

Abstract
BACKGROUND
Intestinal epithelial cell (IEC) Stat3 is required for wound healing following acute Dextran Sodium Sulfate (DSS) injury. We hypothesized that loss of IEC STAT3 would promote the development of chronic colitis following acute DSS injury.
METHODS
Colitis was induced in IEC-specific Stat3 deficient mice (Stat3ΔIEC) and littermate controls (Stat3Flx/Flx) with 4%DSS for 7 days, followed by water consumption for 21 days. Epithelial and immune mediators and severity of colitis were determined.
RESULTS
Survival, colon length, and histologic injury were significantly worse at day 28 in Stat3ΔIEC mice. IEC proliferation and apoptosis did not vary by genotype at day 14 or day 28. The colonic lamina propria frequency of pSTAT3+ cells was increased at day 28 and correlated with histologic injury in Stat3ΔIEC mice. The frequency of colonic F480+pSTAT3+ macrophages and CD3+pSTAT3+ T-lymphocytes were increased in Stat3ΔIEC mice as compared to Stat3Flx/Flx controls. In Stat3ΔIEC mice, colonic expression of Stat3 target genes Reg3β and Reg3γ which mediate epithelial restitution were significantly decreased, while expression of IL-17a, IFNγ, CXCL2, CXCL10, and CCL2 were significantly increased and correlated with the increase in histologic severity at Day 28(p<.05). IL-17a expression also correlated with the increased lamina propria frequency of CD3+pSTAT3+ T-lymphocytes.
CONCLUSIONS
Loss of intestinal epithelial Stat3 leads to more severe chronic inflammation following acute injury which is not accounted for by a sustained defect in epithelial proliferation or apoptosis 7 or 21 days after one cycle of DSS but rather defective REG3 expression and expansion of pSTAT3+ lymphocytes and IL-17a expression.
Keywords: Dextran Sodium Sulfate (DSS), Chronic Colitis, Signal Transducer and Activator of Transcription 3 (STAT3), Interleukin 17A
INTRODUCTION
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic relapsing and remitting inflammatory disorders of the gastrointestinal tract commonly referred to as the inflammatory bowel diseases (IBDs). The precise etiology remains unclear but evidence suggests that it involves the deregulation of the host immune response to luminal flora. Large scale genome wide association studies (GWASs) have now identified multiple genetic factors that confer IBD susceptibility (1-3). Genotypic variation in STAT3 and JAK2 is linked to IBD risk (1). We have recently reported that carriage of the STAT3 rs744166 risk allele is associated with increased cellular STAT3 activation and up-regulation of chemokines expressed on 4q12-13 which promote neutrophil recruitment to the gut (4). Conversely, we have reported in abstract form that patients carrying the JAK2 rs10758669 risk allele have decreased STAT3 activation in IL-6-stimulated PB CD3+/CD4+ lymphocytes and reduced colonic expression of the STAT3 target gene REG3α (5). Prager et al has also recently shown that the JAK2 rs10758669 risk allele is associated with increased intestinal permeability(6). As the biochemical basis for these observations are still unclear it suggests that reduced STAT3 activation may also be associated with increased risk for IBD.
Signal transducers and activators of transcription (STATs) mediate cytokine signaling. Constitutive activation of STATs, especially STAT3, has been reported in several inflammatory and malignant disorders. STAT3 is a pleiotropic transcription factor that displays tissue specific differences in function. STAT3 is activated by multiple cytokines and growth factors and roles for IL-6, IL-11, IL-17, IL-22, IL-27, growth hormone and leptin in experimental colitis have been elucidated(7-16). In animal models, Stat3 activation in intestinal epithelial cells is required for acute wound healing responses via induction of Reg3 family members, but also promotes development of colitis-associated cancer during chronic inflammation (17, 18). Stat3 activation in myeloid cells mediates anti-inflammatory effects of IL-10; targeted deletion of Stat3 in this cell type leads to severe entero-colitis (19). Conversely, Stat3 activation in CD4+ T cells is required for differentiation of Th17 effector lymphocytes, and blockade of IL-6:Stat3 signaling ameliorates both ileitis and colitis in animal models (7, 10). Tyrosine phosphorylation of STAT3 induced by IL-6 has also been linked to effector lymphocyte and granulocyte activation in IBD (20). We have previously shown that STAT3 activation was increased in PB granulocytes, IL-6-stimulated CD3+/CD4+ lymphocytes, and affected colon biopsies of pediatric IBD patients at diagnosis and during therapy (21). We identified an IL-6:STAT3 biological network that drives leukocyte recruitment and thereby mucosal inflammation in this setting. Previous studies in IEC Stat3 deficient mice have examined the effects of either acute DSS induced colon injury, or multiple rounds of DSS administration together with injection of the carcinogen Azoxymethane (AOM) to induce colitis associated cancer. These studies have shown that while Stat3 activation promotes survival and proliferation of IECs in response to acute injury, during chronic colitis under tumorigenic conditions epithelial Stat3 drives the development of colitis associated cancer (17, 18). While these prior studies suggested that loss of Stat3 within the epithelial compartment would likely increase risk for the development of chronic inflammation following an acute self-limited gut injury, this had not been formally tested. This model of acute injury and either resolution or development of chronic colitis in a genetically susceptible host would be expected to be more applicable to the current concept of IBD pathogenesis in humans. We therefore sought to define the functional consequences of intestinal epithelial specific deletion of Stat3 in a model of chronic colitis in which mice are exposed to DSS for seven days, and then assessed during recovery over the next twenty-one days. We hypothesized that loss of IEC Stat3 would promote the development of chronic colitis following acute DSS injury. We found that loss of intestinal epithelial Stat3 leads to more severe inflammation following acute injury, which is not accounted for by a sustained defect in epithelial proliferation or apoptosis at 7 or 21 days after one cycle of DSS but rather defective REG3 expression and STAT3 activation in innate and adaptive immune cells with expansion of pSTAT3+ lymphocytes and IL-17A expression.
MATERIALS AND METHODS
Animals
To delete Stat3 in intestinal epithelial cells (IEC), we crossed Stat3 lox/lox mice (termed Stat3Flx/Flx) (22) obtained with permission from Takeda via Dr. Jeffrey Whitsett at CCHMC and backcrossed for five generations on a C57BL/6 background before intercrossing to villin-Cre (B6.SJL-Tg(Vil-Cre)997Gum/J, Jackson Laboratories, Bar Harbor, Maine) to generate compound mutant villin-Cre/Stat3lox/lox mice (termed Stat3ΔIEC). Cre-negative littermates were used as controls (termed Stat3Flx/Flx). We confirmed deletion of intestinal epithelial STAT3 using an in-vivo model of LPS exposure and PCR for total STAT3 in isolated enterocytes (Suppl. Figure 1). Stat3ΔIEC mice were born in mendelian ratios, developed normally and showed no obvious spontaneous phenotype as has been previously published (11, 18). All mice were maintained in conventional housing at CCHMC and all experiments were performed in accordance with CCHMC and Institutional Animal Care and Use Committees of CCHMC.
DSS Colitis
Intestinal inflammation was induced by 4% DSS water (m.w. 36,000-50,000):MP Biomedical, LLC, Solon, OH) for 7 days in acute studies designated (day 7) or 7 days followed by 7 days of water designated (day 14) or 7 days followed by 21 days of water designated (day 28) to study progression from acute phase to chronic inflammation and recovery. Healthy control animals received water only designated (day 0). The dosing of DSS was established by initial experiments within our colony to induce moderate to severe colitis while minimizing mortality. Body weight change during DSS protocol was calculated by dividing body weight on the specified day by body weight at day 0 (starting body weight) and expressed in percentage and recorded on Day 0, 3, 5, 7, 10, 12, 18, 20, 22, 26, and 28. In all experiments, littermate controls were used to assure comparison of mice on the same genetic background. DSS injury occurs throughout the colon at varying levels and the area involved increases from the proximal to the distal colon(23). Therefore, we utilized the anal verge as the most distal reference point from which to perform histopathological analysis.
Tissue sampling
Mice were sacrificed and the colon from the ileocecal valve to the anal verge was removed. Colon length was determined from the cecocolic junction to the anal verge. The cecum was removed at the cecocolic junction and the whole colon flushed with ice-cold PBS. The colon was then cut into two equal segments one labeled as distal and the other as proximal. The distal and proximal segments were fileted open and cut into two equal portions the length of the segment. One portion of distal and proximal colon was laid flat and fixed in 10% neutral buffered formalin overnight, transferred to 70% ethanol, processed and embedded, sectioned at 5-μm, and utilized for immunohistochemistry. The remaining distal and proximal portions were Swiss-rolled at the base of a vinyl cryomold that was subsequently filled with O.C.T compound (Tissue Tek®, Sakura Finetek USA, Inc) frozen on dry ice, and stored at -80°C for later use in immunohistochemical analyses and RNA extraction (24). All sections from paraffin embedded and Swiss-rolled OCT fixed colons were processed so that the two segments had continual villus crypt architecture from the anal verge to the cecocolic junction for representation of the whole colon.
Histo-pathological assessment of DSS-induced colitis
After staining with hematoxylin-eosin colon sections were scored as previously described with the following alternations (25). It is known that C57BL/6 mice upon DSS administration develop distal and middle colonic disease with little disease in the most proximal portion (23). To orient a starting point for analysis we developed a scoring system to account for total colonic disease by using the anal verge to mark the most distal starting point for scoring and the following four consecutive longitudinal 5x High Powered Fields (HPF) scored as the distal colon and the following four, 5x HPF scored as the proximal colon. With the observer blinded to genotype, a disease score was determined separately for the distal and proximal portions by adding the scores for area involved, erosion/ulceration, and crypt loss scored as: 0 to 4 as follows: 0, normal; 1, less than 10%; 2, 10%-25%; 3, 25% to 50%; and 4, greater than 50% with edema and infiltration of immune cells into the mucosa scored as 0, absent; 1, weak; 2, moderate; 3, severe. The total disease score was calculated as the sum of distal and proximal disease score divided by two.
Immunohistochemical Analyses (IHC)
For IHC, paraffin-embedded slides were deparaffinized and antigen unmasking was done by boiling for 10 minutes in 10mM sodium citrate (pH 6) for cleaved caspase-3 (Cell Signaling, Danvers, MA), and 1mM EDTA (pH 8) for tyrosine phosphorylation state specific (Tyr705) STAT3 (pSTAT3-XP) (Cell Signaling, Danvers, MA). Endogenous peroxide was quenched with 3% hydrogen peroxide for 15min at RT and tissue was permeabilized with .3% Triton X-100 for 15 min at RT. Slides were subsequently blocked with 3% serum and then incubated overnight at 4°C with primary antibodies. Biotinylated secondary anti-rabbit antibodies (Vector Laboratories, Burlingame, CA) were added and incubated at room temperature for 1hr. Detection and visualization of stained cells was achieved using the R.T.U kit (Vector Laboratories, West Grove, PA) with diaminobenzidine (DAB, brown) or nickel-enhanced DAB (DAB-Ni, black) as the chromogen. Quantification of cleaved caspase-3 staining in epithelial cells of the distal and proximal colon was performed, with observer blinded to genotype, by counting positive cells in three 400x high-powered fields (HPFs) in the mucosa that bordered ulcers of the distal and proximal colon or in the corresponding area of the colon in untreated controls. BrdU staining was performed as suggested by the manufacturer (BrdU staining kit; Invitrogen, Carlsbad, CA). Mice were injected with BrdU labeling reagent (Zymed®, Invitrogen, Carlsbad, CA) per manufacturer instructions 2hr before sacrifice and processed for immunohistochemistry as above. Quantification of pSTAT3 or BrDU positive epithelial cells from properly oriented crypts defined as those with clear cell column lining both sides of an open luminal area, were counted within .5cm (or one 50x HPF) of the mucosa that bordered ulcers in the distal and proximal colon or in the corresponding segment of the colon in untreated controls. The number of pSTAT3 or BrdU-positive cells per crypt divided by the total number of cells per crypt was determined with the observer blinded to genotype and data are expressed as a labeling index. Histological analysis and microphotographs were attained on an Olympus BX51 microscope utilizing the Olympus DP71 digital camera system and analyzed using DP-BSW ver.03.02 software.
Immuno-fluorescence
5-μm-thick colonic sections were fixed with 3.7% formaldehyde in phosphate-buffered saline for 15 min and washed with phosphate-buffered saline once followed by permeabilization with ice-cold methanol at -20 for 10 min. The cells were then blocked with 5% bovine serum albumin for 60 min. To double label, sections were incubated with rabbit anti-mouse pSTAT3-XP antibody overnight at 4°c. The following day sections were incubated with Rhodamine (TRITC) conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) and then sequentially incubated with either rat anti-mouse F4/80 (eBioscience, San Diego, CA) to stain macrophages, or goat anti-mouse CD3-ε (Santa Cruz Biotechnology, Santa Cruz, CA.) to stain T lymphocytes for 1 hour at room temperature and finally incubated with DyLight™ 488-conjugated anti-goat or Fluorescein (FITC)-conjugated anti-Rat secondary antibodies (Jackson ImmunoResearch Laboratories Inc.). Cell nuclei were labeled with DAPI diluted in the aqueous mounting media, Fluoromount-G (Southern Biotech), to prevent fluorochrome quenching during analysis and for a semi-permanent seal. Images were captured using Olympus BX51 microscope utilizing the Olympus DP71 digital camera system and analyzed using DP-BSW version.03.02 software.
Real-time RT-PCR
Swiss Rolled colonic tissues embedded in O.C.T compound were cross sectioned into four to five 30-μm-thick sections, giving approximately 20mg of total colonic tissue. The sections were rinsed in RNAlater® RNA stabilization reagent (Ambion, Austin, TX) to remove O.C.T medium and RNA was extracted using RNeasy Plus Kit (Qiagen, Valencia, CA). RNA integrity was validated on a denaturing gel with proper 2:1 ratio (28S:18S) and quantified using NanoDrop® 1000 Spectrophotometer (ThermoScientific, Wilmington, DE). After DNase I treatment (Invitrogen, Carlsbad, CA), RNA was reverse transcribed with Oligo-dT using SuperScript III 1st Strand cDNA Synthesis kit. SYBR Green based detection (Stratgene) utilizing the Stratagene Mx3000P PCR machine was used to determine gene expression. Expression data were normalized to β-actin mRNA expression and presented as relative message level. Primer sequences are listed in Table 1.
Table 1.
Real-time quantitative reverse transcription- PCR (RT-PCR) Primers
| Gene | Forward | Reverse |
|---|---|---|
| STAT3 Signaling | ||
| IL-6 | 5′-CAAAGCCAGAGTCCTTCAGAGAGATAC-3′ | 5′-ATTGGATGGTCTTGGTCCTTAGC-3′ |
| IL-10 | 5′-GGT TGCCAAGCCTTATCGGA-3′ | 5′-ACCTGCTCCACTGCCTTATCGGA-3′ |
| SOCS3 | 5′-GCTCCAAAAGCGAGTACCAGC-3′ | 5′-AGTAGAATCCGCTCTCCTGCAG-3′ |
| RegIIIβ | 5′-TCCCAGGCTTATGGCTCC TA-3′ | 5′-GCAGGCCAGTTCTGC ATC A-3′ |
| RegIIIγ | 5′-TTCCTGTCCTCCATGATCAAAA-3′ | 5′-CATCCACCTCTGTTGGGTTCA-3′ |
| p65 Signaling | ||
| IκBα | 5′-TGGCGAGGTCTGACTGTTGTGG-3′ | 5′-GCTCATCAAAAAGTTCCCTGTGC-3′ |
| Cytokines | ||
| IL-4 | 5′-TGTACCAGGAGCCATATCCAC-3′ | 5‘-GTTCTTCGTTGCTGTGAGGAC-3′ |
| IL-1β | 5′-CAGAGTTCCCCAACTGGTACAT-3′ | 5′-AAGGAGGAAACACAGGCTCTCT-3′ |
| IFNγ | 5′-CATTCATGAGTATTGCCAAGTTTG-3′ | 5′-GCTGGATTCCGGCAACAG-3′ |
| TNFα | 5′-AGGCAGGTTCTGTCCCTTTCA-3′ | 5′-AGATAGCAAATCGGCTGACGG-3′ |
| IL-17A | 5′- GCTCCAGAAGGCCCTCAGA-3′ | 5′-AGCTTTCCCTCCGCATTGA-3′ |
| Chemokines | ||
| CXCL1/KC | 5′-CCCAAACCGAAGTCATAGCCACAC-3′ | 5′-TTGTCAGAAGCCAGCGTTCACCAG-3′ |
| CXCL2 / MIP-2 | 5′-GGCTGTTGTGGCCAGTGAA-3′ | 5′-TGTTCAGTATCTTTTGGATGATTTTCTG-3′ |
| CXCL5 / Lix | 5′-AGCTGCGTTGTGTTTGCTTAACCG-3′ | 5′-TTGAACACTGGCCGTTCTTTCCAC-3′ |
| CXCL10 | 5′-GCCGTCATTTTCTGCCTCAT-3′ | 5′-GCTTCCCTATGGCCCTCATT-3′ |
| CCL2/MCP-1 | 5′-CACCCCATTACATCTCTTCCCC-3′ | 5′-TGTTTCCCTCTCACTTCACTCTGT-3′ |
| CCL4/MIP-1β | 5′-GCCCTCTCTCTCCTCTTGCT-3′ | 5′-GAGGGTCAGAGCCCATTG-3′ |
| CCL5 | 5′-TTGCCTACCTCTCCCTCGCGC-3′ | 5′-GGTTCCTTCGAGTGACAAACA-3′ |
| CCL20 | 5′-GGTGGCAAGCGTCTGCTC-3′ | 5′-GCCTGGCTGCAGAGGTGA-3′ |
| S100 products | ||
| S100A8 | 5′- AGTGTCCTCAGTTTGTGCAG-3′ | 5′-ATCCCTTGTGGCTGTCTTTG-3′ |
| S100A9 | 5′-GTTGATCTTTGCCTGTCATGAG-3′ | 5′-AGCCATTCCCTTTAGACTTGG-3′ |
| Receptors | ||
| CXCR2 | 5′-AGCAAACACCTCTACTACCCTCTA-3′ | 5′-GGGCTGCATCAATTCAAATACCA-3′ |
Statistical analysis
All values are presented as mean ± SEM. Total histology scores, colon length, and histology sub-scores were compared using an analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. %Body Weight was compared using a 2 way Analysis of Variance (ANOVA). Comparisons were made between Stat3Flx/FLx littermate controls and Stat3ΔIEC mice at day 0, day 7, day 14 or day 28 using un-paired Student t test. Association between two variables was measured by Pearson’s correlation coefficient. Statistical analyses were performed using GraphPad PRISM© Version 4.01. A p- value <0.05 was considered statistically significant.
RESULTS
Stat3ΔIEC mice exhibit more severe acute and chronic colitis following DSS administration
Stat3Flx/Flx littermate controls and Stat3ΔIEC mice were administered 4%DSS for 7 days to induce acute colitis (day 7) followed by water consumption alone for 7 days (day 14) or 21 days (day 28) with untreated mice used as controls (day 0) (Figure 1A). Colitis was not apparent at day 0 in either genotype. Stat3ΔIEC mice at day 7 exhibited a more severe total histology score of 12.6 ± .55 compared to 9.1 ±1.5 for Stat3Flx/Flx littermate controls (p<.0001) (Figure 1B). Day 28 analysis revealed that both genotypes had sustained chronic colitis however the Stat3ΔIEC mice had more total histology scores of 8.6 ±.44 compared to 5.3 ±.38 for Stat3Flx/Flx littermate controls (p<.0001) (Figure. 1B). Kaplan-Meyer analysis demonstrated increased mortality in Stat3ΔIEC mice which became apparent by day 12 (Figure. 1C). Both genotypes began to lose weight after the 7th day of DSS administration and by day 10 the body weight of the Stat3ΔIEC mice was reduced by 10.6% compared with 4.3% in Stat3Flx/Flx littermate controls (p<.001) (Figure 1D). Colon length was shortened to a similar extent in both genotypes at day 7, but the length of the colon was significantly reduced at day 28 to 8 ±.2cm in Stat3ΔIEC mice compared to 9 ±.2cm in Stat3Flx/Flx littermate controls (p<.005) (Figure 1E).
Figure 1. Stat3ΔIEC mice exhibit more severe acute and chronic colitis after DSS administration.
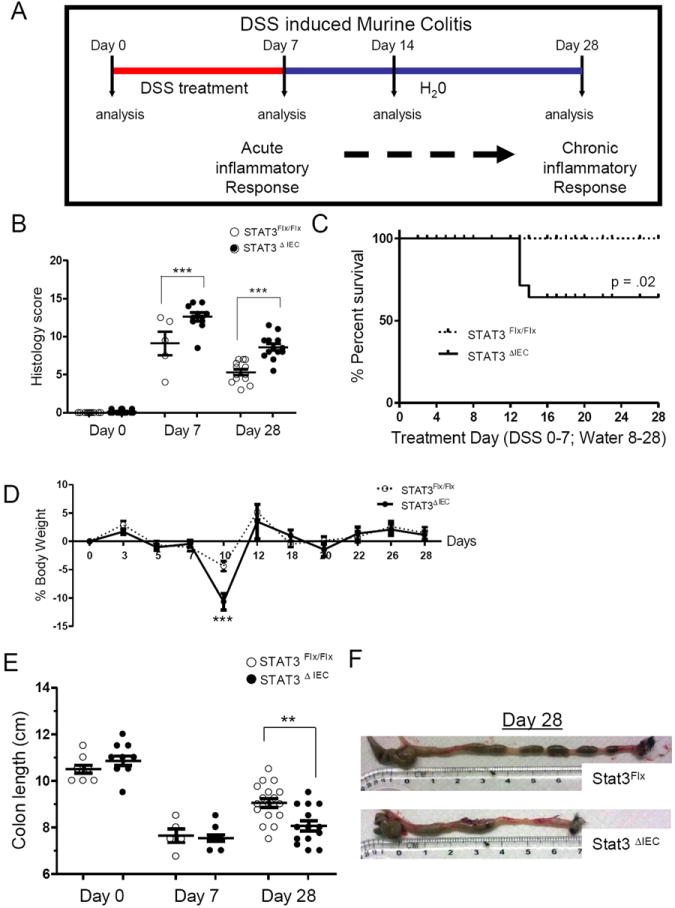
(A) Schematic representation of the DSS-induced colitis model. Mice lacking intestinal epithelial Stat3 (Stat3ΔIEC) and littermate controls were given 4% DSS for 7 days followed by consumption of water alone for 7 or 21 days. (B) Colon histologic injury was determined as shown. (C) Survival and (D) weight loss were determined and are shown (n>21 mice per genotype). (E) Colon length was measured as shown with (F) representative images of the gross appearance. Data are shown as mean ± SEM. Differences between genotypes were analyzed by (B and E) one-way ANOVA or (D) two-way ANOVA, with Bonnferroni post-test (*, p < .05; ** p < .005; *** p < .0001).
The histologic sub-scores for overall area involved at day 7 and day 28 were significantly worse in Stat3ΔIEC mice at 3.4 ±.2 and 2.8 ±.2 compared to 2.6 ±.4 and 2.0 ±.2 in Stat3Flx/Flx littermate controls (p<.001) (Figure 2B). There was substantial specific injury to the epithelial cell layer as measured by ulceration and crypt loss that persisted within the Stat3ΔIEC deficient animals. At day 7 Stat3ΔIEC mice had almost twice the amount of ulceration with a score of 1.7 ±.2 compared to .8 ±.3 in Stat3Flx/Flx littermate controls (p<.0001) (Figure 2C). The ulceration persisted in the Stat3ΔIEC mice at day 28 and was almost four times more severe at .73 ±.1 compared to .15 ±.1 in the Stat3Flx/Flx littermate controls (p<.0001) (Figure 2C). Crypt loss at day 7 was not significantly different between genotypes but at day 28 Stat3ΔIEC mice had almost double the amount of crypt loss at 2.46 ±.2 compared to 1.7 ±.13 in the Stat3Flx/Flx littermate controls (p<.05)(Figure 2D). The immune cell sub-score referring to the infiltration of immunce cells into the lamina propria and submucosa at day 7 in Stat3ΔIEC mice was also significantly higher at 2.4 ±.1 compared to 1.5 ±.3 in Stat3Flx/Flx littermate controls (p<-001) (Figure 2E). At day 28 both genotypes still had infiltration of immune cells but in Stat3ΔIEC mice it was more significant at 2.1 ±.1 compared to 1.3 ±.1 in Stat3Flx/Flx littermate controls (p<.001) (Figure 2E).
Figure 2. All histologic sub-scores are increased in Stat3ΔIEC mice compared to Stat3Flx/Flx littermate controls at day 28.
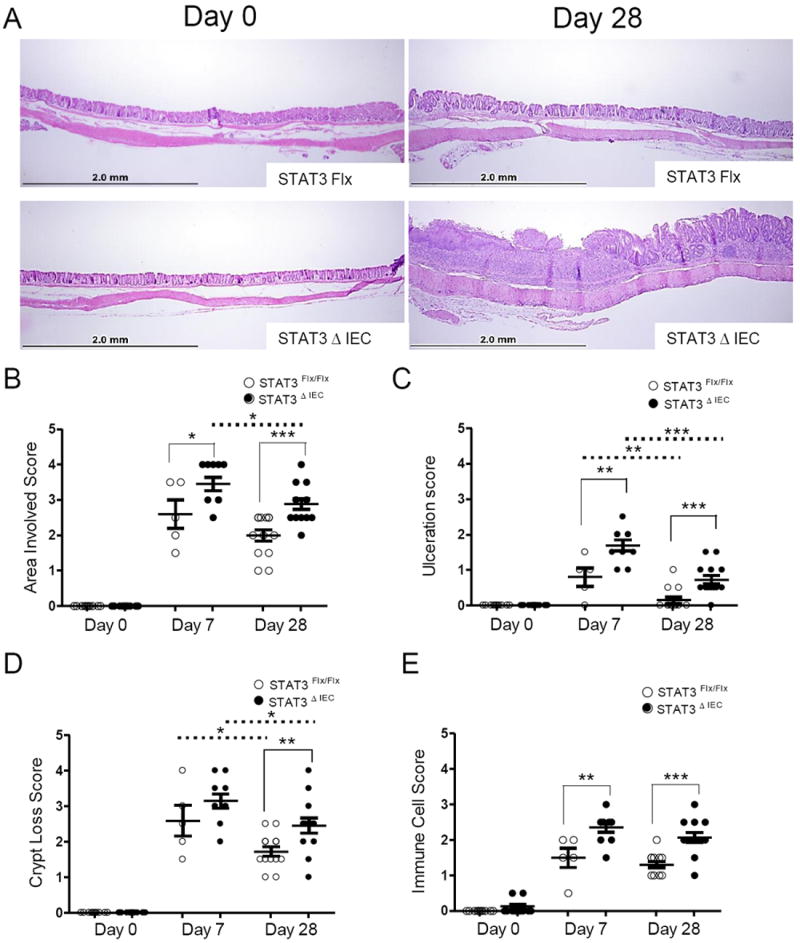
(A) Representative images from the same segment of the colon at day 0 and day 28 utilizing the anal verge as an anatomical landmark. Original magnification was 100x. (B) Area involved, (C) epithelial monolayer ulceration (D) crypt loss and (E) extent of immune cell infiltration are shown at day 7 and day 28 (n>5 mice per group, per genotype). Data are shown as mean ± SEM. Differences within (dotted line) and between (solid line) genotypes were analyzed by one-way ANOVA with Bonnferroni post-test (* p < .05; ** p < .005; *** p < .0001).
IEC proliferation and survival does not differ from controls at Day 28 in Stat3ΔIEC mice
IEC Stat3 deficiency in DSS induced acute colitis has shown that Stat3 functions to induce intestinal wound healing responses, proliferation, and protection from apoptosis during acute injury (11, 17, 18). We confirmed Stat3 activation in response to DSS in colonic enterocytes at day 7 in the littermate controls (Figure 3A). The frequency of nuclear pSTAT3 positive IEC per crypt was equal to 4.8 ±1.6 percent at day 0, which was significantly increased to 15.2 ±6 percent at day 7, and returned to basal levels of 1.6 ±.77 percent at day 28 (p<0.05) (Figure 3B). Stat3ΔIEC mice as expected had no nuclear IEC pSTAT3 staining. To examine proliferation of IECs we injected untreated control, acute and chronic phase mice with BrdU, and sacrificed the animals 2 hours later. At day 7 we found a significant reduction in basal crypt proliferation rates as the percentage of BrdU positive cells per crypt was 3.8 ±.6 in Stat3ΔIEC mice compared to 7.3 ±.6 in Stat3Flx/Flx littermate controls (p<.005) (Figure 3D). There was no difference at day 14 or day 28 between genotypes, however there was an increase in the BrdU positive cells from day 7 to day 14 in the Stat3ΔIEC mice (p<.001). To examine whether IEC-specific deletion of Stat3 renders enterocytes more susceptible to apoptosis at baseline and during acute and chronic phases of colitis we quantified immunohistochemical staining for cleaved caspase-3 positive cells per HPF at day 0, day 7, day 14, and day 28. We found that at day 7 Stat3ΔIEC mice had a trend for increased caspase-3 positive IECs per HPF but this comparison was not significant and there was no difference between genotypes at day 0, day 14 or day 28 (Figure 3F).
Figure 3. IEC proliferation and apoptosis do not differ at day 28 between Stat3ΔIEC mice and littermate controls.
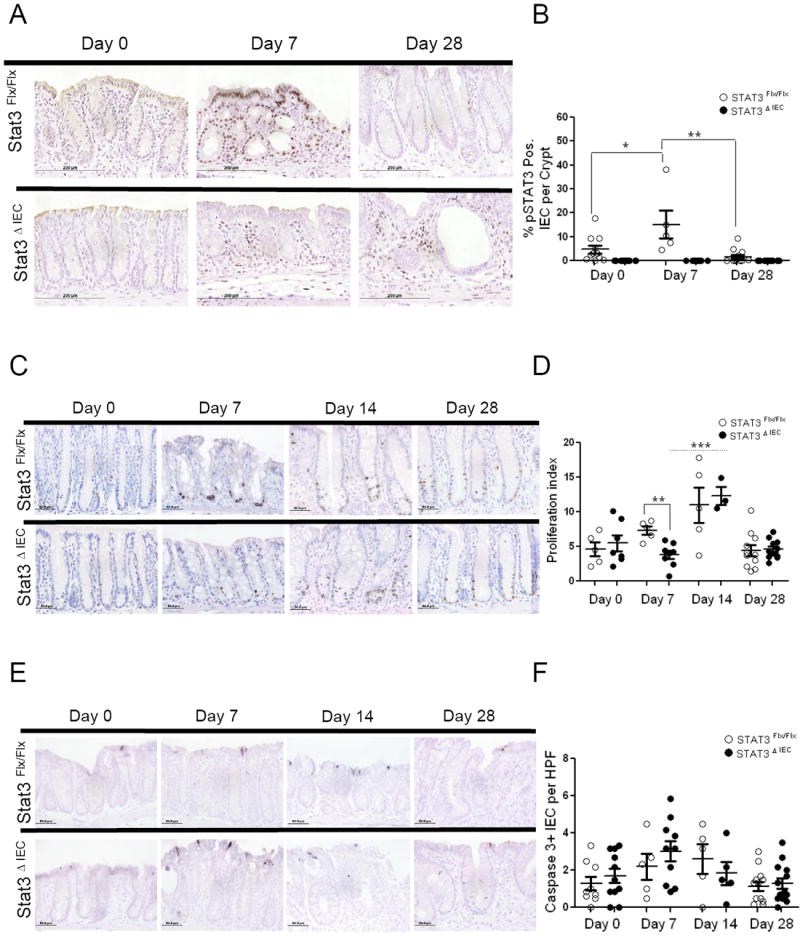
(A) Imuno-histochemical analysis of phophos-STAT3 (pSTAT3) quantified as (B) the percentage of IECs positive for pSTAT3 per crypt at day 0, day 7 and day 28, as an average of 12 properly oriented crypts/mouse. (C) Immuno-histochemical analysis of proliferation measured by BrDU incorporation expressed as (D) a proliferation index at day 0, day 7, day14 and day 28 as an average of 12 properly oriented crypts/mouse. (E) Apoptosis measured by cleaved caspase-3 quantified as (F) the number of positive cells per HPF at day 0, day 7, day 14 and day 28, as an average of six fields counted per mouse. Analysis was performed in .5 cm area of mucosa that bordered ulcers in the distal and middle colon or in corresponding areas of the colon if no ulcer was found. Data are shown as mean + SEM (n>5 mice per genotype). Differences within and between genotypes were compared by un-paired t-test (*p < .05 and ** p < .005).
We therefore asked if other signaling pathways that control the expression of anti-apoptotic, pro-proliferative and immune response genes known to overlap and have redundant functions to Stat3, such as the NFκB signaling pathway, might mediate wound healing, proliferation and enhanced survival in the IEC Stat3 deficient animals (26-34). We stained for nuclear phosphorylated Rela(p65) but did not observe a difference in the percentage of nuclear RelA(p65) positive IEC per crypt at day 0, day 7 or day 28 between the genotypes (Suppl. Figure 2B). As expected, the frequency of RelA(p65) positive IEC per crypt was increased in both genotypes at day 7. We also did not observe a difference in the expression of IκBα, which is a direct downstream target of activated RelA (p65), between the genotypes at day 7 or day 28 (Suppl. Figure 2C) (35-38). STAT1 or STAT5 activation might also promote a survival signal, which could compensate for the loss of STAT3 (39-41). Immuno-histochemical analysis of consecutive colonic sections for pSTAT1 and pSTAT3 showed no activation of pSTAT1 in the epithelial compartment of either genotype at day 0, day 7 (data not shown) or day 28 (Suppl. Figure 2D). Similarly, we observed no nuclear IEC staining for pSTAT5 in either genotype at day 28 (Suppl. Figure 2E). Collectively, these data suggested that the increased severity of colitis at day 28 in Stat3ΔIEC mice was not accounted for by differences in IEC survival or proliferation, or induction of compensatory IEC signaling pathways.
Development of chronic colitis involves Stat3 activation in the non-epithelial compartment in Stat3ΔIEC mice
In order to identify the lamina propria cellular populations involved during DSS induced chronic colitis, colonic sections of DSS-induced chronic colitis (day 28) mice were examined for the frequency of pSTAT3 positive enterocytes and lamina propria cells, F4/80+ MΦ and CD3+ T-lymphocytes, and double positive pSTAT3+/F4/80+ MΦ and pSTAT3+/CD3+ T-lymphocytes per high powered field (HPF). In Stat3Flx/Flx littermate controls the number of IEC that were positive for nuclear pSTAT3 staining at day 28 was equal to 4.5 ±2 cells per HPF with no IEC nuclear pSTAT3 detectable in the Stat3ΔIEC mice (Figure 4A). In the lamina propria, the Stat3ΔIEC mice exhibited a significant increase in pSTAT3+ cells per HPF at 23.5 ±3, compared to 9.2 ±2 cells per HPF in the Stat3Flx/Flx littermate controls (p<.001) (Figure 4B). The frequency of total F4/80+ MΦ was significantly increased from16 ±2 cells per HPF in Stat3Flx/Flx littermate controls to 39 ±4 cells per HPF in Stat3ΔIEC mice (p<.005) (Figure 4C). The frequency of cells in which surface staining for F4/80 co-localized with nuclear pSTAT3 was significantly increased as well from 2 ±.3 cells per HPF in Stat3Flx/Flx mice to 8.7 ±1 cell per HPF in Stat3ΔIEC mice (p<.001) (Figure 4C). The frequency of total CD3+ cells per HPF was also significantly increased from 4 ±1 cells per HPF in Stat3Flx/Flx littermate controls to 8 ±2 cells per HPF in Stat3ΔIEC mice (p<.005) (Figure 4D). The frequency of cells in which surface staining for CD3 co-localized with nuclear pSTAT3 was also significantly increased in Stat3ΔIEC mice at 1 ±.3 cells per HPF compared to .1 ±.02 cells per HPF in Stat3Flx/Flx mice (p<.05)(Figure 4D). Finally, the overall frequency of lamina propria cells expressing nuclear pSTAT3 was significantly associated with histology scores at day 28 in the Stat3ΔIEC mice, but not in littermate controls (r=.77, p =.03) (Figure 4E).
Figure 4. Chronic colitis in Stat3ΔIEC mice involves Stat3 activation in the non-epithelial compartment.
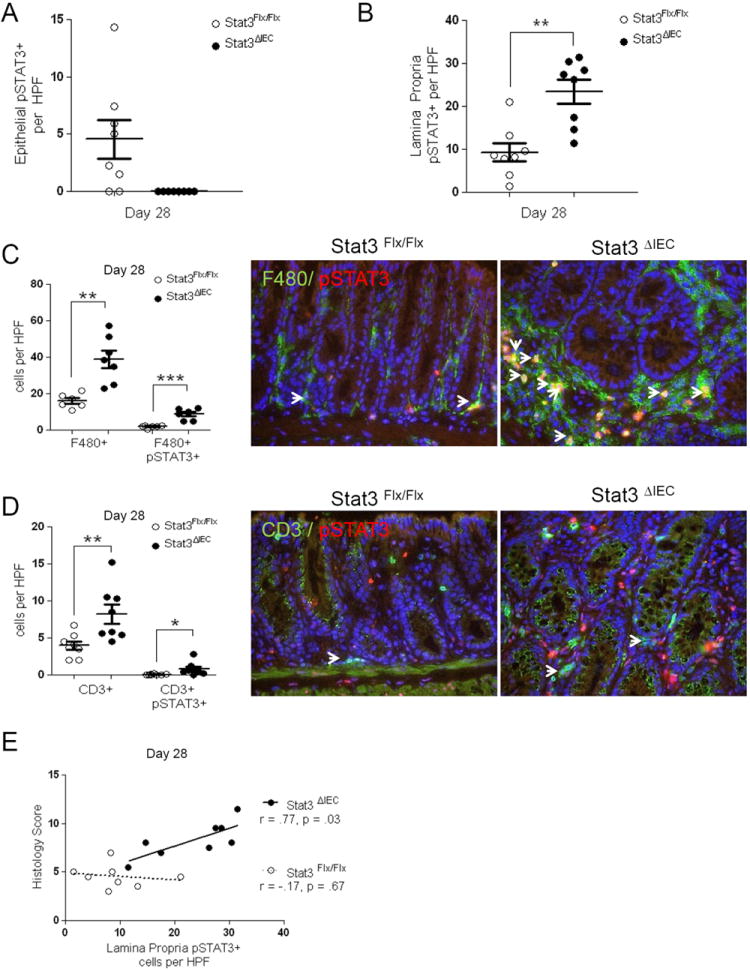
Colon sections were quantified by fluorescent microscopy for phospho-Stat3 and the frequency of (A) enterocyte pSTAT3 positive cells and (B) lamina propria pSTAT3 positive cells in 10 random high powered fields (HPF; 400x magnification) was determined in Stat3Flx/Flx littermate controls and Stat3ΔIEC mice at day 28 (n > 6 mice per group). Immunofluorescent labeling of (C) F4/80+ Mϕ and (D) CD3+ T-lymphocytes along with colocalization with phospho-STAT3 was used to determine the frequency of single and double positive cells in 10 random high powered fields (HPF; 400x magnification) of Stat3Flx/Flx littermate controls and Stat3ΔIEC mice at day 28 (n > 6 mice per group, positive cells are marked by arrowhead). (E) Pearson’s regression analysis was used to test for an association between the frequency of lamina propria pSTAT3+ cells per HPF and histological severity measured by the histology score at day 28 in Stat3Flx/Flx littermate controls (r = .-17, p = .67) and Stat3ΔIEC mice (r = .77, p = .03). Data are shown as mean ± SEM. Differences between genotypes were compared by un-paired t-test (*, p < .05; ** p < .005; *** p < .0001).
Neutrophils did not seem to be playing a major role in the chronic colitis at day 28. We assessed mucosal S100 proteins, S100A8/S100A9, involved in phagocyte chemotaxis and function and CXCR2, a receptor known to mediate neutrophil migration to sites of inflammation in Stat3Flx/Flx littermate controls and Stat3ΔIEC mice at day 0, day 7, and day 28 (42, 43). We found a trend for increased S100A8 and S100A9 mRNA expression at day 28 in STAT3ΔIEC mice compared to STAT3Flx/Flx littermate controls but found no differences for CXCR2 expression (Suppl. Figure 3A). However, when we stained for neutrophil elastase, secreted by neutrophils during inflammation, the frequency of lamina propria neutrophil elastase positive cells per HPF was decreased in Stat3ΔIEC mice compared to Stat3Flx/Flx littermate controls at day 28 (p<.001) (Suppl. Figure 3B). The frequency of cells in which neutrophil elastase staining co-localized with nuclear pSTAT3 was significantly decreased as well from 1.2 ±.3 cells per HPF in Stat3Flx/Flx mice to .08 ±1 cell per HPF in Stat3ΔIEC mice (p<.05) (Suppl. Figure 3B).
Colonic expression of Stat3 target genes that mediate epithelial restitution is significantly decreased while SOCS3, IL-6, IFN-γ and IL-17A expression are increased in Stat3ΔIEC mice at day 28
Since mice deficient in IEC Stat3 exhibited a more significant lamina propria infiltrate at day 28 we next investigated colonic expression of genes involved in epithelial restitution and mucosal inflammation. We performed the expression analysis from the same colonic sections for which we determined the composition of cellular infiltrate, which allowed us to correlate colonic gene expression with immune cell population frequencies and histology scores. Using quantitative RT-PCR we found that mRNA expression for the Stat3 dependent epithelial restitution genes Reg3β and Reg3γ was significantly decreased at day 0, day 7, and day 28 in Stat3ΔIEC mice compared to Stat3Flx/Flx littermate controls (p<.005) (Figure 5A). Conversely, colonic expression of Socs3, a direct downstream target of Stat3 activation, was significantly increased in Stat3ΔIEC mice at day 28, likely reflecting the increased lamina propria Stat3 activation observed at this time point (p<.04) (Figure 5A). Expression of IL-6 and IL-10 was not different between genotypes at day 0 or day 28 (Figure 5B). IL-17A expression was significantly increased at day 28 and did not decrease in expression from day 7 in Stat3ΔIEC mice compared to Stat3Flx/Flx littermate controls (p<.05) (Figure 5B). IFNγ expression was significantly increased at day 0 and day 28 in Stat3ΔIE C mice compared to Stat3Flx/Flx littermate controls (p<.05) (Figure 5C). We did not observe a significant difference in colonic expression of IL-4, IL-1β, or TNFα between genotypes at day 0, day 7, or day 28 (Figure 5C), suggesting that the differences in IFNγ and IL-17A expression were specific.
Figure 5. Colonic expression of Stat3 target genes that mediate epithelial restitution is decreased while SOCS3, IFN-γ, and IL-17A expression are increased in Stat3ΔIEC mice at Day 28.
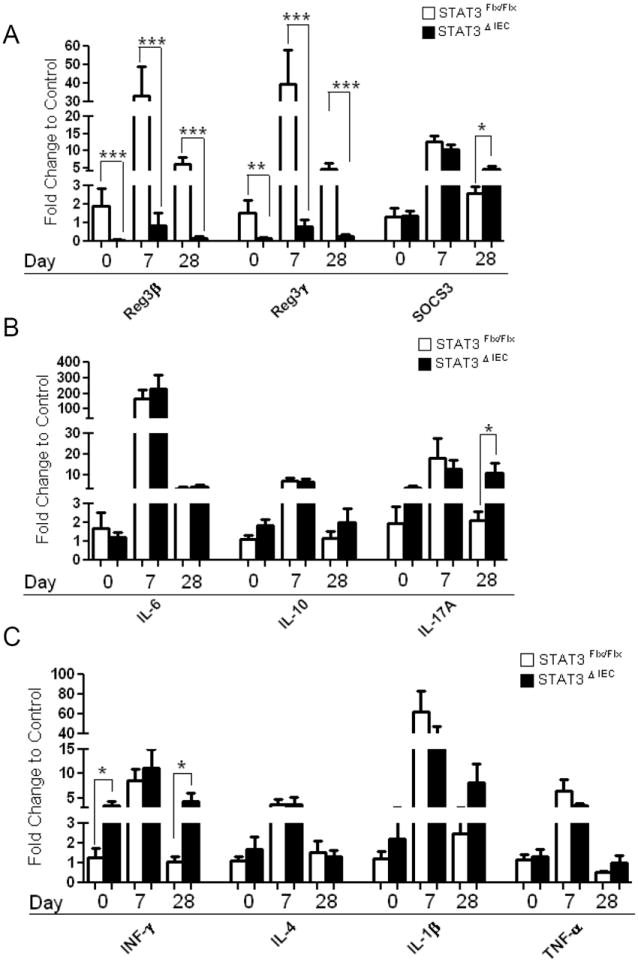
Real-time RT-PCR was performed on mRNA isolated from whole colonic serial sections for (A) Reg3β, Reg3γ, SOCS3 (B) IL-6, IL-10, IL-17A (C) IFN-γ, IL-4, IL-1β, and TNF-α expression in Stat3Flx/Flx littermate controls and Stat3ΔIEC mice at day 0, day 7, and day 28 (n>5 mice per genotype). Data are normalized to β-actin mRNA expression and expressed as fold change to Stat3Flx/Flx day 0 littermate controls shown as mean + SEM. Differences between genotypes at day 0 and day 28 were compared by unpaired t-test (*, p < .05; ** p < .005; *** p < .0001).
Colonic expression of chemokines involved in leukocyte recruitment is increased in Stat3ΔIEC mice at Day 28
We have found that the frequency of colonic pSTAT3+/F4/80+ MΦ, pSTAT3+/CD3+ T-lymphocytes, and colonic expression of IFNγ and IL-17A were increased at day 28 in Stat3ΔIEC mice compared to littermate controls. We therefore next asked whether there would be differences in expression of chemokines, which mediate trafficking of these cells. Colonic expression of CXCL5 was increased in both genotypes at day 28 (Figure 6A). By comparison, day 28 CXCL2 and CXCL10 expression was increased significantly more in Stat3ΔIEC mice compared Stat3Flx/Flx littermate controls (p<.03) (Figure 6A). We also found significant increase in colonic expression CCL2 and with a trend for increased CCL4 at day 28 in Stat3ΔIEC mice compared to Stat3Flx/Flx littermate controls (p<.05) (Figure 6B). By comparison, we did not observe differences between genotypes at day 28 for colonic expression of CXCL1, CCL5, or CCL20, supporting the specificity of the differences observed (Figure 6A&B).
Figure 6. Colonic expression of CXCL2, CXCL10, and CCL2 is significantly increased in Stat3ΔIEC mice at Day 28.
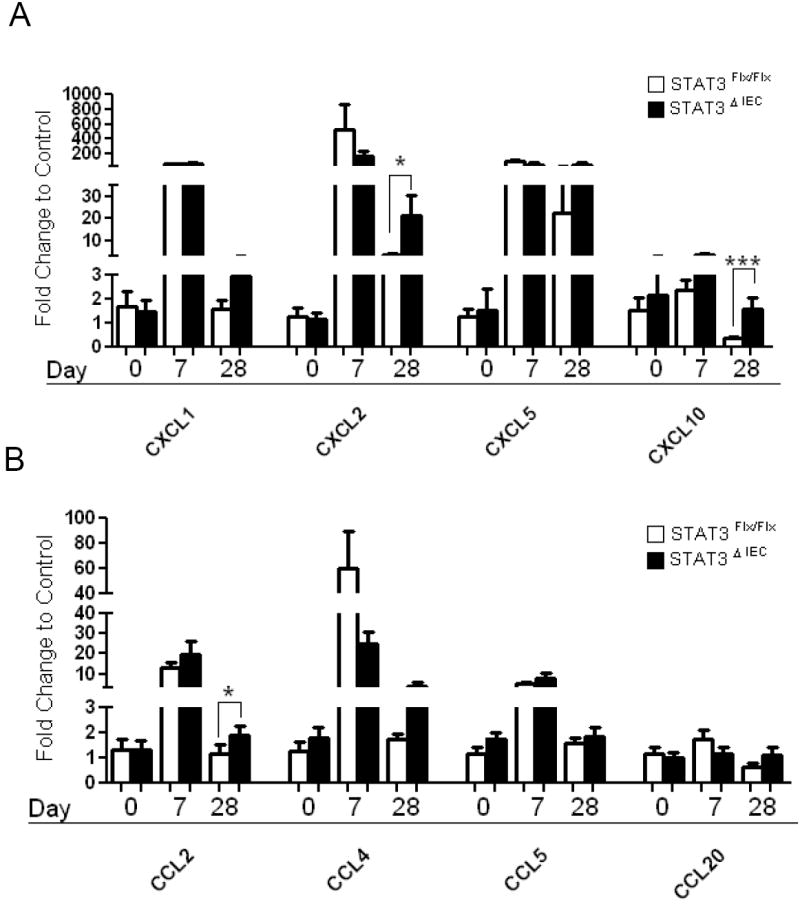
Colon mRNA expression was measured by quantitative real time PCR for (A) CXCL1, CXCL2, CXCL5, CXCL10 (B) CCL2, CCL4, CCL5, and CCL20 in Stat3Flx/Flx littermate controls and Stat3ΔIEC mice at day 0, day 7 and day 28 of the colitis model (n>5 mice per genotype). Data were normalized to β-actin mRNA expression and expressed as fold change to Stat3Flx/Flx day 0 littermate controls shown as the mean ±SEM (*, p < .05; ** p < .005; *** p < .0001 by unpaired t-test).
Colonic expression of IL-6, IL-10, IFNγ and IL-17A correlates with histologic severity and colonic expression of IL-17A correlates with the frequency of pSTAT3+CD3+ T-lymphocytes in Stat3ΔIEC mice at day 28
We then used linear regression analysis to determine whether the colonic expression of IL-6, IL-10, IFNγ, and/or IL-17A was associated with the histology scores. We found that colonic expression of IL-6 was associated with increased histologic score in Stat3ΔIEC mice (r=.76, p=.03) as was IL-10 (r=.77, p=.02), IFNγ (r=.82, p=.01), and IL-17A (r=.77, p=.03) (Figure 7 A, B, C, & D). However there was no significant correlation in the StatFlx/Flx littermate controls for any of these cytokines (Figure 7 A, B, C, & D). We next asked whether the frequency of CD3+/pSTAT3+ T-lymphocytes per HPF correlated with IL-17A colonic mRNA expression, and found that this was the case in the Stat3ΔIEC mice (r=.90, p=.002) but not in the Stat3Flx/Flx littermate controls (Figure 7E).
Figure 7. Colonic expression of IL-6, IL-10, IFN-y and IL-17A correlates with histologic severity and IL-17A expression correlates with the frequency of colon pSTAT3+CD3+ T-lymphocytes in Stat3ΔIEC mice at day 28.
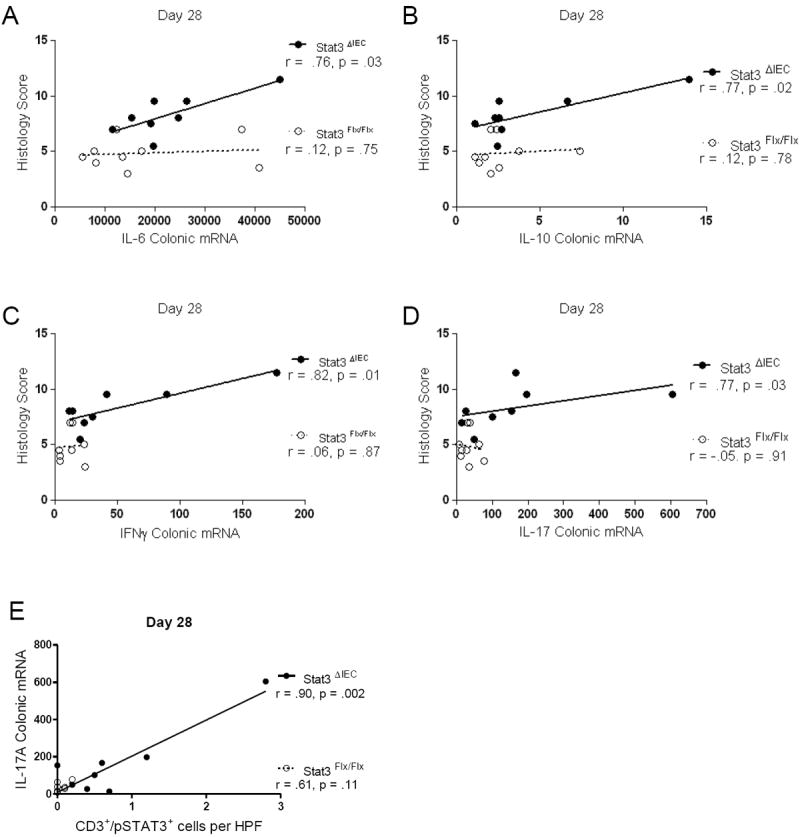
Pearson regression analysis was used to test for an association between normalized colonic expression of (A) IL-6 (B) IL-10 (C) IFNγ and (D) IL-17A and histologic severity score. E) Pearson regression analysis was used to test for an association between IL-17A colonic expression and the frequency of CD3+/pSTAT3+ T-lymphocytes per 400x high-powered field (r=.90, p<.002) (n>7 mice per genotype). Data are shown as mean + SEM (*, p < .05; ** p < .005; *** p < .0001 by unpaired t-test).
DISCUSSION
Development of chronic colitis after DSS administration is strain dependent and we have investigated responses in the well characterized and more susceptible C57BL/6 mouse that progress to chronicity after a single round of DSS (44-47). In the present study we have demonstrated that the loss of IEC Stat3 promotes the development of a chronic inflammatory response after a single round of DSS administration for 7 days, in terms of increased degree of histologic severity, reduced survival, and reduced colon length. Body weight was significantly reduced in Stat3ΔIEC mice as compared to Stat3Flx/Flx littermate controls after removal of DSS at day 10 which was followed by a rapid recovery by day 12 in both genotypes. This is not surprising in that C57BL/6J mouse strain has been shown to develop chronic colitis returning to their original weight 3 to 4 weeks after removal of DSS(44). Since some of the Stat3ΔIEC mice died around day 12 it is likely that those animals would have had a significant negative impact on the body weight, colon length, and histological score parameters, however we nevertheless observed significant differences at day 28 in the Stat3ΔIEC cohort.
IEC Stat3 deficient mice exhibited more severe histologic injury at day 7, recapitulating what has previously been published, and here we show that they continue to exhibit more severe disease through three weeks of water recovery at day 28. The severity was worse in all areas scored including area involved, ulceration, crypt loss, and inflammatory infiltrate. Over the course of the 21 day water consumption Stat3Flx/Flx littermate controls and Stat3ΔIEC mice were able to significantly reduce ulceration and crypt loss when comparing day 7 to day 28 within genotypes. This is thought to be mediated by intestinal epithelial cell proliferation, survival and wound healing responses. However, the immune cell score remained high in both genotypes from day 7 to day 28. This suggests that those Stat3ΔIEC mice that survived were able to induced regenerative responses to the initial DSS induced injury regardless of a loss in Stat3 activation within the epithelium however these mice still had more severe colonic injury compared to Stat3Flx/Flx littermate controls at day 28.
Upon evaluation of pSTAT3 within the epithelial crypts we observed that at day 7 more Stat3 activation was induced than at day 28 in the Stat3Flx/Flx littermate controls. We did not evaluate this at the biochemical level, as epithelial preps from ulcerated and colitic tissue can be contaminated by other cell populations. Secondly, we elected to stain for the activated phosphorylation site of Stat3 within the IEC cellular niche. This allowed us to gain better insight into the relative area in which Stat3 was induced and how it relates to active or disease free areas of the colon and the patchy nature of the DSS colitis model. Thus we concentrated our efforts within 0.5cm from the ulcer border or similar areas of the colon if no ulcer was present. Our studies at day 7 revealed, as previously published, that the Stat3Flx/Flx mice had a greater epithelial proliferative response, however at day 14 during the predominant restitution phase both genotypes had comparable increases in proliferative rates that then similarly decreased at day 28 with no differences between genotypes at day 14 or day 28. Epithelial cell survival was not reduced in Stat3ΔIEC mice at day 7 or day 14, however it has been suggested that the most significant portion of apoptosis occurs within the first 2 or 3 days of DSS administration and therefore we may have missed this difference between genotypes at day 7 (48). Furthermore, there was also no difference for IEC apoptosis at day 28 between genotypes. Both genotypes exhibited an improvement in crypt loss and ulceration between day 7 and day 28, although these remained more severe in the Stat3ΔIEC mice. We did not observe a compensatory increase in the frequency of crypt epithelial cells expressing the activated forms of p65, pSTAT1, or pSTAT5 at day 7 or day 28 in the Stat3ΔIEC mice. However, we did observe a persistent defect in expression of the Stat3 dependent epithelial restitution genes Reg3β and Reg3γ in the Stat3ΔIEC mice. This likely contributed to the relative defect in healing of the epithelial barrier independent of proliferation and apoptosis promoting the increased STAT3 activation in innate and adaptive immune cells seen in the Stat3ΔIEC mice at day 28 (11). We did not assess a longer lag phase of recovery after DSS administration since no difference in proliferation or apoptosis were seen at day 14 or day 28 between genotypes. Our data and others suggest that an extended period of recovery would not show improvement in colitic phenotype but only intensified inflammatory responses (44, 45).
Chemokines are small chemotactic cytokines that regulate the trafficking and migration of leukocytes (49). Previous reports in C57BL/6 mice have determined that expression of CXCL1, CXCL2, CXCL10, CCL2, CCL3 and CCL4 remain elevated during the chronic phase of DSS induced colitis(45). Our results were consistent with these prior reports in that we observed increased expression of CXCL2, CXCL10, CCL2 and CCL4 in Stat3ΔIEC mice in the setting of more severe chronic colitis which is likely contributing to the differences in the inflammatory infiltrate observed at day 28. It will be important in future studies to determine whether the Stat3 deficient epithelium is the source of these chemokines, and if so, whether Stat3 directly regulates their expression. The cellular infiltrate in Stat3ΔIEC mice remarkable showed an increased frequency within the lamina propria of pSTAT3+ cells including pSTAT3+ MΦ and T-lymphocytes along with increased frequency of total MΦ and T-lymphocytes compared to Stat3Flx/Flx littermate controls. The frequency of pSTAT3+ cells within the lamina propria was positively correlated with a more severe histologic score suggesting that these cells are either induced in response to the healing process or are directly mediating worse disease. We realize our limitation in utilizing only immunofluorescence and immunohistochemistry to evaluate these parameters but have had difficulty with the reliability of the flow for pSTAT3 after lamina propria cell isolation and therefore wanted to evaluate these cell types in the cellular niche.
SOCS3, a direct downstream target of activated Stat3, was increased in expression in the Stat3ΔIEC mice which we believe is consistent with the pronounced pSTAT3+ infiltrate within the lamina propria. Colonic IFNγ expression was increased at both day 0 and day 28 in the Stat3ΔIEC mice. We did not observe qualitative differences in pSTAT1 activation in the epithelial cell compartment at these time points. However, as IFNγ is produced by Th1 cell types, and IL-4 expression did not differ between genotypes, this likely represents a basal Th1 skewing in the Stat3ΔIEC mice (50). Colonic IL-17A expression was increased only at day 28 in Stat3ΔIEC mice, in the setting of chronic colitis. IL-17A is released by Th17 inflammatory T-cells, shown to involve direct binding of Stat3 to the IL-17A promoter, and known to be involved in many chronic autoimmune disorders(51-53). We did not detect a difference between genotypes for expression of IL-6, IL-10, IL-1β or TNFα, suggesting that there was not a global difference in activation of mucosal inflammatory pathways in the Stat3ΔIEC mice. Despite no difference between the genotypes in the overall level of IL-6 and IL-10 expression, we did however observe a correlation between IL-6 and IL-10 expression and histologic injury in the Stat3ΔIEC mice. IL-6 is a mediator of inflammation that also serves a protective role in maintaining adequate immune response to infection. Our data suggest that in the intestinal epithelial STAT3 deficient mice IL-6 and IL-10 may have differing effector functions compared to the littermate control cohort. This could explain the more severe colitis at day 28 and the difference in the colitis phenotype. Consistent with an effector function, we observed a strong correlation between the frequency of pSTAT3+ T lymphocytes and IL-17A expression, and between IL-17A expression and histologic injury. Future studies employing Th17 blockade will be required to determine whether this will ameliorate chronic colitis in this setting.
In summary, we have shown that loss of IEC Stat3 leads to more severe chronic inflammation following acute injury, which is not accounted for by a sustained defect in epithelial proliferation or apoptosis 7 or 21 days after one cycle of DSS, but rather defective REG3 gene expression and expansion of pSTAT3+ T-lymphocytes and up-regulation of IL-17A expression. Our data suggests that decreased Reg3 gene expression may implicate impaired healing of the epithelium promoting lamina propria cell stimulation though epithelial barrier defects and activation of STAT3 signaling in immune cells. Moreover, loss of Stat3 signaling in the IEC may help explain why patients carrying the JAK2 risk allele have reduced colonic expression of REG3α, increased intestinal permeability and association with greater risk for development of IBD (5, 6). Future studies are aimed at exploring the differences in intestinal permeability and bacterial translocation as a function of epithelial Stat3 in both steady state and DSS induced acute and chronic colitis conditions. Finally, the current concept of IBD pathogenesis in a genetically susceptible host more closely resembles a single cycle of DSS that progresses to chronic colitis. Future studies to interrogate the biochemical basis associated with carriage of the STAT3 and JAK2 risk alleles may be well suited for this murine preclinical model of colitis.
Supplementary Material
Acknowledgments
We would like to thank Eleana Laws, MS and Erin Molden, MS for excellent technical assistance. This work was supported by Cincinnati Children’s Hospital Research Foundation Digestive Health Center (1P30DK078392-01), and NIH grants R01 DK078683 and DK068164 (LAD).
Grant Support: This work was supported by the Cincinnati Children’s Hospital Research Foundation Digestive Health Center (1P30DK078392-01), and NIH grants R01 DK078683 and DK068164.
Abbreviations
- DSS
Dextran Sodium Sulfate
- STAT3
Chronic Colitis, Signal Transducer and Activator of Transcription 3
- IL-17
Interleukin-17
- AOM
Azoxymethane
- MΦ
macrophage
Footnotes
Financial disclosures: The authors have no financial arrangement(s) with a company whose product figures prominently in the submitted manuscript or with a company making a competing product.
Competing Interest: None to declare
Writing assistance: not applicable
References
- 1.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imielinski M, Baldassano RN, Griffiths A, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathew CG. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat Rev Genet. 2008;9:9–14. doi: 10.1038/nrg2203. [DOI] [PubMed] [Google Scholar]
- 4.Willson TA, Kuhn BR, Jurickova I, et al. STAT3 Genotypic Variation and Cellular STAT3 Activation and Colon Leukocyte Recruitment in Pediatric Crohn Disease. J Pediatr Gastroenterol Nutr. 2011 doi: 10.1097/MPG.0b013e318246be78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurickova I, Willson T, Gerad S, et al. M1791 The Inflammatory Bowel Disease JAK2 and STAT3 Risk Alleles are Associated With Divergent Effects Upon IL-6 Signaling in Lymphocytes. Gastroenterology. 2010;138:S–420. [Google Scholar]
- 6.Prager M, Buttner J, Haas V, et al. The JAK2 variant rs10758669 in Crohn’s disease: altering the intestinal barrier as one mechanism of action. Int J Colorectal Dis. 2012;27:565–573. doi: 10.1007/s00384-011-1345-y. [DOI] [PubMed] [Google Scholar]
- 7.Atreya R, Mudter J, Finotto S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto M, Yoshizaki K, Kishimoto T, et al. IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol. 2000;164:4878–4882. doi: 10.4049/jimmunol.164.9.4878. [DOI] [PubMed] [Google Scholar]
- 9.Kiessling S, Muller-Newen G, Leeb SN, et al. Functional expression of the interleukin-11 receptor alpha-chain and evidence of antiapoptotic effects in human colonic epithelial cells. J Biol Chem. 2004;279:10304–10315. doi: 10.1074/jbc.M312757200. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhry A, Rudra D, Treuting P, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickert G, Neufert C, Leppkes M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugimoto K, Ogawa A, Mizoguchi E, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honda K, Nakamura K, Matsui N, et al. T helper 1-inducing property of IL-27/WSX-1 signaling is required for the induction of experimental colitis. Inflamm Bowel Dis. 2005;11:1044–1052. doi: 10.1097/01.mib.0000191611.05466.1f. [DOI] [PubMed] [Google Scholar]
- 14.Troy AE, Zaph C, Du Y, et al. IL-27 regulates homeostasis of the intestinal CD4+ effector T cell pool and limits intestinal inflammation in a murine model of colitis. J Immunol. 2009;183:2037–2044. doi: 10.4049/jimmunol.0802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. 2002;122:2011–2025. doi: 10.1053/gast.2002.33631. [DOI] [PubMed] [Google Scholar]
- 16.Williams KL, Fuller CR, Dieleman LA, et al. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–937. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- 17.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bollrath J, Phesse TJ, von Burstin VA, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 20.Mudter J, Weigmann B, Bartsch B, et al. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am J Gastroenterol. 2005;100:64–72. doi: 10.1111/j.1572-0241.2005.40615.x. [DOI] [PubMed] [Google Scholar]
- 21.Carey R, Jurickova I, Ballard E, et al. Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:446–457. doi: 10.1002/ibd.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda K, Kaisho T, Yoshida N, et al. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- 23.Mahler M, Bristol IJ, Leiter EH, et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol. 1998;274:G544–551. doi: 10.1152/ajpgi.1998.274.3.G544. [DOI] [PubMed] [Google Scholar]
- 24.Moolenbeek C, Ruitenberg EJ. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Lab Anim. 1981;15:57–59. doi: 10.1258/002367781780958577. [DOI] [PubMed] [Google Scholar]
- 25.Steinbrecher KA, Harmel-Laws E, Sitcheran R, et al. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 26.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 28.Joyce D, Albanese C, Steer J, et al. NF-kappaB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001;12:73–90. doi: 10.1016/s1359-6101(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 29.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 30.Wang CY, Mayo MW, Korneluk RG, et al. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Bonizzi G, Seagroves TN, et al. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell. 2001;107:763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- 32.Tergaonkar V, Pando M, Vafa O, et al. p53 stabilization is decreased upon NFkappaB activation: a role for NFkappaB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 33.Egan LJ, Sandborn WJ. Advances in the treatment of Crohn’s disease. Gastroenterology. 2004;126:1574–1581. doi: 10.1053/j.gastro.2004.01.062. [DOI] [PubMed] [Google Scholar]
- 34.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arenzana-Seisdedos F, Thompson J, Rodriguez MS, et al. Inducible nuclear expression of newly synthesized I kappa B alpha negatively regulates DNA-binding and transcriptional activities of NF-kappa B. Mol Cell Biol. 1995;15:2689–2696. doi: 10.1128/mcb.15.5.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Bail O, Schmidt-Ullrich R, Israel A. Promoter analysis of the gene encoding the I kappa B-alpha/MAD3 inhibitor of NF-kappa B: positive regulation by members of the rel/NF-kappa B family. EMBO J. 1993;12:5043–5049. doi: 10.1002/j.1460-2075.1993.tb06197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun W, Snyder M, Levy DE, et al. Regulation of Stat3 transcriptional activity by the conserved LPMSP motif for OSM and IL-6 signaling. FEBS Lett. 2006;580:5880–5884. doi: 10.1016/j.febslet.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 38.Sun SC, Ganchi PA, Ballard DW, et al. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 39.Ernst M, Najdovska M, Grail D, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan H, Liddle FJ, Mahajan S, et al. IL-6-induced survival of colorectal carcinoma cells is inhibited by butyrate through down-regulation of the IL-6 receptor. Carcinogenesis. 2004;25:2247–2255. doi: 10.1093/carcin/bgh246. [DOI] [PubMed] [Google Scholar]
- 41.Costa-Pereira AP, Tininini S, Strobl B, et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc Natl Acad Sci U S A. 2002;99:8043–8047. doi: 10.1073/pnas.122236099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryckman C, McColl SR, Vandal K, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum. 2003;48:2310–2320. doi: 10.1002/art.11079. [DOI] [PubMed] [Google Scholar]
- 43.Srikrishna G, Turovskaya O, Shaikh R, et al. Carboxylated glycans mediate colitis through activation of NF-kappa B. J Immunol. 2005;175:5412–5422. doi: 10.4049/jimmunol.175.8.5412. [DOI] [PubMed] [Google Scholar]
- 44.Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1328–1338. doi: 10.1152/ajpgi.00467.2004. [DOI] [PubMed] [Google Scholar]
- 45.Melgar S, Drmotova M, Rehnstrom E, et al. Local production of chemokines and prostaglandin E2 in the acute, chronic and recovery phase of murine experimental colitis. Cytokine. 2006;35:275–283. doi: 10.1016/j.cyto.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Hall LJ, Faivre E, Quinlan A, et al. Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig Dis Sci. 2011;56:79–89. doi: 10.1007/s10620-010-1240-3. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki K, Sun X, Nagata M, et al. Analysis of intestinal fibrosis in chronic colitis in mice induced by dextran sulfate sodium. Pathol Int. 2011;61:228–238. doi: 10.1111/j.1440-1827.2011.02647.x. [DOI] [PubMed] [Google Scholar]
- 48.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 49.Olson TS, Ley K. Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol. 2002;283:R7–28. doi: 10.1152/ajpregu.00738.2001. [DOI] [PubMed] [Google Scholar]
- 50.Bradley LM, Dalton DK, Croft M. A direct role for IFN-gamma in regulation of Th1 cell development. J Immunol. 1996;157:1350–1358. [PubMed] [Google Scholar]
- 51.Ma CS, Chew GY, Simpson N, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z, Laurence A, Kanno Y, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ochs HD, Oukka M, Torgerson TR. TH17 cells and regulatory T cells in primary immunodeficiency diseases. J Allergy Clin Immunol. 2009;123:977–983. doi: 10.1016/j.jaci.2009.03.030. quiz 984-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.