

Abstract
The discovery of enzyme inhibitors relies on synthetic methods that enable rapid and modular construction of small molecules. Heterocyclic fragments designed to maximize enthalpic interactions with their protein targets represent a particularly desirable class of molecules. Here we describe a reagent that enables straightforward construction of “borofragments”, in which a heterocycle is separated from the boron center by two or three rotatable bonds. The stability of these molecules depends on the MIDA group which likely acts as a slow-release element under biological conditions. Borofragments can be used to discover inhibitors of enzymes that use catalytic oxygen nucleophiles. We have employed this method to identify inhibitors of ABHD10 and the predicted carboxypeptidase CPVL. This technique should be applicable to other classes of targets.
Organoboron compounds have received considerable attention as valuable building blocks in chemical synthesis.1 Boron-containing molecules have also been used as enzyme inhibitors.2 Both of these applications result from the accessibility of boron’s empty p-orbital, which enables reversible interaction with synthetic and biological nucleophiles. Our recent interest in this area has led to an ongoing exploration of boron-based electrophiles that engage nucleophilic residues in active sites of proteins. In contrast to aldehydes, acrylates and epoxides, which are widely used as “baits” for nucleophilic residues in active sites, small boron-containing electrophiles have received relatively little attention. This is mainly due to their lack of stability and relative difficulty of preparation, especially when it comes to molecules with C(sp3)-B bonds.1 Because the corresponding synthetic protocols often depend on organometallic reactions with low functional group tolerance, we recently developed a boron-containing isocyanide reagent that enabled us to synthesize novel boromorpholinones and boropeptides under mild conditions.3 The N-methyliminodiacetic acid (MIDA) residue on boron4,5 was the main factor that contributed to the stability of boropeptides and their successful one-step synthesis.
Building on our approach to boropeptides, we became interested in advancing the concept of MIDA-containing borofragments, which are chimeric molecules where a boron-containing group is connected to a recognition unit, most commonly a heterocycle. As many biologically relevant heterocycles with high ligand efficiency have conjugate acids with relatively low pKa’s,6 we were motivated to develop a mild method for connecting a diverse set of heterocyclic nucleophiles to the smallest possible C-B unit equipped with the MIDA substituent (furnishing the corresponding borofragments). To facilitate navigation of the accessible chemistry space in enzyme active sites, we were particularly intrigued by the borofragments in which a heterocycle is separated from the boron center by two or three rotatable bonds. In this paper, we present our efforts to synthesize electrophilic borofragments and their evaluation as serine hydrolase inhibitors using competitive activity-based protein profiling (ABPP). We show that the MIDA group is compatible with the synthesis of a diverse collection of borofragments and that select members of this class inhibit several poorly characterized serine hydrolases, including α/β hydrolase domain-containing protein 10 (ABHD10) and predicted serine carboxypeptidase (CPVL). These findings should encourage broader campaigns to append C(sp3)-B bonds in their MIDA forms to various molecular recognition components.
In order to convert readily accessible nucleophilic small molecules into borofragments, we pursued hydroxymethyl-containing building block 5 with the goal of evaluating its reactivity in redox driven condensations. Despite the relative bulk of the MIDA group, the mild and neutral conditions 7 of the Mitsunobu process were particularly attractive to us. Compound 5 was synthesized according to Scheme 1. Starting with dibromomethane (1), bromomethylpinacolborate (2) was prepared according to literature procedure.8 Treatment of 1 with n-butyllithium in the presence of triisopropoxyborate followed by transesterification of the resulting bromomethyldiisopropoxyborane with pinacol furnished intermediate 2. Subsequent treatment of crude product 2 with sodium benzyloxide in DMSO afforded benzyloxymethylpinacolboronic ester (3),9 which was converted to benzyloxymethyl(MIDA)boronate (4) in situ by heating in the presence of MIDA. Most importantly, purification of compound 4 was achieved by trituration with Et2O, which eliminated the need for chromatography. Hydrogenolysis of 4 with Pd/C afforded hydroxymethyl(MIDA)boronate (5) as a bench-stable white solid in pure form (49% isolated yield over 4 steps, >10g).
Scheme 1.
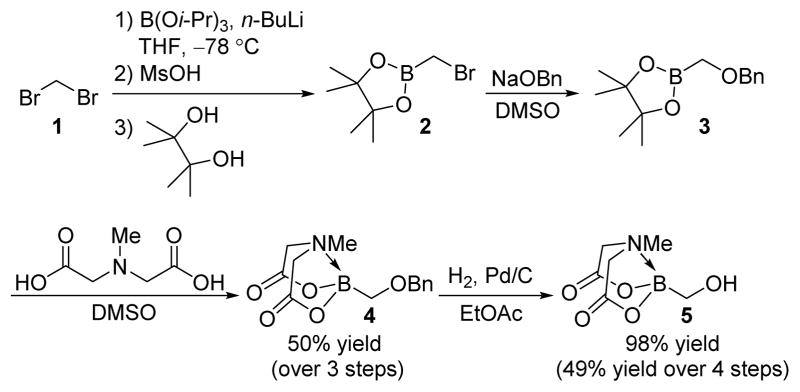
Preparation of hydroxymethyl(MIDA)boronate
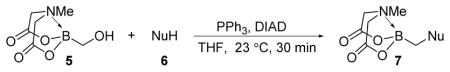
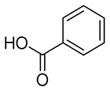
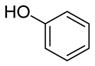
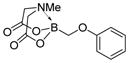
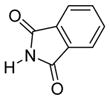
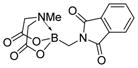
We were pleased to observe that, despite the steric bulk of the B-MIDA substituent, hydroxymethyl(MIDA)boronate (5) reacted with various acidic pro-nucleophiles (NuH) in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosphine to produce a variety of α-functionalized alkyl(MIDA)boronates in excellent yields (Table 1). Ester formation via coupling of benzoic acid (6a) or cinnamic acid (6b) with 5 afforded the corresponding products 7a and 7b, respectively (entries 1 and 2). N-hydroxyphthalimide (6c) was easily converted into the corresponding heterocyclic product 7c (entry 3). Etherification of 5 was also carried out. Thus, phenol 6d was used to produce 7d, although 2.0 equivalents of 6d were required in this case (entry 4). Nitrogen-containing acidic pro-nucleophiles were also employed in this methodology (entries 5 and 6). The reaction of phthalimide (6e) with 5 gave the corresponding product 7e (entry 5). N,O-bis(phenoxycarbonyl)hydroxylamine (6f)10 was converted to 7f (entry 6). Purine derivatives 6g and 6h11 were found to react with 5 to afford N(9)-alkylated products 7g and 7 h, whose structures were confirmed by X-ray crystallography (entries 7 and 8). Sulfur-containing pro-nucleophiles 6i,j reacted with 5 to afford the corresponding products 7i,j (entries 9 and 10). For compounds 7a–e,g,j purification was achieved by trituration with Et2O due to the low solubility of the products in non-polar solvents. We note that Molander and co-workers have reported the nucleophilic substitution of α-halomethyltrifluoroborates with nucleophiles such as amines and organolithium/organosodium reagents.12 Their work also includes nucleophilic substitutions of α-halomethylboronic esters with amines followed by addition of KHF2 to synthesize a number of α-functionalized alkyl trifluoroborates.13 However, those substitutions have been conducted under basic conditions and often at high temperatures. In contrast, our reactions are carried out at neutral pH and room temperature, thus expanding the scope of compatible functional groups, which includes biologically relevant heterocycles with low pKa’s.
Table 1.
Synthesis of α-functionalized alkyl(MIDA)boronatesa
| |||
|---|---|---|---|
| entry | NuH | product | yield (%) |
| 1b |
6a
|
7a
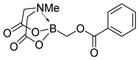
|
99 |
| 2b |
6b
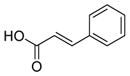
|
7b
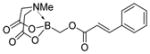
|
94 |
| 3 |
6c
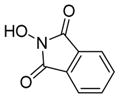
|
7c
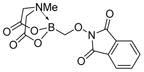
|
97 |
| 4c |
6d
|
7d
|
91 |
| 5 |
6e
|
7e
|
99 |
| 6 |
6f
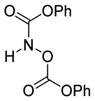
|
7f
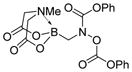
|
78 |
| 7 |
6g
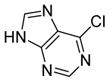
|
7g
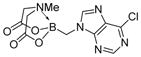
|
98 |
| 8 |
6h
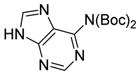
|
7h
|
96 |
| 9 |
6i
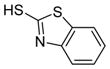
|
7i
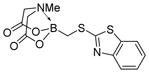
|
85 |
| 10 |
6j
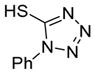
|
7j
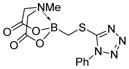
|
92 |
Unless otherwise noted, reactions were conducted with alcohol (1.0 equiv.), 1.0 equiv. of acidic pronucleophile, 1.2 equiv. of triphenylphosphine, and 1.2 equiv. of DIAD in THF at 23 °C for 30 min.
1.5 equiv. of acidic pronucleophile, 1.5 equiv. of triphenylphosphine, and 1.5 equiv. of DIAD were used. Reaction time: 30 min. Temp.: 23 °C.
2.0 equiv. of acidic pronucleophile, 1.5 equiv. of triphenylphosphine, and 1.5 equiv. of DIAD were used. Reaction time: 12 h. Temp.: 23 °C.
We were also able to incorporate the azide function via a modified Mitsunobu reaction of hydroxymethyl(MIDA)boronate (5) with diphenylphosphorylazide (DPPA) (8). This reaction produced azidomethyl(MIDA)boronate (9) in 90% yield (Scheme 2). 14 Compound 9 was also synthesized through an alternate route in 75% yield from the reaction of sodium azide with iodomethyl(MIDA)boronate (10), which was in turn prepared by treatment of 5 with iodine and imidazole. The presence of an azide in compound 9, which can be elaborated via click chemistry,15 further expands the synthetic versatility of borofragment construction.
Scheme 2.
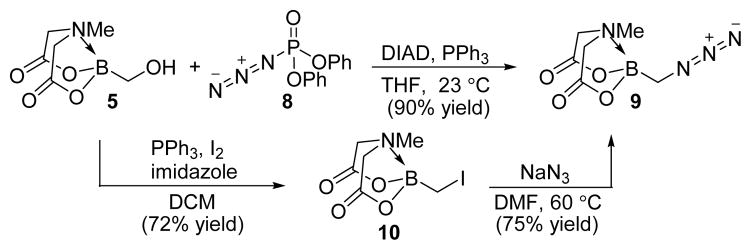
Synthesis of azidomethyl(MIDA)boronate
While boronic acids and their derivatives have been explored as enzyme inhibitors for decades,2 their potency and selectivity across mechanistically conserved enzyme classes remains poorly understood. In large part this is due to the poor stability of free boronic acids under biological conditions and the consequential lack of compounds for analysis. We have found that the MIDA ligand enables preparation of compounds unavailable in their free boronic acid form, yet in a biological milieu would likely be released to form active boronic acids. With this in mind, we wondered if broadly profiling the activity of serine hydrolases in the presence of our (MIDA)boronate fragments would elucidate novel inhibitor-enzyme pairs that could aid functional annotation of poorly characterized enzymes within this class. Using competitive activity-based protein profiling (ABPP) with the serine hydrolase-directed activity-based probe fluorophosphonate-rhodamine (FP-Rh),16 we first assayed 10 library members (Table 1) across the serine hydrolases natively expressed in a human prostate cancer cell line (PC3). Briefly, a soluble PC3 cell proteomic lysate was treated with either DMSO or a (MIDA)boronate (10 μM) for 30 min at 37 °C and then with FP-Rh (1.0 μM) for 30 min. The enzymes labeled by FP-Rh were then resolved by SDS-PAGE and their degree of labeling was measured by fluorescence gel imaging. This initial screen revealed that, while most of these compounds did not affect FP-labeling for any detectable serine hydrolase, two thioether boronates (7i and 7j) displayed moderate inhibition of a ~30 kDa band which we have previously identified17 as ABHD10 (Figure S1). Considering that enzyme inhibition might necessitate decomposition of the (MIDA)boronate fragments to an active boronic acid, we retested each compound at 20 μM with an extended 2 h preincubation time prior to adding FP-Rh, allowing more time for the formation of the putative boronic acid (Figure 1A). Notably, this preincubation time was sufficient to produce >95% conversion of 7j to its corresponding boronic acid in DPBS (see Supporting Information). Using this new assay protocol, 7i and 7j displayed enhanced activity, completely inhibiting ABHD10 with few observable off-targets. We and others have recently shown that ABHD10, a poorly characterized serine hydrolase, is also potently inhibited by β-lactones17,18 and aza-β-lactams19, but to our knowledge, targeting this enzyme with boron-based inhibitors has not been reported. To further assess the potency and selectivity of 7j, we next tested this compound across a broad concentration range using gel-based ABPP (Figure 1B). From this analysis, we observed near complete inhibition of ABHD10 at 10 μM and acyl-coenzyme A thioesterase 1/2 (ACOT1/2)20 (acyl-CoA thioesterases which is involved in fatty acid metabolism21) at 100 μM. Although 7j is less potent for ABHD10 relative to previously described inhibitors,17, 19a it should be emphasized that the ABPP platform used herein, which relies on an irreversible FP probe to report target occupancy, may underestimate the degree of enzyme inhibition for reversible inhibitors, such as boronates/boronic acids, unless the kinetics of FP probe reactivity is carefully monitored while competitive ABPP experiments. This question can be addressed in future studies using established protocols.19b
Figure 1.

Competitive ABPP of (MIDA)boronates in PC3 cell lysates. A, Gel-based ABPP analysis of (MIDA)boronates in the soluble proteome of PC3 cells. The proteome was treated with either DMSO or compounds 7a–j (20 μM) for 2 h, then treated with FP-Rh (1.0 μM) for 30 min. Labeled serine hydrolases were resolved by SDS-PAGE and analyzed by fluorescence gel imaging. B, Gel-based ABPP analysis of 7j (1.0 nM–100 μM) and DMSO in the soluble fraction of PC3 cells showing dose-dependent inhibition of ABHD10 and ACOT1/2. C, ABPP-SILAC analysis of 7j at 25 μM (heavy) versus DMSO (light) in whole PC3 cell proteomic lysates showing inhibition of ABHD10, CPVL and, to a lesser extent, ACOT1/2. Data are presented as the mean ± standard deviations of heavy/light ratios for multiple unique peptides from each serine hydrolase.
To definitively identify the targets of 7j, we used a quantitative mass spectrometry (MS)-based platform termed ABPP-SILAC.22 Following previously described protocols, cell lysates were prepared from PC3 cells cultured in the presence of isotopically light and heavy lysine and arginine. These lysates were then treated with either DMSO (light) or 25 μM 7j (heavy) for 2 h and subsequently treated with a biotinylated FP probe (FP-biotin)23 for 30 min. After mixing the heavy and light proteomes in equal proportions, the biotinylated proteins were enriched by streptavidin chromatography, trypsinized on bead and analyzed by LC-MS/MS to quantify the extent of inhibition for the identified serine hydrolases. Among ~30 serine hydrolases only two targets were maximally inhibited (>95%): ABHD10 and the predicted carboxypeptidase CPVL. While the former target was inferred from the gel profiles, we had not detected CPVL by gel-based profiling. Given that CPVL remains poorly annotated with regard to its endogenous substrates and physiological function, there is a clear need for the development of selective inhibitors of this enzyme, which to our knowledge are still lacking. Our data suggest that the (MIDA)boronates described herein are promising leads for CPVL inhibitor development and may facilitate studying this enzyme’s role in human biology.
In summary, the hydroxymethyl (MIDA)boronate reagent disclosed in this paper allows for straightforward conjugation of boron with heterocycles of biological significance. Significantly, the MIDA group stabilizes borofragments against premature decomposition and is likely released under biological conditions to unmask an active boronic acid. This chemistry allowed us to generate new boron-containing inhibitors of ABHD10 and the predicted carboxypeptidase CPVL. Given the diversity of heterocycles that are either commercially available or exist in the historical collections of the pharmaceutical industry, our straightforward method of linking molecular recognition units with stabilized boron electrophiles should enable facile exploration of previously uncharted covalent inhibitor space.
Supplementary Material
Scheme 3.
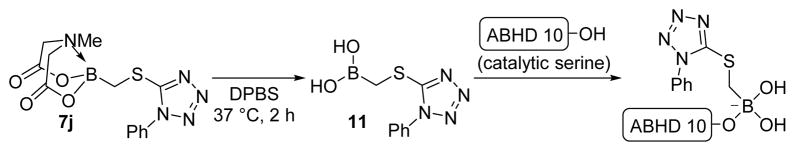
In situ hydrolysis of 7j to the boronic acid 11 and its covalent reaction with the active size nucleophile of ABHD 10
Acknowledgments
We thank NSERC and NIH (DA033760) for financial support and Dr. A. Lough for the X-ray structural analysis of 7f, 7g, and 7 h.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/c000000x/
Notes and references
- 1.[a] Kaufmann DE, Matteson DS, editors. Science of Synthesis: Vol. 6, Boron Compounds. Georg Thieme Verlag; Stuttgart-New York: 2004. [Google Scholar]; [b] Hall DG, editor. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. 2. Wiley-VCH; Weinheim: 2011. [Google Scholar]
- 2.[a] Baker SJ, Tomsho JW, Benkovic SJ. Chem Soc Rev. 2011;40:4279–4285. doi: 10.1039/c0cs00131g. [DOI] [PubMed] [Google Scholar]; [b] Smoum R, Rubinstein A, Dembitsky VM, Srebnik M. Chem Rev. 2012;112:4156–4220. doi: 10.1021/cr608202m. [DOI] [PubMed] [Google Scholar]
- 3.Zajdlik A, Wang Z, Hickey JL, Aman A, Schimmer AD, Yudin AK. Angew Chem Int Ed. 2013;52:8411–8415. doi: 10.1002/anie.201302818. [DOI] [PubMed] [Google Scholar]
- 4.[a] Gillis EP, Burke MD. J Am Chem Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]; [b] Lee SJ, Gray KC, Paek JS, Burke MD. J Am Chem Soc. 2008;130:466–468. doi: 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]; [c] Gillis EP, Burke MD. J Am Chem Soc. 2008;130:14084–14085. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]; [d] Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]; [e] Ballmer SG, Gillis EP, Burke MD. Org Synth. 2009;86:344–359. [Google Scholar]; [f] Gillis EP, Burke MD. Aldrichimica Acta. 2009;42:17–27. [PMC free article] [PubMed] [Google Scholar]; [g] Uno BE, Gillis EP, Burke MD. Tetrahedron. 2009;65:3130–3138. [Google Scholar]; [h] Struble JR, Lee SJ, Burke MD. Tetrahedron. 2010;66:4710–4718. [Google Scholar]; [i] Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314–2317. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]; [j] Lee SJ, Anderson TM, Burke MD. Angew Chem Int Ed. 2010;49:8860–8863. doi: 10.1002/anie.201004911. [DOI] [PMC free article] [PubMed] [Google Scholar]; [k] Woerly EM, Cherney AH, Davis EK, Burke MD. J Am Chem Soc. 2010;132:6941–6943. doi: 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]; [l] Fujii S, Chang SY, Burke MD. Angew Chem Int Ed. 2011;50:7862–7864. doi: 10.1002/anie.201102688. [DOI] [PMC free article] [PubMed] [Google Scholar]; [m] Li J, Burke MD. J Am Chem Soc. 2011;133:13774–13777. doi: 10.1021/ja205912y. [DOI] [PMC free article] [PubMed] [Google Scholar]; [n] Woerly EM, Struble JR, Palyam N, O’Hara SP, Burke MD. Tetrahedron. 2011;67:4333–4343. doi: 10.1016/j.tet.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; [o] Dick GR, Woerly EM, Burke MD. Angew Chem Int Ed. 2012;51:2667–2672. doi: 10.1002/anie.201108608. [DOI] [PMC free article] [PubMed] [Google Scholar]; [p] Woerly EM, Miller JE, Burke MD. Tetrahedron. 2013;69:7732–7740. doi: 10.1016/j.tet.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; [q] Woerly EM, Roy J, Burke MD. Nat Chem. 2014;6:484–491. doi: 10.1038/nchem.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.[a] He Z, Yudin AK. J Am Chem Soc. 2011;133:13770–13773. doi: 10.1021/ja205910d. [DOI] [PubMed] [Google Scholar]; [b] He Z, Zajdlik A, St Denis JD, Assem N, Yudin AK. J Am Chem Soc. 2012;134:9926–9929. doi: 10.1021/ja304173d. [DOI] [PubMed] [Google Scholar]; [c] He Z, Trinchera P, Adachi S, StDenis JD, Yudin AK. Angew Chem Int Ed. 2012;51:11092–11096. doi: 10.1002/anie.201206501. [DOI] [PubMed] [Google Scholar]
- 6.[a] Erlanson DA, McDowell RS, O’Brien T. J Med Chem. 2004;47:3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]; [b] Bamborough P, Brown MJ, Christopher JA, Chung CW, Mellor GW. J Med Chem. 2011;54:5131–5143. doi: 10.1021/jm200349b. [DOI] [PubMed] [Google Scholar]; [c] Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. J Am Chem Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.[a] Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]; [b] Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 8.Murai N, Yonaga M, Tanaka K. Org Lett. 2012;14:1278–1281. doi: 10.1021/ol300149b. [DOI] [PubMed] [Google Scholar]
- 9.Singh RP, Matteson DS. J Org Chem. 2000;65:6650–6653. doi: 10.1021/jo005522b. [DOI] [PubMed] [Google Scholar]
- 10.Stewart AO, Brooks DW. J Org Chem. 1992;57:5020–5023. [Google Scholar]
- 11.Dey S, Garner P. J Org Chem. 2000;65:7697–7699. doi: 10.1021/jo000983i. [DOI] [PubMed] [Google Scholar]
- 12.Molander GA, Ham J. Org Lett. 2006;8:2031–2034. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.[a] Molander GA, Hiebel MA. Org Lett. 2010;12:4876–4879. doi: 10.1021/ol102039c. [DOI] [PMC free article] [PubMed] [Google Scholar]; [b] Molander GA, Shin I. Org Lett. 2012;14:4458–4461. doi: 10.1021/ol301955s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lal B, Pramanik BN, Manhas MS, Bose AK. Tetrahedron Lett. 1977;18:1977–1980. [Google Scholar]
- 15.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Lajkiewicz NJ, Cognetta AB, III, Niphakis MJ, Cravatt BF, Porco JA. J Am Chem Soc. 2014;136:2659–2664. doi: 10.1021/ja412431g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.List A, Zeiler E, Gallastegui N, Rusch M, Hedberg C, Sieber SA, Groll M. Angew Chem Int Ed. 2014;53:571–574. doi: 10.1002/anie.201308567. [DOI] [PubMed] [Google Scholar]
- 19.[a] Zuhl AM, Mohr JT, Bachovchin DA, Niessen S, Hsu K-L, Berlin JM, Dochnahl M, López–Alberca MP, Fu GC, Cravatt BF. J Am Chem Soc. 2012;134:5068–5071. doi: 10.1021/ja300799t. [DOI] [PMC free article] [PubMed] [Google Scholar]; [b] Leung D, Hardouin C, Boger DL, Cravatt BF. Nat Biotech. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 20.We have previously suggested that this band corresponds to either ACOT1, ACOT2 or both (see ref 17), by correlating gel-based to MS-based ABPP profiling along with the predicted molecular weight and gel migration profile of these enzymes. However, the high sequence identity between these two distinct enzymes has hampered a definitive assignment.
- 21.Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK. Prog Lipid Res. 2010;49:366–377. doi: 10.1016/j.plipres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 22.[a] Adibekian A, Martin BR, Wang C, Hsu KL, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Nat Chem Biol. 2011;7:469–478. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]; [b] Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schürer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Proc Natl Acad Sci USA. 2011;108:6811– 6816. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Patricelli MP, Cravatt BF. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.