity of 1 nmol of the native GLP-1 vanished in a few minutes.
Deacon et al. investigated the substitution of Ala8 with Thr, Gly, Ser, and 2-aminoisobutyric acid (Aib).37 They found that only [Aib8]- and [Gly8]-GLP-1(7–36)-NH2 exhibited comparable receptor binding to GLP-1 (IC50 of 0.45 and 2.8 nM, respectively, compared to 0.78 nM of GLP-1). While the other three analogs showed slow degradation, [Aib8]-GLP-1(7–36)-NH2 was found to be completely resistant to DPP-IV and exerted the highest insulinotropic effect. The stereochemical configuration at position 8 appeared to have little effect on receptor recognition, as replacement with d-Ala retained comparable receptor binding (IC50 = 0.15 nM compared to 0.26 nM of GLP-1) and biological activity (EC50 = 0.8 nM compared to 0.8 nM of GLP-1).32
Substitution of Ala8 with Ser reduced receptor binding affinity (11-fold) and receptor stimulation potency (19-fold), presumably resulting from the introduction of a polar side chain group.32 The lack of tolerance of polar residues at this position was supported by an experiment replacing Ala with Val and led to only a marginal reduction in biological activity (3-fold). However, steric factor becomes important when the size of the amino acid at position 8 exceeds a certain threshold, as substitution with Leu lowered the potency by 220-fold.
Glu9
This position is conserved for negatively charged amino acids like Glu and Asp among the secretin family (class B GPCRs) except glucagon.32 This suggests that the carboxylate side chain of Glu9 is required for receptor interaction, which was supported by Glu9 being substituted with Asp without losing receptor binding affinity and potency examined on RIN T3 cells. Interestingly, Xiao et al. reported a contradicting result that substitution with Asp led to a 30-fold potency decrease, although the comparable binding affinity was determined on CHO cells.38 Despite the speculation, hydrophobic amino acids like Leu or Met successfully replaced Glu9 while retaining the same potency of GLP-1, suggesting that the negatively charged side chain of Glu9 may not form an ion pair with a basic residue in the receptor or the peptide.32
Introduction of Lys at position 9 showed a significant loss of binding and potency (830- and 160-fold, respectively) when the resulting peptide was tested on RIN T3 cells.32 In another experiment, [Lys9]-GLP-1 led to no cAMP production on BRIN-BD11 cells, although a moderate loss of binding affinity (17-fold decrease) was observed on CHL fibroblast cells.39 Even though the peptide remained intact when incubated with DPP-IV for 12 h, it showed no insulinotropic action or glucose-lowering capability in ob/ob mice. Substitution with Ala, Ser, and Val resulted in reduced potency (30-, 4-, and 130-fold, respectively).32,38 The stereochemical configuration of Glu9 appears to be crucial, since replacement with d-Glu led to a 130-fold decrease in potency, whereas binding affinity was moderately affected (33-fold decrease).
When Pro, Phe, and Tyr were introduced in lieu of Glu9, the resulting peptides exhibited a comparable or marginally increased insulinotropic effect compared to GLP-1 even though their binding affinity and potency were compromised.40 In particular, [Pro9]-GLP-1 showed a remarkable capability of moderating plasma glucose excursion and enhancing the circulating insulin level in severely insulin-resistant obese diabetic mice. It also substantially reduced the susceptibility to DPP-IV degradation (t1/2 > 12 h compared to 5.2 h of GLP-1).
Gly10
The alanine scanning studies indicate that Gly10 is important for receptor interaction, particularly receptor activation. Meng et al. placed a hexafluoroleucine in the sequence of GLP-1, aiming to improve bioactivity and bioavailability.41 However, the substitution at position 10 only led to a dramatic decrease in binding affinity and potency (60- and 67-fold, respectively).
Asp15
Substitution of Asp15 for Arg was found to significantly damage receptor binding and activity (710- and 90-fold, respectively), indicating the importance of the negatively charged residue at this position.42 While a severe loss of activity (10000-fold) was reported when it was substituted for Ala,29 replacement with Glu led to only a moderate loss in affinity (31-fold), confirming the requirement of a negatively charged amino acid at position 15.32 The stereochemical configuration of this residue appears to be critical, since replacement with d-Asp resulted in a substantial loss of binding affinity (570-fold).43
Val16, Ser18, Glu21
It was found that Val16 can be substituted with a bulky hydrophobic amino acid without significant impact on receptor interaction. For example, substitution with Tyr resulted in a similar affinity and activity.42 Likewise, alanine substitution at Val16 did not show any drastic change either. Whereas substitution of Ser18 for Ala did not affect receptor binding and activation, replacement with a charged amino acid Lys resulted in a 4-fold decrease in binding affinity.42 Substitution of Glu21 for Ala moderately affected receptor binding (15-fold decrease), but replacement with a smaller amino acid Gly severely reduced the affinity (60-fold decrease).42
Gly22
As an amino acid without any side chain, Gly at position 22 has been replaced with various amino acids bearing side chain groups. It appears that Gly22 can be substituted for Aib with retention of comparable binding affinity and potency to GLP-1.44 Azad et al. used this position to attach a bulky metal chelator DOTA through Lys22 for β-cell imaging.45 Two analogs were prepared, Lys22-In-DOTA-GLP-1(7–36) and Lys22-AEEA-In-DOTA-GLP-1(7–36); the latter showed a moderate loss of binding affinity and potency (19- and 6-fold, respectively), remarkably tolerating such a bulky substituent placed in the middle of the GLP-1 sequence.
Lys26
Green and co-workers attached a fatty acid chain at the ε-amino group of Lys26 to avoid renal clearance for prolonging half-life and increasing stability over DPP-IV.46 Whereas [Lys(palmitoyl)26]-GLP-1 has a weak affinity and potency compared to GLP-1 (86- and 23-fold, respectively), all of the palmitoylated Lys26-containing peptides showed a marginal increase (1.3- to 3.4-fold) in dose-dependent stimulation of insulin secretion on insulin-secreting BRIN-BD11 cells. A stable GLP-1 receptor agonist, 2, was created by attaching a palmitoyl group to Lys26 through a γ-glutamyl spacer together with substitution of Lys34 for Arg.25 This peptide demonstrated a potent agonist activity (EC50 = 61 nM compared to 55 nM of GLP-1 on BHK cells, IC50 = 0.11 nM) and superior stability against proteolytic degradation (half-life of 13 h compared to 2 min of GLP-1).
Lys26 was also exploited to attach bulky metal chelators like DOTA for constructing β-cell imaging agents.25,45 However, coupling DOTA either directly to the side chain of Lys or through a linker significantly diminished binding affinity (200- and 500-fold, respectively). On the other hand, various carbohydrate moieties were introduced at position 26 in an attempt to increase proteolytic stability and in vivo glucose-lowering potency.47 Substitution of Lys26 for Asn bearing N-acetylglucosamine (GlcNAc), N-acetyllactosamine (LacNAc), or α-2,6-sialyl-N-acetyl-lactosamine (sialyl LacNAc) resulted in a moderate loss of binding affinity and potency (2- to 8-fold) but higher resistance to the DPP-IV degradation compared to GLP-1. Similarly, PEGylation of Lys26 with mPEG2K was found to have a negligible effect on receptor activation.35
Glu27
Alanine scanning studies suggested that Glu27 is not an important residue for receptor interaction. In agreement with this, it was found that Glu27 can be even substituted with an oppositely charged Lys without losing binding affinity and potency (2-fold decrease).42
Phe28
Phe28 was identified as a critical residue for both receptor binding and activation. Substitution for Ala brought in greater than 1000-fold decrease in binding affinity and potency.28,29 While Phe28 is essential for affinity and activity, it is interesting that replacement with a synthetic residue hexafluoroleucine resulted in only a moderate loss in binding affinity and biological activity (10- and 5-fold, respectively).41
Ile29, Leu32
The alanine scanning studies revealed that Ile29 is critical for receptor binding. When a hexafluoroleucine was introduced at position 29 or 32, a moderate loss in binding affinity and biological activity was observed.41 Its substitution at position 29 showed a moderate potency and affinity (4- and 7-fold decrease, respectively) as well as a marginal reduction in the rate of DPP-IV degradation. Similar results were observed with the substitution at position 32 (2- and 28-fold decrease in potency and affinity, respectively).
Lys34
Connecting DOTA to Lys34 directly or through a linker was reported for imaging pancreatic β-cells.45 However, these bulky substituents resulted in a substantial loss in binding affinity (50- to 100-fold). Replacement of Lys34 with an N-glycosylated Asn34 (e.g., GlcNAc, LacNAc, and sialyl LacNAc) resulted in similar binding affinity and potency to GLP-1.47 Additionally, PEGylation on the side chain of Lys34 with mPEG2K resulted in comparable potency to GLP-1.35 This Lys34-PEG-GLP-1 displayed 93% insulinotropic effect in rat pancreatic islets and higher glucose lowering effect as well as high stability to DPP-IV. Compared to 2, a 2-fold decrease in potency was observed when Lys34 was palmitoylated with a γ-glutamyl spacer together with substitution of Lys26 for Arg.25
Gly35,37
Substitution of Gly35 for Aib, together with introduction of Aib at position 8, resulted in a potent analog called taspoglutide (3).48 It showed outstanding resistance to DPP-IV (half-life of 13 h in human plasma upon subcutaneous administration).49 It is interesting that the double Aib substitutions led to comparable receptor binding and potency (IC50 = 1.1 nM, EC50 = 0.06 nM compared to GLP-1 (IC50 = 1.5 nM, EC50 = 0.08 nM)), whereas a single Aib at position 8 brought in a 4-fold decrease in potency and 2-fold increase in binding affinity.
Direct conjugation of DOTA to the C-terminus of GLP-1 through Lys37 resulted in a moderate loss of binding affinity and potency (12- and 6-fold, respectively).45 Among four positions that have been examined for DOTA conjugation, positions 22 and 37 appear to tolerate the bulky substituent with only a moderate decrease in receptor interaction (5- to 10-fold) compared to positions 26 and 34 (50- to 500-fold). Conversely, glycosylated Asn37 was found to have little impact on receptor interaction.47 Taken together, conjugation of macromolecules at the C-terminus of GLP-1 appears to be well accepted by the receptor.
Truncated GLP-1 Analogs
GLP-1 appears to be quite sensitive to peptide chain truncation, since most of truncated peptides suffer from substantially low or negligible binding affinity. It was found that just one amino acid deletion from the N-terminus (i.e., His7) led to a 100-fold decrease in binding affinity and nearly a complete loss of biological activity.43 Removal of a dipeptide unit (i.e., His7-Ala8) resulted in a complete loss of activity along with a 1000-fold decrease in binding affinity, highlighting the significance of the DPP-IV degradation. Truncation of more residues from the N-terminus did not reinstate receptor binding or activation. On the other hand, truncation of two to three amino acids from the C-terminus did not abolish the biological activity, although binding affinity decreased by 4- to 5-fold. However, further deletion of Val33 resulted in a complete loss of binding affinity and potency. Taken together, these results indicate that the full length of GLP-1 seems to be required for strong receptor interaction.
Because they do not activate GLP-1R, metabolites resulting from GLP-1 degradation have been considered as pharmacologically inactive. However, several metabolites, especially GLP-1(9–36)-NH2, GLP-1(28–36)-NH2, and GLP-1(32–36)-NH2, were reported to exert GLP-1R independent biological activity, such as cardioprotective and glucoregulatory effects.20,50−53 While these metabolites present a new direction to the incretin therapy, there is also a growing concern about pancreatitis and pancreas and thyroid cancers as unwanted side effects.54
Eleven amino acid containing GLP-1R agonists were reported by Mapelli et al.55 They introduced two bulky unnatural amino acids at the C-terminus of a nonapeptide GLP-1(7–15) and demonstrated that receptor binding affinity of these short GLP-1 analogs greatly depend on the structure of these two unnatural residues. The most potent undecapeptide contains two substituted biphenylalanines at the C-terminus and showed surprisingly high potency as a truncated GLP-1 analog (200-fold decrease). Subsequent optimization of the undecapeptide by replacing Ala8 and Phe12 with other unnatural phenylalanine analogs further increased potency, comparable to the native GLP-1 (3-fold decrease). It is a small GLP-1R agonist with high potency in insulin secretion and glucose reduction in vivo.
3. Conformational Studies of GLP-1
The structure of GLP-1 was first reported by Thornton and Gorenstein using 2D NMR in an aqueous solution containing perdeuterated dodecylphosphocholine (DPC) micelles that were used to provide a membrane-like environment.56 Bound to the DPC micelles, GLP-1 was found to adopt two helical segments (residues 13–20 and 24–35) that were connected by a short linker region (residues 21–23). The C-terminal helix contains a larger number of hydrophobic residues and is more stable than the amphiphilic N-terminal one, evidenced by amide proton exchange experiments. This study also suggested that Gly22 plays an important role in separating the two helices and allows the peptide to present a single hydrophobic surface to the membrane; this may facilitate membrane association and receptor binding. However, the structure of the N-terminal region (residues 7–13) that strongly contributes to receptor activation was not clearly elucidated because of high conformational flexibility. The predominant helical structure of GLP-1 was also confirmed by Chang et al. using 2D NMR in 35% aqueous TFE.57 In this study, GLP-1 was found to adopt an extended helical structure from Thr7 to Lys28 with a less well-defined region around Gly22.
Neidigh et al. compared structures of GLP-1 and exendin-4 in the solution and micelle-bound states.58 Although both peptides showed a helix spanning from Thr13 to Lys34, GLP-1 is less helical than exendin-4 because of Gly at position 22. In contrast, the micelle-bound exendin-4 adopts a single helix between residues 11–27, presumably aided by a helix-favoring Glu at position 16, the corresponding position 22 in GLP-1. It is interesting that the C-terminal region of exendin-4 forms a compact tertiary fold called Trp-cage in aqueous TFE or glycol, shielding the side chain of Trp25 from solvent exposure. However, this tertiary structure disappeared when exendin-4 was bound to DPC micelles, since the indole ring of Trp is energetically more favored to associate with the hydrophobic micelles.
Agonist-induced activation of the GLP-1 receptor has been suggested involving a two-step mechanism where the C-terminal region of the peptide binds to the N-terminal domain (NTD) of the receptor while the N-terminal part of the peptide interacts with the core region of the receptor, triggering signal transduction.59,60 Receptor binding and activation of GLP-1 appear to rely on interaction with the NTD of the receptor as well as with the receptor core comprising transmembrane (TM) domains and extracellular loops (ECLs).61 This is also emphasized by the findings that the ligand binding to the full length receptor is due to not only autonomous binding to the NTD but also contacts with the juxtamembrane part.62 It is further supported by photoaffinity labeling studies that determined spatial approximation between the ligand and receptor residues (vide infra).63−65
Exendin-4 displayed a higher binding affinity (IC50 = 6 nM) to the isolated NTD of GLP-1R compared to GLP-1 (IC50 > 500 nM). And GLP-1 is more sensitive to truncation in the N-terminal region compared to exendin-4. For instance, N-terminally truncated GLP-1 analogs showed a significantly higher loss in binding affinity and potency compared to truncated exendin-4 analogs.17 Furthermore, mutagenesis studies at the TM domains of GLP-1R resulted in a decrease in receptor interaction.66−68 X-ray crystal structures of exendin-4 and GLP-1 bound to the isolated NTD of GLP-1R confirmed the multidomain binding model in detail.61,69 However, the interaction between the N-terminal segment of the peptide and the core region of the receptor could not be explained by the X-ray studies either. Whereas both exendin-4 and GLP-1 were found to bind to the same site in the receptor, the crystal structures alone did not adequately explain the reason why exendin-4 has a higher affinity to the isolated NTD of GLP-1R compared to GLP-1. However, a recent biophysical study suggests that higher helical propensity of exendin-4 contributes to the high affinity of the peptide to the isolated NTD of the receptor compared to GLP-1.59 It was found that binding of exendin-4 to GLP-1R resulted in a small increase of helical content, while a significant increase was observed with GLP-1 upon binding to the receptor. This indicates that transition from the solution to the receptor-bound state is more substantial with GLP-1 than exendin-4.
The crystal structure of GLP-1 bound to the NTD of GLP-1R revealed that GLP-1 becomes α-helical when it interacts with the receptor and the C-terminal part of the peptide associates to the NTD of the receptor.61 The crystal structure of GLP-1 showed a continuous α-helix from Thr13 to Val33 with a kink around Gly22. The residues between Ala24 and Val33 were found to interact with the NTD of the receptor, such as Ala24, Ala25, Lys26, Phe28, Ile29, Leu32, and Val33. The C-terminal helix appears to be amphiphilic and has a hydrophilic surface containing polar residues like GIn23, Lys26, Glu27, and Lys34, while nonpolar residues like Ala24, Ala25, Phe28, Ile29, Leu32, and Val33 constitute a hydrophobic one.
The crystal structure of exendin-4(9–39)-NH2 in complex with the isolated NTD of GLP-1R showed that exendin-4 also adopts an α-helical conformation except the C-terminal Trp-cage part.69 The Trp-cage was assumed to account for the superior binding affinity of exendin-4 compared to GLP-1. However, the crystal structure of exendin-4 pointed out that the Trp-cage was not involved in binding to the NTD of the receptor. It was suggested that binding affinity of exendin-4 solely depends on its interaction with the NTD of GLP-1R, not with TM helices and ECLs because the binding affinity of exendin-4 did not alter when it bound to the isolated NTD of GLP-1R.70 Also, the N-terminally truncated exendin-4 analogs still have very similar binding affinity to the native exendin-4. The crystal structure of exendin-4(9–39)-NH2 showed a well-defined helix from Leu10 to Asn28, and the residues between Glu15 and Ser32 were found to interact with the NTD of the receptor. Like GLP-1, the C-terminal segment of exendin-4 also adopts an amphiphilic helix that presents a hydrophilic surface comprising Glu16, Glu17, Arg20, Glu24, and Lys27 and a hydrophobic one containing Ala18, Val19, Phe22, Ile23, Trp25, Leu26, and Pro31.
Understanding the interaction between a ligand and its receptor has been a topic of interest, as it can provide useful insights for designing more effective drugs. To investigate how GLP-1 interacts with the receptor, Miller and co-workers conducted spatial approximation studies by using photolabile probes and molecular modeling.63−65 It was found that the C-terminal residues Ala24 and Gly35 in the peptide were docked in a close proximity to Glu133 and Glu125 of the extracellular domain (ECD) of the receptor, respectively. This supports a previous finding that GLP-1 binds to its receptor via an α-helix formed by the sequence spanning from Thr13 to Val33 with the residues between Ala24 and Val33 directly interacting with ECD. This study also showed that Phe12 and Val16 are positioned near Tyr145 and Leu141 of ECD, respectively, yet Leu20 landed near Trp297 in ECL2. To obtain a clue on less characterized N-terminal residues, a photolabile probe was placed at position 6 and it labeled Tyr205 in ECL1.
4. Conformationally Constrained GLP-1 Analogs
The structures of GLP-1 have been studied by 2D NMR56−58 and found to have two α-helical segments between residues 13–20 and 24–35 that are connected by a short linker region comprising residues 21–23. However, these NMR structures were determined without the presence of the GLP-1 receptor and may be different from the receptor-bound conformation, since micelles and helix-stabilizing TFE cannot mimic specific interactions between the peptide and receptor.
To investigate on the presence and locations of α-helical structures in the receptor-bound conformation, Ahn and co-workers have designed and synthesized a series of conformationally constrained GLP-1 analogs.71 In this study, a lactam bridge between Lysi/Glui+4 was introduced and stabilized a helical conformation at various positions from the N- to C-terminus. Eight cyclic peptides were constructed along with eight corresponding linear counterparts. The receptor binding affinity and potency of the cyclic and linear peptides were compared, and the results clearly suggest the presence of two helical segments (residues 11–21 and 23–34) in the N- and C-terminal regions of the receptor-bound conformation.
For instance, a lactam bridge between Lys16 and Glu20 led to comparable receptor binding and activation to GLP-1, suggesting that a helical structure fixed at this position is a part of the receptor-bound conformation. The significance of this N-terminal helix was further demonstrated by deliberately distorting the helical structure by forming a smaller size ring via a disulfide bridge between Cys16,20, leading to a substantial loss in receptor interaction. In the same manner, the C-terminal helix was confirmed in the receptor-bound conformation. However, a helical structure fixed in the linker region made the corresponding cyclic peptides lose binding affinity and potency dramatically, clearly indicating the presence of a break between the two helices in the bioactive conformation.
While this lactam scanning study does not provide spatially well-defined models like the NMR structures, this method is unique in elucidating structural requirements in receptor-bound conformation of peptides.71,72 The two helices in the N- and C-terminal regions in the NMR solution structures appear to be critical binding determinants for receptor interaction. The C-terminal helix was later confirmed by the X-ray structure of GLP-1 cocrystallized with the NTD of the receptor.61 However, the formation of the N-terminal helix upon receptor binding was only confirmed by this lactam scanning study.
Lactam-containing cyclic GLP-1 analogs were also reported by Miranda and co-workers.44 They placed a lactam bridge between Glui and Lysi+4 at the C-terminal region and showed similar results. In particular, the lactam bridges introduced between positions 18–22, 22–26, and 23–27 were well accepted by the receptor, showing comparable receptor binding and activation to GLP-1. These cyclic peptides have Gly at position 8 and showed higher plasma stability. The lactam bridge between position 18–22 together with Gly8, Aib22, and C-terminal PEGylation offered a sustained in vivo efficacy in blood glucose reduction and body weight decrease.
On the basis of these results, Ahn et al. have designed and synthesized highly constrained GLP-1 analogs containing two or three lactam bridges between Glui and Lysi+4 that were placed simultaneously to stabilize the both helical structures in the N- and C-terminal regions.73 These multiple lactam bridges not only improved potency (up to 4-fold increase) by stabilizing the two helical binding determinants as evidenced by CD spectroscopy but also enhanced resistance to proteolytic degradation. Whereas the DPP-IV degradation can be avoided by substituting Ala8 with various amino acids like Gly, Ser, d-Ala, and Aib,32,37 the degradation by NEP 24.11 is hard to address in a similar manner because of the multiple sites that NEP 24.11 targets (Figure 1). It is remarkable that these multiple lactam bridges offered outstanding protection to the enzyme. Bicyclic and tricyclic GLP-1 analogs, c[Glu18, Lys22]-c[Glu30, Lys34]-GLP-1(7–36)-NH2 and c[Glu16, Lys20]-c[Glu22, Lys26]-c[Glu30, Lys34]-GLP-1(7–36)-NH2, respectively, showed 4.6- and 2.6-fold increase in potency, respectively, as well as half-life of greater than 96 h when incubated with recombinant NEP 24.11. This exceptionally high enzyme stability of the bicyclic and tricyclic GLP-1 analogs would be of interest in achieving a long-acting in vivo activity and molecular imaging of pancreatic β-cells via the GLP-1 receptor (vide infra).
5. Stable GLP-1 Analogs as Therapeutic Intervention
Diabetes mellitus is a chronic disease in which the body does not either produce enough insulin or use it inadequately, leading to the hallmarked symptom of high glucose level. The former is classified as type 1 diabetes (T1D), also known as juvenile-onset diabetes, resulting from an autoimmune attack on insulin-producing pancreatic β-cells.74,75 Type 2 diabetes (T2D), also called as adult-onset diabetes, is composed of multiple metabolic abnormalities that lead to progressive hyperglycemia caused by dysfunctional insulin secretion and impaired insulin utilization (i.e., insulin resistance).76 In addition to high glucose concentrations, diabetic patients are prone to develop other complications, such as heart disease, stroke, hypertension, blindness and eye problems, kidney failure, nervous system related diseases, amputations, and so on. It was estimated that total costs of diabetic treatment and care in the United States were $174 billion in 2007, and diabetes was the seventh leading cause of death based on U.S. death certificates in 2007.77
Treatment of T1D is centered on increasing insulin availability mainly by direct insulin administration, whereas therapeutic interventions for T2D are focused on improving insulin sensitivity along with improving β-cell function by use of oral hypoglycemics like sulfonylureas. Despite advances in options for treating diabetes, optimal glycemic control may often be difficult to achieve, and there is a growing demand for novel therapeutic agents that not only lower plasma glucose levels but also manage diabetic complications and prevent the disease progression. An entirely new therapeutic option for treating T2D became available: incretin-based therapies that mainly involve in the GLP-1 axis (Table 1).
Table 1. Stable GLP-1 Receptor Agonists.
| manufacturer/developer | dosage | plasma half-life | status | ref | |
|---|---|---|---|---|---|
| Peptide GLP-1R Agonists | |||||
| exenatide (1) | AstraZeneca | 5–10 μg b.i.d. sc | 2.4 h | approved in U.S. (2005) | (24) |
| liraglutide (2) | Novo Nordisk | 1.2–1.8 mg once daily sc | 11–15 h | approved in U.S. (2009) | (25) |
| taspoglutide (3) | Ipsen/Roche | 165 h | (49) | ||
| lixisenatide (4) | Sanofi-Aventis | 20 μg once daily | 2.7–4.3 h | approved in European Union, Mexico, Australia, and Japan | (94) |
| semaglutide (5) | Novo Nordisk | 160 h | phase 3 | (97) | |
| constrained GLP-1 analogs (6) | Ahn et al. | (73) | |||
| fluorinated GLP-1 analogs (7) | Kumar et al. | (41) | |||
| bile acid conjugated exendin-4 analogs (8) | Lee et al. | (99) | |||
| truncated GLP-1R agonists (9) | Mapelli et al. (Bristol-Myers Squibb) | (55) | |||
| β3-peptide analog (10) | Schepartz et al. | (100) | |||
| Protein GLP-1R Agonists | |||||
| albiglutide (11) | GlaxoSmithKline | 30 mg/week | 6–8 days | approved in Europe (2013) and U.S. (2014) | (97, 101) |
| CJC-1134-PC (12) | ConjuChem | 8 days | phase 2 | (102) | |
| dulaglutide | Eli Lilly | once weekly | 90 h | phase 3 | (103) |
| langlenatide | Hanmi | once weekly | 180 h | phase 2 | (104) |
| VRS-859 | Diartis | once monthly | 139 h | phase 1 | (105) |
The major physiological role of GLP-1 is to connect the consumption of nutrients with glucose metabolism through a network of regulatory pathways.78 Stimulation of insulin release from pancreatic β-cells is one of them. Since the insulinotropic action of GLP-1 depends on plasma glucose content, it is associated with a reduced risk of hypoglycemia that often comes along with current antidiabetic agents like sulfonylureas and glinides.79,80 GLP-1 has also shown superiority to thiazolidinedione (e.g., rosiglitazone) because GLP-1 therapy does not accompany side effects like peripheral edema, weight gain, congestive heart failure, and osteoporosis, which are common in thiazolidinedione therapy.79 In addition, it has shown favorable cardiovascular benefits, such as lowering blood pressure, beneficial effects on serum lipids, and a potential to improve myocardial contractility and endothelial function.80 Furthermore, GLP-1 is found to have effects on β-cell proliferation and regeneration, which may be of great help in preventing the disease.4,79 Although there are speculations of asymptomatic pancreatitis as a side effect to incretin therapy, it is not clearly studied yet.81,82
The endogenous GLP-1 is difficult agent for therapeutic use because of the rapid cleavage of the dipeptide His7-Ala8 by DPP-IV, which leads to a loss of receptor interaction and insulinotropic action.83 This limitation can be overcome by either metabolically stable GLP-1 analogs or DPP-IV inhibitors.79,84−86 A number of methods and strategies have been developed for protecting peptides from proteolytic degradation. These include introduction of d-amino acids, α,α-dialkylated amino acids like Aib, N-methylated amino acids, β-amino acids, amide bond surrogates,87 PEGylation,88 glycosylated amino acids, halogenated amino acids,41,72,73 and conformational restrictions.72,73
As briefly described in the structure–activity studies, a variety of substitutions have been made in the N-terminal region, particularly Ala8, and examined on receptor interaction and metabolic stability. Discovered in the saliva of the Gila monster,89 exendin-4 is a 39 amino acid containing GLP-1R agonist with comparable binding affinity and potency to GLP-1. It possesses Gly at position 2 instead of Ala8 in GLP-1, offering outstanding resistance to DPP-IV (Figure 2).901 is a synthetic form of exendin-4 and the first incretin-based therapeutic approved by FDA.24 It is recommended to be administered subcutaneously twice daily and found to lower plasma glucose and HbA1c levels and body weight.91 However, several side effects were observed, such as accumulation of antibodies and acute pancreatitis, despite low incidences reported.92
Figure 2.

Structures of selected GLP-1 receptor agonists.
On the other hand, lixisenatide (4, Lyxumia, formerly known as AVE0010, Sanofi-Aventis) is a 44-mer peptide and has a hexalysine extension at the C-terminus of exendin-4.93 It reportedly binds to the receptor more tightly than GLP-1 and exendin-4 (4-fold increase). It showed a median half-life ranging from 2.7 to 4.3 h for a dose of 20 μg, compared to 1–2 min for the native GLP-1.94 It protects pancreatic β-cells from apoptosis and exhibited a higher insulinotropic effect compared to GLP-1. It has been approved in the European Union, Mexico, Australia, and Japan as a once-daily therapeutic (20 μg) to be used in combination with basal insulin.
Approved in 2009, 2 is a GLP-1(7–37) analog containing a palmitoyl chain at Lys26 through a γ-glutamyl spacer together with Arg34 (Figure 2).79 This fatty acid chain facilitates binding to albumin and consequently increases the half-life to 11–15 h after parenteral administration in human.95 Despite this modification, 2 exhibited comparable receptor activation potency to GLP-1.96 Semaglutide (5, NN9535) is an analog of 2 containing Aib8 and a different fatty acid chain connected to Lys26 through a miniPEG spacer (Figure 2).97 It is a once-weekly human GLP-1 analog currently being developed by Novo Nordisk. Another potent and stable GLP-1 analog 3 was developed by Ipsen and Roche by substituting Ala8 and Gly35 for Aib (Figure 2).48 It showed significantly higher proteolytic resistance (half-life of 165 h for a dose of 30 mg).98 However, Roche decided to halt the development because of safety issues involving various gastrointestinal side effects and allergic reactions.
Academic laboratories have focused their research on the quest of more stable and potent GLP-1R agonists as well as pharmaceutical industries. Ahn and co-workers, for instance, have prepared a series of conformationally constrained GLP-1 analogs by introducing multiple lactam bridges between Glui and Lysi+4 to stabilize two α-helices at the N- and C-terminal regions.73 One of the bicyclic peptides, [d-Ala8]-c[Glu18, Lys22]-c[Glu30, Lys34]-GLP-1(7–36)-NH2 (6), was found to show outstanding metabolic stability as well as higher potency compared to GLP-173 (Figure 2). It is remarkable that this highly constrained GLP-1 analog demonstrated high in vivo efficacy matching the gold standards (1 and 2), as examined by intraperitoneal glucose tolerance test (unpublished results).
Kumar et al. incorporated a hexafluoroleucine at various positions, aiming to modulate peptide–receptor interaction and improve resistance to proteolytic cleavage.41 Hexafluoroleucine containing peptide (7) has Glu9 replaced with hexafluoroleucine and showed binding affinity and potency (IC50 = 5.1 nM, EC50 = 2.0 nM) comparable to GLP-1 (IC50 = 1.9 nM, EC50 = 1.0 nM).41 However, it displayed only 1.5-fold increase in stability over DPP-IV compared to GLP-1. Lee and co-workers reported exendin-4 analogs (8) with a bile acid (e.g., cholic, deoxycholic, and lithocholic acid) conjugated to Lys27.998 showed high binding affinity (IC50 = 0.48 nM) compared to exendin-4 (IC50 = 0.14 nM) without loss of noticeable insulinotropic activity. The increased hydrophobicity resulting from the conjugation of the bile acid appeared to elevate albumin-binding property. These bile acid modified exendin-4 analogs exhibited extended antidiabetic effect with prolonged restoration of normoglycemia in T2D mice.
Truncated GLP-1 analogs have been reported as potent GLP-1R agonists. Mapelli and co-workers developed 11 amino acid containing GLP-1R agonists (9) that comprise structurally optimized N-terminal nine residues of GLP-1 and a substituted biphenylalanine (BIP) dipeptide at the C-terminus (Figure 2).55 Despite the significantly shortened length of the peptide, 9 showed a high potency (EC50 = 0.087 nM) compared to GLP-1 (0.034 nM) and a longer pharmacokinetic half-life. It was also found to be highly selective to GLP-1R over other class B GPCRs. As another approach, Schepartz and co-workers developed a β3-peptide analog of GLP-1 (10).100 It was constructed by incorporating the epitopes of Ala18, Phe22, and Trp25 in a 14-helical β3-peptide and extending the sequence at the N-terminus with a short PEG chain followed by a short α-peptide corresponding to the N-terminal nine residues of exendin-4. The β3-peptide analog adopts a 14-helical conformation that is stabilized by hydrogen bonds between an amide proton of the ith residue and a carbonyl of the i + 2 residue. However, 10 showed substantially weak potency (EC50 = 1200 nM) compared to GLP-1 (EC50 = 0.003 nM).
As peptides are quickly cleared out of the body, fusion proteins have been developed to achieve longer plasma circulation. Albiglutide (11, GlaxoSmithKline) is a dimer of modified GLP-1 sequence linked to human serum albumin (Figure 2).101 It gained resistance to DPP-IV by both replacing Ala8 with Gly and being a part of a macromolecule, allowing 5 days of plasma half-life, suitable for once-weekly dosing.97 As a fusion protein, it showed a remarkably high binding affinity (IC50 = 0.61 nM compared to 0.02 nM of GLP-1). It was recently approved by the European Commission under the name of eperzan and very recently (April 2014) in the United States as well. CJC-1134-PC (12, ConjuChem) is another albumin-fused GLP-1R agonist.102 It has the sequence of exendin-4 and similarly showed a long circulation half-life (approximately 8 days) as 11.
Another long acting GLP-1R agonist, dulaglutide (LY2189265, Eli Lilly, structure not shown), is a recombinant fusion protein with two analogs of GLP-1(7–37) covalently linked by a synthetic 16 amino acid linker to a Fc fragment of human IgG4.103 Conversely, langlenatide (HM11260C, Hanmi Pharmaceuticals, structure not shown) is a long acting exendin-4 analog conjugated to a nonglycosylated Fc region via a short PEG linker.104 VRS-859 (exenatide-Xten, Diartis Pharmaceuticals, structure not shown) is a different kind of fusion protein comprising exanatide and a long chain of all natural hydrophilic amino acids referred to as Xten.105
Despite the striking similarity between the two peptides, GLP-1 and glucagon have opposing roles in glucose homeostasis. GLP-1 induces glucose-dependent insulin secretion and reduces the plasma glucose level, whereas glucagon triggers glycogenolysis and gluconeogesis in the liver, increasing the glucose concentration. Since a glucagon antagonist can inhibit hepatic glucose output, creating a bifunctional molecule that is an agonist to the GLP-1 receptor (GLP-1R) and an antagonist to the glucagon receptor (GR) simultaneously may be pharmacologically significant for modulating the overall glucose in the body in multiple ways. Pan et al. developed a balanced GLP-1 agonist/glucagon antagonist, ANC7K2 (structure not shown; GLP-1R, IC50 = 3.5 nM and EC50 = 13 nM; GR, IC50 = 120 nM and 36% inhibition of the receptor activity).106 In addition, a 43 kDa PEG chain was conjugated to position 31, promoting a prolonged glucose lowering activity.
Conversely, DiMarchi and co-workers have reported bifunctional molecules that are GR antagonist/GLP-1R agonist and GR/GLP-1R coagonist.107,108 A glucagon antagonist [PLA6, Asp28]-c[Glu16, Lys20]-glucagon(6–29) that has an IC50 value of 14 nM on GR showed a potent agonism on GLP-1R (EC50 = 11 nM). They also investigated novel multifunctional ligands that show coagonism on both GR and GLP-1R in order to normalize glucose and lipid metabolism.108 A glucagon-based peptide comprising Aib at position 2, a lactam bridge between Glu16 and Lys20, and a 40 kDa PEG chain at position 24 showed a potent agonistic activity on both receptors (EC50 of 0.67 and 0.059 nM on GR and GLP-1R, respectively). This coagonist resulted in a significant decrease in body weight and fat mass, demonstrating that balanced simultaneous activation of the both receptors lowered body adiposity and improved glucose tolerance. Extending these works further, DiMarchi and co-workers also developed a coagonist on GLP-1R and gastric inhibitory polypeptide (GIP) receptor to achieve a greater weight loss effect than GLP-1 alone.109
In collaboration with Boehringer Ingelheim, Zealand Pharma recently developed a GR/GLP-1R dual agonist (ZP2929, structure not shown).110 It is an oxyntomodulin-based peptide drug candidate in preclinical development for the treatment of T2D and obesity. TT401 (Transition Therapeutics, structure not shown) is another dual agonist on GLP-1R and GR under development for once-weekly dosing.111 AC164204 and AC164209 (structures not shown) (Amylin Pharmaceuticals) are analogs of 1 covalently linked to a second generation amylin analog, davalintide, another example of coagonists.112 Both molecules showed full agonism to their cognate receptors in vitro but with a reduced potency to GLP-1R.
Another approach to prolong GLP-1 action in vivo is DPP-IV inhibitors, since DPP-IV is one of the main causes of GLP-1 degradation. DPP-IV inhibitors can be used either alone or as combination therapy for reducing fasting and postprandial glucose levels with a low incidence of hypoglycemia and weight gain. To date, many DPP-IV inhibitors have been developed, including sitagliptin (13), saxagliptin, vildagliptin, and linagliptin (structures not shown). 13 (Januvia, Merck) reduces glucose and HbA1c levels but has no effect on body weight.86 Unlike stable GLP-1 analogs, it can be orally administered once daily and has a half-life of 12–14 h. DPP-IV inhibitors have been extensively reviewed elsewhere.113−116
Despite low potency, nonpeptidic GLP-1R agonists have been reported and showed a potential to achieve orally active GLP-1 mimetics.117 Developing small molecules mimicking peptide hormones is not a trivial task because peptides often have a large surface area required for optimal receptor interaction. In addition, residues important for receptor binding and activation are frequently dispersed across their sequences and structures. Whereas GPCRs have been activated by nonpeptidic ligands (e.g., morphine activating opioid receptors), they have been mainly limited to class A GPCRs that are smaller and more extensively studied than class B receptors to which GLP-1R belongs. However, small molecules have been reported of activating GLP-1R, offering a hope for this challenging quest.
In 2007, Knudsen et al. reported substituted quinoxalines (14) discovered from an extensive screening campaign with 500 000-membered library (Figure 3).118,11914 compounds were found to be allosteric agonists selective for human GLP-1R. Whereas these compounds showed toxicity at high concentrations with an unusual bell-shaped dose–response curve, they induced glucose-dependent insulin release from normal mouse islets but not from GLP-1R knockout mice. On the other hand, Chen and co-workers also reported their discovery of GLP-1R agonists identified from screening 50 000 compounds.120 Two substituted cyclobutanes (15) were found to be orthosteric agonists with weak binding affinity and efficacy.120 These cyclobutane compounds demonstrated oral activity lowering glucose and HbA1c levels.
Figure 3.

Structures of selected non-peptide GLP-1 receptor agonists.
Pyrimidine-based small molecules (16) were identified as GLP-1R agonists by cell-based screening and insulin secretion assays with rodent and human islets (Figure 3).12116 induced GLP-1R-mediated cAMP signaling and stimulated glucose-dependent insulin secretion both in vitro and in vivo, although these molecules did not show competitive binding to the receptor. Sexton and co-workers developed a series of quercetin-like flavonoids (17) with positive allosteric modulator activity on GLP-1R.12217 selectively modulated Ca2+ responses induced by the high-affinity agonists GLP-1 and exendin-4 without altering cAMP signaling. Similarly, Gong and co-workers identified imidazopyridines (18, 19) after screening 10 000 heterocyclic small molecules by a functional assay on GLP-1R overexpressed on HEK293 cells.123 Researchers at Dong-A Pharmaceutical developed 2-thioquinoxaline derivatives (20) as small molecule agonists on GLP-1R.124 They were found to have EC50 values ranging from 50 to 1000 nM in cAMP response element (CRE) luciferase reporter assays. When orally administered, 20 promoted glucose-stimulated insulin secretion in a mouse intravenous glucose tolerance test.
Taking a different approach, Ahn and co-workers developed a new strategy for rationally designing GLP-1 peptidomimetics by using helix-mimicking small molecules.125,126 As they demonstrated with a series of conformationally constrained GLP-1 analogs, the helical segments at the N- and C-terminal regions of the peptide appear to be important for receptor interaction.71,73 To mimic helical peptides, they designed a rigid and preorganized tris-benzamide scaffold that placed three side chain groups of amino acids found at the i, i + 4, and i + 7 positions of a helix, reproducing one side of a helix.127−129 They designed GLP-1 peptidomimetics (21) by replacing a helical peptide segment in GLP-1 with a corresponding tris-benzamide-based helix mimetic (e.g., SH3), which showed promising receptor binding and potency (EC50 = 130 nM; Figure 3).125 Despite weak potency, these are the first examples of rationally designed, not screened, GLP-1 peptidomimetics that are currently under investigation.
Improving efficacy and pharmacokinetic profiles of natural products and therapeutics can be achieved in many ways, including pharmaceutical drug delivery systems and dosage forms. One of the successful approaches in the field of GLP-1 is exenatide LAR, a slow drug releasing subcutaneous depot form of 1.261 is entrapped noncovalently into a biodegradable poly(d,l-lactide-co-glycolide) (PLGA) microsphere from which the drug is slowly released as the polymeric matrix breaks down in vivo, increasing the half-life up to 6 days. It is the first once-weekly injectable formulation approved by the FDA for the treatment of T2D (January 2012). Besides, other formulations are currently under development for sustained release of GLP-1R agonists as summarized in Table 2.
Table 2. Long Acting Pharmaceutical Formulations of GLP-1 Receptor Agonists.
| formulation | manufacturer/developer | description | ref |
|---|---|---|---|
| exenatide LAR | AstraZeneca | slow drug releasing subcutaneous depot with 1 entrapped noncovalently into a biodegradable PLGA microsphere | (26) |
| microsphere | Na et al. | polysaccharide-based microsphere formulation of 1 | (130) |
| hydrogel | Ding et al. | triblock copolymer PLGA-PEG-PLGA-based hydrogel formulation of 1 | (131) |
| hydrogel | Joabsson et al. | lipid-based hydrogel preparation of GLP-1 and 1 for subcutaneous injection | (132) |
| protease-operated depot (POD) | Chilkoti et al. | thermoresponsive depot forming elastin-like polymers containing protease-cleavable oligomers of [Gly8]-GLP-1(7–36) with a dipeptide (Gly-Ala) added at the N-terminus | (133) |
| implantable device | Kuzma et al. | polymeric hydrogel-based implantable devices containing 1 | (134) |
| formulation for pulmonary delivery | Youn et al. | albumin-coated porous hollow PLGA microparticles bound with palmitylated exendin-4 | (135) |
| chewing gum tablet | Andersen et al. | chewing gum containing GLP-1 and exendin-4 | (136) |
| formulation for gene therapy | Kim et al. | exenatide plasmid in an arginine-grafted bioreducible polymer (ABP) poly-cystaminebisacrylamidediaminohexane as the gene carrier | (137) |
6. GLP-1 Analogs as Molecular Imaging Agents
Noninvasive in vivo assessment of pancreatic β-cell mass (BCM) in humans has a tremendous potential as a clinical tool for diagnosing and treating diabetes. While sweet smell or taste of urine is a classical and historical sign of diabetes, fasting plasma glucose (FPG), oral glucose tolerance test (OGTT), glycated hemoglobin A1c (HbA1c), insulin, and C-peptide levels are currently used to examine functional pancreas.138−141 However, these clinical manifestations detect the progress of the disease too late, at which time the majority of the β-cells are destroyed and the disease is already too advanced for any preventive measures to intervene.142 For example, fasting plasma glucose (FPG) test and oral glucose tolerance test (OGTT) are two major methods for clinical diagnosis of diabetes. If either test indicates a positive result (higher than 126 mg/dL (FPG) or 200 mg/dL (OGTT) of glucose levels), patients are diagnosed as diabetic. At the time of diagnosis, T1D patients typically have their BCM reduced by 70–80%.143 However, because the remaining β-cells compensate the loss of insulin production from β-cell death, abnormalities in blood glucose levels are unnoticed by these tests until BCM is diminished by more than 50%.144 Thus, noninvasive imaging of BCM would provide accurate and in-time status as well as useful prediction. Such an approach can be used to monitor the disease progression, examine the loss of BCM, evaluate efficacy of therapies in preserving and restoring BCM, and follow up β-cell replacement therapies like islet transplantation.145
Imaging modalities, such as positron emission tomography (PET), magnetic resonance imaging (MRI), other nuclear imaging techniques like single-photon emission computed tomography (SPECT), and optical absorption or fluorescence spectroscopy and imaging have been promising for visualizing and quantifying various molecular targets.144 However, assessing BCM in vivo is still challenging, since the β-cells exist in pancreatic islets that are small (50–300 μm in diameter), scarce (1–2% of pancreatic mass), and scattered throughout the pancreas.146 Each islet is too small to be spatially resolved by current noninvasive in vivo imaging techniques. In addition, it is not trivial to correlate signals from imaging experiments with changes in BCM.
Many investigators are currently searching for and evaluating β-cell specific biomarkers.144,147−151 A number of potential candidates for β-cell imaging have been reported, such as vesicular monoamine transporter (VMAT2), glucagon-like peptide-1 receptor (GLP-1R), sulfonylurea receptor (SUR1), glucose transporter 2 (GLUT2), glycogen, zinc transporters, fluorodithizone, and monoclonal antibodies. However, many fell short because of either low expression levels or insufficient β-cell specificity.147,152,153 Among them, 11C- and 18F-labeled dihydrotetrabenazine (DTBZ) derivatives (structures not shown) have demonstrated promising results.144,154,155 As a ligand to VMAT2 that is expressed at dopamine nerve ends in the central nerve system and pancreatic β-cells, they were identified from screening an extensive library of radiotracers. However, recent findings of nonspecific binding of [11C]-DTBZ in human pancreas overcasts its clinical applications.156
Compared to other β-cell biomarkers, GLP-1R shows a high promise because of its specific expression in the β-cells and strong interaction with its ligands.157 Thus, GLP-1 analogs are of great interest not only in treating diabetes but also in determining BCM. However, its endogenous ligand GLP-1 cannot be employed as an effective imaging agent because of its extremely short half-life (less than 5 min) resulting from rapid metabolic degradation. This indicates that β-cell imaging via GLP-1R as a biomarker requires GLP-1 analogs with high metabolic stability as well as strong binding affinity. Owing to inherent resistance to DPP-IV degradation, stable GLP-1R agonists, exendin-3 and exendin-4, have been developed for β-cell imaging.158−165 On the other hand, bicyclic GLP-1 analogs developed by Ahn and co-workers have also demonstrated a promising potential in determining BCM.166,167 These stable GLP-1 analogs were conjugated to a metal chelator like DOTA, NOTA, and DTPA, which coordinates a radiotracer (e.g., 64Cu, 68Ga, 99mTc, and 111In) for PET and SPECT imaging modalities.
[Lys40(Ahx-DTPA-111In)NH2]-exendin-4 (22) was reported as a SPECT probe to target GLP-1R for imaging insulinoma in Rip1Tag2 transgenic mice with high tumor uptake (Figure 4).162,168 Whereas ratios of tumor to pancreas (9.71 at 48 h) and other organs were high, a low ratio of the tumor to kidney (1.27 at 48 h) was observed. Despite such a high renal uptake, the potential of GLP-1R in visualizing the β-cells has been demonstrated by PET and SPECT imaging of insulinoma by using exendin-3 or exendin-4-based radioligands.161,163,164 Similarly, a 64Cu-labeled exendin-4, [Lys40(DOTA-64Cu)NH2]-exendin-4 (23) showed high binding specificity to rodent β-cells by ex vivo autoradiography.158 Another 64Cu-labeled exendin-4 analog, 64Cu-DO3A-VS-Cys40-exendin-4 (24), demonstrated a feasibility of in vivo imaging of intraportally transplanted islets in mice by virtue of a high and specific uptake in INS-1 tumors despite high renal uptake.165 It is exciting that 22 was successfully used to highlight autologous islets that have been transplanted into human muscle, proving a clinical potential of human β-cell imaging via GLP-1R.169 Exendin-4 based near-infrared fluorescent imaging agent E4×12-VT750 (structure not shown) was reported as well.170 In addition to the GLP-1R agonists described above, antagonists like exendin(9–39) were also employed as a β-cell imaging probe.171 Radiolabeled with the Bolton–Hunter reagent at lysine residues, exendin(9–39) was examined for receptor specificity in vitro and selectivity in vivo. It showed radioactive signals in pancreatic β-cells although the resolution of the imaging technique was low.
Figure 4.

Structures of selected exendin-4 and GLP-1 based imaging agents for monitoring pancreatic β-cells.
Whereas the majority of β-cell imaging studies have been carried out with exendin-4 analogs, Ahn and co-workers have developed outstanding GLP-1 based PET imaging agents (25, 26) to quantitate BCM.166,167,172 As the endogenous peptide suffers from rapid metabolic degradation, they introduced multiple modifications, such as epimerization at position 8 and two lactam bridges, and produced highly constrained GLP-1 analogs. Strategically placed two lactam bridges made the bicyclic peptides resistant to the proteolytic cleavages by NEP 24.11, and d-Ala8 protected them from the DPP-IV degradation, significantly enhancing metabolic stability. Dynamic PET scans over 60 min showed a high pancreatic uptake of a bicyclic GLP-1 analog labeled with 64Cu (26) as shown in Figure 5.173 The pancreas uptake disappeared in STZ-induced type 1 diabetic mice or competitively displaced by co-injection of unlabeled exendin-4, indicating outstanding selectivity to the β-cells. Ex vivo PET scans and histology were also carried out to confirm the in vivo PET imaging findings (unpublished results). This bicyclic GLP-1 analog is currently under investigation with Ossawbaw mini-pigs for longitudinal PET imaging of BCM in the course of the disease progression.174
Figure 5.
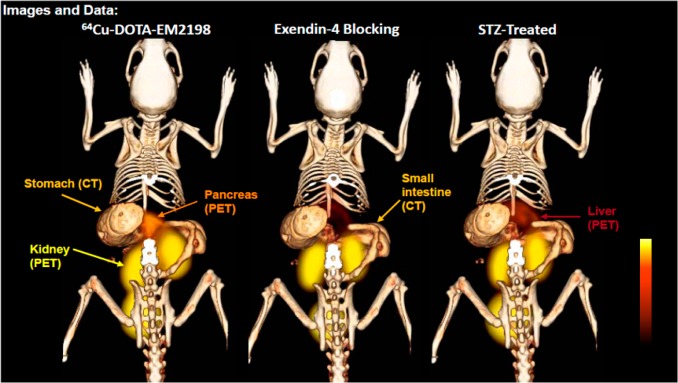
In vivo PET/CT images of pancreas of healthy mice, healthy mice co-injected with a blocking dose of cold exendin-4, and STZ-induced diabetic mice. Reproduced with permission from Peptides across the Pacific, Proceedings of the 23rd American Peptide Symposium.173 Copyright 2013 American Peptide Society.
7. Conclusion and Future Directions
Since its discovery, GLP-1 has attracted tremendous attention because of unique functions that are beneficial for managing diabetes. It not only stimulates insulin secretion in a glucose-dependent manner, preventing hypoglycemic episodes, but also restores pancreatic β-cell mass and functions. Intensive studies over the past decade revealed elements required for strong receptor interaction and many players in its metabolic degradation. These facilitated the development of a new class of incretin-based therapeutics. However, many fundamental features of the peptide and its receptor interaction have not yet been completely explained, although such discovery would be a vehicle to design nonpeptidic GLP-1 receptor agonists possessing long-pursued oral activity. Also, recent advances in molecular imaging have been useful in monitoring changes of pancreatic β-cells, playing a significant role in diagnosing the disease and evaluating effectiveness of therapeutic interventions and islet transplantation. While promising results have been reported, several issues like specific β-cell uptake and high accumulation in nearby organs need to be addressed to become an effective clinical tool.
Acknowledgments
The authors thank the Welch Foundation (Grant AT-1595), American Diabetes Association (Grant 7-07-JF-02), National Institutes of Health (Grant 1 R56 DK080128-01A1), and Juvenile Diabetes Research Foundation (Grant 37-2011-20) for their financial support.
Glossary
Abbreviations Used
- GLP-1
glucagon-like peptide-1
- GPCR
G-protein-coupled receptor
- GLP-1R
glucagon-like peptide-1 receptor
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- DPP-IV
dipeptidyl peptidase IV
- NEP 24.11
neutral endopeptidase 24.11
- Aib
aminoisobutyric acid
- DOTA
1,4,7,10-tetraazacyclododecanetetraacetic acid
- AEEA
aminoethylethanolamine
- GlcNAC
N-acetylglucosamine
- LacNAC
N-acetyllactosamine
- PEG
polyethylene glycol
- DPC
dodecylphosphocholine
- TFE
2,2,2-trifluoroethanol
- NTD
N-terminal domain
- TM
transmembrane
- ECL
extracellular loop
- CD
circular dichroism
- BIP
biphenylalanine
- GR
glucagon receptor
- GIP
gastric inhibitory polypeptide
- LAR
long-acting release
- PLGA
poly(lactic-co-glycolic acid)
- POD
protease-operated depot
- ABP
arginine-grafted bioreducible polymer
- BCM
β-cell mass
- FPG
fasting plasma glucose
- OGTT
oral glucose tolerance test
- HbA1c
glycated hemoglobin
- PET
positron emission tomography
- MRI
magnetic resonance imaging
- SPECT
single-photon emission computed tomography
- VMAT2
vesicular monoamine transporter 2
- SUR1
sulfonylurea receptor 1
- GLUT2
glucose transporter 2
- DTBZ
dihydrotetrabenazine
- NOTA
1,4,7-triazacyclononanetriacetic acid
- DTPA
diethylenetriaminepentaacetic acid
- DO3A
1,4,7-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecane
- STZ
streptozocin
Biographies
Bikash Manandhar completed his Bachelor of Pharmacy (B.Pharm.) at the Kathmandu University, Nepal, in 2007. He worked in pharmaceutical industries in the field of formulation R&D until 2010 and then moved to the United States. He is now a graduate student at the University of Texas at Dallas working towards his Ph.D. in Chemistry under the supervision of Prof. Jung-Mo Ahn. The main focus of his research is to develop peptidic PET imaging agents using novel GLP-1 constructs for noninvasive assessment of pancreatic β-cells.
Jung-Mo Ahn obtained B.S. and M.S. in Chemical Engineering at the Seoul National University, Seoul, South Korea. He then received Ph.D. in Chemistry at the University of Arizona, Tucson, AZ, where he worked with Prof. Victor J. Hruby for developing glucagon antagonists. He went on to pursue postdoctoral studies in organic synthesis and combinatorial chemistry at the Scripps Research Institute, La Jolla, CA, working with Prof. Kim D. Janda. He is currently an Associate Professor in the Department of Chemistry at the University of Texas at Dallas, where his research focuses on constrained GLP-1 analogs for pancreatic β-cell imaging and structure-based design of helix-mimicking small molecules for inhibiting protein–protein interactions.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Gutniak M.; Ørkov C.; Holst J. J.; Ahrén B.; Efendić S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [DOI] [PubMed] [Google Scholar]
- Holst J. J.; Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am. J. Physiol.: Endocrinol. Metabol. 2004, 287, E199–E206. [DOI] [PubMed] [Google Scholar]
- MacDonald P. E.; El-kholy W.; Riedel M. J.; Salapatek A. M. F.; Light P. E.; Wheeler M. B. The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 2002, 51, S434–S442. [DOI] [PubMed] [Google Scholar]
- Brubaker P. L.; Drucker D. J. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004, 145, 2653–2659. [DOI] [PubMed] [Google Scholar]
- Deacon C. F. Therapeutic strategies based on glucagon-like peptide 1. Diabetes 2004, 53, 2181–2189. [DOI] [PubMed] [Google Scholar]
- Kieffer T. J.; Habener J. F. The glucagon-like peptides. Endocr. Rev. 1999, 20, 876–913. [DOI] [PubMed] [Google Scholar]
- Campos R. V.; Lee Y. C.; Drucker D. J. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 1994, 134, 2156–2164. [DOI] [PubMed] [Google Scholar]
- Bullock B. P.; Heller R. S.; Habener J. F. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology 1996, 137, 2968–2978. [DOI] [PubMed] [Google Scholar]
- Tornehave D.; Kristensen P.; Rømer J.; Knudsen L. B.; Heller R. S. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J. Histochem. Cytochem. 2008, 56, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marinis Y. Z.; Salehi A.; Ward C. E.; Zhang Q.; Abdulkader F.; Bengtsson M.; Braha O.; Braun M.; Ramracheya R.; Amisten S.; Habib A. M.; Moritoh Y.; Zhang E.; Reimann F.; Rosengren A. H.; Shibasaki T.; Gribble F.; Renström E.; Seino S.; Eliasson L.; Rorsman P. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010, 11, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G. I.; Santerre R. F.; Mullenbach G. T. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 1983, 302, 716–718. [DOI] [PubMed] [Google Scholar]
- Lund P. K.; Goodman R. H.; Dee P. C.; Habener J. F. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T.; Greig N. H. The glucagon-like peptides: a double-edged therapeutic sword?. Trends Pharmacol. Sci. 2003, 24, 377–383. [DOI] [PubMed] [Google Scholar]
- Holst J. J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [DOI] [PubMed] [Google Scholar]
- Hupe-Sodmann K.; Göke R.; Göke B.; Thole H. H.; Zimmermann B.; Voigt K.; McGregor G. P. Endoproteolysis of glucagon-like peptide (GLP)-1(7-36) amide by ectopeptidases in RINm5F cells. Peptides 1997, 18, 625–632. [DOI] [PubMed] [Google Scholar]
- Mentlein R.; G B.; Schmidt W. E. Dipeptidyl peptidase-IV hydrolyzes gastric-inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 1993, 214, 829–835. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C.; Yang H.; Rodgers B. D.; Beday A.; Pritchette L. A.; Eng J. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J. Biol. Chem. 1997, 272, 21201–21206. [DOI] [PubMed] [Google Scholar]
- Deacon C. F.; Nauck M. A.; Toft-Nielsen M.; Pridal L.; Willms B.; Holst J. J. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995, 44, 1126–1131. [DOI] [PubMed] [Google Scholar]
- Vahl T. P.; Paty B. W.; Fuller B. D.; Prigeon R. L.; D’Alessio D. A. Effects of GLP-1-(7–36)NH2, GLP-1-(7–37), and GLP-1- (9–36)NH2 on intravenous glucose tolerance and glucose-induced insulin secretion in healthy humans. J. Clin. Endocrinol. Metab. 2003, 88, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Elahi D.; Egan J. M.; Shannon R. P.; Meneilly G. S.; Khatri A.; Habener J. F.; Andersen D. K. GLP-1 (9-36) amide, cleavage product of GLP-1 (7-36) amide, is a glucoregulatory peptide. Obesity 2008, 16, 1501–1509. [DOI] [PubMed] [Google Scholar]
- Deacon C. F.; Plamboeck A.; Møller S.; Holst J. J. GLP-1-(9–36) amide reduces blood glucose in anesthetized pigs by a mechanism that does not involve insulin secretion. Am. J. Physiol.: Endocrinol. Metabol. 2002, 282, E873–E879. [DOI] [PubMed] [Google Scholar]
- Nikolaidis L. A.; Elahi D.; Shen Y.-T.; Shannon R. P. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am. J. Physiol.: Heart Circ. Physiol. 2005, 289, H2401–H2408. [DOI] [PubMed] [Google Scholar]
- Plamboeck A.; Holst J.; Carr R.; Deacon C. Neutral endopeptidase 24.11 and dipeptidyl peptidase IV are both mediators of the degradation of glucagon-like peptide 1 in the anaesthetised pig. Diabetologia 2005, 48, 1882–1890. [DOI] [PubMed] [Google Scholar]
- Davidson M. B.; Bate G.; Kirkpatrick P. Fresh from the pipeline: exenatide. Nat. Rev. Drug Discovery 2005, 4, 713–714. [DOI] [PubMed] [Google Scholar]
- Knudsen L. B.; Nielsen P. F.; Huusfeldt P. O.; Johansen N. L.; Madsen K.; Pedersen F. Z.; Thøgersen H.; Wilken M.; Agersø H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J. Med. Chem. 2000, 43, 1664–1669. [DOI] [PubMed] [Google Scholar]
- Kim D.; MacConell L.; Zhuang D.; Kothare P. A.; Trautmann M.; Fineman M.; Taylor K. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007, 30, 1487–1493. [DOI] [PubMed] [Google Scholar]
- Kim D.; Wang L.; Beconi M.; Eiermann G. J.; Fisher M. H.; He H.; Hickey G. J.; Kowalchick J. E.; Leiting B.; Lyons K.; Marsilio F.; McCann M. E.; Patel R. A.; Petrov A.; Scapin G.; Patel S. B.; Roy R. S.; Wu J. K.; Wyvratt M. J.; Zhang B. B.; Zhu L.; Thornberry N. A.; Weber A. E. (2R)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin- 7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2005, 48, 141–151. [DOI] [PubMed] [Google Scholar]
- Gallwitz B.; Witt M.; Paetzold G.; Morys-Wortmann C.; Zimmermann B.; Eckart K.; Folsch U. R.; Schmidt W. E. Structure/activity characterization of glucagon-like peptide-1. Eur. J. Biochem. 1994, 225, 1151–1156. [DOI] [PubMed] [Google Scholar]
- Adelhorst K.; Hedegaard B. B.; Knudsen L. B.; Kirk O. Structure–activity studies of glucagon-like peptide-1. J. Biol. Chem. 1994, 269, 6275–6278. [PubMed] [Google Scholar]
- Hareter A.; Hoffmann E.; Bode H. P.; Goke B.; Goke R. The positive charge of the imidazole side chain of histidine7 is crucial for GLP-1 action. Endocr. J. 1997, 44, 701–705. [DOI] [PubMed] [Google Scholar]
- Gallwitz B.; Ropeter T.; Morys-Wortmann C.; Mentlein R.; Siegel E. G.; Schmidt W. E. GLP-1-analogues resistant to degradation by dipeptidyl-peptidase IV in vitro. Regul. Pept. 2000, 86, 103–111. [DOI] [PubMed] [Google Scholar]
- Sarrauste de Menthiere C.; Chavanieu A.; Grassy G.; Dalle S.; Salazar G.; Kervran A.; Pfeiffer B.; Renard P.; Delagrange P.; Manechez D.; Bakes D.; Ktorza A.; Calas B. Structural requirements of the N-terminal region of GLP-1-[7-37]-NH2 for receptor interaction and cAMP production. Eur. J. Med. Chem. 2004, 39, 473–480. [DOI] [PubMed] [Google Scholar]
- Green B. D.; Mooney M. H.; Gault V. A.; Irwin N.; Bailey C. J.; Harriott P.; Greer B.; O’Harte F. P.; Flatt P. R. N-terminal His(7)-modification of glucagon-like peptide-1(7-36) amide generates dipeptidyl peptidase IV-stable analogues with potent antihyperglycaemic activity. J. Endocrinol. 2004, 180, 379–388. [DOI] [PubMed] [Google Scholar]
- O’Harte F. P.; Mooney M. H.; Lawlor A.; Flatt P. R. N-terminally modified glucagon-like peptide-1(7-36) amide exhibits resistance to enzymatic degradation while maintaining its antihyperglycaemic activity in vivo. Biochim. Biophys. Acta 2000, 1474, 13–22. [DOI] [PubMed] [Google Scholar]
- Youn Y. S.; Chae S. Y.; Lee S.; Jeon J. E.; Shin H. G.; Lee K. C. Evaluation of therapeutic potentials of site-specific PEGylated glucagon-like peptide-1 isomers as a type 2 anti-diabetic treatment: insulinotropic activity, glucose-stabilizing capability, and proteolytic stability. Biochem. Pharmacol. 2007, 73, 84–93. [DOI] [PubMed] [Google Scholar]
- Burcelin R.; Dolci W.; Thorens B. Long-lasting antidiabetic effect of a dipeptidyl peptidase IV-resistant analog of glucagon-like peptide-1. Metabolism 1999, 48, 252–258. [DOI] [PubMed] [Google Scholar]
- Deacon C. F.; Knudsen L. B.; Madsen K.; Wiberg F. C.; Jacobsen O.; Holst J. J. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia 1998, 41, 271–278. [DOI] [PubMed] [Google Scholar]
- Xiao Q.; Giguere J.; Parisien M.; Jeng W.; St-Pierre S. A.; Brubaker P. L.; Wheeler M. B. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry 2001, 40, 2860–2869. [DOI] [PubMed] [Google Scholar]
- Green B. D.; Mooney M. H.; Gault V. A.; Irwin N.; Bailey C. J.; Harriott P.; Greer B.; Flatt P. R.; O’Harte F. P. Lys9 for Glu9 substitution in glucagon-like peptide-1(7-36)amide confers dipeptidylpeptidase IV resistance with cellular and metabolic actions similar to those of established antagonists glucagon-like peptide-1(9-36)amide and exendin (9-39). Metabolism 2004, 53, 252–259. [DOI] [PubMed] [Google Scholar]
- Green B. D.; Gault V. A.; Irwin N.; Mooney M. H.; Bailey C. J.; Harriott P.; Greer B.; Flatt P. R.; O’Harte F. P. Metabolic stability, receptor binding, cAMP generation, insulin secretion and antihyperglycaemic activity of novel N-terminal Glu9-substituted analogues of glucagon-like peptide-1. Biol. Chem. 2003, 384, 1543–1551. [DOI] [PubMed] [Google Scholar]
- Meng H.; Krishnaji S. T.; Beinborn M.; Kumar K. Influence of selective fluorination on the biological activity and proteolytic stability of glucagon-like peptide-1. J. Med. Chem. 2008, 51, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Kawai K.; Ohashi S.; Yokota C.; Suzuki S.; Yamashita K. Structure–activity relationships of glucagon-like peptide-1(7-36)amide: insulinotropic activities in perfused rat pancreases, and receptor binding and cyclic AMP production in RINm5F cells. J. Endocrinol. 1994, 140, 45–52. [DOI] [PubMed] [Google Scholar]
- Fehmann H. C.; Goke B.. The insulinotropic gut hormone glucagon like peptide-1. In Frontiers in Diabetes; Belfiore F., Ed.; Karger: Basel, Switzerland, 1997; Vol. 13, pp 1–99. [Google Scholar]
- Miranda L. P.; Winters K. A.; Gegg C. V.; Patel A.; Aral J.; Long J.; Zhang J.; Diamond S.; Guido M.; Stanislaus S.; Ma M.; Li H.; Rose M. J.; Poppe L.; Véniant M. M. Design and synthesis of conformationally constrained glucagon-like peptide-1 derivatives with increased plasma stability and prolonged in vivo activity. J. Med. Chem. 2008, 51, 2758–2765. [DOI] [PubMed] [Google Scholar]
- Behnam Azad B.; Rota V. A.; Breadner D.; Dhanvantari S.; Luyt L. G. Design, synthesis and in vitro characterization of glucagon-like peptide-1 derivatives for pancreatic beta cell imaging by SPECT. Bioorg. Med. Chem. 2010, 18, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Green B. D.; Gault V. A.; Mooney M. H.; Irwin N.; Harriott P.; Greer B.; Bailey C. J.; O’Harte F. P.; Flatt P. R. Degradation, receptor binding, insulin secreting and antihyperglycaemic actions of palmitate-derivatised native and Ala8-substituted GLP-1 analogues. Biol. Chem. 2004, 385, 169–177. [DOI] [PubMed] [Google Scholar]
- Ueda T.; Tomita K.; Notsu Y.; Ito T.; Fumoto M.; Takakura T.; Nagatome H.; Takimoto A.; Mihara S.; Togame H.; Kawamoto K.; Iwasaki T.; Asakura K.; Oshima T.; Hanasaki K.; Nishimura S.; Kondo H. Chemoenzymatic synthesis of glycosylated glucagon-like peptide 1: effect of glycosylation on proteolytic resistance and in vivo blood glucose-lowering activity. J. Am. Chem. Soc. 2009, 131, 6237–6245. [DOI] [PubMed] [Google Scholar]
- Sebokova E.; Christ A. D.; Wang H.; Sewing S.; Dong J. Z.; Taylor J.; Cawthorne M. A.; Culler M. D. Taspoglutide, an analog of human glucagon-like peptide-1 with enhanced stability and in vivo potency. Endocrinology 2010, 151, 2474–2482. [DOI] [PubMed] [Google Scholar]
- Dong J. Z.; Shen Y.; Zhang J.; Tsomaia N.; Mierke D. F.; Taylor J. E. Discovery and characterization of taspoglutide, a novel analogue of human glucagon-like peptide-1, engineered for sustained therapeutic activity in type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 19–25. [DOI] [PubMed] [Google Scholar]
- Tomas E.; Habener J. F. Insulin-like actions of glucagon-like peptide-1: a dual receptor hypothesis. Trends Endocrinol. Metab. 2010, 21, 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J. J.; Gethmann A.; Nauck M. A.; Götze O.; Schmitz F.; Deacon C. F.; Gallwitz B.; Schmidt W. E.; Holst J. J. The glucagon-like peptide-1 metabolite GLP-1-(9–36) amide reduces postprandial glycemia independently of gastric emptying and insulin secretion in humans. Am. J. Physiol.: Endocrinol. Metabol. 2006, 290, E1118–E1123. [DOI] [PubMed] [Google Scholar]
- Mundil D.; Beca S.; Cameron-Vendrig A.; El-Mounayri O.; Momen A.; Backx P. H.; Husain M. GLP-1[28–36] exerts direct cardioprotective effects, activating pro-survival kinases and soluble adenylyl cyclase. Circulation 2012, 126, 13657. [Google Scholar]
- Elahi D.; Angeli F. S.; Vakilipour A.; Carlson O. D.; Tomas E.; Egan J. M.; Habener J. F.; Shannon R. P. GLP-1(32–36)amide, a novel pentapeptide cleavage product of GLP-1, modulates whole body glucose metabolism in dogs. Peptides 2014, 59, 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taing M. W.; Rose F. J.; Whitehead J. P. GLP-1(28-36)amide, the glucagon-like peptide-1 metabolite: friend, foe, or pharmacological folly?. Drug Des. Dev. Ther. 2014, 8, 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli C.; Natarajan S. I.; Meyer J. P.; Bastos M. M.; Bernatowicz M. S.; Lee V. G.; Pluscec J.; Riexinger D. J.; Sieber-McMaster E. S.; Constantine K. L.; Smith-Monroy C. A.; Golla R.; Ma Z.; Longhi D. A.; Shi D.; Xin L.; Taylor J. R.; Koplowitz B.; Chi C. L.; Khanna A.; Robinson G. W.; Seethala R.; Antal-Zimanyi I. A.; Stoffel R. H.; Han S.; Whaley J. M.; Huang C. S.; Krupinski J.; Ewing W. R. Eleven amino acid glucagon-like peptide-1 receptor agonists with antidiabetic activity. J. Med. Chem. 2009, 52, 7788–7799. [DOI] [PubMed] [Google Scholar]
- Thornton K.; Gorenstein D. G. Structure of glucagon-like peptide(7-36) amide in a dodecylphosphocholine micelle as determined by 2D NMR. Biochemistry 1994, 33, 3532–3539. [DOI] [PubMed] [Google Scholar]
- Chang X.; Keller D.; Bjørn S.; Led J. J. Structure and folding of glucagon-like peptide-1-(7–36)-amide in aqueous trifluoroethanol studied by NMR spectroscopy. Magn. Reson. Chem. 2001, 39, 477–483. [Google Scholar]
- Neidigh J. W.; Fesinmeyer R. M.; Prickett K. S.; Andersen N. H. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry 2001, 40, 13188–13200. [DOI] [PubMed] [Google Scholar]
- Runge S.; Schimmer S.; Oschmann J.; Schiødt C. B.; Knudsen S. M.; Jeppesen C. B.; Madsen K.; Lau J.; Thøgersen H.; Rudolph R. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry 2007, 46, 5830–5840. [DOI] [PubMed] [Google Scholar]
- Mann R. J.; Nasr N. E.; Sinfield J. K.; Paci E.; Donnelly D. The major determinant of exendin-4/glucagon-like peptide 1 differential affinity at the rat glucagon-like peptide 1 receptor N-terminal domain is a hydrogen bond from SER-32 of exendin-4. Br. J. Pharmacol. 2010, 160, 1973–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood C. R.; Garibay P.; Knudsen L. B.; Hastrup S.; Peters G. H.; Rudolph R.; Reedtz-Runge S. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J. Biol. Chem. 2010, 285, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder-Tittmann K.; Bosse-Doenecke E.; Reedtz-Runge S.; Ihling C.; Sinz A.; Tittmann K.; Rudolph R. Recombinant expression, in vitro refolding, and biophysical characterization of the human glucagon-like peptide-1 receptor. Biochemistry 2010, 49, 7956–7965. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Pinon D. I.; Miller L. J.; Dong M. Molecular basis of glucagon-like peptide 1 docking to its intact receptor studied with carboxyl-terminal photolabile probes. J. Biol. Chem. 2009, 284, 34135–34144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Pinon D. I.; Miller L. J.; Dong M. Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor. J. Biol. Chem. 2010, 285, 24508–24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. J.; Chen Q.; Lam P. C.-H.; Pinon D. I.; Sexton P. M.; Abagyan R.; Dong M. Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modeling. J. Biol. Chem. 2011, 286, 15895–15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Maturana R.; Treece-Birch J.; Abidi F.; Findlay J. B.; Donnelly D. Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues. Protein Pept. Lett. 2004, 11, 15–22. [DOI] [PubMed] [Google Scholar]
- Al-Sabah S.; Donnelly D. The positive charge at Lys-288 of the glucagon-like peptide-1 (GLP-1) receptor is important for binding the N-terminus of peptide agonists. FEBS Lett. 2003, 553, 342–346. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R.; Donnelly D. The glucagon-like peptide-1 receptor binding site for the N-terminus of GLP-1 requires polarity at Asp198 rather than negative charge. FEBS Lett. 2002, 530, 244–248. [DOI] [PubMed] [Google Scholar]
- Runge S.; Thogersen H.; Madsen K.; Lau J.; Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 2008, 283, 11340–11347. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R.; Willshaw A.; Kuntzsch A.; Rudolph R.; Donnelly D. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J. Biol. Chem. 2003, 278, 10195–10200. [DOI] [PubMed] [Google Scholar]
- Murage E. N.; Schroeder J. C.; Beinborn M.; Ahn J.-M. Search for alpha-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg. Med. Chem. 2008, 16, 10106–10112. [DOI] [PubMed] [Google Scholar]
- Ahn J.-M.; Gitu P. M.; Medeiros M.; Swift J. R.; Trivedi D.; Hruby V. J. A new approach to search for the bioactive conformation of glucagon: positional cyclization scanning. J. Med. Chem. 2001, 44, 3109–3116. [DOI] [PubMed] [Google Scholar]
- Murage E. N.; Gao G.; Bisello A.; Ahn J.-M. Development of potent glucagon-like peptide-1 agonists with high enzyme stability via introduction of multiple lactam bridges. J. Med. Chem. 2010, 53, 6412–6420. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Polychronakos C. Immunogenetics of type 1 diabetes. Horm. Res. 2005, 64, 180–188. [DOI] [PubMed] [Google Scholar]
- Steele C.; Hagopian W. A.; Gitelman S.; Masharani U.; Cavaghan M.; Rother K. I.; Donaldson D.; Harlan D. M.; Bluestone J.; Herold K. C. Insulin secretion in type 1 diabetes. Diabetes 2004, 53, 426–433. [DOI] [PubMed] [Google Scholar]
- Kahn C. R. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994, 43, 1066–1084. [DOI] [PubMed] [Google Scholar]
- National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, 2011. [Google Scholar]
- Mojsov S. Glucagon-like peptide-1 (GLP-1) and the control of glucose metabolism in mammals and teleost fish. Am. Zool. 2000, 40, 246–258. [Google Scholar]
- Drucker D. J.; Dritselis A.; Kirkpatrick P. Liraglutide. Nat. Rev. Drug Discovery 2010, 9, 267–268. [DOI] [PubMed] [Google Scholar]
- Mudaliar S.; Henry R. R. Effects of incretin hormones on beta-cell mass and function, body weight, and hepatic and myocardial function. Am. J. Med. 2010, 123, S19–S27. [DOI] [PubMed] [Google Scholar]
- Drucker D. J.; Sherman S. I.; Gorelick F. S.; Bergenstal R. M.; Sherwin R. S.; Buse J. B. Incretin-based therapies for the treatment of type 2 diabetes: evaluation of the risks and benefits. Diabetes Care 2010, 33, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler P. C.; Dry S.; Elashoff R. GLP-1-based therapy for diabetes: What you do not know can hurt you. Diabetes Care 2010, 33, 453–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui H.; Zhao X.; Perfetti R. Structure and function studies of glucagon-like peptide-1 (GLP-1): the designing of a novel pharmacological agent for the treatment of diabetes. Diabetes Metab. Res. Rev. 2005, 21, 313–331. [DOI] [PubMed] [Google Scholar]
- Lovshin J. A.; Drucker D. J. Incretin-based therapies for type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 262–269. [DOI] [PubMed] [Google Scholar]
- Russell-Jones D. Molecular, pharmacological and clinical aspects of liraglutide, a once-daily human GLP-1 analogue. Mol. Cell. Endocrinol. 2009, 297, 137–140. [DOI] [PubMed] [Google Scholar]
- Drucker D.; Easley C.; Kirkpatrick P. Sitagliptin. Nat. Rev. Drug Discovery 2007, 6, 109–110. [DOI] [PubMed] [Google Scholar]
- Ahn J.-M.; Boyle N. A.; MacDonald M. T.; Janda K. D. Peptidomimetics and peptide backbone modifications. Mini-Rev. Med. Chem. 2002, 2, 463–473. [DOI] [PubMed] [Google Scholar]
- Kim T. H.; Jiang H. H.; Lee S.; Youn Y. S.; Park C. W.; Byun Y.; Chen X.; Lee K. C. Mono-PEGylated dimeric exendin-4 as high receptor binding and long-acting conjugates for type 2 anti-diabetes therapeutics. Bioconjugate Chem. 2011, 22, 625–632. [DOI] [PubMed] [Google Scholar]
- Eng J.; Kleinman W. A.; Singh L.; Singh G.; Raufman J. P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [PubMed] [Google Scholar]
- Thum A.; Hupe-Sodmann K.; Goke R.; Voigt K.; Goke B.; McGregor G. Endoproteolysis by isolated membrane peptidases reveal metabolic stability of glucagon-like peptide-1 analogs, exendins-3 and -4. Exp. Clin. Endocrinol. Diabetes 2002, 110, 113–118. [DOI] [PubMed] [Google Scholar]
- Ahren B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat. Rev. Drug Discovery 2009, 8, 369–385. [DOI] [PubMed] [Google Scholar]
- Gallwitz B. Benefit-risk assessment of exenatide in the therapy of type 2 diabetes mellitus. Drug Saf. 2010, 33, 87–100. [DOI] [PubMed] [Google Scholar]
- Werner U.; Haschke G.; Herling A. W.; Kramer W. Pharmacological profile of lixisenatide: A new GLP-1 receptor agonist for the treatment of type 2 diabetes. Regul. Pept. 2010, 164, 58–64. [DOI] [PubMed] [Google Scholar]
- Christensen M.; Knop F. K.; Holst J. J.; Vilsboll T. Lixisenatide, a novel GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus. IDrugs 2009, 12, 503–513. [PubMed] [Google Scholar]
- Elbrond B.; Jakobsen G.; Larsen S.; Agerso H.; Jensen L. B.; Rolan P.; Sturis J.; Hatorp V.; Zdravkovic M. Pharmacokinetics, pharmacodynamics, safety, and tolerability of a single-dose of NN2211, a long-acting glucagon-like peptide 1 derivative, in healthy male subjects. Diabetes Care 2002, 25, 1398–1404. [DOI] [PubMed] [Google Scholar]
- Madsen K.; Knudsen L. B.; Agersoe H.; Nielsen P. F.; Thøgersen H.; Wilken M.; Johansen N. L. Structure–activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J. Med. Chem. 2007, 50, 6126–6132. [DOI] [PubMed] [Google Scholar]
- Madsbad S.; Kielgast U.; Asmar M.; Deacon C.; Torekov S. S.; Holst J. J. An overview of once-weekly GLP-1 receptor agonists—available efficacy and safety data and perspectives for the future. Diabetes Obes. Metab. 2011, 13, 394–407. [DOI] [PubMed] [Google Scholar]
- Kapitza C.; Heise T.; Birman P.; Jallet K.; Ramis J.; Balena R. Pharmacokinetic and pharmacodynamic properties of taspoglutide, a once-weekly, human GLP-1 analogue, after single-dose administration in patients with type 2 diabetes. Diabet. Med. 2009, 26, 1156–1164. [DOI] [PubMed] [Google Scholar]
- Son S.; Chae S. Y.; Kim C. W.; Choi Y. G.; Jung S. Y.; Lee S.; Lee K. C. Preparation and structural, biochemical, and pharmaceutical characterizations of bile acid-modified long-acting exendin-4 derivatives. J. Med. Chem. 2009, 52, 6889–6896. [DOI] [PubMed] [Google Scholar]
- Denton E. V.; Craig C. J.; Pongratz R. L.; Appelbaum J. S.; Doerner A. E.; Narayanan A.; Shulman G. I.; Cline G. W.; Schepartz A. A β-peptide agonist of the GLP-1 receptor, a class B GPCR. Org. Lett. 2013, 15, 5318–5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M. A.; Matthews J. E.; De Boever E. H.; Dobbins R. L.; Hodge R. J.; Walker S. E.; Holland M. C.; Gutierrez M.; Stewart M. W. Safety, tolerability, pharmacodynamics and pharmacokinetics of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in healthy subjects. Diabetes Obes. Metab. 2009, 11, 498–505. [DOI] [PubMed] [Google Scholar]
- Baggio L. L.; Huang Q.; Cao X.; Drucker D. J. An albumin-exendin-4 conjugate engages central and peripheral circuits regulating murine energy and glucose homeostasis. Gastroenterology 2008, 134, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Grunberger G.; Chang A.; Garcia Soria G.; Botros F. T.; Bsharat R.; Milicevic Z. Monotherapy with the once-weekly GLP-1 analogue dulaglutide for 12 weeks in patients with type 2 diabetes: dose-dependent effects on glycaemic control in a randomized, double-blind, placebo-controlled study. Diabetic Med. 2012, 29, 1260–1267. [DOI] [PubMed] [Google Scholar]
- Kang J.; Kim S.-E.; Choi I. Y.; Bae S.; Yoon Y.-K.; Lee K. H.; Park K.-M.; Kwon S. C.; Son J.. Pharmacokinetic and pharmacodynamic effects of a single dose of the long-acting GLP-1R agonist HM11260C in subjects with type 2 diabetes mellitus. Presented at the 72nd Scientific Sessions American Diabetes Association, Philadelphia, PA, 2012.
- Diartis presents VRS-859 positive clinical results for type 2 diabetes at 48th EASD annual meeting. http://www.marketwired.com/press-release/diartis-presents-vrs-859-positive-clinical-results-type-2-diabetes-48th-easd-annual-1708016.htm (accessed Aug 21, 2013).
- Pan C. Q.; Buxton J. M.; Yung S. L.; Tom I.; Yang L.; Chen H.; MacDougall M.; Bell A.; Claus T. H.; Clairmont K. B.; Whelan J. P. Design of a long acting peptide functioning as both a glucagon-like peptide-1 receptor agonist and a glucagon receptor antagonist. J. Biol. Chem. 2006, 281, 12506–12515. [DOI] [PubMed] [Google Scholar]
- DiMarchi R. D.; Yang B.; Ouyang C.. Compounds exhibiting glucagon antagonist and GLP-1 agonist activity. WO2009058734A1, 2009.
- Day J. W.; Ottaway N.; Patterson J. T.; Gelfanov V.; Smiley D.; Gidda J.; Findeisen H.; Bruemmer D.; Drucker D. J.; Chaudhary N.; Holland J.; Hembree J.; Abplanalp W.; Grant E.; Ruehl J.; Wilson H.; Kirchner H.; Lockie S. H.; Hofmann S.; Woods S. C.; Nogueiras R.; Pfluger P. T.; Perez-Tilve D.; DiMarchi R.; Tschop M. H. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [DOI] [PubMed] [Google Scholar]
- DiMarchi R. D.; Tao M.. GIP-based mixed agonists for treatment of metabolic disorders and obesity. WO/2010/011439, 2010.
- Fosgerau K.; Skovgaard M.; Larsen S. A.; Baek C. A.; Meier E.; Groendahl C.; Bak H. H. Combination of long-acting insulin with the dual GluGLP-1 agonist ZP2929 causes improved glycemic control without body weight gain in db/db mice. Diabetes 2011, 60, A418. [Google Scholar]
- Transition Therapeutics announces results of clinical study of type 2 diabetes drug candidate TT-401. http://www.transitiontherapeutics.com/media/archive.php# (archived Apr 30, 2013) (accessed Aug 21, 2013).
- Trevaskis J. L.; Mack C. M.; Sun C.; Soares C. J.; D’Souza L. J.; Levy O. E.; Lewis D. Y.; Jodka C. M.; Tatarkiewicz K.; Gedulin B.; Gupta S.; Wittmer C.; Hanley M.; Forood B.; Parkes D. G.; Ghosh S. S. Improved glucose control and reduced body weight in rodents with dual mechanism of action peptide hybrids. PLoS One 2013, 8, e78154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilsbøll T.; Knop F. K. Review: DPP IV inhibitors—current evidence and future directions. Br. J. Diabetes Vasc. Dis. 2007, 7, 69–74. [Google Scholar]
- Green B. D.; Flatt P. R.; Bailey C. J. Dipeptidyl peptidase IV (DPP IV) inhibitors: a newly emerging drug class for the treatment of type 2 diabetes. Diabetes Vasc. Dis. Res. 2006, 3, 159–165. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Walunj S. S.; Tokala R. K.; Parsa K. V.; Singh S. K.; Pal M. Emerging drug candidates of dipeptidyl peptidase IV (DPP IV) inhibitor class for the treatment of type 2 diabetes. Curr. Drug Targets 2009, 10, 71–87. [DOI] [PubMed] [Google Scholar]
- Dicker D. DPP-4 inhibitors. Diabetes Care 2011, 34, S276–S278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K. G.; Bloom S. R. Nonpeptidic glucagon-like peptide 1 receptor agonists: A magic bullet for diabetes?. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 689–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen L. B.; Kiel D.; Teng M.; Behrens C.; Bhumralkar D.; Kodra J. T.; Holst J. J.; Jeppesen C. B.; Johnson M. D.; de Jong J. C.; Jorgensen A. S.; Kercher T.; Kostrowicki J.; Madsen P.; Olesen P. H.; Petersen J. S.; Poulsen F.; Sidelmann U. G.; Sturis J.; Truesdale L.; May J.; Lau J. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N.; Flatt P. R.; Patterson S.; Green B. D. Insulin-releasing and metabolic effects of small molecule GLP-1 receptor agonist 6,7-dichloro-2-methylsulfonyl-3-N-tert-butylaminoquinoxaline. Eur. J. Pharmacol. 2010, 628, 268–273. [DOI] [PubMed] [Google Scholar]
- Chen D.; Liao J.; Li N.; Zhou C.; Liu Q.; Wang G.; Zhang R.; Zhang S.; Lin L.; Chen K.; Xie X.; Nan F.; Young A. A.; Wang M.-W. A nonpeptidic agonist of glucagon-like peptide 1 receptors with efficacy in diabetic db/db mice. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloop K. W.; Willard F. S.; Brenner M. B.; Ficorilli J.; Valasek K.; Showalter A. D.; Farb T. B.; Cao J. X.; Cox A. L.; Michael M. D.; Gutierrez Sanfeliciano S. M.; Tebbe M. J.; Coghlan M. J. Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes 2010, 59, 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Simms J.; Koole C.; Woodman O. L.; Summers R. J.; Christopoulos A.; Sexton P. M. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J. Pharmacol. Exp. Ther. 2011, 336, 540–550. [DOI] [PubMed] [Google Scholar]
- Gong Y.-D.; Cheon H. G.; Lee T.; Kang N. S. A novel 3-(8-chloro-6-(trifluoromethyl)imidazo[1,2-a]pyridine-2-yl)phenyl acetate skeleton and pharmacophore model as glucagon-like peptide 1 receptor agonists. Bull. Korean Chem. Soc. 2010, 31, 3760–3764. [Google Scholar]
- Moon H.-S.; Yang J.-S.; Kim M.-K.; Kim J.-K.; Lee J.-Y.; Kang S.-M.; Kim H.-D.; Shin C.-Y.; Cheong Y.-H.; Yang E.-K.; Choi S.-H.; Park Y.-S.; Kim E.-J.; Chae Y.-N.; Cho E.-J.; Son M.-H.; Kim S.-H.. Novel quinoxaline derivatives. WO/2011/122815, 2011.
- Kumarasinghe I. R.; Murage E.; Ahn J.-M.. Solution structure of a GLP-1 receptor peptidomimetic agonist, Ac-SH3-GLP-1(22-36)-NH2. In Peptides:Building Bridges, Proceedings of the 22nd American Peptide Symposium; Lebl M., Ed.; Prompt Scientific Publishing: San Diego, CA, 2011; pp 182–183. [Google Scholar]
- Ahn J.-M.; Han S.-Y.; Murage E.; Beinborn M.. Rational design of peptidomimetics for class B GPCRs: potent non-peptide GLP-1 receptor agonists. In Peptides: Chemistry, Structure and Biology, Proceedings of the 20th American Peptide Symposium; Escher E., Lubell W. D., Valle S. D., Eds.; Springer: New York, NY, 2009; pp 125–126. [DOI] [PubMed] [Google Scholar]
- Ravindranathan P.; Lee T.-K.; Yang L.; Centenera M. M.; Butler L.; Tilley W. D.; Hsieh J.-T.; Ahn J.-M.; Raj G. V. Peptidomimetic targeting of critical androgen receptor–coregulator interactions in prostate cancer. Nat. Commun. 2013, 4, 1923. 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- Ahn J.-M.; Han S.-Y. Facile synthesis of benzamides to mimic an a-helix. Tetrahedron Lett. 2007, 48, 3543–3547. [Google Scholar]
- Lee T.-K.; Ahn J.-M. Solid-phase synthesis of tris-benzamides as α-helix mimetics. ACS Comb. Sci. 2011, 13, 107–111. [DOI] [PubMed] [Google Scholar]
- Yang H. J.; Park I. S.; Na K. Biocompatible microspheres based on acetylated polysaccharide prepared from water-in-oil-in-water (W1/O/W2) double-emulsion method for delivery of type II diabetic drug (exenatide). Colloids Surf. Physicochem. Eng. Aspects 2009, 340, 115–120. [Google Scholar]
- Li K.; Yu L.; Liu X.; Chen C.; Chen Q.; Ding J. A long-acting formulation of a polypeptide drug exenatide in treatment of diabetes using an injectable block copolymer hydrogel. Biomaterials 2013, 34, 2834–2842. [DOI] [PubMed] [Google Scholar]
- Joabsson F. C.; Johnsson M. C.; Tiberg F. C.. GLP-1 analogue formulations. EP1888031A1, 2008.
- Amiram M.; Luginbuhl K. M.; Li X.; Feinglos M. N.; Chilkoti A. Injectable protease-operated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 2792–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker S.; Quandt H.; Kuzma P.. Sustained delivery of exenatide and other polypeptides. EP2303226A2, 2011.
- Kim H.; Lee J.; Kim T.; Lee E.; Oh K.; Lee D.; Park E.-S.; Bae Y.; Lee K.; Youn Y. Albumin-coated porous hollow poly(lactic-co-glycolic acid) microparticles bound with palmityl-acylated exendin-4 as a long-acting inhalation delivery system for the treatment of diabetes. Pharm. Res. 2011, 28, 2008–2019. [DOI] [PubMed] [Google Scholar]
- Andersen C.; Lorenzen G.; Arent N.; Thorengaard B.; Wittorff H.. Compressed chewing gum tablet. US20100255063A1, 2010.
- Kim P.-H.; Lee M.; Kim S. W. Delivery of two-step transcription amplification exendin-4 plasmid system with arginine-grafted bioreducible polymer in type 2 diabetes animal model. J. Controlled Release 2012, 162, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADA. Standards of Medical Care in Diabetes—2010. Diabetes Care 2010, 33, S11–S61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn S. E.; Carr D. B.; Faulenbach M. V.; Utzschneider K. M. An examination of β-cell function measures and their potential use for estimating β-cell mass. Diabetes Obes. Metab. 2008, 10, 63–76. [DOI] [PubMed] [Google Scholar]
- Gomis R.; Figuerola D.; Micalo T.; Millan M.; Peig M. Glycosylated hemoglobin as an index of diabetes mellitus control (author’s transl). Med. Clin. 1981, 76, 251–254. [PubMed] [Google Scholar]
- Berger B.; Stenstrom G.; Sundkvist G. Random C-peptide in the classification of diabetes. Scand. J. Clin. Lab. Invest. 2000, 60, 687–693. [DOI] [PubMed] [Google Scholar]
- Matveyenko A. V.; Butler P. C. Relationship between β-cell mass and diabetes onset. Diabetes Obes. Metab. 2008, 10, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M.; Welsh N.; Jonas J.-C.; Jörns A.; Lenzen S.; Eizirik D. L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005, 54Suppl. 2S97–107. [DOI] [PubMed] [Google Scholar]
- Souza F.; Freeby M.; Hultman K.; Simpson N.; Herron A.; Witkowsky P.; Liu E.; Maffei A.; Harris P. E. Current progress in non-invasive imaging of beta cell mass of the endocrine pancreas. Curr. Med. Chem. 2006, 13, 2761–2773. [DOI] [PubMed] [Google Scholar]
- Halban P. A. Cellular sources of new pancreatic beta cells and therapeutic implications for regenerative medicine. Nat. Cell Biol. 2004, 6, 1021–1025. [DOI] [PubMed] [Google Scholar]
- Ueberberg S.; Meier J. J.; Waengler C.; Schechinger W.; Dietrich J. W.; Tannapfel A.; Schmitz I.; Schirrmacher R.; Köller M.; Klein H. H.; Schneider S. Generation of novel single-chain antibodies by phage-display technology to direct imaging agents highly selective to pancreatic β- or α-cells in vivo. Diabetes 2009, 58, 2324–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. Advances in beta-cell imaging. Eur. J. Radiol. 2009, 70, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudek F.; Brogren C. H.; Manohar S. Imaging the beta-cell mass: why and how. Rev. Diabetic Stud. 2008, 5, 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichise M.; Harris P. E. Imaging of β-cell mass and function. J. Nucl. Med. 2010, 51, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaisse W. J.; Louchami K.; Sener A. Noninvasive imaging of pancreatic beta cells. Nat. Rev. Endocrinol. 2009, 5, 394–400. [DOI] [PubMed] [Google Scholar]
- Shiue C. Y.; Schmitz A.; Schirrmacher R.; Shiue G. G.; Alavi A. Potential approaches for beta cell imaging with PET and SPECT. Curr. Med. Chem.: Immunol., Endocr. Metab. Agents 2004, 4, 271–280. [Google Scholar]
- Schneider S. Efforts to develop methods for in vivo evaluation of the native β-cell mass. Diabetes Obes. Metab. 2008, 10, 109–118. [DOI] [PubMed] [Google Scholar]
- Murthy R.; Harris P.; Simpson N.; Van Heertum R.; Leibel R.; Mann J.; Parsey R. Whole body [11C]-dihydrotetrabenazine imaging of baboons: biodistribution and human radiation dosimetry estimates. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 790–797. [DOI] [PubMed] [Google Scholar]
- Kung M. P.; Hou C.; Lieberman B. P.; Oya S.; Ponde D. E.; Blankemeyer E.; Skovronsky D.; Kilbourn M. R.; Kung H. F. In vivo imaging of beta-cell mass in rats using 18F-FP-(+)-DTBZ: a potential PET ligand for studying diabetes mellitus. J. Nucl. Med. 2008, 49, 1171–1176. [DOI] [PubMed] [Google Scholar]
- Harris P.; Ferrara C.; Barba P.; Polito T.; Freeby M.; Maffei A. VMAT2 gene expression and function as it applies to imaging β-cell mass. J. Mol. Med. 2008, 86, 5–16. [DOI] [PubMed] [Google Scholar]
- Goland R.; Freeby M.; Parsey R.; Saisho Y.; Kumar D.; Simpson N.; Hirsch J.; Prince M.; Maffei A.; Mann J. J.; Butler P. C.; Van Heertum R.; Leibel R. L.; Ichise M.; Harris P. E. 11C-Dihydrotetrabenazine PET of the pancreas in subjects with long-standing type 1 diabetes and in healthy controls. J. Nucl. Med. 2009, 50, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornehave D.; Kristensen P.; Romer J.; Knudsen L. B.; Heller R. S. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J. Histochem. Cytochem. 2008, 56, 841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly B.; Vanko A.; McQuade P.; Guenther I.; Meng X.; Rubins D.; Waterhouse R.; Hargreaves R.; Sur C.; Hostetler E. Ex vivo imaging of pancreatic beta cells using a radiolabeled GLP-1 receptor agonist. Mol. Imaging Biol. 2012, 14, 79–87. [DOI] [PubMed] [Google Scholar]
- Selvaraju R. K.; Velikyan I.; Johansson L.; Wu Z.; Todorov I.; Shively J.; Kandeel F.; Korsgren O.; Eriksson O. In vivo imaging of the glucagon like peptide 1 receptor in the pancreas with 68Ga-labeled DO3A-exendin-4. J. Nucl. Med. 2013, 54, 1458–1463. [DOI] [PubMed] [Google Scholar]
- Reiner T.; Kohler R. H.; Liew C. W.; Hill J. A.; Gaglia J.; Kulkarni R. N.; Weissleder R. Near-infrared fluorescent probe for imaging of pancreatic β cells. Bioconjugate Chem. 2010, 21, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brom M.; Oyen W. J.; Joosten L.; Gotthardt M.; Boerman O. C. 68Ga-labelled exendin-3, a new agent for the detection of insulinomas with PET. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1345–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild D.; Béhé M.; Wicki A.; Storch D.; Waser B.; Gotthardt M.; Keil B.; Christofori G.; Reubi J. C.; Mäcke H. R. [Lys40(Ahx-DTPA-111In)NH2]Exendin-4, a very promising ligand for glucagon-like peptide-1 (GLP-1) receptor targeting. J. Nucl. Med. 2006, 47, 2025–2033. [PubMed] [Google Scholar]
- Gotthardt M.; Lalyko G.; van Eerd-Vismale J.; Keil B.; Schurrat T.; Hower M.; Laverman P.; Behr T. M.; Boerman O. C.; Göke B.; Béhé M. A new technique for in vivo imaging of specific GLP-1 binding sites: first results in small rodents. Regul. Pept. 2006, 137, 162–167. [DOI] [PubMed] [Google Scholar]
- Wild D.; Wicki A.; Mansi R.; Béhé M.; Keil B.; Bernhardt P.; Christofori G.; Ell P. J.; Mäcke H. R. Exendin-4-based radiopharmaceuticals for glucagon like peptide-1 receptor PET/CT and SPECT/CT. J. Nucl. Med. 2010, 51, 1059–1067. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Todorov I.; Li L.; Bading J. R.; Li Z.; Nair I.; Ishiyama K.; Colcher D.; Conti P. E.; Fraser S. E.; Shively J. E.; Kandeel F. In vivo imaging of transplanted islets with 64Cu-DO3A-VS-Cys40-exendin-4 by targeting GLP-1 receptor. Bioconjugate Chem. 2011, 22, 1587–1594. [DOI] [PubMed] [Google Scholar]
- Ahn J.-M.; Murage E. N.; Lo S.-T.; Lin M.; Sun X.. Highly constrained GLP-1 analogues as non-invasive PET imaging agents for the assessment of pancreatic beta-cell mass. In Peptides:Building Bridges, Proceedings of the 22nd American Peptide Symposium; Lebl M., Ed.; Prompt Scientific Publishing: San Diego, CA, 2011; pp 220–221. [Google Scholar]
- Ahn J.-M.; Sun X.. Cyclic peptide analogues for non-invasive imaging of pancreatic beta-cells. WO2010118034A2, 2010.
- Wicki A.; Wild D.; Storch D.; Seemayer C.; Gotthardt M.; Behe M.; Kneifel S.; Mihatsch M. J.; Reubi J.-C.; Mäcke H. R.; Christofori G. [Lys40(Ahx-DTPA-111In)NH2]-exendin-4 is a highly efficient radiotherapeutic for glucagon-like peptide-1 receptor-targeted therapy for insulinoma. Clin. Cancer Res. 2007, 13, 3696–3705. [DOI] [PubMed] [Google Scholar]
- Pattou F.; Kerr-Conte J.; Wild D. GLP-1–receptor scanning for imaging of human beta cells transplanted in muscle. N. Engl. J. Med. 2010, 363, 1289–1290. [DOI] [PubMed] [Google Scholar]
- Reiner T.; Thurber G.; Gaglia J.; Vinegoni C.; Liew C. W.; Upadhyay R.; Kohler R. H.; Li L.; Kulkarni R. N.; Benoist C.; Mathis D.; Weissleder R. Accurate measurement of pancreatic islet β-cell mass using a second-generation fluorescent exendin-4 analog. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12815–12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai E.; Toyoda K.; Kimura H.; Kawashima H.; Fujimoto H.; Ueda M.; Temma T.; Hirao K.; Nagakawa K.; Saji H.; Inagaki N. GLP-1 receptor antagonist as a potential probe for pancreatic beta-cell imaging. Biochem. Biophys. Res. Commun. 2009, 389, 523–526. [DOI] [PubMed] [Google Scholar]
- Gao H.; Niu G.; Yang M.; Quan Q.; Ma Y.; Murage E. N.; Ahn J.-M.; Kiesewetter D. O.; Chen X. PET of insulinoma using 18F-FBEM-EM3106B, a new GLP-1 analogue. Mol. Pharmaceutics 2011, 8, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar B.; Lo S.-T.; Murage E.; Lin M.; Sun X.; Ahn J.-M.. Targeting GLP-1R for non-invasive assessment of pancreatic beta-cell mass. In Peptides across the Pacific, Proceedings of the 23rd American Peptide Symposium; Lebl M., Ed. Prompt Scientific Publishing: San Diego, CA, 2013; pp 172–173. [Google Scholar]
- Silvers W.; Ramezani S.; Lo S.-T.; Kumar A.; Ahn J.-M.; Manandhar B.; Lingvay I.; Anderson J. A.; Sun X.; Öz O. K.. A pilot study on glucagon-like peptide 1 receptor (GLP-1R) targeted PET imaging of beta-cell mass (BCM) in Ossawbaw mini-pigs. Presented at the World Molecular Imaging Congress, Sep 19, 2013; Presentation LBAP 022.