


Abstract
Sequence-specific nucleases such as TALEN and the CRISPR/Cas9 system have so far been used to disrupt, correct or insert transgenes at precise locations in mammalian genomes. We demonstrate efficient ‘knock-in’ targeted replacement of multi-kilobase genes in human induced pluripotent stem cells (iPSC). Using a model system replacing endogenous human genes with their mouse counterpart, we performed a comprehensive study of targeting vector design parameters for homologous recombination. A 2.7 kilobase (kb) homozygous gene replacement was achieved in up to 11% of iPSC without selection. The optimal homology arm length was around 2 kb, with homology length being especially critical on the arm not adjacent to the cut site. Homologous sequence inside the cut sites was detrimental to targeting efficiency, consistent with a synthesis-dependent strand annealing (SDSA) mechanism. Using two nuclease sites, we observed a high degree of gene excisions and inversions, which sometimes occurred more frequently than indel mutations. While homozygous deletions of 86 kb were achieved with up to 8% frequency, deletion frequencies were not solely a function of nuclease activity and deletion size. Our results analyzing the optimal parameters for targeting vector design will inform future gene targeting efforts involving multi-kilobase gene segments, particularly in human iPSC.
INTRODUCTION
Targeted gene replacements have been essential tools for exploring gene function; however, the low frequency of homologous recombination (10−3–10−7) limits this technique even when using antibiotic selection markers and homology arms of up to 14 kb (1–5). This low efficiency meant screening hundreds (or sometimes thousands) of clones to obtain a successfully targeted event. Since only one allele could reasonably be targeted, ‘knock-in’ animals had to be intercrossed to obtain a homozygous model system, which took extra time, and such intercrosses are not possible for human cells. Generating biallelic gene targeting events in human tumor cell lines was cumbersome and typically necessitated two different antibiotic selection markers or sequential rounds of gene targeting and selection (3,6–10). Custom-engineered nuclease systems, such as zinc finger nucleases (ZFN) (3,5,9,11–14), transcription activator-like effector nucleases (TALEN) (15–18) or CRISPR/Cas9 nucleases (3,11,19–20), create double-stranded DNA (dsDNA) breaks at specific sequences and allow efficient genome modification, even in cell types or species previously resistant to gene editing.
The CRISPR/Cas9 nuclease system has been favored due to its easy construction and multiplexability. The Streptococcus pyogenes Cas9 nuclease targets a 20 bp sequence specified by a single guide RNA (sgRNA) that binds to the nuclease. When the Cas9–sgRNA complex binds to a complementary 20 bp sequence of target genomic DNA in position next to a 3 bp protospacer adjacent motif (PAM)—NGG for S. pyogenes Cas9—it generates a blunt-ended dsDNA break three base pairs upstream of the PAM (19–21). Multiple sgRNAs, each complementary to a different sequence, can be co-introduced with the Cas9 nuclease to simultaneously cleave multiple genomic loci (13,22–24).
Once a dsDNA break is generated at a specific target site, gene repair using the non-homologous end joining (NHEJ) pathway (1,25–26) can mutate and disrupt genes (2,4,8,19,27). Two dsDNA breaks can also be used to excise the intervening portion of the genome (2,4,9,12,28–32) or generate inversions (9,12,33–34), duplications (33) or translocations (16,18,35–36).
Alternatively, the dsDNA break can be repaired by homologous recombination (HR). Creating a dsDNA break increases the HR frequency over 1000-fold (2,4,19–20,37–39). Homologous recombination with an ssODN donor (6–7,15,17,19–20) or plasmid targeting vector (9,12,15,19,22) can introduce specific small changes at the cut site. Entire transgenes can be inserted into a single cut site using flanking homology arms (1,3,5,16,18) although the efficiency of gene insertion declines with larger insertions (3,8–10,19–20). For transgene insertions into a single cut site, introducing a dsDNA break reduces the necessary homology arm length to ∼0.2–0.8 kb (3,5,11,13–14,19–20). However, the dsDNA break must occur near the mutation point for optimal targeting efficiency—within ∼40 bp for ssODN donors (15,17,22) and ∼200 bp for plasmid donors in mammalian cells (1,3,11).
For generating larger multi-kilobase targeted gene replacements, conventional gene targeting methods (without nucleases) involve HR crossovers between a set of flanking homology arms. We wanted to test whether dsDNA breaks needed to be generated near both ends of the gene replacement for optimal targeting efficiency. Whereas transgene insertion vectors simply contain homology arms flanking the single cut site, the use of potential multiple cut sites introduces further design possibilities for the targeting vector. Earlier attempts comparing the use of single versus multiple nuclease sites for gene targeting could only measure a limited set of parameters (8,19,21). Other attempts at targeted gene replacement have used microhomology-mediated end joining (MMEJ) between the short single-stranded overhangs resulting from ZFN cleavage (2,4,13,23–24,29–32). To examine the extent and frequency of multi-kilobase nuclease-mediated targeted gene replacements, we developed a new model system of replacing human genes with their mouse counterpart. We focused our studies on human iPSC, as this cell type is especially relevant for future tissue engineering approaches, whereas immortalized tumor cell lines have abnormally high HR rates while primary somatic cells have lower HR rates compared to embryonic stem (ES) cells (9,12,25–26,33–34).
Here, we report that homozygous multi-kilobase gene replacements can be generated in human iPSC at efficiencies high enough to not require selection and identify the critical parameters for targeting vector design. With two nuclease sites, we observed a high rate of excision and inversion mutations, such that the highest rate of homozygous gene replacement was achieved with a single nuclease site. In addition, we examine the relationship between deletion frequency and size for heterozygous and homozygous gene deletions and potential off-target mutation rates. As these studies were done in iPSC, they are especially relevant for constructing gene replacements for future tissue engineering or animal models.
MATERIALS AND METHODS
Single guide RNA target sequences
A computational algorithm was used to identify sgRNA sequences at various positions around the human THY1 (CD90) and (CD147) genes based on their uniqueness in the human genome (2,4,27,33). Sequences are listed in Supplementary Table S1. Potential off-target sites in Supplementary Figure S7 were identified using the CasOT searching tool (9,12,16,18,28,35–36). Off-target sites were ranked first by the number of mismatches in the 12 bp ‘seed’ sequence, then by the total number of mismatches (adding 1 if there is an alternate NAG PAM and subtracting 1 if there is a mismatch in the base pair at the 5′ end), and finally by mismatches farthest from the PAM. The Open Chromatin Code for each region was taken from the DNaseI / FAIRE / ChIP Synthesis track from the ENCODE project for the H1 human ES cell line. (16,18,40). The H7 and H9 human ES cell lines and three other human iPSC lines in the ENCODE database showed a similar chromatin pattern.
Cas9, sgRNA and TALEN plasmid construction
The U6 promoter and sgRNA backbone sequence were synthesized as described (19,20) (IDT) and cloned using isothermal assembly into a minimal plasmid backbone containing the Ampicillin resistance gene and pBR322 ori PCR amplified from pUC19 using primers 5′ CTTTCTTGTACAAAGTTGGCATTATTAGACGTCAGGTGGCACTTTTC 3′ and 5′ CCTTTAAAGCCTGCTTTTTTGTACA GTTTGCGTATTGGGCGCTCTTC 3′. Various sgRNA sequences were cloned into this vector using isothermal assembly. The overlapping segments for isothermal assembly are underlined. The first bp of each sgRNA was made a G to enable expression from the U6 promoter. All primers were from IDT; all PCR reactions were done with the KAPA HiFi HotStart PCR kit. Plasmids were maintained in either TOP10 or Stbl3 bacteria (Invitrogen).
A human codon-optimized Cas9 nuclease gene (19,20) was PCR amplified using primers 5′ GCCACCATGGACAAGAAGTACTCC 3′ and 5′ TCACACCTTCCTCTTCTTCTTGGG 3′ and cloned using isothermal assembly between an EF1α promoter and a bGH polyadenylation sequence on a pCDNA3 plasmid backbone. The overlapping segments for isothermal assembly are underlined. The EF1α promoter was PCR amplified from pEGIP (Addgene #26777) using primers 5′ CCGAAAAGTGCCACCTGACGTCGACGGA TGAAA GGAGTGGGAATTGGC 3′ and 5′ GGAGTACTTCTTG TCCATGGTGGC GGCC AACTAGCCAGCTTGGGTCTCCC 3′. The bGH polyadenylation sequence was PCR amplified from pST1374 (Addgene #13426) (22,41) using primers 5′ GCTGACCCCAAGAAGAAGAGGAAGGTGTGA CATCACCATTGAGTTTAAACCCGC 3′ and 5′ CCAAGCTCTAGCTAGAGGTCGACGGTAT C GAGCCCCAGCTGGTTC 3′. The plasmid backbone was PCR amplified from pCDNA3 (Invitrogen) using primers 5′ ATACCGTCGACCTCTAGCTAG 3′ and 5′ TCCGTCGACGTCAGGTGG 3′.
TALE pairs (16.5mer) targeting the human Thy1 gene were assembled using Iterative Capped Assembly (1,42) and are further described in the Supplementary Methods.
Targeting plasmid construction
All targeting plasmids were cloned using PCR and isothermal assembly and verified by Sanger sequencing. All homology arm sequences were amplified from PGP1 genomic DNA. Details, including primer sequences, are further described in the Supplementary Methods.
Human iPSC culture
Verified human iPSC from Personal Genome Project donors PGP1 and PGP4 (8,19,43–44) were obtained through Coriell. Genome sequences for PGP1 and PGP4 can be obtained from the Personal Genomes Project website at my.personalgenomes.org. Cell lines were maintained on Matrigel-coated plates (BD) and grown in mTesr1 (Stem Cell Technologies) according to manufacturer's instructions. The ROCK inhibitor Y-27632 (10 μM, Millipore) was added to the culture before, during, and after passaging with Accutase (Millipore).
Pluripotency of iPSC cultures was verified by flow cytometry staining using anti-human Tra-1/60 PE (TRA-1–60, BD), anti-human Oct3/4 eFluor 660 (EM92, eBioscience) and anti-human Nanog Alexa 488 (N31–355, BD) with the BD Cytofix and Phosflow Perm/Wash buffers. Human g-band karyotype analysis was done by Cell Line Genetics. Teratoma assays were performed by the Induced Pluripotent Stem Cell Core at the Joslin Diabetes Center.
Viable cell counts (Figure 2B) were measured with the Muse Count & Viability Assay kit (EMD Millipore).
Figure 2.
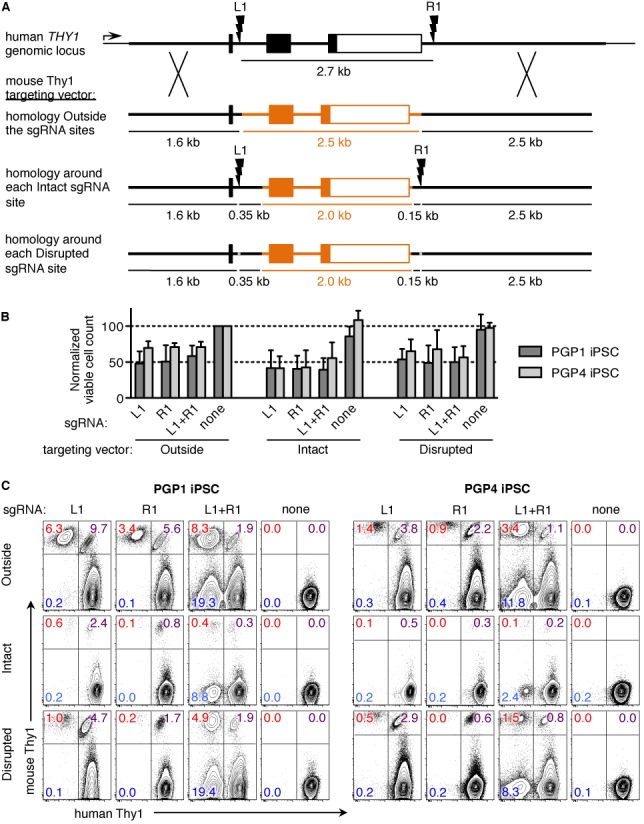
Targeted gene replacement with homology on either site of each cut site. (A) The mThy1 targeting vector from Figure 1 (outside) was modified such that the human Thy1 homology arms extend inside the L1 and R1 sgRNA sites. While mouse Thy1 exons 2 and 3 (orange) are completely retained in this targeting vector, 350 bp of mouse Thy1 intron 1 and 150 bp of mouse Thy1 sequence after the polyadenylation site was replaced with the corresponding human sequence. The resulting targeting vector contains intact L1 and R1 sgRNA sites (Intact). Next, a single base pair was deleted from each sgRNA site in the targeting vector to develop a alternate version with similar homology arms but disrupted sgRNA sites (Disrupted). (B and C) PGP1 or PGP4 iPSC were nucleofected with one of the mouse Thy1 targeting vectors (outside, Intact, or disrupted) along with plasmids encoding the Cas9 nuclease, and L1, R1, both, or no sgRNAs. (B) Two days post-nucleofection, the viable cells in each condition were counted. Viable cell counts were normalized to that of the Outside mouse Thy1 targeting vector with no sgRNA (100). Error bars show mean ± S.E.M. of at least three independent experiments. (C) Five days post-nucleofection, cells were analyzed by flow cytometry. The percentage of cells that have gained expression of mouse Thy1 and / or lost expression of human Thy1 are indicated. Results are representative of three (PGP1) and two (PGP4) independent experiments.
Human iPSC transfection
All plasmids were purified using the Qiagen Endo-free Plasmid Maxiprep kit. Plasmids were nucleofected into iPSC cells using the Lonza 4D-Nucleofector X unit (Buffer P3, Program CB-150) according to manufacturer's instructions. For each 20 μl nucleofection reaction, 0.2–0.5 × 106 iPSC were transfected with up to 4 μg of plasmid DNA. Post-nucleofection, iPSC were plated onto 24- and 96-well Matrigel-coated plates containing mTesr1 media plus 10 μM Y-27632.
For CRISPR-based nucleofections with a targeting vector (Figures 1 and 2, and Supplementary Figures S2–S7), 2 μg of targeting vector plasmid, 0.5 μg of Cas9 plasmid, and 1.5 μg of total sgRNA plasmid were used. When two sgRNAs were used, 0.75 μg of each plasmid was combined. When no sgRNAs were used, 1.5 μg of pUC19 was used instead.
Figure 1.
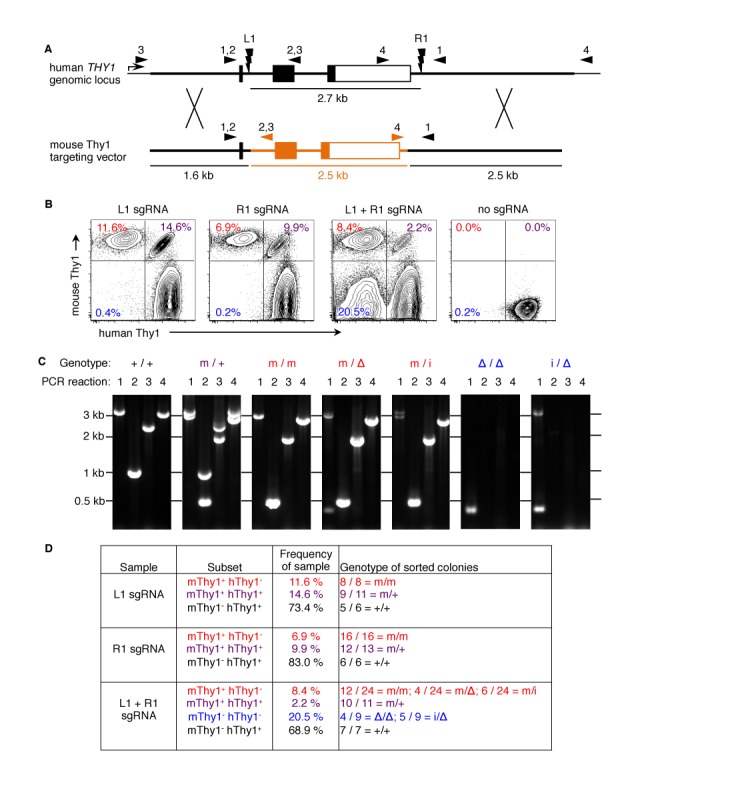
Homozygous targeted gene replacement using one or two CRISPR sgRNAs. (A) Two Crispr sgRNAs target hThy1 within intron 1 (L1) or after the polyadenylation sites (R1). The mThy1 targeting vector plasmid contains mThy1 exons 2 and 3 (orange), flanked by hThy1 homology arms outside the sgRNA sites—coding exon 1 (which encodes the signal peptide) is retained but the sgRNA sites are disrupted. Small triangles indicate the primer sites for the four genotyping PCR reactions. (B) PGP1 iPSC were nucleofected with plasmids encoding the mThy1 targeting vector, the Cas9 nuclease, and L1, R1, both, or no sgRNAs. Five days later, cells were analyzed by flow cytometry. The percentage of cells that have gained mThy1 expression and/or lost hThy1 expression are indicated. (C) Single iPSC were FACS sorted from each quadrant, cultured in individual wells, and genotyped using the four PCR reactions. Alleles were identified based on the size and Sanger sequencing of the PCR products: native human (+); recombined mouse (m); excised between the two sgRNA sites (Δ); and inverted between the two sgRNA sites (i). Representative gels from +/+ wild type, m/+ heterozygous, m/m homozygous, m/Δ heterozygous, m/i heterozygous, Δ/Δ homozygous, and i/Δ heterozygous colonies are shown. (D) Frequency of genotypes among FACS-sorted iPSC colonies. Results are representative of three independent experiments.
For CRISPR-based nucleofections without a targeting vector (Figure 3 and Supplementary Figures S10 and S11), 2 μg of total plasmid was used: 0.5 μg of Cas9 plasmid with 0.75 μg of each sgRNA plasmid or pUC19.
Figure 3.
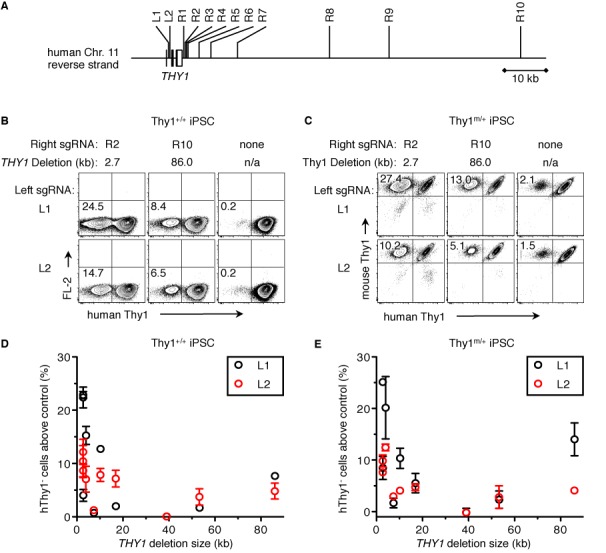
Frequency of CRISPR-generated homozygous and heterozygous deletions. (A) Crispr sgRNAs were generated targeting the human Thy1 gene: two within intron 1 (left: L1 and L2), and 10 at various distances after hThy1 (right: R1 through R10). (B and C) Pairs of one left and one right sgRNA were nucleofected into either (B) PGP1 iPSC or (C) a Thy1m/+ PGP1 iPSC clone. As a negative control, only a left sgRNA was nucleofected (right column). Five days later, cells were analyzed by flow cytometry for either (B) homozygous deletion of both human Thy1 alleles or (C) heterozygous deletion of the remaining human Thy1 allele and retention of the mouse Thy1 allele. The distance between the sgRNA sites (Thy1 Δ) and the frequency of hThy1− cells is indicated. (D and E) The percentage of hThy1− cells from each sgRNA pair minus that from the left sgRNA-only control is plotted against the size of the Thy1 deletion. sgRNA pairs that included L1 or L2 are shown in black or red, respectively. Error bars show mean ± S.E.M. of two independent experiments.
For TALEN-based nucleofections with a targeting vector (Supplementary Figure S5), 2 μg of targeting vector plasmid plus 2 μg total of TALEN plasmid was used. For one dsDNA break using one TALEN pair, 1 μg of each TALEN-expressing heterodimer plasmid was used. For two dsDNA breaks using two TALEN pairs, 0.5 μg of each TALEN-expressing heterodimer plasmid was used.
FACS staining
iPSC were dissociated using TrypLE Express (Invitrogen) and washed in FACS buffer: PBS (Invitrogen) with 0.2% bovine serum albumin (Sigma). Cells were stained in FACS buffer with 10% fetal calf serum for 30 min at 4°C. The following antibodies were purchased from eBioscience: Anti-human Thy1 APC (eBio5E10), anti-mouse Thy1.2 PE (30-H12), anti-human CD147 APC (8D12), anti-mouse CD147 PE (RL73), isotype control mouse IgG1 κ APC (P3.6.2.8.1), isotype control mouse IgG2b PE (eBMG2b). Cells were washed twice in FACS buffer, and then resuspended in FACS buffer with the viability dye SYTOX Blue (Invitrogen). Samples were collected on a BD LSR Fortessa flow cytometer with a High Throughput Sampler (HTS) and analyzed using FlowJo software (Tree Star).
Single-cell iPSC FACS sorting
For FACS sorting, iPSC were sorted one cell per well into 96-well plates containing feeder cells. 96-well flat-bottom tissue culture plates were coated with gelatin (Millipore) and cultured with irradiated CF-1 mouse embryonic fibroblasts (106 MEFs per plate; Global Stem) the night before. Before sorting, media in the plates was changed to hES cell maintenance media (29–32,45) with 100 ng/ml bFGF (Millipore), SMC4 inhibitors (33–34,46) (BioVision) and 5 μg/ml fibronectin (Sigma).
For at least 2 h, before FACS sorting, iPSC were pre-treated with mTesr1 containing SMC4 inhibitors. Cells were dissociated with Accutase, and stained as described above. iPSC were sorted using a BD FACS Aria into the MEF-coated 96-well plates. Established iPSC colonies were then mechanically passaged onto new MEF-coated wells.
PCR genotyping
Genomic DNA from sorted iPSC clones was purified from the 96-well plates (35–36,47). Each Thy1 edited clone was genotyped using four sets of PCR primers (Figure 1) using the KAPA HiFi HotStart polymerase and run on a 0.8% agarose gel. Reaction 1: 5′ AGGGACTTAGATGACTGCCATAGCC 3′ and 5′ ATGTTGGCAGTAAGCATGTTGTTCC 3′. Wild type Thy1 (+) or inverted (i) allele: 3129 bp; targeted mouse Thy1 (m) allele: 2904 bp; excised (Δ) allele: 387 bp. Reaction 2: 5′ AGGGACTTAGATGACTGCCATAGCC 3′, 5′ CTCACCTCTGAGCACTGTGACGTTC 3′, and 5′ ACTGAAGTTCTGGGTCCCAACAATG 3′. Wild type (+) allele: 993 bp; targeted mouse (m) allele: 490 bp; excised (Δ) or inverted (i) allele: no PCR product. Reaction 3: 5′ ATGAATACAGACTGCACCTCCCCAG 3′, 5′ CTCACCTCTGAGCACTGTGACGTTC 3′, and 5′ CCATCAATCTACTGAAGTTCTGGGTCCCAACAATG 3′. Wild-type (+) allele: 2393 bp; targeted mouse (m) allele: 1891 bp; excised (Δ) or inverted (i) allele: no PCR product. Reaction 4: 5′ TGAAGTGAAACCCTAAAGGGGGAAG 3′, 5′ AAACCACACACTTCAACCTGGATGG 3′, and 5′ GTTTGGCCCAAGTTTCTAAGGGAGG 3′. Wild-type (+) allele: 3064 bp; targeted mouse (m) allele: 2707; excised (Δ) or inverted (i) allele: no PCR product.
Each Bsg edited clone was genotyped using five sets of PCR primers (Supplementary Figure S4). Reaction 1: 5′ GATCGGGAAGGGATTACCGTTCTTC 3′ and 5′ AAAACATGCCGAGAGGAGTAACAGG 3′. Wild-type Bsg (+) or inverted (i) allele: 10 404 bp; targeted mouse (m) allele: 6452 bp; excised (Δ) allele: 617 bp. Reaction 2: 5′ GATCGGGAAGGGATTACCGTTCTTC 3′, 5′ CAGGTGGCCATTACAGGGATACAG 3′, and 5′ TTGTTCCCAACACATCGGTCAC 3′. Wild-type (+) allele: 1149 bp; targeted mouse (m) allele: 328 bp; excised (Δ) or inverted (i) allele: no PCR product. Reaction 3: 5′ CATTGCGACTCCGAGTTTAACTTCCAAC 3′, 5′ CAGGTGGCCATTACAGGGATACAG 3′, and 5′ TTGTTCCCAACACATCGGTCA 3′. Wild-type Bsg (+) allele: 3248 bp; targeted mouse (m) allele: 2429 bp; excised (Δ) or inverted (i) allele: no PCR product. Reaction 4: 5′ CCTTGCCCTTTGTGGAAAGTCACAG 3′, 5′ TGTCACTACTTGAAACTTCAGGACCCC 3′, and 5′ CAATTTGCCCAGGACAGGAATAGG 3′. Wild-type Bsg (+) allele: 2663 bp; targeted mouse (m) allele: 2412 bp; excised (Δ) or inverted (i) allele: no PCR product. Reaction 5: 5′ CTGTGATGGCAGCTCCTGGAAAAC 3′ and 5′ CGTTCTTGCCTTTGTCATTCTGGTG 3′. Wild-type Bsg (+) or inverted (i) allele: 533 bp; targeted mouse (m) or excised (Δ) allele: no PCR product.
Sequencing
Genomic DNA from sorted individual iPSC clones was PCR amplified and Sanger sequenced using the primers in Thy1 genotyping Reaction 1, which span outside the sgRNA nuclease sites (Supplementary Figure S2). When the PCR genotyping bands were of different lengths (the excision-and-inversion alleles in Supplementary Figure S2C), each PCR band was extracted on an agarose gel (Qiagen) and individually Sanger sequenced. However, when the PCR genotyping bands were the same length (double excisions in Supplementary Figure S2B), the Sanger sequencing chromatogram of the single PCR band showed a uniform set of single peaks before the Crispr cut site, but then changed to a mixed trace series of superimposed double-peaks, reflecting sequence differences in the two alleles. Deconvolution of double peaks in a Sanger sequencing chromatogram is an established method to detect heterozygous indels (37–39,48). The sequence of double-peaks was mapped back to the genome sequence at two places, reflecting the different size indels formed. All of the PCR genotyping bands were Sanger sequenced from both directions, to confirm the indel.
The percent of indel mutations at a given sgRNA site from a population of targeted iPSC was assayed using next-generation sequencing (15,17) (Supplementary Table S1 and Supplementary Figures S10 and S11). Each target site was PCR amplified using primers with the requisite MiSeq adaptor sequences appended to the 5′ end, as listed in Supplementary Table S2. A second round of nested Nextera index primers (Illumina) was used to add barcodes and remaining adaptor sequences. One hundred fifty paired-end Illumina MiSeq reactions were performed by the Molecular Biology Core Facilities at Dana-Farber Cancer Institute and analyzed by the CRISPR Genome Analyzer (15,19,49).
RESULTS
Targeted gene replacements
To study the optimal targeting vector design for large gene replacements, we developed a model system to replace the 2.7 kb human THY1 gene (hThy1) with its mouse homologue (mThy1) in human iPSC derived from Personal Genome Project (PGP) donors (3,5,43). The surface marker and immunoglobulin superfamily member THY1 (CD90) was chosen because it is expressed on the surface of human iPSC, not essential for iPSC survival in vitro, and species-specific staining antibodies are available (3,9–10,50). Human and mouse Thy1 share 70% amino acid homology, but have heterologous genome sequences that do not align with more than 75% identity (Supplementary Figure S1), below levels needed for HR (11,51). Two single guide RNAs (sgRNA) were designed that target human THY1 after coding exon 1 or after the polyadenylation sequence. The mThy1 targeting vector plasmid contained exons 2 and 3 of mouse Thy1 flanked by human THY1 homology arms outside of the cut sites—hThy1 exon 1 is retained but the sgRNA sites are disrupted (Figure 1A and Supplementary Figure S1). Unlike human Thy1, mouse Thy1 is not expressed in mouse ES cells. Coding exon 1 encodes the beginning of the signal peptide, which is proteolytically cleaved off during protein export to the cell surface (3,5,11,13–14,52).
When the mThy1 targeting vector was transfected along with plasmids expressing the Cas9 nuclease and both sgRNAs, 8.4% of iPSC became mThy1+ hThy1− (Figure 1B). Subsequent cloning by single-cell FACS sorting and PCR genotyping revealed that mThy1+ hThy1− iPSC were a mixture of homozygous targeted replacement (m/m), replacement of one human allele with excision of the other (m/Δ), and replacement with inversion (m/i) (Figure 1C and D). In addition, 2.2% of cells were mThy1+ hThy1+ double positive (m/+; heterozygous targeted replacement) (Figure 1B). While most of the sorted iPSC genotypes could be fully confirmed by PCR and sequencing, a few colonies showed aberrant or absent PCR products in some reactions (Figure 1D). These rare instances may encompass: partial targeted gene replacements where HR has occurred at one homology arm, but not the other; incorporation of plasmid backbone sequences; or segmental duplications.
Finally, 20.5% of cells were mThy1− hThy1− double negative: PCR and Sanger sequencing of single cell FACS-sorted clones revealed a mixture of homozygous excision (Δ/Δ) and heterozygous inversion and excision (i/Δ) between the sgRNA sites (Figure 1). We did not observe any segmental duplications resulting from two dsDNA breaks (33). While a few indels and inserted bases were observed at the excision and inversion sites, the largest indel was only 15 bp, and most alleles were re-joined exactly between the cut sites (Supplementary Figure S2). Previous reports generating two dsDNA breaks with ZFN or TALEN observed indels in most excision and inversion alleles, up to 200 bp (15,17,30,33,35). In contrast to the short 5′ DNA overhangs produced by ZFN and TALEN, CRISPR nucleases produce blunt-end dsDNA breaks (3,11,20), which may contribute to the increased fidelity of re-joining, which was also observed for other CRISPR-mediated excisions from 19 bp to over 1 Mb (13,19,22–24,34).
Surprisingly, when only a single sgRNA was used, mThy1 homozygous replacement occurred in up to 11.6% of cells (mThy1+ hThy1−; m/m) and heterozygous replacement occurred in up to 14.6% of cells (mThy1+ hThy1+; m/+). Very few mThy1− hThy1− double negative cells and no excised hThy1 alleles (Δ) were observed with a single sgRNA (Figure 1). These efficiencies were achieved without selection and with overall transfection efficiencies around 70%. Human iPSC clones expanded from individual sorted cells after successful gene targeting retained expression of pluripotency markers, normal karyotypes and the ability to form all three germ layers in teratoma assays (Supplementary Figure S3 and data not shown).
A similar pattern of results occurred when replacing the 9.8 kb human basigin (BSG, CD147) gene with 5.8 kb of its mouse homologue (Supplementary Figure S4). Like THY1, BSG is an immunoglobulin superfamily marker expressed on the surface of human iPSC, not essential for iPSC survival in vitro, and staining antibodies are available. In addition, the human THY1 gene could be replaced with a 1.9 kb fluorescent mCherry transgenic reporter in either a forward or reverse orientation—a single dsDNA break made using either CRISPR/Cas9 or TALEN was also sufficient for targeted gene replacement (Supplementary Figure S5).
Conventional gene targeting vectors are typically transfected as linearized plasmids (2,25–26). With ZFN, circular plasmid targeting constructs produced higher rates of HR-mediated gene insertion than linearized plasmids, although linear constructs were more effective at MMEJ-mediated gene insertion (13,23). A linearized mThy1 targeting vector produced far less gene targeting compared to the circular plasmid (Supplementary Figure S6), which may be due to the reduced nucleofection efficiency of linearized plasmids (Amaxa Technote) or increased DNA degradation (23).
Homology arm length and position with respect to nuclease site
For conventional HR-mediated gene targeting, targeting frequency increased with homology arm length up to ∼14 kb (4), although additional homology arm length (up to ∼70 kb) using bacterial artificial chromosomes could improve weak or non-isogenic targeting vectors (53,54). However, a dsDNA break reduces the necessary homology arm length to ∼0.2–0.8 kb for transgene insertions into a single cut site (3,5,11,13–14). To examine the effect of homology length on targeted gene replacements in human iPSC, versions of the mThy1 targeting vector were constructed with various length homology arms (Table 1 and Supplementary Figure S7A). In addition to the ∼2 kb homology arms from the original targeting vector (long, L), shorter lengths of ∼800 bp (medium, M) or ∼100 bp (short, S) were chosen, as these lengths are often used for HR-mediated gene insertion (3,5) or ssODN correction (15,17). Longer homology arms of ∼5 kb (extra-long, X) were also tested. With two dsDNA breaks, targeting frequencies were highest with ∼2 kb homology arms (LL and ML), and generally declined with less homology, although frequencies of >1% were achieved down to ∼1.5 kb total homology (MM, LS and SL). Extra-long homology arms did not improve gene targeting efficiency (XX versus LL).
Table 1. Effect of homology arm length on recombination efficiency.
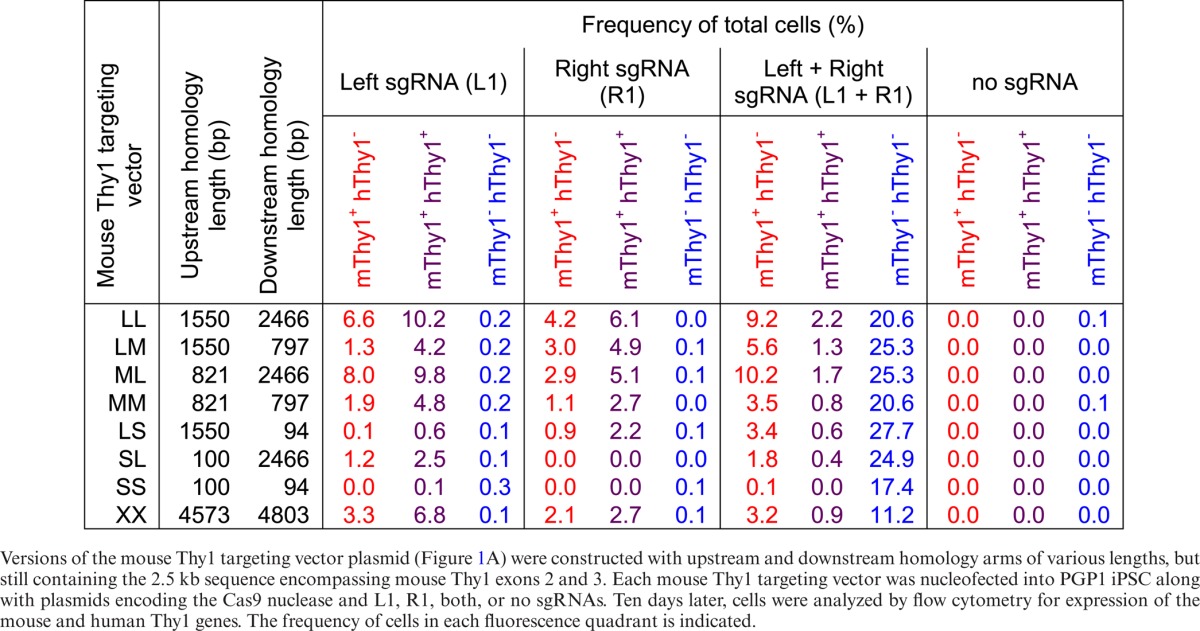
When only one dsDNA break was used, homology length was most important on the arm opposite the cut site. The LM and LS vectors showed higher gene targeting with the right sgRNA than the left; the ML and SL vectors showed higher gene targeting with the left sgRNA than the right (Table 1). Such asymmetric homology requirements may allow gene targeting vectors to be designed with a shorter homology arm, for easier PCR-based genotyping using primers spanning the shorter homology arm. The percentage of GC content throughout the human THY1 locus generally ranged between 25–75% for a 100 bp window size (Supplementary Figure S8). While some repetitive elements were present in the homology arm sequences, particularly in the extra-long (XX) vector, it is not clear whether this had an effect on targeting efficiency beyond increased homology arm length (Table 1 and Supplementary Figure S8).
In these mThy1 targeting vectors, the homology arm sequences only extend outside of the dsDNA break sites, with the foreign replacement sequence present on the inside. Previous reports determined that the dsDNA break must be generated within ∼200 bp for efficient gene correction (3). Extending the homology arms to span either side of each cut site (intact vector) sharply reduced targeting efficiency (Figure 2). The Intact targeting vector retains the sgRNA cut sites, so the homology arms could simply be cleaved off the targeting vector (Supplementary Figure S9), and targeted replacements could potentially be re-cut. These increased dsDNA breaks did not result in greater iPSC death (Figure 2B). Previous reports have shown that cleaving the targeting vector in addition to the genomic DNA can increase targeting efficiency (23); however, these reports used ZFN, which leave single-stranded DNA overhangs at the dsDNA breaks that can drive MMEJ. When the sgRNA sites in the targeting vector were disrupted by a single bp deletion (disrupted vector), targeting efficiency was still reduced compared to the original mThy1 targeting vector (Figure 2 and Supplementary Figure S9). The single bp deletions in the disrupted targeting vector completely ablated Cas9 nuclease activity at the mutated L1 sgRNA site, although some residual Cas9 nuclease activity remained at the mutated R1 sgRNA site, as measured by in vitro Cas9 nuclease assays (Supplementary Figure S9). Nevertheless, our results show that extending the homology arms in the targeting vector beyond the dsDNA break sites reduced targeting efficiency, even though these vectors possessed greater homology overall.
Similar results were observed in iPSC derived from a different donor (PGP4). Although PGP4 iPSC showed lower overall gene targeting efficiencies than PGP1 iPSC, the same pattern of homologous recombination occurred with respect to homology arm length and the position of nuclease cut sites (Supplementary Figure S7b and Figure 2). This difference was not due to mismatches in the homology arm sequences—analysis of the sequenced PGP genomes revealed that PGP1 and PGP4 share the same two heterozygous SNPs in the upstream Thy1 homology arm and have no identified SNPs in the downstream Thy1 homology arm. Instead, the differences in HR activity may be due to genetic and epigenetic heterogeneity among these reprogrammed iPSC lines (55).
Frequency of multi-kilobase gene deletions
Our results using two sgRNAs showed a high frequency of alleles that were deleted between the two cut sites (Figure 1). Multi-kilobase deletions in at least one allele have been achieved using nucleases in tumor cell lines, although the deletion frequency generally declines with larger deletion sizes (17,30,34). To delineate the relationship between the frequency and size of CRISPR-mediated deletions in human iPSC, we designed 2 left and 10 right sgRNAs that target the human Thy1 gene at various distances apart (2.7–86 kb, Figure 3A). Since no targeting vector was used, cells that have both hThy1 alleles disrupted become hThy1− (Figure 3B). Cells nucleofected with only the Left sgRNA were used to determine the background level of hThy1− cells (Figure 3B, right column). While the frequency of homozygous deletion above the background level tended to be higher for shorter distances—up to 24% for a 2.7 kb deletion and 8% for a 86 kb deletion—other sgRNA sites produced much lower deletion frequencies, which did not always correspond to size (Figure 3D).
The frequency of monoallelic deletions was determined using the same set of sgRNAs on a mThy1+ hThy1+ clonal line of iPSC generated as described in Figure 1. Since the mThy1 allele does not contain the two Left sgRNA sites, only the single remaining hThy1 allele was subject to deletion. While the frequency of heterozygous deletions was occasionally higher than that for homozygous deletions with the same sgRNA pair, they were usually within a few percentage points, producing a similar pattern for each sgRNA pair (Figure 3C and E).
Other reports have described nuclease activity to vary among sgRNA sites, even though they match the genome sequence exactly. These variations may be due to differences in the melting temperature of the sgRNA sequence, or the gene expression and chromatin environment at the target site (15,56). Our individual sgRNAs varied in activity (indel mutation rates from 1.7–27%) and this activity did not necessarily correlate with open chromatin regions listed in the ENCODE database for H1 human ES cells (Supplementary Table S1). All sgRNA sequences had between 40–70% GC content and did not fall within the high (>80%) or low (<20%) GC content range shown to be less effective (56). While the rate of Thy1 deletion was a function of the individual sgRNA activities, these differences do not completely explain our observed variations in gene deletion frequency, as neither the L1 nor L2 sgRNA consistently produced more deletions when paired with the same right sgRNA. Pair-specific variables, such as microhomologies between the two dsDNA cut sites, may also influence the deletion frequency.
DISCUSSION
Our results here examine various designs for HR-mediated targeted replacement of multi-kilobase gene segments. Our recommendations for optimal gene targeting are summarized in Table 2. These replacements were efficient enough for homozygous targeting at frequencies around 10−2, which can then be screened without a selection marker. Targeted gene replacements using a single cut site may be a preferable design as this reduces the diversity of genotypes formed (e.g. just m alleles instead of m, Δ and i alleles). Current techniques for HR-mediated insertions require that the dsDNA break must be made close to the mutation or insertion site (optimally within 100 bp), which limits the potentially available sgRNA sites (11,15). Now that large gene replacements can be made using a wider range of nuclease sites, more unique sgRNA sites may be available at one end, thus avoiding conserved coding sequences within a gene family. Flanking homology arms of up to 2 kb showed the optimal gene targeting efficiency (Table 1), although the increased length may complicate PCR-based genotyping approaches. Additional homology of up to 5 kb reduced gene targeting efficiency, possibly because the extra homology was outweighed by the extra size of the plasmid. This optimal homology arm length may reflect the length of gene conversion tracts in human iPSC.
Table 2. Recommendations for targeted gene replacement.
| • Flanking homology arms of around 2 kb |
| • Homology arms extending outside of the dsDNA break sites |
| • No regions of extensive DNA homology within the gene replacement area |
| • A single dsDNA break positioned at one side of the gene replacement helps avoid excised / inverted alleles |
| • If possible, test multiple sgRNAs for activity and specificity |
| • Circular plasmids (versus linear) for transient transfection |
| • Do not cleave plasmid targeting vector |
| • If using two dsDNA breaks, check genotypes for excisions and inversions |
The most effective targeting vectors had homology arms extending only outside the nuclease sites, even though vectors with homology arms extending within the nuclease sites had greater total homology (Figure 2). These results are consistent with a model of Synthesis-Dependent Strand Annealing (SDSA) (3,9,57): the resected chromosome outside the dsDNA break anneals to the corresponding sequence in the targeting vector plasmid. The foreign sequence in the targeting vector is extended, forming a D-loop, until human sequence on the opposite homology arm is reached. Sufficient length of this homology arm is crucial for its subsequent homology search and re-annealing to the corresponding part of the chromosome for resolution of the D-loop. According to this model, the homology arm sequences should only extend outside of the dsDNA break sites, with the non-homologous sequence present throughout the replacement.
This agrees with previous literature that SDSA is the predominant HR mechanism for dsDNA break repair in somatic cells, including mouse ES cells (58,59). Our results argue against a HR mechanism of double Holliday junctions, where both ends around each dsDNA break would anneal to homologous sequences on both sides of each the sgRNA site in the targeting vector, forming separate cross-overs (57). This contradicts a recent report that double Holliday junctions are instead the predominant mechanism for nuclease-mediated gene replacement in human tumor cell lines (60). One explanation may be that the nuclease sites in that study were >250 bp inside the homology arms, and any SDSA events that repaired the dsDNA break but did not incorporate the antibiotic selection marker may have been missed. If the desired gene replacement contains sequences with significant matching homology to the genome, HR may instead resolve at these areas and produce partial targeted replacements.
Mutations at off-target sgRNA sites by the Cas9 nuclease have been reported to occur with high efficiency in tumor cell lines (61–64), although not in stem cells (65). We detected minimal off-target mutations at genomic sequences closest to the sgRNA sites (Supplementary Figure S10). Off-target nuclease activity is known to be a dose-dependent function of the on-target activity (61)—the lower on-target Cas9 nuclease activity in iPSC may thus explain the low frequency of off-target mutations. Using a mutated Cas9 to generate two single-stranded DNA nicks to form a dsDNA break improved specificity (66), although this reduced Thy1 gene targeting rates (data not shown).
The precise rejoining of the excision and inversion junctions from blunt-ended Cas9-generated dsDNA breaks (Supplementary Figure S2) is consistent with previous reports (19,22,34) and supports the notion that NHEJ may not be inherently error-prone, as commonly thought (67). Notably, when Cas9/sgRNA and TALEN pairs were designed to cleave at the same sites within human Thy1 exon 2 or after the polyadenylation sequence, the highest rates of gene disruption were achieved using two (blunt-ended) cut sites for the Cas9 nuclease, but a single (5′ overhang) cut site for TALEN (Supplementary Figure S5). A perfectly rejoined sgRNA site may be continually re-cleaved until the target site is destroyed by either an indel mutation or an excision / inversion. The rate of homozygous gene deletions (hThy1− iPSC) sometimes surpassed the rate of indels among the remaining non-deleted alleles (Supplementary Figure S11). This occurred with smaller gene deletions (2.7 kb) and more active sgRNA sites, although not always. While we observed an inverse relationship between deletion size and frequency in human iPSC (Figure 3), consistent with earlier reports in tumor cell lines (17,30,34), deletion frequency could not be fully predicted by deletion size and individual sgRNA activity.
Our results examining the optimal design for gene replacement vectors complements current efforts in improving the activity, accuracy, toxicity, and delivery of custom-engineered nuclease systems, as it is not particular to which nuclease system generates the dsDNA break. We have achieved efficient multi-kilobase gene replacements from a single dsDNA break using ZFN, TALEN or Cas9 nucleases (Supplementary Figure S5 and data not shown). Considering the wide variability in activity among CRISPR sgRNAs (Supplementary Table S1), multiple sgRNAs may need to be tested empirically for a particular region. We also observed consistent differences of a few-fold in gene targeting rates between two human iPSC lines (derived in the same manner from different donors) (Figure 2). This underscores the influence of cell type in genome editing studies.
Making efficient targeted gene replacements, not just gene disruptions or insertions, will expand the use of ZFN, TALEN and CRISPR/Cas nucleases for genome editing. These homozygous multi-kilobase gene replacements were achieved using standard plasmid engineering and nucleofection methods, similar to those used for conventional gene targeting, but will be enhanced by future gene delivery technologies. The high targeting efficiencies allow successfully targeted clones to be isolated by screening alone—without selection. However, genes could also be replaced with a fluorescent protein marker so successfully targeted stem cells could be selected and cloned by FACS. Gene replacements with heterologous sequences will be particularly beneficial for generating ‘knock-in’ animals or human cell lines for general science or disease models. Particular applications include: placing reporter constructs under endogenous promoters, replacing an endogenous human gene with a recoded transgene, comparative genomics across different species, or generating humanized animal models.
Supplementary Material
Footnotes
Present address: Prashant Mali, Bioengineering Department, University of California San Diego, La Jolla, CA 92093, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ellison Medical Foundation [AG-SS-2084–80]; National Institutes of Health (National Human Genome Research Institute) [1P50 HG005550]; National Science Foundation Synthetic Biology Engineering Research Center (SynBERC) [SA5283-11210]. Funding for open access charge: National Institutes of Health [P50 HG005550].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kass E.M., Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett. 2010;584:3703–3708. doi: 10.1016/j.febslet.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bollag R.J., Waldman A.S., Liskay R.M. Homologous recombination in mammalian cells. Annu. Rev. Genet. 1989;23:199–225. doi: 10.1146/annurev.ge.23.120189.001215. [DOI] [PubMed] [Google Scholar]
- 3.Moehle E.A., Rock J.M., Lee Y.-L., Jouvenot Y., DeKelver R.C., Gregory P.D., Urnov F.D., Holmes M.C. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng C., Capecchi M.R. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol. Cell. Biol. 1992;12:3365–3371. doi: 10.1128/mcb.12.8.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B., et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J.P., Sedivy J.M., Kinzler K.W., Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 7.Di Nicolantonio F., Arena S., Gallicchio M., Zecchin D., Martini M., Flonta S.E., Stella G.M., Lamba S., Cancelliere C., Russo M., et al. Replacement of normal with mutant alleles in the genome of normal human cells unveils mutation-specific drug responses. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20864–20869. doi: 10.1073/pnas.0808757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urnov F.D., Rebar E.J., Holmes M.C., Zhang H.S., Gregory P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 10.Guye P., Li Y., Wroblewska L., Duportet X., Weiss R. Rapid, modular and reliable construction of complex mammalian gene circuits. Nucleic Acids Res. 2013;41:e156. doi: 10.1093/nar/gkt605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott B., Richardson C., Winderbaum J., Nickoloff J.A., Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell. Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bibikova M., Carroll D., Segal D.J., Trautman J.K., Smith J., Kim Y.G., Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol. Cell. Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orlando S.J., Santiago Y., DeKelver R.C., Freyvert Y., Boydston E.A., Moehle E.A., Choi V.M., Gopalan S.M., Lou J.F., Li J., et al. Zinc-finger nuclease-driven targeted integration into mammalian genomes using donors with limited chromosomal homology. Nucleic Acids Res. 2010;38:e152. doi: 10.1093/nar/gkq512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beumer K.J., Trautman J.K., Mukherjee K., Carroll D. Donor DNA utilization during gene targeting with zinc-finger nucleases. G3 Genes|Genomes|Genetics. 2013;3:657–664. doi: 10.1534/g3.112.005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L., Guell M., Byrne S., Yang J.L., De Los Angeles A., Mali P., Aach J., Kim-Kiselak C., Briggs A.W., Rios X., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J., et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2010;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 17.Chen F., Pruett-Miller S.M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T.N., Frodin M., Davis G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joung J.K., Sander J.D. TALENs: a widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2012;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donoho G., Jasin M., Berg P. Analysis of gene targeting and intrachromosomal homologous recombination stimulated by genomic double-strand breaks in mouse embryonic stem cells. Mol. Cell. Biol. 1998;18:4070–4078. doi: 10.1128/mcb.18.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cristea S., Freyvert Y., Santiago Y., Holmes M.C., Urnov F.D., Gregory P.D., Cost G.J. In vivo cleavage of transgene donors promotes nuclease-mediated targeted integration. Biotechnol. Bioeng. 2013;110:871–880. doi: 10.1002/bit.24733. [DOI] [PubMed] [Google Scholar]
- 24.Maresca M., Lin V.G., Guo N., Yang Y. Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 2013;23:539–546. doi: 10.1101/gr.145441.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xia S.J., Shammas M.A., Shmookler Reis R.J. Elevated recombination in immortal human cells is mediated by HsRAD51 recombinase. Mol. Cell. Biol. 1997;17:7151–7158. doi: 10.1128/mcb.17.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kass E.M., Helgadottir H.R., Chen C.-C., Barbera M., Wang R., Westermark U.K., Ludwig T., Moynahan M.E., Jasin M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5564–5569. doi: 10.1073/pnas.1216824110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aach J., Mali P., Church G.M. CasFinder: flexible algorithm for identifying specific Cas9 targets in genomes. bioRxiv. 2014 doi:10.1101/005074. [Google Scholar]
- 28.Xiao A., Cheng Z., Kong L., Zhu Z., Lin S., Gao G., Zhang B. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics. 2014;30:1180–1182. doi: 10.1093/bioinformatics/btt764. [DOI] [PubMed] [Google Scholar]
- 29.Sollu C., Pars K., Cornu T.I., Thibodeau-Beganny S., Maeder M.L., Joung J.K., Heilbronn R., Cathomen T. Autonomous zinc-finger nuclease pairs for targeted chromosomal deletion. Nucleic Acids Res. 2010;38:8269–8276. doi: 10.1093/nar/gkq720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H.J., Kim E., Kim J.-S. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L., et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran F.A., Hsu P.D., Lin C.-Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee H.J., Kweon J., Kim E., Kim S., Kim J.S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res. 2012;22:539–548. doi: 10.1101/gr.129635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canver M.C., Bauer D.E., Dass A., Yien Y.Y., Chung J., Masuda T., Maeda T., Paw B.H., Orkin S.H. Characterization of genomic deletion efficiency mediated by CRISPR/Cas9 in mammalian cells. J. Biol. Chem. 2014 doi: 10.1074/jbc.M114.564625. doi:10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piganeau M., Ghezraoui H., De Cian A., Guittat L., Tomishima M., Perrouault L., Rene O., Katibah G.E., Zhang L., Holmes M.C., et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013;23:1182–1193. doi: 10.1101/gr.147314.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres R., Martin M.C., Garcia A., Cigudosa J.C., Ramirez J.C., Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- 37.Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- 38.Porteus M.H., Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 39.Bibikova M., Beumer K., Trautman J.K., Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- 40.Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.-K., Sheffield N.C., Gräf S., Huss M., Keefe D., et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21:1757–1767. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maeder M.L., Thibodeau-Beganny S., Sander J.D., Voytas D.F., Joung J.K. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat. Protoc. 2009;4:1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briggs A.W., Rios X., Chari R., Yang L., Zhang F., Mali P., Church G.M. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012;40:e117. doi: 10.1093/nar/gks624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ball M.P., Thakuria J.V., Zaranek A.W., Clegg T., Rosenbaum A.M., Wu X., Angrist M., Bhak J., Bobe J., Callow M.J., et al. A public resource facilitating clinical use of genomes. Proc. Natl. Acad. Sci. U.S.A. 2012;109:11920–11927. doi: 10.1073/pnas.1201904109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee J.-H., Park I.-H., Gao Y., Li J.B., Li Z., Daley G.Q., Zhang K., Church G.M. A robust approach to identifying tissue-specific gene expression regulatory variants using personalized human induced pluripotent stem cells. PLoS Genet. 2009;5:e1000718. doi: 10.1371/journal.pgen.1000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lerou P.H., Yabuuchi A., Huo H., Miller J.D., Boyer L.F., Schlaeger T.M., Daley G.Q. Derivation and maintenance of human embryonic stem cells from poor-quality in vitro fertilization embryos. Nat. Protoc. 2008;3:923–933. doi: 10.1038/nprot.2008.60. [DOI] [PubMed] [Google Scholar]
- 46.Valamehr B., Abujarour R., Robinson M., Le T., Robbins D., Shoemaker D., Flynn P. A novel platform to enable the high-throughput derivation and characterization of feeder-free human iPSCs. Sci. Rep. 2012;2:213–213. doi: 10.1038/srep00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramírez-Solis R., Rivera-Pérez J., Wallace J.D., Wims M., Zheng H., Bradley A. Genomic DNA microextraction: a method to screen numerous samples. Anal. Biochem. 1992;201:331–335. doi: 10.1016/0003-2697(92)90347-a. [DOI] [PubMed] [Google Scholar]
- 48.Dmitriev D.A., Rakitov R.A. Decoding of superimposed traces produced by direct sequencing of heterozygous indels. PLoS Comput. Biol. 2008;4:e1000113. doi: 10.1371/journal.pcbi.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guell M., Yang L., Church G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA) Bioinformatics. 30:2968–2970. doi: 10.1093/bioinformatics/btu427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Draper J.S., Pigott C., Thomson J.A., Andrews P.W. Surface antigens of human embryonic stem cells: changes upon differentiation in culture. J. Anat. 2002;200:249–258. doi: 10.1046/j.1469-7580.2002.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waldman A.S., Liskay R.M. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol. Cell. Biol. 1988;8:5350–5357. doi: 10.1128/mcb.8.12.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seki T., Spurr N., Obata F., Goyert S., Goodfellow P., Silver J. The human Thy-1 gene: structure and chromosomal location. Proc. Natl. Acad. Sci. U.S.A. 1985;82:6657–6661. doi: 10.1073/pnas.82.19.6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y., Seed B. Site-specific gene targeting in mouse embryonic stem cells with intact bacterial artificial chromosomes. Nat. Biotechnol. 2003;21:447–451. doi: 10.1038/nbt803. [DOI] [PubMed] [Google Scholar]
- 54.Valenzuela D.M., Murphy A.J., Frendewey D., Gale N.W., Economides A.N., Auerbach W., Poueymirou W.T., Adams N.C., Rojas J., Yasenchak J., et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 55.Cahan P., Daley G.Q. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat. Rev. Mol. Cell Biol. 2013;14:357–368. doi: 10.1038/nrm3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chapman J.R., Taylor M.R.G., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 58.Heyer W.-D., Ehmsen K.T., Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LaRocque J.R., Jasin M. Mechanisms of recombination between diverged sequences in wild-type and BLM-deficient mouse and human cells. Mol. Cell. Biol. 2010;30:1887–1897. doi: 10.1128/MCB.01553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kan Y., Ruis B., Lin S., Hendrickson E.A. The mechanism of gene targeting in human somatic cells. PLoS Genet. 2014;10:e1004251. doi: 10.1371/journal.pgen.1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 65.Wu X., Scott D.A., Kriz A.J., Chiu A.C., Hsu P.D., Dadon D.B., Cheng A.W., Trevino A.E., Konermann S., Chen S., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bétermier M., Bertrand P., Lopez B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014;10:e1004086. doi: 10.1371/journal.pgen.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.