


Abstract
In various organisms, an efficient RNAi response can be triggered by feeding cells with bacteria producing double-stranded RNA (dsRNA) against an endogenous gene. However, the detailed mechanisms and natural functions of this pathway are not well understood in most cases. Here, we studied siRNA biogenesis from exogenous RNA and its genetic overlap with endogenous RNAi in the ciliate Paramecium tetraurelia by high-throughput sequencing. Using wild-type and mutant strains deficient for dsRNA feeding we found that high levels of primary siRNAs of both strands are processed from the ingested dsRNA trigger by the Dicer Dcr1, the RNA-dependent RNA polymerases Rdr1 and Rdr2 and other factors. We further show that this induces the synthesis of secondary siRNAs spreading along the entire endogenous mRNA, demonstrating the occurrence of both 3′-to-5′ and 5′-to-3′ transitivity for the first time in the SAR clade of eukaryotes (Stramenopiles, Alveolates, Rhizaria). Secondary siRNAs depend on Rdr2 and show a strong antisense bias; they are produced at much lower levels than primary siRNAs and hardly contribute to RNAi efficiency. We further provide evidence that the Paramecium RNAi machinery also processes single-stranded RNAs from its bacterial food, broadening the possible natural functions of exogenously induced RNAi in this organism.
INTRODUCTION
Small RNAs (sRNAs) are important regulators of transcript processing in most eukaryotes. They are produced from single-stranded (ss) or double-stranded (ds) precursors homologous to their target transcripts. If long dsRNA is the initial trigger, it is processed into 21–28 nt short interfering RNAs (siRNAs) by Dicer-mediated endonucleolytic cleavage (1). Loaded onto the Argonaute subunit of effector complexes, siRNAs can target a variety of complementary RNAs, such as messenger RNAs, non-coding or nascent transcripts (2,3). RNA interference (RNAi)-related mechanisms have diversified into complex networks of interconnected pathways. They control gene expression either post-transcriptionally, through mRNA cleavage or translational inhibition, or transcriptionally, through the deposition of epigenetic modifications on chromatin (1,4).
In addition to their functions in the regulation of cellular genes, RNAi mechanisms can provide efficient control of parasitic sequence elements, such as transposons and viruses. Environmental RNAi, though relying on different mechanisms, is widespread; organisms known to respond to exogenous dsRNA include plants (5,6), various groups of metazoans (7), fungi (8) and some ciliates (9–11). In nematodes, RNAi can be induced by feeding the worms with dsRNA-producing bacteria (12,13), which appears to rely on an antiviral defence pathway (14,15). This shares components with endogenous RNAi pathways (16), is negatively regulated by the endogenous Eri pathway (17) and competes with endogenous RNAi targets (18).
Dicer-mediated cleavage of dsRNA generates ‘primary’ siRNAs that can target complementary transcripts. In plants, nematodes and fungi, the targeting of an mRNA by primary siRNAs induces a second round of siRNA synthesis through the recruitment of RNA-dependent RNA polymerases (RdRPs), leading to amplification of the silencing response (for reviews see 5,6 and 19). In Arabidopsis thaliana, Rdr6 synthesizes long dsRNA molecules from mRNA cleavage products, providing new substrates for the same Dicer (5,6). This leads to 5′-to-3′ and 3′-to-5′ transitivity (20), i.e. production of secondary siRNAs beyond the region of the transcript matching the initial dsRNA trigger. In Caenorhabditis elegans, transitivity is mechanistically different: secondary siRNAs are synthesized in a Dicer-independent manner by an unprimed, discontinuous mode of RdRP activity (Rrf-3; Ego-1) which produces short antisense molecules with 5′-triphosphate ends (21–24). These secondary siRNAs are in excess to primaries and are the major effectors of silencing (22,25,26). As in plants, C. elegans secondary siRNAs are a common feature of endogenously and exogenously induced RNAi. The biosynthesis and function of secondary siRNAs has not been thoroughly investigated in other organisms.
Remarkable complexity of sRNA pathways has been observed in ciliates, unicellular eukaryotes separating their genome into somatic functions (macronucleus (MAC)), responsible for all gene expression, and a germline lineage (micronucleus). Paramecium tetraurelia mounts an RNAi response to exogenous dsRNA produced by food bacteria (9). A forward genetic screen for mutants deficient in dsRNA-induced silencing, yielding a collection of complete and partial loss-of-function alleles (27), showed that this pathway is non-essential and that it involves the non-essential genes RDR1 (RdRP), CID1 (nucleotidyl-transferase) and PDS1 (unknown function), and the essential genes DCR1 (Dicer), RDR2 and CID2 (27–29). In addition, three proteins of the Piwi subclade of Argonaute proteins (PTIWI12, PTIWI15 and PTIWI13) have been implicated in dsRNA-induced RNAi (30). Small-scale sRNA sequencing in conjugating cells suggested that two distinct siRNA species are produced within the dsRNA target region, one accumulating on both strands and likely representing primary siRNAs and another, showing a strong antisense bias, suggestive of secondary siRNAs (28). Intriguingly, two distinct RdRPs, Rdr1 and Rdr2, were implicated in exogenously triggered primary siRNA production (29). However, secondary siRNA synthesis has not yet been confirmed in this or any other ciliate. Silencing by dsRNA in P. tetraurelia seems to operate at the post-transcriptional level, resulting in mRNA cleavage within the dsRNA target region (31). However, the natural function of this pathway is unknown; apart from antiviral defence (no Paramecium viruses are known so far), it may target other non-self RNAs (32) to which bacterial feeders are typically exposed.
Another constitutively expressed RNAi pathway can be experimentally induced by high-copy, non-translatable transgenes (33,34). Transgene-induced silencing requires some of the genes involved in dsRNA-induced RNAi, such as RDR2, CID2, DCR1 and PTIWI13 (27,28,30), but also additional genes, such as RDR3 and PTIWI14. In addition, RDR2 and RDR3 are required for the production of endogenous siRNAs from an intergenic locus of unknown function (27,29).
In this study, we have deep-sequenced the sRNAs produced in wild-type (WT) and mutant Paramecium strains subjected to dsRNA feeding with the aims (i) to test the previous hypothesis that a transitivity mechanism produces secondary siRNAs, (ii) to shed light on the mechanisms of primary and secondary siRNA synthesis by dissecting their genetic requirements and (iii) to provide new insight into the natural functions of these pathways. Using dsRNA triggers that contain either an intron or a substitution to distinguish it from the endogenous target mRNA, we show that although the vast majority of siRNAs are primary siRNAs processed from the dsRNA, a minor fraction of secondary siRNAs are indeed produced from the entire mRNA, clearly demonstrating the existence of transitivity for the first time in any ciliate. Paramecium tetraurelia secondary siRNAs do not seem to serve as amplificators of the silencing response and may have a different function. Further, we show that some of the factors implicated in exogenously triggered are involved in endogenous RNAi and provide evidence that siRNAs are also produced from single-stranded bacterial RNAs, suggesting possible natural functions for RNAi processing of food-derived RNAs in P. tetraurelia.
MATERIALS AND METHODS
Paramecium strains, cultivation, induction of RNAi and phenotypic analyses
Experiments were carried out with wild type (WT) strain 51 of P. tetraurelia and strain 51-derived RNAi-deficient mutants (27), backcrossed at least once. For standard cultures, cells were grown at 27°C in wheat grass powder (Pines International Co., Lawrence, KS, USA) infusion medium bacterized with Klebsiella pneumoniae the day before use and supplemented with 0.8 μg/ml β-sitosterol. ND169, a non-essential, single-copy gene required for the discharge of secretory granules (trichocysts) was used as RNAi reporter gene. Silencing of ND169 results in a quantitative phenotype that allows distinguishing full and partial RNAi deficiencies. Production of dsRNA in Escherichia coli strain HT115 DE3, feeding to Paramecium cells and monitoring of trichocyst discharge phenotypes were carried out as described previously (27,29,35). To reveal partial loss-of-function phenotypes of previously uncharacterized mutant alleles, ND169 dsRNA producing E. coli were typically mixed in 1:5 ratio with ICL7a dsRNA producing bacteria, as described (27). For dsRNA feeding experiments aiming at sequencing sRNA pools, the respective food bacteria were used in an undiluted form, unless otherwise stated. In order to reduce the amount of RNAi-unrelated sRNA fragments possibly present within dsRNA-producing E. coli food bacteria or Paramecium food vacuoles at the time of sampling, cells were first allowed to eat the majority of food bacteria. Then, prior to harvest for RNA preparation, cells were washed for 30 min in Klebsiella-conditioned, exhausted medium (free of bacteria).
Plasmid constructs
To induce silencing by dsRNA feeding, fragments of the coding region had been cloned into the plasmid L4440. Plasmid constructs of the genes ND169, ICL7a, RDR3, PTIWI08 and PTIWI14 were previously described (29,30). DsRNA was synthesized in E. coli HT115 DE3 from T7 promotors as described (29,35).
sRNA analysis by northern blot and high-throughput sequencing
Total RNA extraction and sRNA northern blots were carried out as described (29). To detect ND169 siRNAs two adjacent 50 nt sense oligonucleotide probes matching to the dsRNA region were used, as most of the siRNAs produced from this region are antisense to the target transcript.
For sRNA library construction, the ∼19–28 nt fraction was purified from 50 μg of total RNA by polyacrylamide gel electrophoresis (15%, 19:1 acrylamide:bisacrylamide) and gel-eluted with 0.3 M sodium chloride, followed by ethanol precipitation. The eluate was used for library construction using standard Illumina protocols relying on 5′-adaptor ligation to sRNAs carrying a 5′ monophosphate, with the minimum recommended number of 11 polymerase chain reaction cycles.
sRNA mapping and data analyses
For the analysis of dsRNA-feeding associated siRNAs trimmed sequence data sets were mapped with the Burrows–Wheeler Alignment tool (36) using default settings. Reads were first aligned to a set of bacterial sequences, as well as to mitochondrial and ribosomal DNA sequences, tRNA and snoRNA, according to the recent P. tetraurelia genome annotation (ParameciumDB, http://paramecium.cgm.cnrs-gif.fr/), allowing one mismatch per read. Then, cleared data sets were aligned to the reference genome of strain 51 (37,38), without mismatch. Read counts of target siRNAs were normalized to the total number of sequence reads obtained per sample, expressed as reads per million (rpm). For determination of siRNA levels mapping within the dsRNA trigger region, the average of 22 and 24 nt normalized sRNA counts was defined as a threshold level to be passed for unambiguous siRNA accumulation. siRNA levels represented in Figure 3D and E are corrected by this method. Outside the dsRNA region, 22, 23 and 24 nt size fractions were considered as siRNAs, as they were detectably dependent on Rdr2. SiRNAs mapping to the L4440 vector sequences (polylinker), to the ICL7a mismatch, to introns and to exon-exon junctions were defined as those overlapping the respective sequence/nucleotide with a minimum of 3 nt. SiRNAs carrying 3′-untemplated nucleotides were identified in sequential rounds of removing the last 3′ nucleotide of each non-mapped sRNA, and re-aligning of these clipped reads to contaminants, and then to the MAC genome, as described above. Non-mapping reads of an alignment round were processed until eight 3′ nucleotides were clipped.
Figure 3.
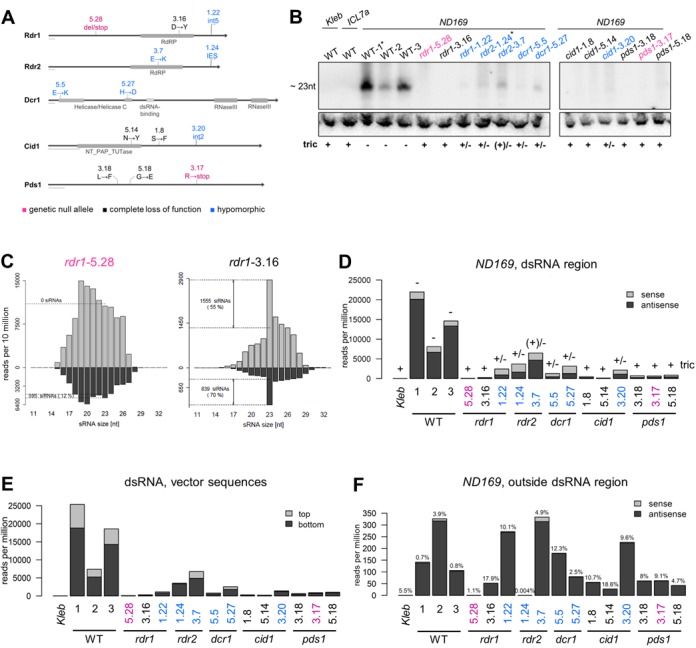
siRNA accumulation is strongly affected in RNAi mutants. (A) Mutant alleles of the genes RDR1, RDR2, DCR1, CID1 and PDS1 used in this study. Non-ambiguous null alleles are represented in purple, other phenotypic complete loss-of-function alleles in black and hypomorphic alleles in blue. The alleles rdr-1.22 and cid1-3.20 retain an intron due to mutations in an intron boundary. The allele rdr2-1.24 retains a micronucleus-specific non-coding fragment (internal eliminated sequence) in the MAC-version of RDR2, leading to a frameshift and premature termination. Each scale bar represents 50 aa. (B) Northern blot analysis of siRNAs produced in vegetative WT cells and RNAi-deficient mutants subjected to ND169 dsRNA feeding. A probe corresponding to a 100-bp fragment of the dsRNA and oriented sense to the mRNA was used to reveal ND169 siRNAs. In the case of WT-1 and rdr2-1.24, the ND169 food bacteria were slightly diluted with ICL7a medium (2.5:1 ND169:ICL7a) (labeled with an asterisk). However, this did not alter the penetrance of the phenotype compared to non-diluted medium. Despite the moderate dilution, ∼2.5-fold more siRNAs mapped to the dsRNA in WT-1 compared to WT-2. Of note, the strongly hypomorphic allele rdr2-3.7 produced nearly WT levels of siRNAs. RNAi phenotypes of the cultures are given below (RNAi-positive: no tric discharge (tric -); RNAi-deficient: complete discharge (tric +)). (C) Normalized size distribution of sRNAs mapping the ND169 dsRNA region in WT (WT-1) and rdr1-5.28. Note that the absolute number of 23 nt sRNAs is higher in the rdr1 null mutant, but no unambiguous siRNAs accumulating above the background threshold level were found. (D) Normalized counts of sense (gray) and antisense (black) 23 nt siRNAs mapping the dsRNA region in WT and mutant samples (in rpm). Counts were corrected by the threshold definition for unambiguous siRNAs described in the text. Corrected siRNA numbers and their percentage relative to the total number of 23 nt sRNA reads are indicated. RNAi phenotypes are given on top of the bars (see A). (E) Normalized counts of 23 nt siRNAs mapping the 5′ and 3′ vector part of the dsRNA sequence. The same correction as described for (D) was used. (F) Normalized counts of 23 nt siRNAs mapping outside the ND169 dsRNA region in WT and mutant samples (in rpm). Read counts were not corrected, as no background level due to experimentally induced dsRNA degradation was expected. The percentage of 23 nt secondary siRNAs (i.e. mapping outside the dsRNA region) relative to the corrected total number of 23 nt siRNAs is indicated on top of the bars. Note that the proportion of secondary siRNAs varied 4-fold between WT samples (0.4% in WT-1 and WT-3; 1.6% in WT-2).
For identification of MAC regions producing endogenous siRNAs, reads were aligned as described above (allowing no mismatch), then a sliding window of 50 bp was applied to identify siRNAs clusters. Among those regions producing predominantly 23 nt siRNAs, only clusters were considered that showed a minimum siRNA coverage of 60× and a minimum size of 250 bp. For identification of siRNAs produced from bacterial sequences, trimmed reads were matched without mismatch to E.coli strain W3110 (AP009048.1), first on its 16S (ECK3750:JWR0082:b3756) and 23S (ECK2587:JWR0052:b2589) rRNA, then on the 4227 protein coding genes (http://bacteria.ensembl.org/escherichia_coli_str_k_12_substr_w3110/Info/Index). The LacZ gene was removed from the sequences prior to mapping. Representations of siRNA distributions were made with R scripts, ggbio and R samtools packages (39,40). Raw sequencing data for ND169- and ICL7a-dsRNA feeding to wild type and mutants and aligned reads of RDR3- and PTIWI08/14-dsRNA feeding to wild type have been submitted to the NCBI Sequence Read Archive (SRA). All data sets are available under the project accession number SRP051377.
RESULTS
A large fraction of sRNAs maps to genes targeted by dsRNA feeding
A previous small-scale analysis of siRNAs associated with dsRNA-induced silencing of the ND7 gene in conjugating cells suggested the existence of two distinct classes: one that appeared to be cleaved from both strands of the dsRNA trigger and another one, exclusively antisense to the mRNA and carrying untemplated 3′ polyA tails, which may represent secondary siRNAs (28).
To characterize dsRNA-induced siRNAs, we first carried out high-throughput sequencing of small (∼15–30 nt) RNAs from 3 cultures of the wild type (WT) strain 51 in which one or two non-essential genes were silenced. The single-copy gene ND169, involved in trichocyst discharge, was targeted either alone (WT-2 or WT-3 samples) or together with ICL7a (WT-1 sample), by feeding cells with a mix of the bacterial strains producing each dsRNA (Figure 1A and Supplementary Figure S1). ICL7a is a centrin gene which has a 93%-identical ohnolog from WGD1, the most recent whole-genome duplication. sRNA libraries were constructed to specifically capture 5′-monophosphorylated molecules, since previous northern blot analyses indicated that detectable siRNAs from the dsRNA sequence are monophosphorylated (29). We obtained between 10 and 26 million reads for each library. After removal of reads derived from bacterial, mitochondrial and structural RNAs, such as rRNA and tRNA (together representing between 54% and 88% of the total), the remaining ones were mapped to the MAC genome of strain 51, allowing no mismatch. This yielded numerous sRNAs matching the genes targeted by dsRNA in the relevant cultures (Supplementary Table S1), but not in a control culture fed with the standard Klebsiella pneumoniae. The numbers of target-gene sRNAs varied between 15 000 and 94 000 rpm, which represents a large fraction of MAC-mapping sRNAs in each case: 36% for ND169 and 20% for ICL7a in sample WT-1 (Supplementary Table S1). To broaden the analysis, we used sRNA sequencing data sets from other dsRNA-feeding experiments: the single silencing of RDR3 or PTIWI14, both specifically involved in transgene- but not in dsRNA-induced RNAi (29,30), PTIWI08 (a WGD1 paralog of PTWI14), or ICL7a (for gene details see Supplementary Figure S1). Similarly, large numbers of sRNAs specifically mapped to the cognate target gene in each case (Supplementary Table S1).
Figure 1.
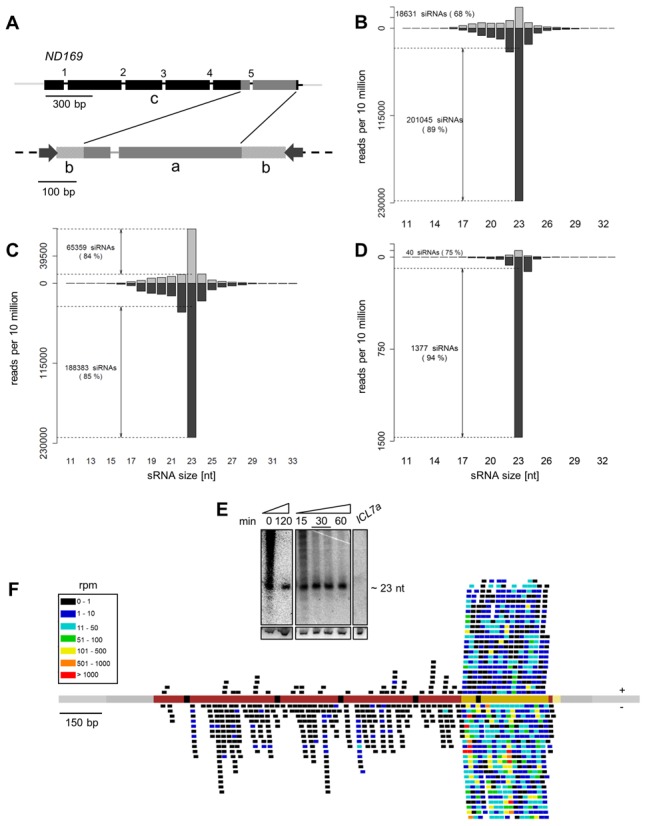
Twenty-three nucleotide siRNAs are produced from both strands of the dsRNA region and spread along the target mRNA. (A) The ND169 gene (top) was silenced using a 411-bp dsRNA fragment, located in the 3′ part of the coding sequence (c). Introns are numbered 1–5. The dsRNA fragment was cloned into the polylinker part of the L4440 vector (b), flanked by convergent T7 promotors (black arrows). The intron 5 sequence was present in the dsRNA (a). (B) Normalized size distribution of sense (gray) and antisense (black) sRNAs mapping to the ND169 dsRNA sequence (region a in subfigure (A) in reads per 10 million. Dashed lines represent threshold levels for accumulation of unambiguous 23 nt siRNAs above the background (see text). Note that 23 nt siRNAs represent 54% of ND169 sRNAs on average. (C) Normalized size distribution of sRNAs mapping to the top (gray) and bottom strand (black) of the L4440 5′ and 3′ vector sequence (b in subfigure A). Note that 57% of vector siRNAs are in the 23-nt peak. (D) Normalized size distribution of sense (gray) and antisense (black) sRNAs mapping outside the ND169 dsRNA sequence (c in subfigure A). No threshold level for unambiguous siRNAs was defined (see text). (E) Total RNA from vegetative WT cells subjected to ND169 dsRNA feeding was hybridized with a sense probe corresponding to the dsRNA region. Smear is detectable when cells were not allowed to complete digestion of dsRNA-containing food bacteria prior to RNA isolation. Samples of cultures washed and incubated for 0–120 min in Klebsiella standard food are shown. ICL7a dsRNA feeding was used as negative control. Hybridization to Gln tRNA served as loading control. (F) Distribution of 23 nt siRNAs on the ND169 genomic sequence (red line; black: introns; light gray: flanking intergenic regions; dark gray: 3′ UTR; yellow: dsRNA target region) (in rpm). Individual siRNAs are represented as trait, the color indicating its abundance. siRNAs represented above and below the line are sense and antisense to the ND169 mRNA, respectively. All subfigures show sample WT-1 (size distributions of other samples see Supplementary Figure S5). 3′U-siRNAs are not included in the figure.
The vast majority of siRNAs map to the dsRNA trigger region of target genes
The 411-bp segment of the ND169 gene used for production of the dsRNA trigger is located near the 3′-end of the coding sequence (Figure 1A). In addition to this segment, the dsRNA trigger contains short sequences derived from the plasmid vector, between the convergent T7 promoters and flanking the restriction sites used for cloning (71 and 114 bp at the left and right ends, respectively). By definition, siRNAs matching these vector sequences, which are not present in the P. tetraurelia genome, can only be primary siRNAs, i.e. siRNAs cleaved by Dicer from the dsRNA trigger.
The mapping of sRNAs perfectly matching ND169 revealed that 99% of them are located in the dsRNA trigger region (details see Supplementary Table S2A), and their size distribution shows a strong peak at 23 nt (Figure 1B). In addition, a large number of sRNAs was found to map to the vector parts of the dsRNA (54% of dsRNA-mapping reads in WT-1) (Figure 1C). sRNAs of other sizes between 16 and 29 nt, as well as a fraction of 23-nt molecules, may not be siRNAs but likely represent random degradation products from the dsRNA being digested in food vacuoles. They were readily detected on northern blots when cells were not allowed to complete digestion of ingested bacteria before RNA extraction (Figure 1E). To count the 23-nt siRNAs in a stringent manner, the number of RNAi-unrelated 23-nt RNAs was estimated to be the mean of the numbers of 22- and 24-nt molecules, and only those in excess of this value were counted as unambiguous siRNAs.
Both sense and antisense siRNAs were found in the ND169 dsRNA region, though an antisense bias was noted (88% of 23-nt siRNAs on average). Strikingly, some antisense siRNAs were highly overrepresented, with more than 560 rpm, whereas most others were represented by less than 30 rpm (Figure 1F). These copy-number variations are highly reproducible for a given sequence since the same ND169 siRNAs were overrepresented in the three WT samples (Supplementary Figure S2), and they fully account for the global antisense bias in the dsRNA region (Figure 1F). Among other genes tested, dramatic overrepresentation of some siRNAs was also found to result in a global strand bias in the PTIWI08 and PTIWI14 dsRNA regions, whereas overrepresented siRNAs occur equally on both strands for RDR3 and ICL7a (Supplementary Figure S3 and Table S2A). Similarly, siRNAs mapped in equivalent numbers to both strands of the right vector part of the ND169 dsRNA trigger, whereas the left vector part showed an excess of reads on the bottom strand (85%) (Supplementary Figure S4). Thus, read-count heterogeneity appears to be a local, sequence-dependent effect that does not necessarily result in any strand bias. No particular feature (nucleotide position biases, GC or purine content) was found to be associated with high-copy reads, and it remains unclear whether the observed heterogeneity is of biological or, as described (41), of technical origin, or both. We conclude that siRNAs accumulate at high levels on both strands of the entire dsRNA.
Secondary siRNAs are produced from the entire target transcript
About ∼1% of ND169 sRNAs mapped outside the dsRNA region, covering the entire mRNA including the 33-nt 3′ UTR (Figure 1F), but none was found in the 500-bp immediately upstream and downstream of the ND169 transcript, even in the genes located there. Thus, dsRNA feeding induces the production of secondary siRNAs that spread beyond the trigger region but seem to be produced exclusively from the target transcript. Their lower abundance and spreading in both directions was also observed for the other target genes, in which the dsRNA trigger regions were located at different positions relative to the coding sequence (Supplementary Figure S3 and Table S2A). Note that 86% of ND169 sRNAs outside the dsRNA region are 23 nt in length (Figure 1D), indicating that they represent bona fide siRNAs, and the 22- and 24-nt molecules (12%) are also likely to be secondary siRNAs since they depend on Rdr2 (see below; Supplementary Figure S5B). Very few molecules were found at other sizes between 16 and 29 nt, confirming that the high background observed in the trigger region is due to dsRNA degradation products.
ND169 secondary siRNAs exhibit a strong antisense bias (97% on average in WT samples) (Figure 1D and Supplementary Table S2A). Unlike the weaker biases seen in the dsRNA region, which can favor either strand, the same strong antisense bias was observed for all genes tested outside the dsRNA region (Supplementary Table S2A). In all cases, some secondary siRNAs were highly overrepresented, as seen in the dsRNA regions (Supplementary Figure S2). The antisense bias was observed both 5′ and 3′ of the dsRNA region and appears to be a characteristic feature of secondary siRNAs, which is reminiscent of the situation in C. elegans (22,23). Yet the small but significant fraction found on the sense strand, which was not observed when cells were grown on Klebsiella or upon feeding of other dsRNAs, suggests that at least some secondary siRNAs may derive from a dsRNA precursor. Both sense and antisense secondaries show a random frequency of each nucleotide at the 5′ end (not shown).
Outside the dsRNA region, many ND169 siRNAs contained exon-exon junctions (213 rpm on average in WT samples), indicating that they were produced from the spliced mRNA. Secondary siRNAs containing intron sequences were much rarer (7 rpm on average) (Table 1). From these analyses we conclude that dsRNA feeding induces the accumulation of predominantly antisense secondary siRNAs over the entire length of the target transcript, at much lower levels than in the dsRNA trigger region, and mostly produced from the spliced mRNA.
Table 1. Number of ND169 siRNAs (in rpm) overlapping exon-exon junctions and mapping introns in WT samples fed with E. coli (ND169 dsRNA) or Klebsiella (no dsRNA).
| Sample | Outside dsRNA region | dsRNA region | ||||||
|---|---|---|---|---|---|---|---|---|
| Intron 1–4 | Junction 1–4 | Intron 5 | Junction 5 | |||||
| Sense | Antisense | Sense | Antisense | Sense | Antisense | Sense | Antisense | |
| WT-1 | 1 | 5 | 0 | 116 | 936 | 3858 | 1 | 1 |
| WT-2 | 0 | 5 | 1 | 135 | 1087 | 8665 | 1 | 2 |
| WT-3 | 0 | 9 | 2 | 385 | 1910 | 2443 | 2 | 2 |
| WT-Kleb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
siRNAs from the dsRNA region are predominantly primary siRNAs
SiRNAs mapping in the dsRNA region may be either primaries or secondaries. To determine the proportions of each, we took advantage of the fact that the ND169 dsRNA contains one 23-nt intron (intron 5) (Figure 1A). In contrast to introns outside the dsRNA region, intron 5 was highly covered by siRNAs (1311 and 4989 rpm on average for sense and antisense strands, respectively), whereas very few siRNAs contained the exon-exon junction (1.3 and 1.7 rpm on average) (Table 1 and Figure 2A). Intron 5 is very efficiently spliced, as determined by the comparison of RNA-Seq data from WT and nonsense-mediated mRNA decay (NMD) knockdown conditions (Saudemont et al., unpublished data), which would suggest that the vast majority of siRNAs in the dsRNA region are primaries. However, the intron 5 region is poorly covered compared to other sequences of the dsRNA trigger. To confirm this result independently of any possible effect of dsRNA feeding on splicing, we counted siRNAs covering a point substitution present in the ICL7a dsRNA fragment, and those containing the endogenous base. 96.7% (11,445 rpm sense and antisense on average in two replicates) of siRNAs contained the substitution versus 2.9% (164 rpm) that matched the endogenous transcript (Figure 2B and C). Taken together, these results indicate that secondary siRNAs are indeed produced from the dsRNA region of the mRNA, where they accumulate at the same relatively low level as over the rest of the transcript; the vast majority of siRNAs mapping in the dsRNA region are primary products of dsRNA cleavage by Dicer.
Figure 2.
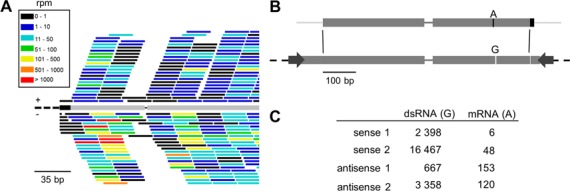
Predominantly primary siRNAs are produced from the dsRNA target region. (A) Very few siRNAs overlap the exon-exon junction 5 (black gap) located in the ND169 dsRNA region (gray bar). The graphic representation is identical to Figure 1F. The black bar represents the 5′ vector. (B) An A to G mismatch is located in the ICL7a dsRNA (lower gray bar; dashed bars correspond to the 5′ and 3′ vector); the dsRNA corresponds to the bulk of the ICL7a cds (upper bar, black parts are not represented in the dsRNA). (C) siRNAs mapping to the mismatch represent primary siRNAs, whereas those overlapping the respective genomic sequence represent secondaires, produced from the endogenous transcript. Numbers in rpm are given for replicate 1 (WT-1) and 2 (ICL7a single dsRNA feeding). Note that the 2.9% of siRNAs mapping to the endogenous nucleotide are 14-fold higher than the average occurrence of sequencing errors (0.2%) at this site. 3′U-siRNAs are not included in the figure.
Primary siRNA accumulation is strongly affected in mutants
We then sequenced sRNA pools in mutants of the dsRNA-induced RNAi pathway fed with ND169 dsRNA (overview of alleles see Figure 3A). Only hypomorphic missense alleles were available for the essential genes DCR1 (dcr1-5.5 and dcr1-5.27) and RDR2 (rdr2-1.24 and rdr2-3.7); these mutations only partially impair silencing efficiency. Of note, the allele rdr2-3.7 produces nearly WT levels of siRNAs and its phenotype can only be revealed clearly by diluting the dsRNA trigger (not applied in this study) (27). For RDR1, we analyzed two strong alleles resulting in complete silencing deficiency (rdr1-5.28, non-sense; rdr1-3.16, catalytic missense) and a hypomorphic allele (rdr1-1.22). We further studied complete-deficiency missense alleles of the nucleotidyl-transferase CID1 and the RNAi factor PDS1 (cid1-1.8, cid1-5.14, pds1-3.18 and pds1-5.18) as well as a non-sense PDS1 allele (pds1-3.17) and a hypomorphic CID1 allele (cid1-3.20) (for the number of raw sequence reads see Supplementary Table S3). Northern blots previously showed that the phenotypic strength of these mutations correlates with the reduction of siRNA levels in the dsRNA region (27), which could be confirmed here (Figure 3B and Supplementary Table S4A).
Accordingly, the 23-nt peak of ND169 siRNAs mapping to the dsRNA region collapsed more or less strongly in the different mutants (Figure 3C and Supplementary Figure S5). Of note, normalized siRNA levels varied about 3-fold among WT samples (Figure 3D), although silencing was fully efficient in all three cases. This may be due to differences in the amount of dsRNA produced by bacterial cultures after isopropyl β-D-1-thiogalactopyranoside (IPTG) induction and/or to differences in the washing and starvation of cells before RNA extraction (see Materials and Methods). Compared to the average of WT samples, 23-nt siRNA counts were strongly reduced in mutants with a full phenotype, i.e. between 17- (pds1-5.18) and 250-fold (rdr1-5.28), and moderately reduced in hypomorphic mutants, i.e. between 2- (rdr2-3.7) and 11-fold (rdr1-5.5) (Figure 3D). Consistent with the conclusion that most dsRNA-region siRNAs are primaries, siRNAs mapping to the vector parts of the dsRNA were reduced to a similar extent in each mutant (Figure 3E). The 2-fold reduction in rdr2-3.7, the most hypomorphic of the two RDR2 alleles, seems not significant given the variations observed among WT samples, consistent with the weak effect of the mutation on silencing efficiency. Overall, these results confirm that all factors are involved in the accumulation of primary siRNAs, including the two RdRPs. The data show a clear correlation of the phenotype with the level of primary siRNAs. We further note that the 23-nt peak is completely abolished in the non-sense RDR1 allele (rdr1-5.28) (Figure 3C), indicating that primary siRNAs cannot be produced in the absence of the Rdr1 protein.
Rdr2 is required for accumulation of secondary siRNAs
In contrast to primary siRNAs, secondary siRNAs were hardly affected in most mutants. In dcr1 mutants (dcr1-5.5 and dcr1-5.27, both missense mutations in the helicase domain), the number of ND169 23-nt secondary siRNAs was not significantly reduced and the antisense bias was not affected. Likewise, in cid1, pds1 and two of the rdr1 alleles (rdr1-3.16 and rdr1-1.22), secondary siRNA levels were not or only moderately (3- to 7-fold) reduced (Figure 3F and Supplementary Table S4A). Thus, these genes may not be directly involved in the synthesis of secondary siRNAs; where observed, the modest reduction may simply be due to a stronger depletion of primary siRNAs, which are needed to trigger production of secondaries. Only in the case of the non-sense mutant rdr1-5.28 were secondary siRNAs completely lost, which may be linked to the complete absence of 23-nt primaries. Under this hypothesis the substantially reduced levels of primary siRNAs in other mutants, though precluding efficient silencing of ND169, would still be sufficient to induce near-WT levels of secondaries.
The strongest allele of RDR2 (rdr2-1.24, retaining a 78-bp micronuclear-specific sequence leading to premature termination) behaved differently: despite a considerable level of primaries, secondary siRNAs were almost completely absent (250-fold compared to the average level of WT samples) (Figure 3F, Supplementary Figures S5B and S6A). The few reads counted in rdr2-1.24 and rdr1-5.28 are within the range of sample contamination, as determined from the control sample WT-K, not fed with ND169 dsRNA (10 reads in WT-K, 9 in rdr1-5.28 and 4 in rdr2-1.24). Thus, Rdr2 appears to be responsible for the synthesis of secondary siRNAs. In turn, the silencing phenotype was only partially impaired, indicating that residual primaries are functional in silencing. Whereas rdr2-1.24 affected accumulation of both primary and secondary siRNAs, the strongly hypomorphic rdr2-3.7 allele was still able to produce WT levels of secondary siRNAs, suggesting that the substitution of the Glu residue to Lys specifically (though weakly) impairs primary siRNAs. Overall, in contrast to primaries, the level of secondary siRNAs does not correlate with the phenotype of the mutants, indicating that primaries are the main effectors of silencing.
The occurrence of 5′-to-3′ transitivity suggests that priming by primary siRNAs is not necessary for the synthesis of a long dsRNA precursor, which is reminiscent of the Dicer-independent, 5′ triphosphate secondaries in C. elegans (21–24,42). Although previous analyses only detected 5′ monophosphate siRNAs in the ND169 dsRNA region (29), we asked whether a significantly larger amount of 5′ triphosphate molecules, which would not be included in our libraries, can be detected in other regions of the mRNA. Northern blots probed with ND169 sequences upstream of the dsRNA region revealed at best trace amounts of siRNAs (Supplementary Figure S7). Although this does not exclude a small amount of 5′-triphosphate siRNA molecules, clearly the total amount of secondaries is much less than that of primaries.
A fraction of primary and secondary siRNAs is 3′-uridylated
To check for possible 3′ addition of untemplated nucleotides, reads that could not be mapped in the above analysis were trimmed at the 3′-end by one nucleotide and mapped again, allowing no mismatch. The procedure was repeated up to eight times. This revealed the presence of up to eight non-genomic 3′ uridines (U) in a small fraction of sRNAs mapping within and outside the ND169 dsRNA, and of sRNAs mapping to dsRNA vector sequences (8%, 4% and 5% on average in WT cells, respectively) (Supplementary Figure S8A–C). More than 50% were modified with a single U. Their size distribution shows a peak at 23 genomic nucleotides (Supplementary Figure S8D–G), indicating that they represent modified primary and secondary siRNAs. Other untemplated nucleotides were observed at much lower frequencies compared to Us, and no significant 3′ modification was detected on the bulk of rRNA-derived sRNAs (e.g. on average 0.08% carrying a 3′U in WT samples), indicating that uridylation is not an artifact due to library construction or sequencing but specifically occurs in a fraction of siRNAs.
Whereas primary and secondary 3′U-siRNAs of ND169 were reproducibly more abundant on the sense strand (77% and 72% of the total 23-nt 3′U primary and secondary siRNAs on average in WT samples, respectively) (Supplementary Figure S8 and Table S4B), 3′U siRNAs from other target genes occurred with a similar strand distribution as non-uridylated siRNAs (Supplementary Table S2B and C), and showed somewhat lower abundances (≤7% of 23-nt siRNAs versus 9.7% on average for ND169) (Supplementary Table S2C), suggesting potential sequence-specific effects.
In mutants, the levels of primary 3′-uridylated siRNAs (ND169 dsRNA region and vector sequences) seemed to be reduced to a similar extent as non-modified primary siRNAs (Supplementary Figure S8H). Likewise, and despite their low absolute numbers, 3′U secondary siRNAs appear to have the same genetic requirements as non-modified ones. 3′ uridylation is a modification catalyzed by nucleotidyl-transferases (43) and promotes siRNA degradation (44–47). Cid1 does not seem to be involved directly in the uridylation of these siRNAs, as the proportion of 3′U siRNAs in the residual fraction of primary and possibly also secondary siRNAs was not reduced in cid1 mutants (Supplementary Figure S8I). This suggests that one or several of the other ≥22 nucleotidyl-transferases encoded in the P. tetraurelia genome (27) are responsible for their uridylation. The very low proportion of 3′ adenylation deviates from the previous observation that a substantial fraction of dsRNA-induced and other siRNAs in conjugating cells (50%) carried 1–7 nt 3′ polyA tails (28).
Endogenous siRNA clusters depend either on Rdr1 and Cid1 or on Rdr2
Rdr2 was previously shown to be involved in the production of endogenous siRNAs of unknown function, clustering in an intergenic region (cluster 22) (27). To gather insight into the endogenous roles of proteins involved in the exogenously induced RNAi pathway we looked for spontaneous production of 23-nt siRNAs from the MAC genome in WT and mutant cells, comparing only samples subjected to dsRNA feeding. We considered only clusters of sRNAs showing a 23-nt peak, regions longer than 250 bp and a minimum coverage of 60× on average in WT samples to allow for comparison with mutants. This revealed a non-exhaustive list of 10 clusters (Table 2, Supplementary Figures S9 and S10) which showed strong reduction of sRNA count in at least one mutant. This, together with their predominant size of 23 nt, indicates that they represent bona fide siRNAs.
Table 2. Genomic cluster loci producing endogenous 23 nt siRNAs.
| Cluster no. | Length [bp] | Gene annotation (ParameciumDB) | Transcript count | Mean siRNA coverage/bp | %siRNAs on strand - | ||
|---|---|---|---|---|---|---|---|
| + | - | strand + | strand - | ||||
| 17 | 982 | GSPATT00007066001 | 516 | 8 | 13 | 443 | 97 |
| 51 | 3021 | GSPATT00017018001 | 474 | 0 | 0 | 20 | 98 |
| 22 | 331 | intergenic | 466 | 2 | 1545 | 3540 | 70 |
| 79 | 440 | intergenic | 28 | 0 | 70 | 106 | 60 |
| 47 | 611 | intergenic | 0* | 0* | 7 | 38 | 84 |
| 121 | 1007 | intergenic | 24 | 0 | 403 | 2567 | 86 |
| 110 | 275 | intergenic | 16 | 0 | 515 | 1801 | 78 |
| 160 | 412 | intergenic | 22 | 0 | 153 | 166 | 52 |
| 112 | 303 | intergenic | 4* | 4* | 56 | 105 | 65 |
| 143 | 545 | intergenic | 128 | 0 | 16 | 18 | 53 |
Overlapping poly(A)-containing transcripts were identified using directional RNA-Seq data sets (normalized values). + and – strands of siRNAs were defined according to these transcripts, but only represent top and bottom strands for the 2 clusters with no clearly defined single-stranded transcripts (*). SiRNA coverages are mean values of WT1-3.
Clusters fell mainly in two categories. In the first one the number of siRNAs was significantly reduced in the strongest rdr2 allele (rdr2-1.24), but not in other mutants (the known cluster 22, as well as clusters 17 and 51 (Figure 4A–C)). Two of these (cluster 17 and 51) overlap exactly annotated mRNAs and consist of siRNAs with a strong antisense bias (>95%), also mapping to exon-exon junctions (Supplementary Table S5), similar to Rdr2-dependent ND169 secondaries. Thus, Rdr2 seems to be able to produce siRNAs independently of Rdr1-dependent primary siRNAs. Clusters of the second group were only located in intergenic regions, and for five of them transcripts were detected by directional RNA-Seq (Table 2). SiRNAs mapped equally to both strands or showed a minor bias to one strand, and some overlapped splice-junctions. SiRNAs from these clusters were less abundant in rdr1 and cid1 mutants (rdr1-5.28, rdr1-3.16, cid1-1.8, cid1-5.14) (clusters 79, 47, 121, 110, 160, 112 and 143) (Figure 4D–J), indicating a concerted action of the two proteins.
Figure 4.
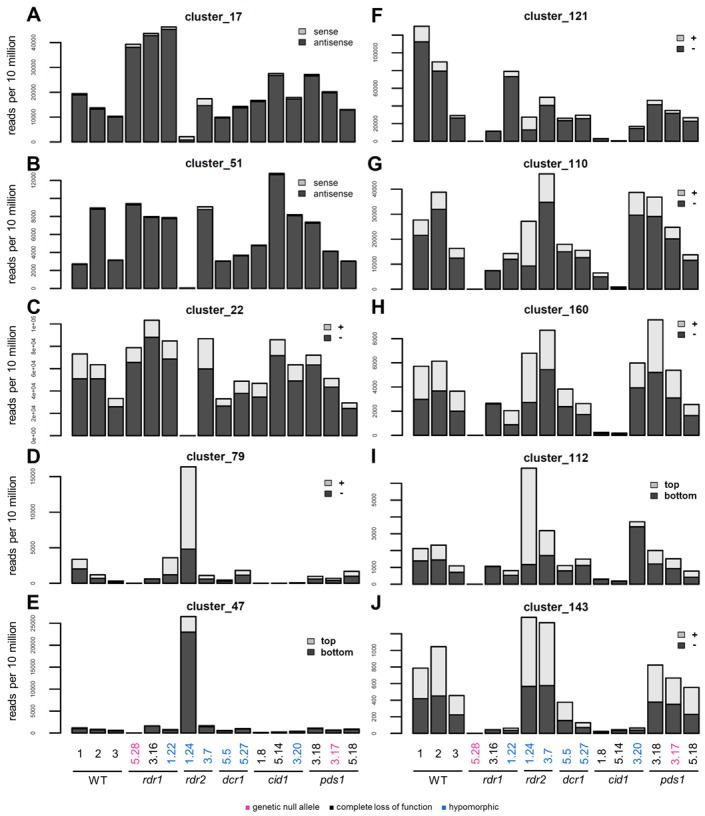
Endogenous siRNA clusters dependent of either Rdr2 or Rdr1 and Cid1. (A and B) Abundance of endogenously produced 23 nt siRNAs mapping to coding genes in WT cells and RNAi-deficient mutants. (C–J) Endogenous 23 nt siRNAs mapping to ncRNAs (see Table 2). SiRNAs oriented sense/+ to mRNAs/non-coding transcripts are represented in gray, those oriented antisense/- are shown in black. SiRNA size distributions for each cluster are shown in Supplementary Figure S9, graphic representations of their distribution on genomic regions are shown in Supplementary Figure S10.
Interestingly, some Rdr1/Cid1-dependent siRNA clusters are clearly overrepresented in the rdr2-1.24 mutant (clusters 79, 47, 112). This may indicate that Rdr2 plays a role antagonistic to that of Rdr1 in the accumulation of these siRNAs.
Similarly to dsRNA-induced siRNAs, a minor fraction of endogenous cluster siRNAs was found to carry untemplated 3′Us (7.7–28% in intergenic clusters and 1.3–2.6% in gene mapping clusters) (Supplementary Figure S9K). We conclude that the genes involved in the dsRNA-induced RNAi mechanism possess endogenous functions in siRNA accumulation at specific cluster regions. Endo-siRNAs exhibit similar properties to experimentally induced ones, as they consist of 3′ unmodified and a minor fraction of 3′U siRNAs, and have overlapping genetic requirements. However, no cluster appeared to be affected by pds1 mutations.
The Paramecium RNAi machinery can process single-stranded bacterial RNA
As a preliminary test to determine whether the uptake of RNA from food vacuoles and its processing by RNAi is restricted to dsRNA, the reads were mapped to E. coli 16S and 23S rRNA sequences as well as to an E. coli transcriptome data set without pre-mapping to other sequences, allowing no mismatch. In WT samples, variable quantities of sRNAs corresponding to the sense rRNA and mRNA transcripts were identified (Figure 5, top). Variations are likely due to differences in cell starvation and washes prior to RNA extraction (see Materials and Methods), leading to variable densities of food bacteria. The size distribution of both sense rRNA-derived and sense mRNA-derived sRNAs did not show any specific pattern, consistent with most of these molecules representing rRNA and mRNA fragments being degraded. Intriguingly, however, a number of rRNA and mRNA matches were of antisense polarity, and their size distribution showed a clear peak at 23 nt (Figure 5, bottom). In both cases, these antisense sRNAs were much less abundant than sense fragments, but they likely represent bona fide siRNAs synthesized by the Paramecium RNAi machinery: indeed, the same analysis performed on the mutant data sets revealed that the antisense rRNA count is strongly reduced in the rdr2-1.24 mutant, and the remaining molecules do not show a peak at 23 nt (Supplementary Figure S11). Although antisense rRNA counts are also reduced in rdr1-5.28 and dcr1-5.27, all mutants except rdr2-1.24 still show a clear peak at 23 nt specifically in the antisense distribution. Therefore, it is more difficult to conclude about the possible effect of the other mutations on rRNA-derived siRNAs. Interestingly, E. coli mRNA-derived antisense siRNAs (23 nt) showed similar genetic requirements as observed for ND169 primary siRNAs (Supplementary Figures S12 and S13). Size distributions did not show the 23-nt peak in the RDR1 null mutant rdr1-5.28 and in the two CID1 mutants cid1-1.8 and cid1-5.14 (Supplementary Figure S12). However, the quantitative variations observed call for more carefully controlled experiments to conclude about the implication of RNAi components in the accumulation of bacterially derived siRNAs.
Figure 5.
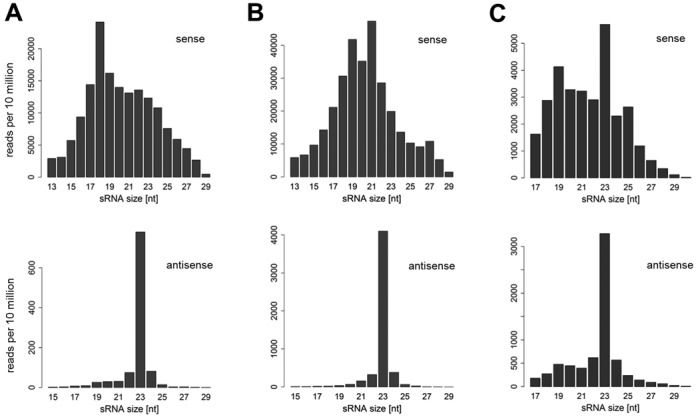
16S and 23S E. coli rRNA- and mRNA-derived siRNAs accumulate in WT P. tetraurelia cells. Size distribution and normalized abundance of sense (top) and antisense (bottom) oriented sRNAs are shown (sample WT-1), in reads per 10 million. (A) 16S-rRNA mapping, (B) 23S-rRNA mapping and (C) mRNA mapping sRNAs (using a complete E. coli W3110 transcriptome as template).
DISCUSSION
The biogenesis and impact of secondary siRNAs induced by exogenous dsRNA have most intensively been studied in plants and C. elegans. Here, we report RdRP-dependent secondary siRNA production and transitivity of the silencing response for the first time in a ciliate, and to our knowledge, in the SAR clade of eukaryotes (Stramenopiles, Alveolates, Rhizaria). Paramecium secondary siRNAs do not seem to serve as amplificators of silencing, as they accumulate at low levels compared to primaries, and, unlike in plants and nematodes, contribute poorly to the instantaneous silencing phenotype. Thus, despite of non-self dsRNA ingested by food being a widespread trigger of RNAi (7), its processing mechanisms and functions have evolved diversely. In Paramecium, as observed in other organisms, this pathway genetically overlaps with endogenous RNAi. Interestingly, single-stranded exogenous RNA species ingested by food are also processed by universal RNAi factors in this bactivorous organism. This provides the first evidence that the control of diverse types of environmental RNA may be a natural function of exogenously induced RNAi in this eukaryote.
Mechanisms of primary siRNA synthesis
The sequence data sets from various mutants, including both complete loss-of-function and hypomorphic alleles, allowed us to confirm previous conclusions from northern blot analyses that siRNAs matching the dsRNA trigger, here shown to be overwhelmingly primaries, require the DCR1, RDR1, RDR2, CID1 and PDS1 genes for their synthesis or accumulation at WT levels.
While it seems reasonable to assume that Dcr1 cleaves the dsRNA to yield 23-nt primary siRNAs, the reason why two different RdRPs should also be required is intriguing. RdRP activity might be required to amplify the dsRNA trigger, if it is imported from the food vacuole in very small amounts. This would be in contrast to C. elegans, where ingested dsRNA is not amplified (42). Alternatively, the bacterial dsRNA could be imported from the food vacuole in single-stranded form; RdRP activity would then be required to produce a genuine dsRNA trigger (48–52).
Primary siRNAs were completely absent in the non-sense mutant rdr1-5.28, indicating that the Rdr1 protein is strictly required, and that Rdr2 alone is not sufficient. The residual amount of primary siRNAs observed in other RDR1 alleles therefore indicates that the mutant proteins are still partially functional in this respect. In the rdr1-3.16 mutant, a highly conserved Asp residue thought to be required for RdRP catalytic activity (53–55) is changed to Tyr. Although some residual catalytic activity cannot be excluded, this raises the possibility that Rdr1 plays a structural role rather than a catalytic one. It could for instance associate with Dcr1, as shown for the homologous proteins in Tetrahymena (56,57), in a complex that would be required for Dcr1 activity, or recruitment to the dsRNA substrate. The requirement for RDR2 in primary siRNA generation may be less stringent compared to RDR1, since only the most compromised allele rdr2-1.24 showed reduced primary siRNA levels.
Primary siRNAs were severely depleted in cid1 full-deficiency mutants. Nucleotidyl-transferases have similarly been implicated in RNAi pathways in S. pombe (58,59), C. elegans (60,61) and T. thermophila (56,62,63). In several cases they have been shown to physically interact with RdRPs in RNA-dependent RNA polymerase complexes (RdRCs) (47,57,58,63). In vitro tests of T. thermophila RdRCs with ssRNA templates indicated that RdRP activity is primed by looping of the 3′-end, and that addition of polyU tails by the Rdn nucleotidyl-transferases enables full-length conversion to dsRNA (57). Such a mechanism might explain the role of Cid1 in primary siRNA synthesis in Paramecium, if RdRP catalytic activity is required. The parallel effects of cid1 and rdr1 mutations on all siRNAs examined, including endogenous siRNAs, strongly suggest that the two proteins are obligate partners in an RdRC (Figure 6).
Figure 6.
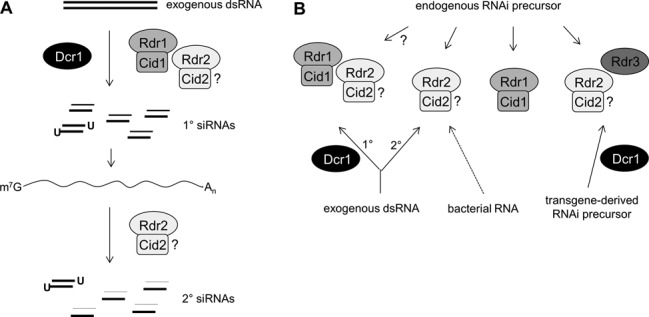
RdRP functions in P. tetraurelia exogenous and endogenous RNAi pathways. (A) Two distinct RdRPs co-operate and act sequentially in the biosynthesis of siRNAs from exogenous dsRNA. Whereas accumulation of both, primary and secondary siRNAs requires Rdr1 and Rdr2, the production of secondaries is only dependent on Rdr2, provided primary siRNAs accumulate above a threshold level. Note that complex formation of RdRPs with Cid nucleotidyl transferases (RdRCs) (see text) is hypothetical. The role of the essential Cid2 was not tested in this study. (B) SiRNAs produced from different types of endogenous RNAi precursors require distinct, but overlapping RdRP activities, creating limiting conditions in the pathway network. Rdr2 and the putatively non-catalytic RdRP Rdr3 act together with Dcr1 in transgene-induced silencing (27,29). Some endo-siRNAs seem to be produced independently of Rdr2 by Rdr1-Cid1, or vice versa. Note that Rdr1-Rdr2 co-operation may also occur in endo-siRNA biogenesis. Only confirmed roles of Dcr1 (27,28) and Rdr3 (29) are shown.
We found that a minor fraction of exogenous and endogenous siRNAs is modified by 3′ uridylation in vegetatively growing cells, however, Cid1 is not obviously involved in this process. The occurrence of 3′U siRNAs in vegetative cells is in contrast to conjugating cells, where 3′ adenylation is the dominant modification (28). Whereas the function of 3′U siRNAs remains unclear, this suggests that nucleotidyl-transferase activity underlies specific constraints at different stages of the life cycle and/or in different physiological conditions.
Mutations in the non-essential Paramecium-specific PDS1 gene also strongly decreased the levels of dsRNA-induced primary siRNAs, but unlike cid1 and rdr1 mutations they did not affect any of the clusters of endogenous siRNAs examined. The Pds1 protein thus appears to be specifically involved in exogenously triggered RNAi, suggesting it may participate in the import of bacterial dsRNA from the food vacuole.
Mechanisms of secondary siRNA synthesis
By definition, secondary siRNAs are synthesized from an mRNA as a consequence of its targeting by primary siRNAs. Any mutation that completely abolishes primary siRNA production should therefore also result in the lack of secondaries, as observed for the non-sense mutant rdr1-5.28. However, most other mutants, including the other two rdr1 alleles, still produce a residual amount of primary siRNAs. This shows that these mutations have little or no impact on secondary siRNA levels: in cases where a small reduction was observed, it may be explained by a stronger depletion of primaries. The only exception is rdr2-1.24: despite a significant amount of residual primaries, secondary siRNAs were completely absent in this mutant. Our results thus implicate only Rdr2 in their production, likely through its catalytic activity (Figure 6A).
Spreading of siRNA production beyond the dsRNA trigger involves RdRP activity in other systems (6,19). In both plants and nematodes, primer-independent RdRP initiation permits 5′-to-3′ transitivity (20,22). Similarly, the occurrence of 5′-to-3′ transitivity in P. tetraurelia implies that the synthesis of antisense RNAs by the Rdr2 protein does not require priming by primary siRNAs. Rdr2 activity may also require association with a nucleotidyl-transferase in an RdRC, such as the essential CID2 (27).
Unlike primary siRNAs, secondary siRNAs show a strong antisense bias for all target genes tested and were not significantly depleted in the two hypomorphic dcr1 alleles tested. This suggests that a large fraction could be produced directly by unprimed RdRP activity as observed in C. elegans (21–23). The small but significant fraction of sense secondary siRNAs could conceivably be due to the same mechanism acting on natural antisense transcripts, which can be detected at low levels for much of the macronuclear genome (64). However, the resulting 5′-triphosphate siRNAs would have to be at least partly converted to 5′-monophosphate by an RNA phosphatase, such as PIR-1 (65).
An alternative model has been proposed for the biosynthesis of predominantly antisense, 5′-monophosphate endogenous siRNAs in the ciliate T. thermophila (62,66). The RdRC, which can synthesize Dicer substrates from ssRNA in vitro, physically interacts with Dicer. This may dictate the polarity of Dicer processing, resulting in preferential loading of the antisense strand onto Piwi proteins (56,57). The reason why secondary siRNA levels were not reduced in the two dcr1 alleles tested could be that the helicase domain is dispensable for the synthesis of secondary, but not primary, siRNAs. Specific requirement of the helicase domain has been observed for C. elegans Dcr-1 (67,68). Alternatively, one of the other Dicer or Dicer-like proteins in P. tetraurelia (28) may be responsible for dsRNA dicing.
The functions of primary and secondary siRNAs in dsRNA-induced silencing
In contrast to the secondary siRNAs of C. elegans induced by dsRNA feeding (22,23,42), we found Paramecium secondary siRNAs to be much less abundant than primaries. Although we sequenced only 5′-monophosphate molecules, our northern blot analyses (29 and this study) indicate that triphosphorylated secondary siRNAs, if present at all, are at best a very minor fraction of the total siRNA pool. Unlike primaries, secondary siRNAs are produced from the target mRNA, so that mRNA abundance may be a limiting factor. Continuous dsRNA import and production of primary siRNAs, together with mRNA slicing (31) and eventually degradation, could thus explain the observed ratio. Furthermore, secondary siRNAs might affect transcription of the target gene in the macronucleus (see below), resulting in a negative feedback. A non-exclusive possibility is that a limiting amount of the Rdr2 protein, which is involved in the synthesis of both primaries and secondaries, is titrated away from its role in secondary siRNA production by continuous dsRNA feeding.
Interestingly, another difference with C. elegans is that in the latter, secondary siRNAs were reported to be the actual silencing effectors (22,25,26), whereas our results suggest that primary siRNAs are the major actors in Paramecium, at least in the laboratory strain we used. Indeed, a considerable silencing deficiency is seen in mutants with significantly reduced amounts of primary siRNAs, but WT or only slightly reduced levels of secondaries (rdr1-3.16, dcr1-5.5). Conversely, complete absence of secondaries and slightly reduced amounts of primaries only lead to a very partial silencing deficiency, as seen in the rdr2-1.24 mutant. Thus, secondary siRNAs do not appear to contribute much to the observed silencing phenotype, suggesting they may have distinct functions.
As in C. elegans (69), primary and secondary siRNAs may be loaded onto different Argonaute proteins. Among the three P. tetraurelia Piwi proteins implicated in dsRNA-induced silencing, Ptiwi13, which contains the typical slicer catalytic triad DDH, was hypothesized to load primary siRNAs (30) and cleave cytoplasmic mRNAs within the dsRNA region (31). In contrast, the closely related ohnologs Ptiwi12 and Ptiwi15 have EDH triads possibly lacking slicer activity, but otherwise resemble the nuclear, scnRNA-specific Piwi09 (30). Thus, a possible function of secondary siRNAs may be to relay information about cytoplasmic RNAi to the nucleus. In C. elegans for instance, some secondary siRNAs are transported to the nucleus by the non-slicing Ago protein Nrde-3 to induce heritable silencing by pre-mRNA-targeting (70–73).
Endogenous siRNA clusters: RdRPs may compete as well as co-operate
Mutants deficient in exogenously triggered RNAi were found to have altered levels of endogenous siRNAs clustering at specific macronuclear loci, suggesting that the corresponding genes normally participate in transcriptome regulation. However, no obvious phenotype was observed during vegetative growth, and only rdr2 mutants showed defects in sexual reproduction (27). Among the 10 siRNA clusters analyzed, 3 were almost completely absent in the strongest rdr2 allele. These mapped to known transcripts (2 protein-coding, spliced mRNAs, 1 ncRNA) with a strong antisense bias and appear to be mechanistically similar to dsRNA-induced secondary siRNAs, although they do not depend on upstream Rdr1 action. One cluster was previously shown to depend on the putatively non-catalytic Rdr3, indicating that Rdr2 and Rdr3 can co-operate for endogenous clusters, as they do in transgene-induced silencing (27,29) (schematized in Figure 6B).
In the other 7 clusters (including 5 that appear to map to ncRNAs with variable antisense biases), siRNA levels were significantly reduced in full-deficiency rdr1 and cid1 mutants. Interestingly, in at least two cases siRNA levels were in fact markedly increased in the strongest rdr2 allele, suggesting that Rdr2 may normally down-regulate transcripts that are used as precursors for Rdr1/Cid1-dependent siRNAs. Thus, Rdr1 and Rdr2 may compete for such transcripts. Alternatively, Rdr2 may compete at these loci with Rdr1 for pathway components, such as unknown RdRC co-factors. If the Rdr1-Rdr2 co-operation on exogenous dsRNA is physically interdependent, loss of Rdr2 during dsRNA feeding may result in increased overall availability of Rdr1.
The natural functions of exogenously triggered RNAi
We have provided evidence that P. tetraurelia synthesizes siRNAs that are antisense to the ribosomal and messenger RNAs of its bacterial food, in an Rdr2-dependent manner. Thus, the coupling of RNA import from food vacuoles to RNAi processing is not restricted to dsRNA. In previous tests, feeding cells with bacteria producing ssRNA homologous to a Paramecium gene did not result in phenotypically detectable silencing (9), but this may be due to the higher stability of dsRNA in the HT115 DE3 strain of E. coli, and the need for high levels of primary siRNAs to completely suppress a Paramecium mRNA.
This finding raises the possibility that single-stranded bacterial RNAs compete with the dsRNA trigger for RNAi factors (Figure 6B), which could at least in part explain the important variations observed in primary siRNA levels between different WT cultures fed the same dsRNA-producing bacterial strain. More fundamentally, the finding considerably broadens the possible natural functions of this mechanism. dsRNA-induced silencing could obviously provide protection against dsRNA viruses, but no virus infecting Paramecium has been described so far; the mechanism may now be envisioned to protect against the horizontal transfer of single-stranded retroelements. Furthermore, it might provide control of bacterial RNAs that could possibly impact the cell's endogenous transcriptome, as observed recently in C. elegans (74). It will be interesting to study how this unicellular eukaryote adapts RNAi responses to competing exogenous and endogenous RNAi triggers, and how this may affect gene expression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Véronique Tanty and all other lab members for continuous support and input, and Marcel Schulz (Max Planck Institute for Informatics, Saarbrücken) for critical discussion of sequencing data. We are thankful to Mireille Bétermier (Centre de Génétique Moléculaire, Gif-sur-Yvette) for providing unpublished RNA-Seq data sets. The sequencing of small RNAs benefited from the facilities and expertise of the high-throughput sequencing platform of IMAGIF (Centre de Recherche de Gif—www.imagif.cnrs.fr). We thank Jasmin Kirch for technical assistance and Karl Nordström for support in bioinformatics (Department of Epigenetics, University of Saarbrücken).
FUNDING
Agence Nationale de la Recherche [‘Investissements d'Avenir’ ANR-10-LABX-54 MEMO LIFE/ANR-11-IDEX-0001-02 Paris Sciences et Lettres* Research University, ANR-08-BLAN-0233 ‘ParaDice’, ANR-12-BSV6-0017 ‘INFERNO’ to E.M.]; Fondation pour la Recherche Médicale [‘Equipe FRM’ to E.M.]; Fondation Pierre Gilles de Gennes [post-doctoral fellowship to S.M.]; Deutsche Forschungsgemeinschaft [DFG SI-1397-2 to M.S.]. CNRS-supported European Research Group ‘Paramecium Genome Dynamics and Evolution’; European COST Action [BM1102]. Funding for open access charge: Agence Nationale de la Recherche (ANR-12-BSV6-0017).
Conflict of interest statement. None declared.
REFERENCES
- 1.Meister G., Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 2.Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ketting R.F. The many faces of RNAi. Dev. Cell. 2011;20:148–161. doi: 10.1016/j.devcel.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Castel S.E., Martienssen R.A. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013;14:100–112. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brodersen P., Voinnet O. The diversity of RNA silencing pathways in plants. Trends Genet. 2006;22:268–280. doi: 10.1016/j.tig.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Voinnet O. Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci. 2008;13:317–328. doi: 10.1016/j.tplants.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Whangbo J.S., Hunter C.P. Environmental RNA interference. Trends Genet. 2008;24:297–305. doi: 10.1016/j.tig.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Nicolas F.E., Torres-Martinez S., Ruiz-Vazquez R.M. Loss and retention of RNA interference in fungi and parasites. PLoS Pathog. 2013;9:e1003089. doi: 10.1371/journal.ppat.1003089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galvani A., Sperling L. RNA interference by feeding in Paramecium. Trends Genet. 2002;18:11–12. doi: 10.1016/s0168-9525(01)02548-3. [DOI] [PubMed] [Google Scholar]
- 10.Sobierajska K., Joachimiak E., Bregier C., Fabczak S., Fabczak H. Effect of phosducin silencing on the photokinetic motile response of Blepharisma japonicum. Photochem. Photobiol. Sci. 2011;10:19–24. doi: 10.1039/c0pp00221f. [DOI] [PubMed] [Google Scholar]
- 11.Slabodnick M.M., Ruby J.G., Dunn J.G., Feldman J.L., DeRisi J.L., Marshall W.F. The kinase regulator mob1 acts as a patterning protein for stentor morphogenesis. PLoS Biol. 2014;12:e1001861. doi: 10.1371/journal.pbio.1001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timmons L., Court D.L., Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- 13.Timmons L., Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854–854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 14.Wilkins C., Dishongh R., Moore S.C., Whitt M.A., Chow M., Machaca K. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature. 2005;436:1044–1047. doi: 10.1038/nature03957. [DOI] [PubMed] [Google Scholar]
- 15.Lu R., Maduro M., Li F., Li H.W., Broitman-Maduro G., Li W.X., Ding S.W. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature. 2005;436:1040–1043. doi: 10.1038/nature03870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee R.C., Hammell C.M., Ambros V. Interacting endogenous and exogenous RNAi pathways in Caenorhabditis elegans. RNA. 2006;12:589–597. doi: 10.1261/rna.2231506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy S., Wang D., Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–649. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 18.Sarkies P., Ashe A., Le Pen J., McKie M.A., Miska E.A. Competition between virus-derived and endogenous small RNAs regulates gene expression in Caenorhabditis elegans. Genome Res. 2013;23:1258–1270. doi: 10.1101/gr.153296.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baulcombe D.C. Molecular biology. Amplified silencing. Science. 2007;315:199–200. doi: 10.1126/science.1138030. [DOI] [PubMed] [Google Scholar]
- 20.Vaistij F.E., Jones L., Baulcombe D.C. Spreading of RNA targeting and DNA methylation in RNA silencing requires transcription of the target gene and a putative RNA-dependent RNA polymerase. Plant Cell. 2002;14:857–867. doi: 10.1105/tpc.010480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aoki K., Moriguchi H., Yoshioka T., Okawa K., Tabara H. In vitro analyses of the production and activity of secondary small interfering RNAs in C. elegans. EMBO J. 2007;26:5007–5019. doi: 10.1038/sj.emboj.7601910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sijen T., Steiner F.A., Thijssen K.L., Plasterk R.H. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science. 2007;315:244–247. doi: 10.1126/science.1136699. [DOI] [PubMed] [Google Scholar]
- 23.Pak J., Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- 24.Maniar J.M., Fire A.Z. EGO-1, a C. elegans RdRP, modulates gene expression via production of mRNA-templated short antisense RNAs. Curr. Biol. 2011;21:449–459. doi: 10.1016/j.cub.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu W., Shirayama M., Conte D., Jr, Vasale J., Batista P.J., Claycomb J.M., Moresco J.J., Youngman E.M., Keys J., Stoltz M.J., et al. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol. Cell. 2009;36:231–244. doi: 10.1016/j.molcel.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claycomb J.M., Batista P.J., Pang K.M., Gu W., Vasale J.J., van Wolfswinkel J.C., Chaves D.A., Shirayama M., Mitani S., Ketting R.F., et al. The Argonaute CSR-1 and its 22G-RNA cofactors are required for holocentric chromosome segregation. Cell. 2009;139:123–134. doi: 10.1016/j.cell.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marker S., Carradec Q., Tanty V., Arnaiz O., Meyer E. A forward genetic screen reveals essential and non-essential RNAi factors in Paramecium tetraurelia. Nucleic Acids Res. 2014;42:7268–7280. doi: 10.1093/nar/gku223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lepère G., Nowacki M., Serrano V., Gout J.F., Guglielmi G., Duharcourt S., Meyer E. Silencing-associated and meiosis-specific small RNA pathways in Paramecium tetraurelia. Nucleic Acids Res. 2009;37:903–915. doi: 10.1093/nar/gkn1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marker S., Le Mouël A., Meyer E., Simon M. Distinct RNA-dependent RNA polymerases are required for RNAi triggered by double-stranded RNA versus truncated transgenes in Paramecium tetraurelia. Nucleic Acids Res. 2010;38:4092–4107. doi: 10.1093/nar/gkq131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouhouche K., Gout J.F., Kapusta A., Bétermier M., Meyer E. Functional specialization of Piwi proteins in Paramecium tetraurelia from post-transcriptional gene silencing to genome remodelling. Nucleic Acids Res. 2011;39:4249–4264. doi: 10.1093/nar/gkq1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaillon O., Bouhouche K., Gout J.F., Aury J.M., Noel B., Saudemont B., Nowacki M., Serrano V., Porcel B.M., Segurens B., et al. Translational control of intron splicing in eukaryotes. Nature. 2008;451:359–362. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- 32.Sarkies P., Miska E.A. RNAi pathways in the recognition of foreign RNA: antiviral responses and host-parasite interactions in nematodes. Biochem. Soc. Trans. 2013;41:876–880. doi: 10.1042/BST20130021. [DOI] [PubMed] [Google Scholar]
- 33.Galvani A., Sperling L. Transgene-mediated post-transcriptional gene silencing is inhibited by 3′ non-coding sequences in Paramecium. Nucleic Acids Res. 2001;29:4387–4394. doi: 10.1093/nar/29.21.4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruiz F., Vayssie L., Klotz C., Sperling L., Madeddu L. Homology-dependent gene silencing in Paramecium. Mol. Biol. Cell. 1998;9:931–943. doi: 10.1091/mbc.9.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon M.C., Marker S., Schmidt H.J. Posttranscriptional control is a strong factor enabling exclusive expression of surface antigens in Paramecium tetraurelia. Gene Expr. 2006;13:167–178. doi: 10.3727/000000006783991809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aury J.M., Jaillon O., Duret L., Noel B., Jubin C., Porcel B.M., Segurens B., Daubin V., Anthouard V., Aiach N., et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- 38.Arnaiz O., Mathy N., Baudry C., Malinsky S., Aury J.M., Wilkes C.D., Garnier O., Labadie K., Lauderdale B.E., Le Mouël A., et al. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 2012;8:e1002984. doi: 10.1371/journal.pgen.1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin T., Cook D., Lawrence M. ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012;13:2012–2013. doi: 10.1186/gb-2012-13-8-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lawrence M., Gentleman R., Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25:1841–1842. doi: 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raabe C.A., Tang T.H., Brosius J., Rozhdestvensky T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014;42:1414–1426. doi: 10.1093/nar/gkt1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pak J., Maniar J.M., Mello C.C., Fire A. Protection from feed-forward amplification in an amplified RNAi mechanism. Cell. 2012;151:885–899. doi: 10.1016/j.cell.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim Y.K., Heo I., Kim V.N. Modifications of small RNAs and their associated proteins. Cell. 2010;143:703–709. doi: 10.1016/j.cell.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 44.Li J., Yang Z., Yu B., Liu J., Chen X. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005;15:1501–1507. doi: 10.1016/j.cub.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren G., Chen X., Yu B. Uridylation of miRNAs by hen1 suppressor1 in Arabidopsis. Curr. Biol. 2012;22:695–700. doi: 10.1016/j.cub.2012.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Y., Yu Y., Zhai J., Ramachandran V., Dinh T.T., Meyers B.C., Mo B., Chen X. The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr. Biol. 2012;22:689–694. doi: 10.1016/j.cub.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Wolfswinkel J.C., Claycomb J.M., Batista P.J., Mello C.C., Berezikov E., Ketting R.F. CDE-1 affects chromosome segregation through uridylation of CSR-1-bound siRNAs. Cell. 2009;139:135–148. doi: 10.1016/j.cell.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 48.Gent J.I., Lamm A.T., Pavelec D.M., Maniar J.M., Parameswaran P., Tao L., Kennedy S., Fire A.Z. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans soma. Mol. Cell. 2010;37:679–689. doi: 10.1016/j.molcel.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vasale J.J., Gu W., Thivierge C., Batista P.J., Claycomb J.M., Youngman E.M., Duchaine T.F., Mello C.C., Conte D., Jr Sequential rounds of RNA-dependent RNA transcription drive endogenous small-RNA biogenesis in the ERGO-1/Argonaute pathway. Proc. Natl. Acad. Sci. U.S.A. 2010;107:3582–3587. doi: 10.1073/pnas.0911908107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee H.C., Aalto A.P., Yang Q., Chang S.S., Huang G., Fisher D., Cha J., Poranen M.M., Bamford D.H., Liu Y. The DNA/RNA-dependent RNA polymerase QDE-1 generates aberrant RNA and dsRNA for RNAi in a process requiring replication protein A and a DNA helicase. PLoS Biol. 2010;8:e1000496. doi: 10.1371/journal.pbio.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalmay T., Hamilton A., Rudd S., Angell S., Baulcombe D.C. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell. 2000;101:543–553. doi: 10.1016/s0092-8674(00)80864-8. [DOI] [PubMed] [Google Scholar]
- 52.Mourrain P., Beclin C., Elmayan T., Feuerbach F., Godon C., Morel J.B., Jouette D., Lacombe A.M., Nikic S., Picault N., et al. Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell. 2000;101:533–542. doi: 10.1016/s0092-8674(00)80863-6. [DOI] [PubMed] [Google Scholar]
- 53.Sugiyama T., Cam H., Verdel A., Moazed D., Grewal S.I. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc. Natl. Acad. Sci. U.S.A. 2005;102:152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Curaba J., Chen X. Biochemical activities of Arabidopsis RNA-dependent RNA polymerase 6. J. Biol. Chem. 2008;283:3059–3066. doi: 10.1074/jbc.M708983200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makeyev E.V., Bamford D.H. Cellular RNA-dependent RNA polymerase involved in posttranscriptional gene silencing has two distinct activity modes. Mol. Cell. 2002;10:1417–1427. doi: 10.1016/s1097-2765(02)00780-3. [DOI] [PubMed] [Google Scholar]
- 56.Lee S.R., Collins K. Physical and functional coupling of RNA-dependent RNA polymerase and Dicer in the biogenesis of endogenous siRNAs. Nat. Struct. Mol. Biol. 2007;14:604–610. doi: 10.1038/nsmb1262. [DOI] [PubMed] [Google Scholar]
- 57.Talsky K.B., Collins K. Initiation by a eukaryotic RNA-dependent RNA polymerase requires looping of the template end and is influenced by the template-tailing activity of an associated uridyltransferase. J. Biol. Chem. 2010;285:27614–27623. doi: 10.1074/jbc.M110.142273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Motamedi M.R., Verdel A., Colmenares S.U., Gerber S.A., Gygi S.P., Moazed D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 59.Volpe T.A., Kidner C., Hall I.M., Teng G., Grewal S.I., Martienssen R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- 60.Chen C.C., Simard M.J., Tabara H., Brownell D.R., McCollough J.A., Mello C.C. A member of the polymerase beta nucleotidyltransferase superfamily is required for RNA interference in C. elegans. Curr. Biol. 2005;15:378–383. doi: 10.1016/j.cub.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 61.Jose A.M., Garcia G.A., Hunter C.P. Two classes of silencing RNAs move between Caenorhabditis elegans tissues. Nat. Struct. Mol. Biol. 2011;18:1184–1188. doi: 10.1038/nsmb.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Couvillion M.T., Lee S.R., Hogstad B., Malone C.D., Tonkin L.A., Sachidanandam R., Hannon G.J., Collins K. Sequence, biogenesis, and function of diverse small RNA classes bound to the Piwi family proteins of Tetrahymena thermophila. Genes Dev. 2009;23:2016–2032. doi: 10.1101/gad.1821209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee S.R., Talsky K.B., Collins K. A single RNA-dependent RNA polymerase assembles with mutually exclusive nucleotidyl transferase subunits to direct different pathways of small RNA biogenesis. RNA. 2009;15:1363–1374. doi: 10.1261/rna.1630309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lepère G., Bétermier M., Meyer E., Duharcourt S. Maternal noncoding transcripts antagonize the targeting of DNA elimination by scanRNAs in Paramecium tetraurelia. Genes Dev. 2008;22:1501–1512. doi: 10.1101/gad.473008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duchaine T.F., Wohlschlegel J.A., Kennedy S., Bei Y., Conte D., Jr, Pang K., Brownell D.R., Harding S., Mitani S., Ruvkun G., et al. Functional proteomics reveals the biochemical niche of C. elegans DCR-1 in multiple small-RNA-mediated pathways. Cell. 2006;124:343–354. doi: 10.1016/j.cell.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 66.Lee S.R., Collins K. Two classes of endogenous small RNAs in Tetrahymena thermophila. Genes Dev. 2006;20:28–33. doi: 10.1101/gad.1377006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavelec D.M., Lachowiec J., Duchaine T.F., Smith H.E., Kennedy S. Requirement for the ERI/DICER complex in endogenous RNA interference and sperm development in Caenorhabditis elegans. Genetics. 2009;183:1283–1295. doi: 10.1534/genetics.109.108134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Welker N.C., Pavelec D.M., Nix D.A., Duchaine T.F., Kennedy S., Bass B.L. Dicer's helicase domain is required for accumulation of some, but not all, C. elegans endogenous siRNAs. RNA. 2010;16:893–903. doi: 10.1261/rna.2122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yigit E., Batista P.J., Bei Y., Pang K.M., Chen C.C., Tolia N.H., Joshua-Tor L., Mitani S., Simard M.J., Mello C.C. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006;127:747–757. doi: 10.1016/j.cell.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 70.Burton N.O., Burkhart K.B., Kennedy S. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 2011;108:19683–19688. doi: 10.1073/pnas.1113310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guang S., Bochner A.F., Burkhart K.B., Burton N., Pavelec D.M., Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gu S.G., Pak J., Guang S., Maniar J.M., Kennedy S., Fire A. Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence-targeted histone H3 lysine 9 methylation footprint. Nat. Genet. 2012;44:157–164. doi: 10.1038/ng.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guang S., Bochner A.F., Pavelec D.M., Burkhart K.B., Harding S., Lachowiec J., Kennedy S. An Argonaute transports siRNAs from the cytoplasm to the nucleus. Science. 2008;321:537–541. doi: 10.1126/science.1157647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu H., Wang X., Wang H.D., Wu J., Ren J., Meng L., Wu Q., Dong H., Kao T.Y., Ge Q. Escherichia coli noncoding RNAs can affect gene expression and physiology of Caenorhabditis elegans. Nat. Commun. 2012;3:1073. doi: 10.1038/ncomms2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.