

Abstract
Background
The Berlin Heart EXCOR® ventricular assist device has been approved for use in the United States as a bridge to heart transplantation in children. We sought to characterize neurological events in children supported with the Berlin Heart EXCOR® device.
Methods and Results
The multicenter prospective cohort consisted of all 204 children implanted with the Berlin Heart EXCOR® device at 47 centers in North America between May 2007 and December 2010. There were 73 neurological events in 59 patients, with 29% of the cohort experiencing ≥1 neurological event. Events included 52 strokes in 43 patients (21% of the cohort). The neurological event rate was 0.51 events per 100 patient‐days. Many of the neurological events occurred early in the course of support, with 30 events recorded during the first 14 days of support. The mortality rate in participants with at least 1 neurological event was 42% (25 of 59), significantly higher than the 18% mortality rate (26 of 145) for those who did not have a neurological event (P=0.0006). Risk‐factor analysis did not identify significant preimplantation predictors of neurological injury.
Conclusions
Of children treated with the Berlin Heart EXCOR® device as a bridge to transplant, 29% experienced at least 1 neurological event. The majority of neurological events were ischemic strokes, and many of those occurred early in the course of support. Neurological injury was the leading cause of death after implantation of the Berlin Heart EXCOR® device. Risk stratification for stroke or neurological injury is not possible based on baseline preimplantation characteristics.
Clinical Trial Registration
URL: www.clinicaltrials.gov. Unique Identifier: NCT00583661.
Keywords: heart failure, cardiomyopathy, stroke, pediatrics
Introduction
Systolic heart failure (HF) is a major cause of death in adults in the United States, and although it is less commonly encountered in children, it is highly lethal, leading to death or transplantation in 46% of children within 5 years of diagnosis.1–2 Children listed for heart transplantation face prolonged waiting times for organ availability and experience mortality of up to 17% while on a 30‐day waiting list among patients requiring mechanical ventilation.3 Over the past decade, ventricular assist devices (VADs) have emerged as a major tool for the treatment of end‐stage HF in adults, but until recently, no comparable devices were approved for use in children.4–6
This situation changed in 2011, when the Berlin Heart EXCOR® pediatric VAD (EXCOR®) was approved by the US Food and Drug Administration for use in the United States as a bridge to heart transplantation for children with severe ventricular dysfunction (ClinicalTrials.gov identifier NCT00583661). The decision by the US Food and Drug Administration was informed to a large degree by a clinical investigational device exemption (IDE) trial using historical controls with extracorporeal membrane oxygenation.7 The IDE trial demonstrated a successful bridge to transplantation or recovery in 88% to 92% of children supported with EXCOR®, but a high incidence of neurological dysfunction was noted in this trial (29%). This finding was substantiated in a subsequent analysis of a cohort of 204 children composed of the IDE cohort and a larger compassionate‐use cohort enrolled contemporaneously.8 In both of these reports, the most important identified morbidity associated with use of EXCOR® was neurological injury. A recent publication of a cohort of 102 children treated at 2 sites in the United Kingdom describes a similarly high incidence of 25.4%.9 Neurological injury was also the most common cause of death in the EXCOR® IDE trial. The purpose of this report is to provide a detailed analysis of the neurological adverse events for all 204 participants in the EXCOR® IDE trial. The major objectives of this analysis are to describe the neurological injury, including frequency, severity, and clinical impact, and to attempt to develop a risk‐stratification model for neurological adverse events during support.
Methods
Study Population and Data Sources
All children who received EXCOR® between May 9, 2007, and December 31, 2010, in the United States and Canada were identified using data collected by the study sponsor for its US regulatory trial. The trial design, inclusion criteria, and end points have been reported previously, as have survival rates on VAD compared with extracorporeal membrane oxygenation and incidence and risk factors for mortality.7,10 The major inclusion criteria for the primary IDE study cohort were age of ≤16 years, weight of 3 to 60 kg, 2‐ventricle circulation, severe HF despite optimized medical treatment, and inclusion on a waiting list for cardiac transplantation. The IDE study was conducted at 17 centers in the United States and Canada. Compassionate‐use access to EXCOR® was available to children at the IDE sites who did not meet the inclusion criteria for the trial. Compassionate‐use access was also available to children who were implanted at any of 27 non‐IDE trial sites in North America. Data were gathered by study coordinators at study sites using standard data definitions and a data platform adapted from the Interagency Registry for Mechanically Assisted Circulatory Support (INTERMACS) database.4 INTERMACS is a federally funded US postmarket VAD registry with data elements and definitions developed jointly by the academic medical community, industry, the National Heart Lung and Blood Institute, the US Food and Drug Administration, and the Centers for Medicare and Medicaid Services. All children were followed from the time of EXCOR® implant until transplant, death, or recovery. At the 17 IDE study sites, a study monitor visited each site regularly; an independent clinical events committee adjudicated the adverse events and causes of death for each patient. For non‐IDE sites, adverse events were reported but were not centrally adjudicated.
An independent data and safety monitoring board monitored the study and reviewed outcome data for the primary study cohorts. The study protocol was approved by each center's institutional review board, and written informed consent was obtained for all patients. Non‐IDE sites followed the IDE protocol when possible and as allowed by their individual compassionate‐use site approvals; these patients were consented under compassionate‐use regulations.
Timing and Assessment of Neurological Function
The trial protocol specified that prior to undergoing implantation of EXCOR®, patients would receive a baseline head computed tomography (CT) scan and a standardized neurological examination, the Pediatric Stroke Outcome Measure (PSOM).10–12 During the period of support, standardized medical evaluations were performed at 1 week, 2 weeks, 4 weeks, 6 weeks, and 3 months after implant and every 3 months while the participant remained on support. PSOM examinations were performed at the 1‐week, 4‐week, and 3‐month evaluations and every 3 months while the participant remained on support. If a neurological event was judged to have occurred by study site personnel, then head CT and PSOM were obtained. PSOM examinations were also repeated 30 and 60 days after a neurological event. After explant, PSOM was obtained at 30 days or at the time of hospital discharge, whichever was longer.
Study Definitions and Outcome Measures
All major adverse events had prespecified definitions. Neurological dysfunction was defined as any new, temporary or permanent, focal or global neurological deficit ascertained by a standard neurological examination (administered by a neurologist or other qualified physician and documented with appropriate diagnostic tests and consultation note).
All neurological events were adjudicated by a committee that included 2 pediatric stroke neurologists and were subcategorized as follows:
Transient ischemic attack: an acute event that resolved completely within 24 hours, with no evidence of infarction
Ischemic stroke: an event that persisted >24 or <24 hours and that was associated with infarction on an imaging study
Hemorrhagic stroke: an event that persisted >24 or <24 hours and that was associated with parenchymal hemorrhage or hemorrhagic infarction on an imaging study
New abnormality of head ultrasound (if patient was aged <6 months)
Electroencephalogram positive for seizure activity with or without clinical seizure (if patient was aged <6 months)
Other: adverse events that did not meet 1 of the other protocol neurological dysfunction categories; this category included subdural and epidural hematomas and asymptomatic or “clinically covert” strokes
Time on the device was defined as the duration of time from EXCOR® implant until transplant, recovery, or death. All clinical and demographic baseline variables were collected within a 48‐hour window prior to the time of EXCOR® implantation.
The PSOM is a validated outcome scale for infants and children with stroke and is based on a conventional clinical neurological examination by a child neurologist, the findings of which are recorded and scored in a standardized manner so as to provide information about the localization, nature, and functional severity of the abnormalities in an age‐appropriate manner.11–12 All children in this trial were assessed by a pediatric neurologist. PSOM subscores are assigned in 5 areas: right sensorimotor, left sensorimotor, expressive language, receptive language, and cognition/behavior. PSOM subscores are graded 0 (no deficit), 0.5 (mild deficit that does not interfere with function), 1 (moderate deficit that interferes with function), and 2 (severe deficit with loss of function). The total score ranges from 0 to 10. For the purposes of this study, a total PSOM of 0.5 to 1.0 was classified as a mild deficit, 1.5 to 2.0 was classified as a moderate deficit, and ≥2.5 was classified as a severe deficit. An unacceptable neurological outcome was defined as any 1 or a combination of the following: comatose state, quadriparesis (PSOM of 3 to 4 on motor scales), severe global aphasia (PSOM of 3 to 4 on language scales), or a severe cognitive deficit (PSOM of 2 on the cognition/behavior subscale).
In this analysis, for those children with neurological events, deidentified head CT scans that were gathered during the study from the IDE sites were reviewed by an independent pediatric neuroradiologist (S.P.) who was blinded to clinical information to determine stroke location, size, and presence of midline shift, tonsillar herniation, and extra‐axial hemorrhage.
Statistical Analysis
Summary statistics are presented as median (interquartile range) or number (percentage). Patient characteristics were compared across study cohorts using the chi‐square test or Fisher exact test when any expected frequency was <5 for categorical variables and the Kruskal–Wallis test for continuous variables. Time from implant to neurological event was estimated using the Kaplan–Meier method. Multivariable analysis was performed using backward stepwise logistic regression with a P value of 0.05 to retain variables in the final model. Analyses were performed using SAS version 9.2 (SAS Institute Inc).
Results
Study Cohort
During the 4‐year study period, 204 children were enrolled in the EXCOR® IDE trial. This included 48 participants (24%) who were enrolled in the original US Food and Drug Administration IDE trial cohort and 20 participants (10%) who met all trial criteria but were implanted after trial enrollment was complete under a continued access protocol. An additional 136 patients (67%) were implanted under compassionate‐use criteria, including 95 who received EXCOR® at 1 of 27 centers not participating in the trial and 41 who were implanted at an IDE center but who met at least 1 clinical trial exclusion criterion.
The demographic summary of these patients is presented in Table 1. Median age for the cohort was 18.6 months; median weight was 10 kg. The recorded diagnosis was cardiomyopathy in 71% (145 participants), specified as dilated cardiomyopathy in 110, myocarditis in 28, restrictive cardiomyopathy in 4, and other cardiomyopathies in 3. At the time of implantation, INTERMACS status was level 1 in 52% (107 participants) and level 2 in 44% (90 participants), indicating very high severity of illness in the study population at the time of EXCOR® implantation.
Table 1.
Characteristics of Study Participants (n=204)
| Characteristics | n (%) |
|---|---|
| Sex (female) | 97 (48) |
| Age median, months (IQR) | 18.6 (6.3 to 64.9) |
| Weight median, kg (IQR) | 10.0 (6.5 to 16.6) |
| BSA median, m2 (IQR) | 0.48 (0.35 to 0.72) |
| Nonwhite race | 74 (37) |
| Cardiomyopathy diagnosis | 145 (71) |
| Previous stroke | 14 (7) |
| ECMO prior to VAD | 83 (41) |
BSA indicates body surface area; ECMO, extracorporeal membrane oxygenation; IQR, interquartile range; VAD, ventricular assist device.
Neurological Event Rates and Timing
In total, 73 neurological events were recorded in 59 participants; therefore, 29% of the cohort experienced ≥1 neurological events. Of these neurological events, 52 strokes occurred in 43 patients (21%). Fourteen participants were known to have experienced a stroke prior to implantation of EXCOR®, of whom only 1 had an additional event during the time on EXCOR® support. Preimplantation strokes were evaluated as a potential risk factor in predicting strokes following implantation of EXCOR® but were not included in the postimplantation neurological event counts reported in this paper. Although all sites underwent training and were provided with an anticoagulation protocol (published as supplementary material to the primary IDE trial),7 event adjudication was performed only for events reported at IDE sites. There was no difference in the neurological event rate based on whether the implanting center was an IDE site (29 affected participants of 109 total, 27%) or a non‐IDE site (30 affected participants of 95 total, 32%, P value not significant).
The daily neurological event rate for the entire cohort was 0.51 events per 100 patient‐days; however, many of the neurological events occurred early in the course of support, with 30 events recorded during the first 14 days of support and an additional 15 events during the next 14 days. The complete distribution of events versus time is depicted in Figure 1. The distribution of events partly reflects the greater number of patients at risk in the first 30 days because the median duration of support for the entire cohort was 38.5 days. A hazard function analysis confirms that the risk per patient‐day is generally higher in the early period of support, decreasing to a comparatively low baseline by day 50. This is illustrated in Figure 2. The late spikes in hazard function seen in Figure 2 are due to isolated neurological events occurring in a small residual sample size.
Figure 1.
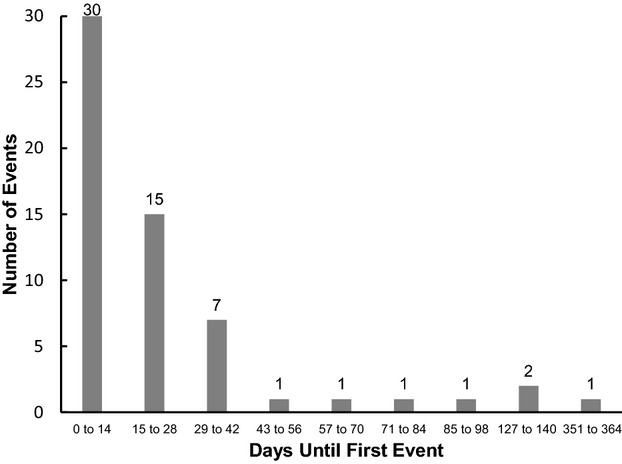
Temporal distribution of neurological events after implantation with the Berlin Heart EXCOR® ventricular assist device.
Figure 2.
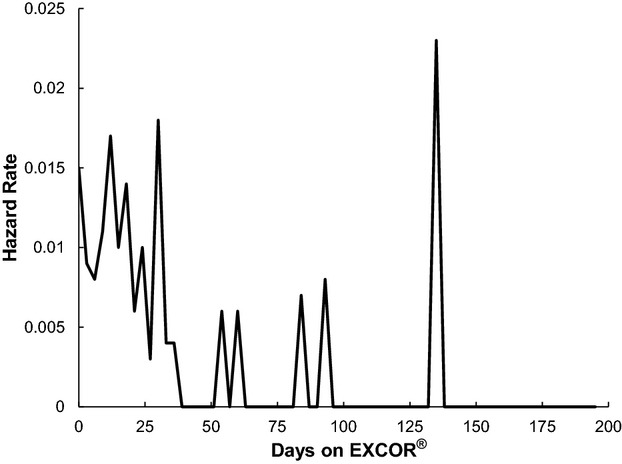
Hazard rate of first neurological event compared with the number of days of EXCOR® support. EXCOR® indicates Berlin Heart EXCOR® ventricular assist device.
The overall freedom from neurological events was 73% at 30 days, falling further to 64% at 90 days, as shown in the Kaplan–Meier analysis in Figure 3.
Figure 3.

Freedom from first neurological event. The Kaplan–Meier curve shows the survival function for the probability of children implanted with EXCOR® to remain free from neurological events. EXCOR® indicates Berlin Heart EXCOR® ventricular assist device.
Event Classification and Neuroimaging Findings
Of the 73 documented neurological events, 12 were defined as transient ischemic attack, 7 were described as a new abnormality of head ultrasound, 1 was an electroencephalogram finding of seizure activity, and 52 events were strokes. Considering only those events described as strokes, the vast majority (n=47, 89%) were ischemic, and there were comparatively few hemorrhagic strokes (n=5, 9%). These data are summarized in Table 2.
Table 2.
Types of Neurological Injuries Reported (n=204)
| Type of Neurological Injury | Number of Events | Patients With Events (%) |
|---|---|---|
| Ischemic stroke | 46 | 41 (20.1) |
| Covert stroke | 1 | 1 (0.5) |
| Hemorrhagic stroke | 5 | 5 (2.5) |
| Transient ischemic attack | 12 | 12 (5.9) |
| New abnormality of head ultrasound | 7 | 6 (2.9) |
| EEG positive for seizure | 1 | 1 (0.5) |
| Global atrophy | 1 | 1 (0.5) |
| Total neurological events | 73 | 59 (28.9) |
| Total strokes | 52 | 43 (21.1) |
EEG indicates electroencephalogram.
Head CTs were available for review for all 29 children with neurological events at IDE sites. Of these children, 59% (17 of 29) had baseline head CT prior to implantation, and 12% (2 of 17) of the baseline head CTs documented prior ischemic stroke. These 29 children had 35 neurological events: 28 were ischemic strokes, including 1 covert stroke (ie, a symptom prompted evaluation with head CT, which demonstrated an ischemic stroke, but the neurological examination was nonfocal); 4 were hemorrhagic strokes; 2 were transient ischemic attacks; and 1 was progressive global cerebral volume loss (atrophy). In 52% of children with ischemic stroke, multiple new infarcts were present on the head CT at the time of the first recorded neurological event. Infarcts were bilateral in 38% of patients. The majority of ischemic infarcts (57%) were in the anterior circulation, 25% were in the anterior and posterior circulation, 3% were in the posterior circulation only, and 14% were border zone or watershed‐type infarcts. In 10% of the strokes, the infarcts were large, occupying more than one third of the middle cerebral artery territory. Hemorrhagic transformation of ischemic strokes occurred in 3% and occurred only in the setting of a large infarct (ie, stroke in more than one third of the middle cerebral artery territory). Cerebral edema causing midline shift was present in 4 children, and tonsillar herniation was seen in 2 children. Extra‐axial hemorrhages were noted in 8 children, all of which were subdural hematomas. Recurrent infarcts were present on follow‐up head CTs in 6 children.
Event Severity and Outcome
Indicators of the severity of the neurological events can be found in the PSOM scores and by considering the impact of the stroke on subsequent likelihood of successful transplantation. Unfortunately, for the majority of participants, a preimplantation baseline PSOM assessment was not performed because the patient was too ill and/or was sedated and intubated. Baseline PSOM scores were obtained in 23 of the 68 patients (34%) at IDE sites who met the primary enrollment criteria and in 6 of the 41 patients (15%) at IDE sites who did not meet enrollment criteria. At the non‐IDE sites, care was provided according to existing standard of care, and preimplantation PSOM examinations were rare (4%).
Of the 59 participants who had neurological events, 29 were implanted at IDE sites, and of those, 25 had at least 1 PSOM completed, whereas 15 had PSOM data collected within 7 days following the event. For each of these participants, the PSOM following the event and the last available PSOM are shown in Table 3. As can be seen, in this subset of 15 patients, PSOM data capture and follow‐up were essentially complete, with follow‐up values lacking only for the 2 participants who died. For this group, the median value of the PSOM within 7 days after the event was 3.0 (interquartile range 1 to 5), whereas the last PSOM obtained for each of these participants had a median value of 1.00 (interquartile range 0 to 3). This change was not statistically significant. The time between the event and the last PSOM was a median of 136 days (interquartile range 79 to 171). The distribution of PSOM scores according to clinical severity is shown in Figure 4 for both the initial and final PSOM scores. This figure includes all PSOM data available, even if no PSOM was administered immediately following the event. The proportion of patients with a PSOM score indicating severe neurological deficit was 53% based on the PSOM within 7 days of the event and 39% based on the last PSOM following the event (P value not significant for change over time).
Table 3.
PSOM Scores at Investigational Device Exemption Sites Following First Neurological Event
| No. | PSOM Within 7 Days of Event | Last Available PSOM | Time From Outcome to Last PSOM (Days) | Time From Event to Last PSOM (Days) | Outcome | ||
|---|---|---|---|---|---|---|---|
| Score | Severity | Score | Severity | ||||
| 1 | 10 | Severe | 10 | Severe | 376 | 538 | Transplant |
| 2 | 7 | Severe | 1 | Mild | 221 | 222 | Transplant |
| 3 | 5 | Severe | 1 | Mild | 27 | 57 | Transplant |
| 4 | 5 | Severe | 0 | Normal | −8 | 169 | Transplant |
| 5 | 4 | Severe | 3.5 | Severe | 68 | 93 | Transplant |
| 6 | 4 | Severe | 3 | Severe | −56 | 145 | Weaned‐Failure |
| 7 | 3.5 | Severe | N/A | Death | |||
| 8 | 3 | Severe | N/A | Death | |||
| 9 | 2 | Moderate | 1 | Mild | 24 | 70 | Transplant |
| 10 | 1.5 | Moderate | 0 | Normal | 50 | 146 | Transplant |
| 11 | 1 | Mild | 3.5 | Severe | 151 | 234 | Transplant |
| 12 | 1 | Mild | 3 | Severe | 111 | 136 | Transplant |
| 13 | 1 | Mild | 0 | Normal | 56 | 63 | Transplant |
| 14 | 0.5 | Mild | 1.5 | Moderate | 17 | 130 | Transplant |
| 15 | 0.5 | Mild | 0 | Normal | 82 | 86 | Transplant |
N/A indicates not applicable due to death; PSOM, Pediatric Stroke Outcome Measure.
Figure 4.
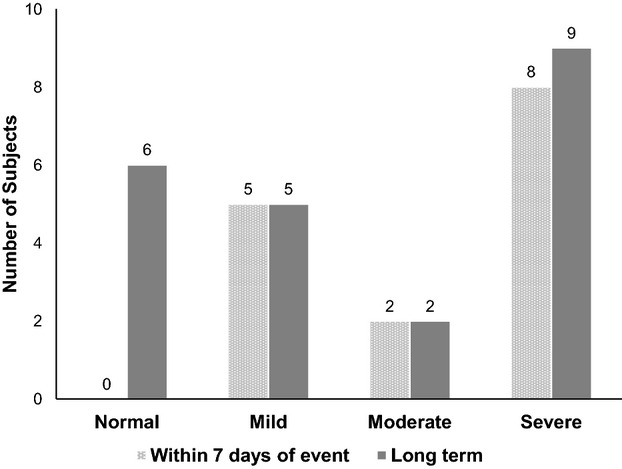
Pediatric Stroke Outcome Measure (PSOM) severity following neurological event. The bar graph shows neurological outcome severity as measured by the PSOM, when available, for children within 7 days of a neurological event and long term, at a median of 136 days (interquartile range 79 to 171) after a neurological event.
Severity of neurological injury was further assessed by evaluating the clinical course subsequent to the injury. Table 4 provides the subsequent clinical outcome for the 59 patients who experienced ≥1 neurological event. Overall, 56% of patients who experienced a neurological event continued to successfully undergo heart transplantation. An additional patient had successful weaning from EXCOR® because of recovery of ventricular function with good neurological outcome. The remaining patients either died (n=23) or were weaned from the device with subsequent mortality (n=2). The mortality rate for patients who had at least 1 neurological event was 42% (25 of 59), significantly greater than the rate of 18% (26 of 145) for those who did not have a neurological event (P=0.0006). As can also be seen in Table 4, the mortality (including weaning failure) associated with neurological events did not appear to be evenly distributed between the study sites, with mortality of 17% at IDE sites compared with 67% at non‐IDE sites (P=0.0002). To determine whether this result was more likely a site effect or a difference in patients, mortality was compared between patients who did and did not meet the primary study enrollment criteria, regardless of site of care. This analysis demonstrated 58% mortality in patients who did not meet primary enrollment criteria compared with 31% for those who did meet these criteria. The findings fell slightly short of achieving the specified statistical significance (P=0.06).
Table 4.
Clinical Outcome Following Neurological Injury
| Outcome | IDE Site (N=109) | Non‐IDE Site (N=95) | Total (N=204) |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Patients with ≥1 neurological event | 29 (27) | 30 (32) | 59 (29) |
| Outcome for all patients that experienced neurological events | |||
| Transplanted | 24 (83) | 9 (30) | 33 (56) |
| Weaned successfully | 0 (0) | 1 (3) | 1 (2) |
| Died (support withdrawn) | 4 (14) | 19 (63) | 23 (39) |
| Weaned failure | 1 (3) | 1 (3) | 2 (3) |
IDE indicates investigational device exemption.
Among the 59 children with neurological events, 42 children had 47 ischemic strokes, and 14 of those had support withdrawn. Five children had hemorrhagic strokes, and 1 had support withdrawn because of the severity of the neurological injury. Surgical intervention for hematoma evacuation was performed in 2 of 29 children (1 parenchymal hematoma evacuation and 1 subdural hematoma evacuation).
Risk Stratification
In view of the clinical importance of the neurological events, we attempted to construct a risk‐stratification model using demographic and clinical variables available at the time of implantation. The variables selected and the univariate test results are shown in Table 5. These included sex, weight, cardiac diagnosis, baseline renal and hepatic function, use of extracorporeal membrane oxygenation prior to EXCOR®, use of left VAD versus bi‐VAD, and pump size. In univariate analysis, sex, diagnosis of congenital heart defect, use of extracorporeal membrane oxygenation prior to EXCOR®, device type (left VAD or bi‐VAD), prior stroke, and any pump replacement because of thrombus all had P<0.25 and were included in the multivariate model. In the multivariate analysis, only sex and pump replacement were significant risk factors for a neurological event, with female participants and those with a pump change because of thrombus more likely to experience a neurological event. Because of the manner in which the data were collected, it was not possible to reconstruct the temporal relationship between the pump exchange and the neurological event.
Table 5.
Potential Risk Factors for Occurrence of Neurological Events in Univariable and Multivariable Analyses
| Variable | With CNS Event* (n=59) | Without CNS Event* (n=145) | Univariable P Value; OR (95% CI) | Multivariable P Value; OR (95% CI) |
|---|---|---|---|---|
| Sex | 0.03* | 0.02 | ||
| Male | 24 (41) | 83 (57) | 0.51 (0.28 to 0.95) | 0.45 (0.23 to 0.86) |
| Female | 35 (59) | 62 (43) | ||
| Weight | 0.54 | |||
| <5 kg | 11 (19) | 22 (15) | 0.78 (0.35 to 1.73) | |
| ≥5 kg | 48 (81) | 123 (85) | ||
| Age, months, median (IQR) | 19.4 (6.2 to 66.5) | 18.1 (6.3 to 64.3) | 0.84 1.00 (0.99 to 1.01) | |
| Primary diagnosis | 0.08* | 0.12 | ||
| CHD | 12 (20) | 47 (32) | 0.53 (0.26 to 1.10) | 0.54 (0.25 to 1.18) |
| Non‐CHD | 47 (80) | 98 (68) | ||
| INTERMACS profile | 0.99 | |||
| 1 | 31 (52) | 76 (52) | 1.00 (0.55 to 1.84) | |
| ≥2 | 28 (48) | 69 (48) | ||
| Creatinine preimplant, dL/mL, median (IQR) | 0.4 (0.3 to 0.6) | 0.4 (0.3 to 0.7) | 0.75 0.94 (0.67 to 1.33) | |
| Bilirubin preimplant, dL/mL, median (IQR) | 1.0 (0.6 to 2.5) | 1.0 (0.5 to 1.9) | 0.30 1.00 (0.92 to 1.08) | |
| ECMO prior to implant | 28 (47) | 55 (38) | 0.21*; 1.48 (0.80 to 2.72) | 0.10; 1.73 (0.89 to 3.33) |
| Stroke prior to implant | 1 (2) | 13 (9) | 0.07*; 0.18 (0.02 to 1.67) | 0.06; 0.13 (0.02 to 1.12) |
| Met IDE entrance criteria | 35 (59) | 88 (61) | 0.86; 1.47 (0.46 to 4.66) | |
| BSA, m2, median (IQR) | 0.47 (0.33 to 0.68) | 0.50 (0.35 to 0.72) | 0.55; 0.73 (0.28 to 1.87) | |
| Device type | 0.20* | 0.38 | ||
| LVAD | 41 (70) | 87 (60) | 1.52 (0.80 to 2.90) | 1.36 (0.68 to 2.70) |
| BVAD | 18 (30) | 58 (40) | ||
| Device size | 0.52 | |||
| 10 mL | 24 (41) | 52 (36) | 1.23 (0.66 to 2.28) | |
| Other | 35 (59) | 93 (64) | ||
| Pump replacement for thrombus | 0.003* | 0.005 | ||
| None | 26 (44) | 97 (67) | 2.56 (1.38 to 4.77) | 2.58 (1.34 to 4.97) |
| ≥1 | 38 (56) | 48 (33) |
BSA indicates body surface area; BVAD, biventricular assist device; CHD, congenital heart defect; CNS, central nervous system; ECMO, extracorporeal membrane oxygenation; IDE, investigational device exemption; INTERMACS, Interagency Registry for Mechanically Assisted Circulatory Support; IQR, interquartile range; LVAD, left ventricular assist device; OR, odds ratio.
Values are number (percentage) except as indicated.
Variable included in multivariable logistic regression model.
Discussion
This analysis of the EXCOR® IDE database represents the most detailed evaluation to date of neurological adverse events associated with use of EXCOR®. The present report includes >200 children treated at multiple centers using a common protocol. The overall incidence of neurological events was high, with 29% of patients experiencing at least 1 neurological event. Half of all patients who would ultimately experience a neurological event did so within the first 14 days of support, with an additional 25% occurring in the next 14 days. Consequently, the initial 28 days of support defined a period of particularly high risk for the development of a neurological event. The freedom from neurological events was 64% after 90 days of support. This report does not answer the question of why the neurological event rate is initially high, but several possibilities may be considered. The perioperative period involves inhibited platelet function and surgical bleeding, both of which complicate the initiation of effective anticoagulation. In addition, given the severity of HF prior to implantation, renal and hepatic function may both be in flux in this period, further complicating the ability to balance thrombotic and hemorrhagic risk. A further contribution may be cardiovascular instability following initiation of VAD support.
In recent pivotal trials of adult continuous‐flow VADs, the reported rates of neurological events range from 8% to 17%.13–14 In comparison, the observed rate of 29% with the pulsatile‐flow EXCOR® is quite high, using a comparable set of data definitions. There are significant differences between continuous‐flow and pulsatile pumps, so a more pertinent comparison might be the neurological injury rate reported for adults supported on pulsatile devices. The Thoratec XVE left VAD is associated with a very low neurological event rate (0% in 1 study), whereas the more recently developed SynCardia Total Artificial Heart has been reported to have a rate of 8%.15–16 The greater stroke risk with EXCOR® likely reflects a combination of device risks such as smaller pump size and inherent difficulties associated with anticoagulation in children, including the lack of adequate normative data and the complex interplay of developmental factors.
Although a small number of events were transient ischemic attacks, and thus were not associated with permanent neurological injury, 71% of neurological events were strokes. With a single exception, these strokes produced overt neurological symptoms. The preponderance of ischemic strokes was striking (47 ischemic compared with 5 hemorrhagic). This raises the question of whether a more aggressive anticoagulation protocol might have reduced the frequency of neurological events. Given that the incidence of major bleeding was 44%, increased intensity of anticoagulation would be likely to carry significant risk of increased hemorrhage. Because the definition of major bleeding included the requirement for blood product transfusion without surgical exploration (but excluded hemorrhagic stroke from the definition), it may be appropriate to accept a higher bleeding risk in exchange for a reduction in ischemic stroke risk; however, if the consequence of more intensive anticoagulation is hemorrhagic stroke, that would be a less desirable exchange.
The clinical severity of the neurological events was difficult to establish because of data limitations. PSOM data were intended to characterize stroke severity, but in many instances, these data were not collected because of the patient's medical condition. The majority of patients were sedated prior to implantation and sometimes up to 2 weeks after implantation, and this precluded a detailed neurological assessment. The available PSOM data demonstrate that the clinical burden from stroke is initially consistent with severe impairment (median PSOM within 7 days following event 3.0 ) but is lower at last available follow‐up (median PSOM at last assessment 1.0), which is indicative of mild to moderate impairment. In addition, as judged by the crude measure of associated mortality rates, the strokes that occurred at IDE trial sites were comparatively well tolerated, with 17% mortality. In contrast, children treated at non‐IDE sites had mortality of 67% after a first neurological event despite a very similar overall rate of neurological events. The reasons for this discrepancy are not clear. Potentially, the differences might relate to differences in patient selection for study enrollment, in severity of stroke, or in site decision making following a neurological event. Our analysis suggests that patient mix is likely a significant contributor to this finding, with patients who did not meet primary study enrollment showing a trend toward higher mortality rates following stroke compared with those patients who met enrollment criteria, regardless of site of treatment. The importance of severity of illness at the time of stroke onset is supported by studies of children with stroke from all causes: The presence of critical illness prior to stroke onset is associated with increased mortality, and the need for intensive care unit care after stroke is associated with higher mortality.17–18 Neither severity of stroke nor decision making following stroke can be compared between IDE and non‐IDE sites, given the available data, although there is no reason to suspect a difference between IDE and non‐IDE sites in this regard. Non‐IDE sites were provided with the anticoagulation protocol, but adherence was not required or tracked, so it is possible that this played a role in modulating outcome after a neurological injury.
The significant neurological injury rate and severity observed in this study led to the question of what clinical care might be indicated following treatment with EXCOR®. A recent American Heart Association scientific statement urged periodic developmental surveillance, screening, evaluation, and reevaluation of children with congenital heart defect throughout childhood.19 In children with HF requiring mechanical circulatory support, this may take on even greater importance. Children who have a stroke should be considered candidates for standardized developmental assessments at least annually until aged 5 years and also should undergo cognitive assessments (usually aged >5 years). Neuropsychological testing should be considered at intervals thereafter to understand the effects of stroke and to allow targeted educational interventions for these children.20–21
In view of the important role of neurological injury in shaping both survival and long‐term outcomes after device support, a risk‐prediction model for development of stroke would be an important clinical tool. We attempted to model risk using many variables widely believed to be influential. Contrary to what many investigators believe, this analysis does not demonstrate a relationship between use of the 10‐mL EXCOR® pump and the risk of neurological injury. In fact, the only variables that emerged as predictive of increased risk of neurological injury are female sex and VAD pump change because of thrombus. Female sex has also emerged as a stroke risk factor in the adult experience reported by INTERMACS, but the reason for this remains unclear.22 Pump exchange as a risk factor is difficult to interpret because the study database does not allow us to determine the temporal relationship between the pump replacement and the neurological injury. Furthermore, although the neurological events had a defined onset time, the recognition of an event may be delayed because of factors associated with critical care, such as systemic sedation for intubation or pain relief. It is possible that visible thrombus precedes neurological injury and represents a therapeutic target, either via pump exchange or alteration of anticoagulation. It is also possible that clinical care is altered by an antecedent neurological injury, leading to more frequent exchange of the VAD. There is also complex interplay between center experience and threshold for VAD pump exchange; there were no established criteria for replacement of the VAD, either at the time of the trial or even today.
This trial provided other important lessons that can be applied to future clinical trials of VADs in children. The adult definitions of neurological events from INTERMACS were difficult to apply to critically ill children in this trial. The accepted definition of an arterial ischemic stroke, for example, specifies a focal deficit that conforms to an arterial territory with a corresponding infarct on brain imaging; however, in a child aged <6 months, brain injury may be associated only with seizures or altered mental status rather than a focal deficit, so the imaging determines the event classification. In addition, seizures as a defined neurological injury were problematic because they are best regarded as a symptom rather than as a specific type of neurological injury. Future trials of mechanical circulatory support might be better supported by a revised set of neurological definitions for pediatric stroke that are consistent with the National Institutes of Health Common Data Elements.23
This study also illustrates the difficulty of obtaining unambiguous neurological baseline information, given the acute and severe presentation of HF in a large number of the children in this study. Imaging data are likely to continue to be required to provide adequate baseline data in future trials. Ongoing assessment of neurological injury also proved problematic, as shown by the erratic collection of PSOM data. This may reflect limited access to skilled pediatric neurologists, even at sites that were able to enroll children in a VAD trial. Solutions to this problem will be difficult to implement. To the degree that is possible, use of instruments that can be completed at the bedside by the primary treating physician will ameliorate that problem, but such instruments have not been validated for assessing neurological injury in children. Finally, preimplantation strokes were recognized in 14 of 204 children (7%). This observation raises several issues. First, there is a need for preimplantation imaging for future device trials in which clarity is needed around timing and attribution of neurological events relative to device use. Second, neurological morbidity is part of the underlying disease process in children with HF and requires attention and research at the preimplant stage of disease, separate from the postimplant stage of disease. Third, the long‐term functional outcome reflects the totality of neurological injury that these children experience, including the strokes that occur before device use and in those children who come close to needing a device but who are eventually managed without device support. If possible, more rigorous assessment of baseline neurological function in children with severe HF is warranted prior to the point of requiring mechanical circulatory support.
Conclusion
Of children treated with EXCOR® as a bridge to transplant, 29% experienced a neurological injury. The majority of neurological injuries were ischemic strokes and occurred in the first 28 days of support. Neurological injury does not preclude successful transplantation in the majority of cases but nonetheless ranks as the leading cause of death after EXCOR® implantation. Currently we cannot effectively risk‐stratify patients based on baseline characteristics.
Sources of Funding
The Berlin Heart IDE study was supported by Berlin Heart, Inc and by a grant from the Food and Drug Administration Office of Orphan Product Development (R01‐FD03357).
Disclosures
Tjossem is an employee of Berlin Heart. Humpl received payments for lectures and/or service on speaker's bureaus from Berlin Heart, Inc. Jordan, Ichord, and Rosenthal served as members of the Clinical Event Committee for Berlin Heart EXCOR® Pediatric IDE trial. Reinhartz and Pruthi have no disclosures.
References
- Lloyd‐Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel‐Smoller S, Wong ND, Wylie‐Rosett JAmerican Heart Association Statistics C and Stroke Statistics S. Executive summary: heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010; 121:948-954. [DOI] [PubMed] [Google Scholar]
- Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006; 296:1867-1876. [DOI] [PubMed] [Google Scholar]
- Almond CS, Thiagarajan RR, Piercey GE, Gauvreau K, Blume ED, Bastardi HJ, Fynn‐Thompson F, Singh TP. Waiting list mortality among children listed for heart transplantation in the United States. Circulation. 2009; 119:717-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirklin JK, Naftel DC, Stevenson LW, Kormos RL, Pagani FD, Miller MA, Ulisney K, Young JB. INTERMACS database for durable devices for circulatory support: first annual report. J Heart Lung Transplant. 2008; 27:1065-1072. [DOI] [PubMed] [Google Scholar]
- Kirklin JK, Naftel DC, Kormos RL, Stevenson LW, Pagani FD, Miller MA, Ulisney KL, Baldwin JT, Young JB. Second INTERMACS annual report: more than 1 000 primary left ventricular assist device implants. J Heart Lung Transplant. 2010; 29:1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirklin JK, Naftel DC, Kormos RL, Stevenson LW, Pagani FD, Miller MA, Ulisney KL, Baldwin JT, Young JB. Third INTERMACS Annual Report: the evolution of destination therapy in the United States. J Heart Lung Transplant. 2011; 30:115-123. [DOI] [PubMed] [Google Scholar]
- Fraser CD, Jr, Jaquiss RD, Rosenthal DN, Humpl T, Canter CE, Blackstone EH, Naftel DC, Ichord RN, Bomgaars L, Tweddell JS, Massicotte MP, Turrentine MW, Cohen GA, Devaney EJ, Pearce FB, Carberry KE, Kroslowitz R, Almond CS. Prospective trial of a pediatric ventricular assist device. N Engl J Med. 2012; 367:532-541. [DOI] [PubMed] [Google Scholar]
- Almond CS, Morales DL, Blackstone EH, Turrentine MW, Imamura M, Massicotte MP, Jordan LC, Devaney EJ, Ravishankar C, Kanter KR, Holman W, Kroslowitz R, Tjossem C, Thuita L, Cohen GA, Buchholz H, St Louis JD, Nguyen K, Niebler RA, Walters HL, III, Reemtsen B, Wearden PD, Reinhartz O, Guleserian KJ, Mitchell MB, Bleiweis MS, Canter CE, Humpl T. Berlin Heart EXCOR pediatric ventricular assist device for bridge to heart transplantation in US children. Circulation. 2013; 127:1702-1711. [DOI] [PubMed] [Google Scholar]
- Cassidy J, Dominguez T, Haynes S, Burch M, Kirk R, Hoskote A, Smith J, Fenton M, Griselli M, Hsia TY, Ferguson L, Van Doorn C, Hasan A, Karimova A. A longer waiting game: bridging children to heart transplant with the Berlin Heart EXCOR device–the United Kingdom experience. J Heart Lung Transplant. 2013; 32:1101-1106. [DOI] [PubMed] [Google Scholar]
- Almond CS, Buchholz H, Massicotte P, Ichord R, Rosenthal DN, Uzark K, Jaquiss RD, Kroslowitz R, Kepler MB, Lobbestael A, Bellinger D, Blume ED, Fraser CD, Jr, Bartlett RH, Thiagarajan R, Jenkins K. Berlin Heart EXCOR Pediatric ventricular assist device Investigational Device Exemption study: study design and rationale. Am Heart J. 2011; 162:e6. [DOI] [PubMed] [Google Scholar]
- Kitchen L, Westmacott R, Friefeld S, MacGregor D, Curtis R, Allen A, Yau I, Askalan R, Moharir M, Domi T, deVeber G. The pediatric stroke outcome measure: a validation and reliability study. Stroke. 2012; 43:1602-1608. [DOI] [PubMed] [Google Scholar]
- deVeber GA, MacGregor D, Curtis R, Mayank S. Neurologic outcome in survivors of childhood arterial ischemic stroke and sinovenous thrombosis. J Child Neurol. 2000; 15:316-324. [DOI] [PubMed] [Google Scholar]
- Miller LW, Pagani FD, Russell SD, John R, Boyle AJ, Aaronson KD, Conte JV, Naka Y, Mancini D, Delgado RM, MacGillivray TE, Farrar DJ, Frazier OH, HeartMate IICI. Use of a continuous‐flow device in patients awaiting heart transplantation. N Engl J Med. 2007; 357:885-896. [DOI] [PubMed] [Google Scholar]
- Aaronson KD, Slaughter MS, Miller LW, McGee EC, Cotts WG, Acker MA, Jessup ML, Gregoric ID, Loyalka P, Frazier OH, Jeevanandam V, Anderson AS, Kormos RL, Teuteberg JJ, Levy WC, Naftel DC, Bittman RM, Pagani FD, Hathaway DR, Boyce SWHeartWare Ventricular Assist Device Bridge to Transplant ATI. Use of an intrapericardial, continuous‐flow, centrifugal pump in patients awaiting heart transplantation. Circulation. 2012; 125:3191-3200. [DOI] [PubMed] [Google Scholar]
- Copeland J, Copeland H, Nolan P, Gustafson M, Slepian M, Smith R. Results with an anticoagulation protocol in 99 SynCardia total artificial heart recipients. ASAIO J. 2013; 59:216-220. [DOI] [PubMed] [Google Scholar]
- Slaughter MS, Sobieski MA, Gallagher C, Dia M, Silver MA. Low incidence of neurologic events during long‐term support with the HeartMate XVE left ventricular assist device. Tex Heart Inst J. 2008; 35:245-249. [PMC free article] [PubMed] [Google Scholar]
- Jordan LC, van Beek JG, Gottesman RF, Kossoff EH, Johnston MV. Ischemic stroke in children with critical illness: a poor prognostic sign. Pediatr Neurol. 2007; 36:244-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox CK, Johnston SC, Sidney S, Fullerton HJ. High critical care usage due to pediatric stroke: results of a population‐based study. Neurology. 2012; 79:420-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, Mussatto KA, Uzark K, Goldberg CS, Johnson WH, Jr, Li J, Smith SE, Bellinger DC, Mahle WTAmerican Heart Association Congenital Heart Defects Committee CoCDitYCoCN and Stroke C. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012; 126:1143-1172. [DOI] [PubMed] [Google Scholar]
- Westmacott R, Askalan R, MacGregor D, Anderson P, Deveber G. Cognitive outcome following unilateral arterial ischaemic stroke in childhood: effects of age at stroke and lesion location. Dev Med Child Neurol. 2010; 52:386-393. [DOI] [PubMed] [Google Scholar]
- Studer M, Boltshauser E, Capone MA, Datta A, Fluss J, Mercati D, Hackenberg A, Keller E, Maier O, Marcoz JP, Ramelli GP, Poloni C, Schmid R, Schmitt‐Mechelke T, Wehrli E, Heinks T, Steinlin M. Factors affecting cognitive outcome in early pediatric stroke. Neurology. 2014; 82:784-792. [DOI] [PubMed] [Google Scholar]
- Boyle AJ, Jorde UP, Sun B, Park SJ, Milano CA, Frazier OH, Sundareswaran KS, Farrar DJ, Russell SD, HeartMate IICI. Pre‐operative risk factors of bleeding and stroke during left ventricular assist device support: an analysis of more than 900 HeartMate II outpatients. J Am Coll Cardiol. 2014; 63:880-888. [DOI] [PubMed] [Google Scholar]
- Saver JL, Warach S, Janis S, Odenkirchen J, Becker K, Benavente O, Broderick J, Dromerick AW, Duncan P, Elkind MS, Johnston K, Kidwell CS, Meschia JF, Schwamm LNational Institute of Neurological D and Stroke Stroke Common Data Element Working G. Standardizing the structure of stroke clinical and epidemiologic research data: the National Institute of Neurological Disorders and Stroke (NINDS) Stroke Common Data Element (CDE) project. Stroke. 2012; 43:967-973. [DOI] [PMC free article] [PubMed] [Google Scholar]