


Abstract
Candida glabrata is the second most common pathogenic Candida species and has emerged as a leading cause of nosocomial fungal infections. Its reduced susceptibility to antifungal drugs and its close relationship to Saccharomyces cerevisiae make it an interesting research focus. Although its genome sequence was published in 2004, little is known about its transcriptional dynamics. Here, we provide a detailed RNA-Seq-based analysis of the transcriptomic landscape of C. glabrata in nutrient-rich media, as well as under nitrosative stress and during pH shift. Using RNA-Seq data together with state-of-the-art gene prediction tools, we refined the annotation of the C. glabrata genome and predicted 49 novel protein-coding genes. Of these novel genes, 14 have homologs in S. cerevisiae and six are shared with other Candida species. We experimentally validated four novel protein-coding genes of which two are differentially regulated during pH shift and interaction with human neutrophils, indicating a potential role in host–pathogen interaction. Furthermore, we identified 58 novel non-protein-coding genes, 38 new introns and condition-specific alternative splicing. Finally, our data suggest different patterns of adaptation to pH shift and nitrosative stress in C. glabrata, Candida albicans and S. cerevisiae and thus further underline a distinct evolution of virulence in yeast.
INTRODUCTION
Although the number of fungal infections has increased over the last decades, their impact on human health remains underestimated (1,2) and with constantly increasing numbers of patients at risk in medical care, fungal infection will likely continue to grow in importance (3,4). In contrast to Aspergillus fumigatus and Cryptococcus neoformans that are linked to environmental sources and cause exogenous infections, Candida spp. are commensals of the human gut and form part of the normal microflora (5,6). Candida albicans and Candida glabrata are the two clinically most important species of the latter group (7).
While C. albicans is part of the CUG clade (8), C. glabrata is more closely related to Saccharomyces cerevisiae than to any other Candida species (9). Moreover, both pathogens have evolved different strategies for adhesion, tissue invasion, nutrient acquisition and interaction with immune cells (6,10). While C. albicans is polymorphic and mostly diploid, C. glabrata is strictly haploid and grows as unicellular yeast (11). In contrast to the aggressive strategy of C. albicans that includes active penetration of host cells and rapid dissemination into deeper parts of the host tissue (12), C. glabrata engages in a combination of immune evasion and persistence (9,10). For example, it uses the uptake by macrophages to evade other immune cells and is able to proliferate within macrophages (11,13,14). Additionally, it was shown that C. glabrata induced only transient pro-inflammatory cytokine responses, especially compared to C. albicans (15,16). Despite this, infections with C. glabrata are associated with longer hospital stays and higher costs (17). This may also be related to the intrinsically reduced susceptibility of C. glabrata to commonly used azole antifungal drugs (6). Knowledge about the molecular mechanisms employed by C. glabrata to infect humans remains limited. Although transcriptome studies are helpful to understand infection biology of pathogenic fungi (18), studies investigating the transcriptome of C. glabrata under relevant conditions are relatively rare (19–23).
Gene structure annotation is a vital approach of genomics, as it predicts the location and structure of all protein-coding and non-protein-coding genes in a genome assembly. Incorrect gene structure annotation may influence conclusions based on bioinformatics research (e.g. functional annotation, comparative genomics, classification) and experimental results (e.g. transcriptomics, gene deletions). Due to the increasing number and diversity of sequenced fungal strains and advances in sequencing technologies, the research community continually gains further insights into complex and diverse fungal gene structures (24). For this reason, new tools are being implemented, existing tools are constantly updated (25–27) and fungal gene structural annotations are being refined (24).
Next-Generation-Sequencing of cDNAs derived from RNA samples (RNA-Seq) (28) does not only help to study gene expression but also to elucidate gene structures. The transcriptional landscape i.e. the location of short sequence fragments derived from RNA molecules (‘reads’) within the genome can hint toward the locations of exons and introns (spliced reads) during gene prediction (29). Moreover, reads can be used to assemble full lengths transcripts (30,31) under different conditions. Since all gene structure annotation approaches have their specific weaknesses and strengths, researchers often combine their information into weighted consensus structures (32).
In yeasts and filamentous fungi, RNA-Seq has been applied to identify transcriptionally active regions (TARs), i.e. regions outside of known genes with mapped reads from RNA-Seq experiments. For C. albicans, 30-bp strand-specific Illumina short reads were used and 602 novel TARs were identified (33). Within the Candida clade there were further studies including Candida parapsilosis (34) and Candida dubliniensis (35). Additional fungal pathogens that have previously been studied include A. fumigatus (36,37), Aspergillus niger (38), Aspergillus flavus (39) and Aspergillus oryzae (40). These studies have contributed to the knowledge base of the respective pathogens by identifying novel TARs, elucidating transcriptional events, e.g. alternative splicing, and prompting further biological experiments. However, in studies regarding transcriptional landscapes in fungi, details of TARs were rarely investigated. In fact a TAR could be the result of noise, wrongly mapped reads, a protein-coding gene or non-protein-coding RNA species. As a consequence, TAR prediction needs to be combined with classical protein-coding gene prediction tools and tools for the prediction of non-protein-coding RNAs.
In order to comprehensively study the transcriptional landscape of the pathogenic fungus C. glabrata, we performed RNA-Seq in conditions that reflect situations potentially encountered in the human host. During contact with the host and throughout infection-associated processes, pathogens must adapt to harsh and dynamic environments, e.g. changing pH (41). In the human host, C. glabrata encounters niches of varying ambient pH, e.g. gastrointestinal tract, vaginal mucosa and bloodstream, reflecting the pathogen's ability to effectively adapt to pH (10,42,43). We therefore sequenced RNA generated from fungal cells cultured in alkaline (pH8) and acidic (pH4) conditions.
As resistance to stress is considered an important virulence factor of C. glabrata and may play a role in drug resistance (44), we exposed fungal cells cultured in yeast extract peptone dextrose (YPD) to S-nitrosoglutathione (GSNO), a potent inducer of nitrosative stress. Finally, C. glabrata cultured in YPD medium served as a control for the GSNO condition was used to identify genes required for standard fungal growth and metabolism and served as a control for the identification of differentially expressed genes (DEGs).
In this study, we updated the gene structure annotation of C. glabrata with the help of RNA-Seq data using a combination of TAR prediction with classical protein-coding gene prediction tools and tools for the prediction of non-protein-coding RNAs. Even though this approach has been used for de novo annotation of fungal pathogens (45,46), this is the first study applying it for re-annotation.
We re-annotated protein-coding genes, identified 5′ untranslated regions (UTRs) and 3′ UTRs for most genes and identified so far unknown non-protein-coding genes and novel protein-coding (NP) genes. Using real time quantitative PCR (RT-PCR), we validated four novel protein-coding genes, two of which were regulated during interaction with human neutrophilic granulocytes. Comparing our gene expression data to data for S. cerevisiae and C. albicans, we observed common and distinct patterns of stress adaptation in the three species. These findings will help to better understand gene expression dynamics and regulatory mechanisms in C. glabrata in the context of virulence evolution in fungi.
MATERIALS AND METHODS
Media and growth conditions
C. glabrata ATC2001 was cultured in YPD medium containing 1% yeast extract, 2% peptone and 2% D-glucose at 37°C. For the following stimuli exponentially grown cells were used. This has been achieved by a short-term induction culture before introduction of any specific stimulus. For GSNO conditions, C. glabrata was cultured overnight in YPD. 1 × 106 cells/ml were suspended in 50-ml fresh medium containing 0.6-mM S-nitrosoglutathione (GSNO) (Sigma Aldrich) for 60 min at 37°C while shaking at 180 rpm.
For the pH shift experiments, C. glabrata was cultured in M199 medium containing 9.8-g/l M199 powder (35.7-g/l HEPES, 2.2-g/l sodium carbonate) and adjusted to the described pH values with either 2-M sodium hydroxide or 12-M hydrochloric acid. The pH shift experiment was performed as previously described (41). Briefly C. glabrata was cultured by two subsequent overnight cultures in M199 pH4 at 37°C. 1 × 106 cells/ml were suspended in either M199 pH4 or pH8 for 60 min, at 37°C while shaking.
RNA isolation
Following 1-h incubation under the conditions described above, RNA was isolated as described previously (41). RNA quantity was determined with the Nanodrop 1000 (Thermo Scientific) and integrity was determined with an Agilent 2100 Bioanalyzer (Agilent Technologies).
RNA sequencing
Library preparation and sequencing of RNA was performed at GATC Biotech (Konstanz, Germany). After Poly-A filtering, libraries were generated for the conditions pH4, pH8, GSNO and YPD. All samples were prepared in biological triplicates and subject to removal of rRNA before cDNA library generation. From these libraries, 100-bp paired-end and strand-specific sequence reads were produced with Illumina HiSeq 2000. As the eukaryotic transcriptome may also contain long RNA molecules without a Poly-A tail, we prepared an additional fourth RNA sample to which no rRNA filter was applied. These libraries were sequenced as single-end, and not strand-specific. All raw RNA-Seq data were uploaded to Gene Expression Omnibus (47) and are publicly available (GSE61606).
Mapping and detection of DEGs and isoforms
For spliced read mapping, TOPHAT2 (48) was used (non-standard parameter: ‘no-mixed’, ‘no-discordant’, ‘b2-very-sensitive’, ‘max-intron-length 10 000′, for strand-specific samples ‘libtype fr-firststrand’). For counting the number of reads within exons and genes, htseq (49) was used (‘-m union’, ‘-t exon’). The expression values of reads were normalized by the number of reads per kilobase of exon region per million mapped reads (RPKM) (50). To identify genes that are differentially expressed, the raw counts were utilized with the tools DeSeq (51) and EdgeR (52) using standard parameters. Since samples without PolyA filter build outliers in RPKM values (most reads map to rRNA), they were not considered for DEG testing (Supplementary Table S1). We identified DEGs using an adjusted P-value cutoff < = 0.01 for EdgeR and DeSeq in combination with an absolute log2-fold-change cutoff < = 1. Functional categories enriched with DEGs were identified with the help of FungiFun2 (53). In order to compare our gene expression data to other available data, pH shift data from C. albicans (60 min only) (41) and S. cerevisiae (30 min) (54) as well as nitrosative stress data for C. albicans (33) and S. cerevisiae (80 min) (55) were obtained. For pH shift, DEGs were obtained with an adjusted P-value < = 0.01 and an absolute fold-change of > = 1.5. For the nitrosative stress condition, average fold-changes were calculated and then used to apply a 2-fold-change filter.
For the detection of significantly differentially expressed isoforms (DEIs), the mapped reads were utilized with the tools MATS (56), SplicingCompass (57) and DiffSplice (58). Results with a false discovery rate value < 0.05 were considered significant. All splicing events were manually checked using Integrative Genomics Viewer (59).
Prediction of protein-coding genes
The complete gene prediction workflow is shown in Figure 1. EVidenceModeler (EVM) (32) was used to combine weighted predictions from 14 different gene structure annotation approaches: From the current annotation at the Candida Genome Database (CGDB) (60) structures of verified open reading frames (ORFs) (weight = 10) and unverified ORFS (weight = 1) were downloaded. Alignments of protein sequences to the reference genome (weight = 1) for C. albicans SC5314, C. albicans WO1, S. cerevisiae, S. castelli and K. polysporus were created with exonerate (61). Hidden-Markov-Models (HMM) for the de novo gene prediction tools SNAP (26) and AUGUSTUS (27) were trained with 207 verified ORFs from GCDB. AUGUSTUS HMM parameters for UTRs were additionally trained with the help of 200 UTR examples from visual examination of the mapping data. SNAP was run with standard parameters, AUGUSTUS with the option to create UTRs.
Figure 1.
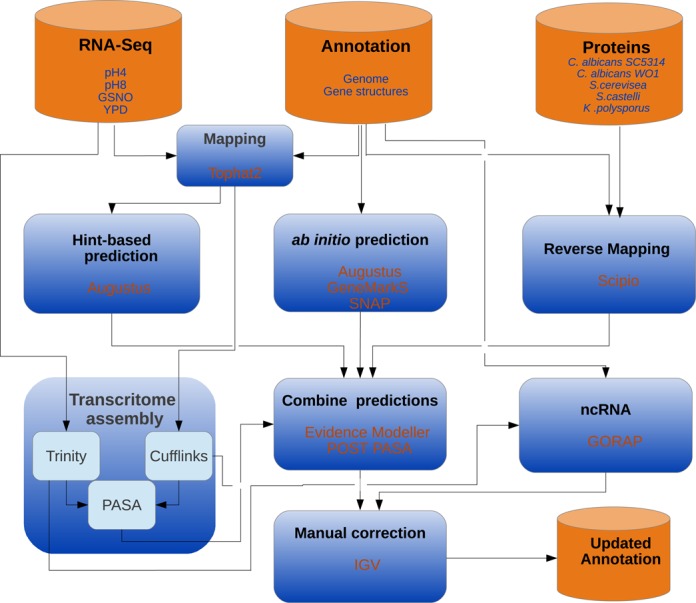
Workflow. Overview of the applied workflow for updating C. glabrata structural gene annotation. RNA-Seq data were used for transcriptome assembly and to generate hints for the existence of introns and exons. The RNA-Seq-based approach was combined with ab initio gene prediction and mapping of proteins from homologous species. Results from different approaches were combined in a weighted manner by EVidenceModeler. Finally, changed genes were manually corrected with the help of the Integrated Genomics Viewer (IGV). For the prediction of non-protein-coding RNAs, the tool GORAP was used. As input for GORAP we used the genome sequence as well as mapping-based transcriptome assembly (CUFFLINKS) and mapping-free transcriptome assembly (TRINITY).
De novo gene prediction tool GeneMarkS (25) (weight = 0.3) does not need any training and was used with standard parameters.
Mapped reads from RNA-Seq data were utilized to create hints for the location of introns and exons (for details and parameters see (29)). Those hints were integrated in a hint-based run of AUGUSTUS (‘AUGUSTUS_RNA’, weight = 5).
Mapping-based transcript assembly was performed with the help of CUFFLINKS (31) following the protocol of the CUFFLINKS-team (62). Mapping-free transcript assembly was performed using TRINITY (30) based on the merged strand-specific RNA-Seq data (alignment option = blat). The Program to Assemble Spliced Alignments (PASA, standard parameters) (63) was used for spliced alignment mapping of the transcriptome assemblies from TRINITY and CUFFLINKS. All PASA gene structures predicted by PASA were integrated (weight = 5). TRANSDECODER was used to identify those gene structures from PASA which code for proteins (weight = 5).
Those genes of the POST-PASA annotation which are different to the CGDB annotation were studied using the Integrative Genomic Viewer (IGV) (59). By visual examination of all gene structures predictions at those loci and the mapped reads from RNA-Seq data, a final annotation was created.
Novel protein-coding genes were further analyzed by BLAST (blastp 2.2.27+) against the non-redundant protein database. To use the latest sequences of Candida spp, Protein-BLAST and protein-to-genome Blast (tblastn) at the CGDB were used online against all available species (60) and S. cerevisiae (64). Finally, Blast2Go (65) and InterproScan (66) were performed with the novel protein-coding genes using standard parameters.
Confrontation of C. glabrata with human polymorphonuclear leukocytes
Venous blood of healthy volunteers was collected in ethylenediaminetetraacetic acid monovettes (Sarstedt). Polymorphonuclears (PMNs) were subsequently purified as described (67,68). Immune cells were suspended in SCGM media (CellGenix), counted with a cellometer X2 (Nexcelom) and immediately subject to confrontation assay.
Confrontation of 3 × 106 C. glabrata cells with 6 × 106 PMN was performed in one well of a 6-well plate (Corning) in a total volume of 3-ml SCGM media. Following the incubation period, 20-U DNase (Invitrogen) were added to the co-incubation to dissolve DNA based structures, which trapped fungal material. Co-incubations were flooded with five volumes of RNAlater (Ambion). The solution was mixed with an equal volume of 4°C H2O to lyse neutrophils. Fungal cells were harvested by centrifugation at maximum g for 5 min and subject to immediate RNA isolation.
Real-time quantitative PCR
RT-PCR was performed using the Precision Onestep qRT-PCR Mastermix containing SYBR green (Pimerdesign) according to the manufacturer's instructions in conjunction with the Stratagene Mx3005P (Agilent Technologies) instrument. Fold changes in gene expression were determined in 25-ng/μl template RNA using the ΔΔCt method (69) and data were normalized against PDC1. This gene was selected for normalization as its expression was stable under various experimental conditions, including the interaction with immune cells (70). Primers used in this study are listed in Supplementary Table S1.
Prediction of UTR
The gene structures of EVM were used as input for POST-PASA (63) in order to generate 3′ and 5′ UTR based on mapped RNA-Seq reads and to identify splice variants. POST-PASA predicted 2454 3′ UTRs and 2428 5′ UTRs. To add further UTR annotation, AUGUSTUS_RNA predictions were used. To this end, gene structures of the POST-PASA prediction without 3′ or 5′ UTR annotation where all exons have the same coordinates as in the AUGUSTUS_RNA predictions were identified. For those genes, the AUGUSTUS_RNA UTR predcitions were copied to the POST-PASA annotation leading to additional annotation of 2490 3′ UTRs and 2498 5′ UTRs.
Prediction of non-protein-coding genes
The tool suite GORAP (www.rna.uni-jena.de/software.php, Riege & Marz) was used to predict non-protein-coding RNAs based on the reference genome and the constructed transcriptome assemblies (TRINITY (30), CUFFLINKS (31)).
GORAP uses BLAST (71), Infernal v1.1 (72), RNAmmer v1.2 (73), tRNAscan-SE v1.3.1 (74) and Bcheck v0.6 (75). Additionally, GORAP screens for known non-protein-coding genes provided by the Rfam database v. 11.0 (76). After running GORAP, all resulted Stockholm alignments, related FASTA and GFF files were manually reviewed. Additional detection of small nucleolar RNAs (snoRNAs) was performed by snoStrip v1.0 (77) followed by manual validation. We accepted predictions at the transcriptomic level only if the sequences could be mapped to the genomic level.
RESULTS
From RNA-Seq data to a re-annotated genome sequence
To comprehensively study the transcriptional landscape of C. glabrata, we cultured fungal cells in nutrient-rich media (YPD) as well as in nitrosative stress (GNSO), pH4 and pH8. Three independent biological replicates were used for sequencing after rRNA filtering. Additionally one fourth library of each condition was sequenced without rRNA filtering (see the Materials and Methods section). 244 million reads mapped to the reference genome that corresponds to theoretical genome coverage of ∼900-fold (Supplementary Table S1).
With the help of the Poly-A filtered samples we were able to identify expression of 5418 coding and non-protein-coding genes, while we did not find evidence of the expression of 64 genes. Out of those 64 genes, 56 were tRNAs, four ORFs and one non-protein-coding RNA (CaglfM01). The non Poly-A filtered libraries added expression values to 12 of the 64 genes. The remaining genes without expression are 48 tRNAs, the mitochondrial ribosomal protein VAR1 (CaglfMp02), the subunit of the mitochondrial ATP synthase ATP8 (CaglfMp08) and the uncharacterized ORF CAGL0G06644g. These genes may not have been detected in our study because they were either not expressed under our conditions or they were previously incorrectly annotated.
In order to update the current gene structure annotation of C. glabrata, we made use of the RNA-Seq data in three different ways (Figure 1): (i) after mapping to the genome, read location allowed the determination of exon and intron locations, which we used as ‘hints’ for AUGUSTUS (27). (ii) We used CUFFLINKS (31) to create a transcriptome assembly based on the aligned reads. (iii) We created an unbiased mapping-free transcriptome assembly with the help of the tool TRINITY (30).
Next, we mapped back TRINITY and CUFFLINKS assemblies to the reference genome with PASA (63), applied the de novo gene prediction tools SNAP (26), AUGUSTUS (without hints) (29), GeneMarkS (25) (see the Materials and Methods section) and aligned 4501 protein sequences of homologous species to the reference genome. Finally, we combined the different gene structure annotations into consensus structures with EVidenceModeler (EVM) (32) and predicted untranslated upstream and downstream regions with PASA (63) and AUGUSTUS (29) (see the Materials and Methods section).
The resulting final protein-coding gene prediction consisted of 5288 protein-coding genes, 5305 transcripts, 5523 exons and 175 introns (Table 1 and Supplementary Table S1). The new annotation contains 38 novel introns, 4906 5′ UTRs, 4923 3′ UTRs, 49 novel protein-coding (NP) genes and 58 novel non-protein-coding genes. With the help of our strand-specific data we identified seven genes that were annotated on the wrong strand. Of these seven genes, four overlap with genes on the opposite strand which have been extended by our annotation and three genes of the old annotation completely overlap with novel genes on the opposite strand of the novel annotation. As an example, Figure 2A visualizes the locus around the gene CAGL0L08470g where only a very few reads map to the minus strand while most reads map to the plus strand and support the gene Novel_protein-coding50 (NP50).
Table 1. Comparison gene structures in old and new annotation.
| Feature | Old | New | Add | Rem | Merg | Split | Ext | Short |
|---|---|---|---|---|---|---|---|---|
| Introns | 137 | 175 | 38 | 0 | 0 | 0 | 2 | 1 |
| 5′UTR | 0 | 4906 | 4906 | 0 | 0 | 0 | 0 | 0 |
| 3′UTR | 0 | 4923 | 4923 | 0 | 0 | 0 | 0 | 0 |
| Proteincoding Genes | 5213 | 5288 | 49 | 7 | 2 | 1 | 123 | 6 |
| Pseudogenes | 22 | 42 | 0 | 0 | 0 | 14 | 0 | 0 |
| tRNAs | 230 | 230 | 0 | 0 | 0 | 0 | 0 | 0 |
| ncRNAs | 10 | 68 | 58 | 0 | 0 | 0 | 6 | 1 |
| rRNAs | 6 | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
This table compares the old and new annotation for different genomic features. As main results, we identified 49 novel protein-coding genes and 58 novel non-protein-coding genes, as well as added UTR annotation to the majority of gene. In this study, we split 14 previously annotated pseudogenes into 32 genes whose translated sequences do not contain a stop codon. Most probably these are not pseudogenes. Add = added, Rem = removed, Merg = merged, Split = splitted, Ext = extended, Short = shortened.
Figure 2.

Genome tracks of novel and changed genes. Genome tracks, including mapped RNA-Seq reads, visualizing novel and changed genes. Reads originating from plus strand are blue; reads from minus strand are red. The histogram at the top indicates the number of reads. Genes from Candida Genome Database annotation are visualized in blue, while our novel annotation is red. For simplicity only coding regions are shown. (A) The reads on chromosome L around CAGL0L08470g mainly originate from the plus strand. For this reason, we removed this ORF from the annotation. Instead, we identified a novel gene on the opposite strand. (B) At the locus around CAGL0A01606g on chromosome A where a number of reads are mapped after being spliced by TOPHAT2, indicating a so far unknown intron within this gene which has been added by our novel annotation. (C) On chromosome G, CAGL0G00110g is annotated as pseudogene but also as putative adhesin containing a GPI anchor. The translated protein sequence contains an in-frame stop codon. Our novel annotation split this gene as supported by the mapped reads. CAGL0G00110g.2 contains a GPI anchor. While CAGL0G00110g.1 is not differentially expressed in the tested conditions, CAGL0G00110g.2 is ∼5-fold upregulated in pH4 compared to pH8. (D) On chromosome M, we identified the so far unknown protein-coding gene that is strongly supported by strand-specific reads.
Introns and pseudogenes
Prior to our study, 138 introns had been identified in the genome sequence of C. glabrata. Based on our RNA-Seq data, we were able to identify 38 new introns. Eight of these were located in previously known genes, including the new intron in CAGL0A01606g, whose homologs have DNA binding activity. A number of reads were spliced during mapping, therefore indicating a previously unknown intron (Figure 2B). Based on the actual structure of assembled transcripts and aligned reads, we modified several known introns (Supplementary Table S1), for example the intron in ASC1 (CAGL0D02090g) was shortened and the intron in CAGL0L02255g was extended. For five genes, our updated annotation detected a so-far unknown second isoform including ANC1 (CAGL0M02739g) which codes for a transcription initiation factor. Since different isoforms of one gene suggest condition-specific alternative splicing, we used the tools MATS (56) and DiffSplice (58) to detect significantly DEIs. Using this approach, we detected 22 DEIs during pH change and 100 when adding GSNO. Five novel genes are DEI. Interestingly, the adhesins EPA6 (CAGL0C00110g) and EPA20 (CAGL0E0275g) express different isoforms in both conditions, while EPA3 (CAGL0E006688g) is differentially spliced in GSNO compared to nutrient-rich medium. Altogether, five of the novel isoforms are DEI. The main alternative splicing event is intron retention, which is generally the most frequent splicing event in fungi (78).
The current C. glabrata annotation contains 22 pseudogenes. Out of these, eight contain a predicted GPI anchor (79) while seven have been experimentally demonstrated to function as adhesins (80). Since the current annotation predicts in-frame stop-codons for these proteins, we took a detailed look at these potential virulence factors. In our updated annotation, we split 14 previous pseudogenes into 32 genes whose translated sequences do not contain a stop codon. Twelve of these genes are identical to the terminal part of the previously predicted pseudogenes, while nine still contain a GPI anchor predicted by the fungal big-Pi tool (79) (also used in (80)). Figure 2C visualizes the genome track of the locus of CAGL0G00110g, an annotated pseudogene that we split into two genes, where the second protein (CAGL0G00110g.2) contains a GPI anchor.
Non-coding RNA species in C. glabrata
We performed a homology-based non-protein-coding RNA (ncRNA) analysis for C. glabrata with the software GORAP (www.rna.uni-jena.de/software.php, Riege & Marz). This software was developed for automated whole genome ncRNA screenings. Based on improved homology search strategies and specially developed filtering procedures GORAP ensures a low prediction of false positives and increased sensitivity. As input, we used the genome sequence, as well as constructed transcriptome assemblies (CUFFLINKS (31), TRINITY (30)).
In total, 68 non-protein-coding RNAs were identified. Out of these, 58 have not been detected previously (Table 1 and Supplementary Table S1). Seven of the novel ncRNAs were identified in the introns of C. glabrata genes, five new ncRNAs were located on the opposite strand of coding sequences and 23 novel ncRNAs were identified to be controlling regions, located in UTRs of different ORFs. In comparison to the CGDB annotation, GORAP failed to identify three ncRNAs which we added manually: the telomerase TLC1 (CAGL0I04700r), which extends telomere ends by a repetitive sequence motif (81), the RNA component of the mitochondrial RNase and RNase P (CaglfM01) (82) and the snRNA H1 (CAGL0L08044r).
With the help of the GORAP software, we were able to identify the evolutionary-related RNase MRP that initiates mitochondrial replication and separation of 18S from 5.8S rRNA in basal eukaryotes (83). We re-annotated the RNA part in the C. glabrata spliceosome: in detail, the U1, U2, U4, U5 and U6 RNAs well as the part of the U2-type complex major spliceosome were extended. Finally, we shortened the ncRNA part of the signal recognition particle SRP (CAGL0K01961r), which guides proteins to the endoplasmic reticulum (84). Of six rRNAs in CGDB, GORAP found three while the other three were added after manually checking their expression in the Genome browser.
Novel protein-coding genes in C. glabrata
A principal aim of this study was the identification of novel protein-coding (NP) genes, i.e. yet unknown loci coding for proteins in the genome of C. glabrata. With the help of our structural annotation approach that is supported by transcriptomic data (Figure 1), we identified 49 NP genes (Supplemental Table S1) which are not annotated in the current assembly of the C. glabrata genome. As an example, we show a predicted novel gene that is strongly supported by mapped reads (Figure 2D).
Out of these 49 NP genes, 17 are located in gene loci that are conserved between C. glabrata and S. cerevisiae (Figure 3A). Potential S. cerevisiae homologs were found for nine of these genes (Figure 3A). The remaining 32 C. glabrata novel protein-coding genes are located in genomic regions that are not conserved between the two yeast species. Nonetheless, this group contains five NP genes that have potential homologs in S. cerevisiae (Figure 3A). One example for a novel gene that is part of a conserved locus is CgNP1. The gene is located at chromosome A in C. glabrata. The highly similar genomic region on chromosome IV of S. cerevisiae is occupied by the yeast gene WIP1 (Figure 3B). By comparing protein sequences (85), we could show that CgNp1 shares 33% identity with Wip1 (Figure 3C). As Wip1 is the baker's yeast homolog of the human CENP-W protein (86), we predict that CgNP1 might have the same function in C. glabrata. Interestingly, no homolog for either CgNp1 or Wip1 could be identified in C. albicans.
Figure 3.
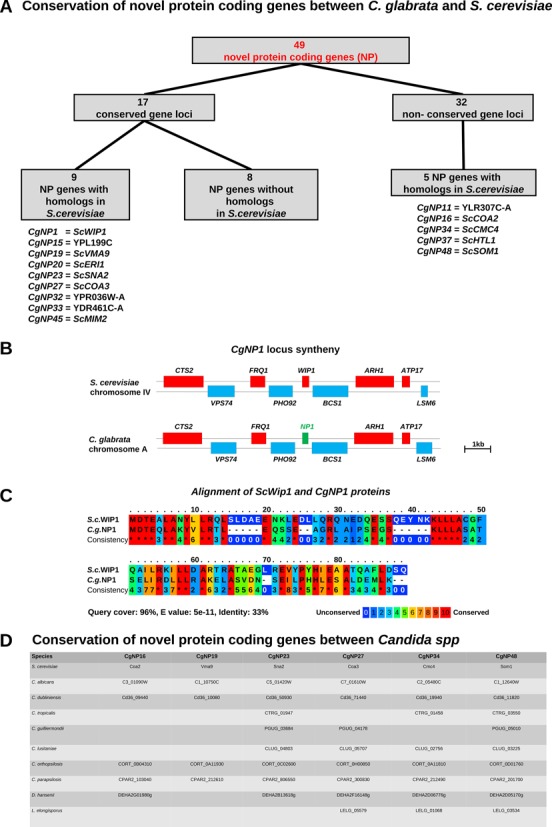
Novel protein-coding genes in C. glabrata. (A) Analysis of novel protein-coding (NP) genes in C. glabrata. For all NPs, protein BLAST search was performed within the NCBI database as well as the Saccharomyces and Candida genome databases. The gene locus of every NP gene was compared to S. cerevisiae by looking for the location of at least four neighbored genes. Seventeen NP genes were found in conserved loci with S. cerevisiae, of which nine are homologs. Of the 32 NP genes in non-conserved loci, five have homologs in S. cerevisiae. (B) The gene locus of CgNP1 is compared to the corresponding locus of S. cerevisiae WIP1. (C) Alignment of the CgNp1 and ScWip1 protein sequences indicates that CgNp1 is the Cenp-W homolog in C. glabrata. (D) Protein IDs of proteins with highest sequence similarity in all Candida species from CGDB and S. cerevisiae. Only highly similar sequences (E-value <0.001) are shown.
In order to systematically analyze which NP genes have potential homologs in pathogenic and non-pathogenic yeasts, we blasted all NP against all proteomes of CGDB and S. cerevisiae (64). For six NP a significant BLAST hit was found (Figure 3D): CgNP16, CgNP19, CgNP23, CgNP27, CgNP34 and CgNP48. All of which were also conserved in S. cerevisiae.
As we were interested if the novel proteins were also missed in the annotation of other yeasts, we blasted the NP sequences against the genomes (tBLASTn; see the Materials and Methods section) of Candida spp and S. cerevisiae. In addition to the six proteins for which we found matches at the protein level, we found exact matches of CgNP40 in the genome of all other Candida spp and S. cerevisiae, indicating that this protein is also missing in their annotation. CgNP40 is located close to the highly conserved 25s ribosomal RNA RDN25.
A total of 11 CgNP genes encode putative transmembrane proteins (Supplementary Table S1). Additionally, one (CgNP25) gene encodes for a putative GPI-anchored transmembrane protein. This gene is very similar to those of the EPA gene family within the C. glabrata genome, and might be member of this gene family (Supplementary Table S1). Interestingly, eight novel genes were localized in subtelomeric regions, close to members of the EPA or PWP gene families (Supplementary Table S1).
We predicted functional Gene Ontology (GO) categories (65,87) for four proteins of the NP genes (Supplementary Table S1). Based on GO categories, we predict that CgNP16 and CgNP27 act in the assembly of mitochondrial respiratory chain complex IV, while CgNP19 might have ATPase activity and CgNP34 is part of an organelle.
As a control of our predictions, we validated the expression of four randomly selected novel genes by quantitative RT-PCR. The genes CgNP4, CgNP11, CgNP32 and CgNP38 were found to be upregulated after the shift from pH4 to pH8 (Figure 4). Under nitrosative stress conditions, CgNP32 was strongly downregulated while the other three genes were not affected (Figure 4). As we are interested if the NP genes play a role in virulence, we tested the expression of these four genes during the confrontation of C. glabrata with human neutrophilic granulocytes. While CgNP11 and CgNP32 were stable in their expression, we observed a downregulation of CgNP4 and CgNP38 in response to confrontation with PMNs (Figure 4). Therefore, we could not only validate the expression of these four genes but also provide hints for putative functions in host–pathogen interaction.
Figure 4.
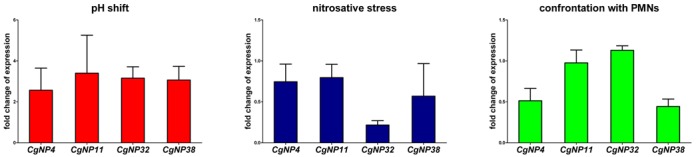
Real-time quantitative PCR of selected NP genes during pH change, nitrosative stress and interaction with immune cells. The novel protein-coding genes NP4, NP11, NP32 and NP38 were randomly selected for a gene expression analysis under three different conditions. For all conditions, cells were incubated at 37°C for 60 min prior to RNA isolation. Total RNA was used for quantitative RT PCR. Shown are the fold changes of NP gene expression in M199 medium with pH8 against M199 medium pH4 (pH shift), YPD with 0.6-mM S-nitrosoglutathione (GNSO) against YPD (nitrosative stress) and for SCGM medium with freshly isolated human neutrophils against SCGM medium alone (confrontation with PMNs). Based on three independent experiments, the expression of the NP genes was normalized against the C. glabrata house keeping gene PDC1 (70).
C. glabrata exerts distinct responses to pH and nitrosative stress
A total of 834 C. glabrata genes (including 15 NP genes) were differentially expressed during the shift from acidic to alkaline pH, with 426 down- and 409 upregulated (Supplementary Table S1). In order to systematically analyze the list of DEGs, we scanned for significantly enriched GO categories (Figure 5A). Within the GO category ‘heme binding’, 11 of 20 genes are DEGs (adjusted P-value = 6.19 × 10−3) indicating that C. glabrata cells reorder their iron homoeostasis in response to pH changes. Furthermore, the enrichment of ‘ubiquinol-cytochrome-c reductase activity’ (adjusted P-value = 2.51 × 10−3) shows that the redox state of the cells is changed.
Figure 5.
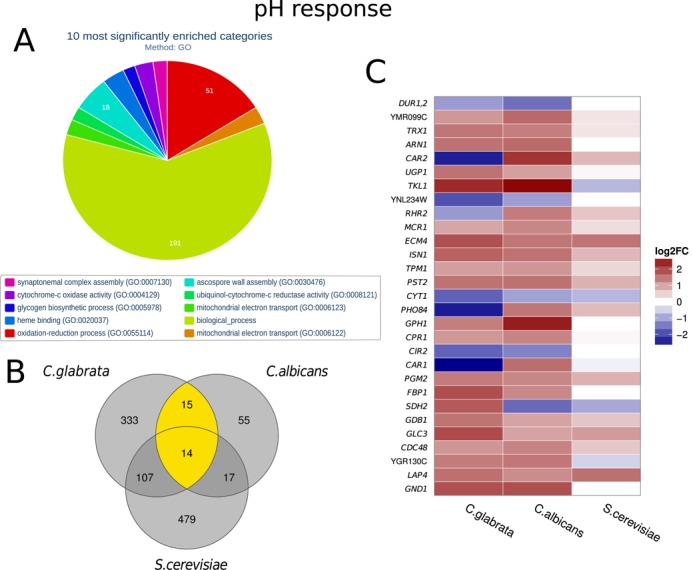
Gene expression of C. glabrata during pH change. (A) Systematic analysis of DEGs with the help of enriched Gene Ontologies categories calculated with FungiFun2. The top significantly enriched molecular functions ore biological processes are shown. (B) Number of differentially expressed genes in response to pH change in C. glabrata compared to C. albicans and S. cerevisiae. Only homologous genes (defined by CGDB) are shown. Twenty nine genes are shared between the pathogenic yeasts, while 14 are shared by all. (C) Heatmap comparing all homologous DEGs shared between C. glabrata and C. albicans (yellow in (B)). Names of S. cerevisiae genes are shown; see Supplementary Table S1 for IDs and names of the other homologs. While the majority of pH responsive genes are similarly regulated, some genes strongly differ. For example, C. glabrata PHO84 homolog was downregulated in response to alkaline pH, while its homologs were upregulated.
C. glabrata and C. albicans are both opportunistic human pathogens, challenged with, among other stresses, pH changes within the host. Considerable attention is garnered by the comparison of these two species. On the other hand, S. cerevisiae is more closely related to C. glabrata than C. albicans but non-pathogenic. In what follows, we compare pH response of C. glabrata to recently published studies of C. albicans (41) and S. cerevisiae (54). For this comparison, we took into account genes that have homologs in all three fungi. As shown in Figure 5B, C. glabrata and S. cerevisiae generally have more pH responsive genes than C. albicans. Interestingly, 15 genes are exclusively shared by the pathogenic fungi, while 14 genes are shared by all three fungi. The majority of these genes showed similar dynamics of increase or decrease of expression in all fungi (Figure 5C). While most of the genes showed similar expression dynamics between the two pathogenic yeasts, four genes, CAR1, CAR2, RHR2, PHO84, were upregulated in C. albicans, but downregulated in C. glabrata and S. cerevisiae (Figure 5C). Only the SDH2 homolog was upregulated in C. glabrata, but downregulated in the other two fungi (Figure 5C). The homologs of the transketolase encoding gene TKL1 were upregulated in both pathogens, but not in S. cerevisiae. Taken together, these results indicate a distinct species-specific pH response in C. glabrata.
During nitrosative stress, 2564 genes were differentially expressed by C. glabrata (Supplementary Table S1) including 23 NP genes. Significantly enriched GO categories are visualized in Figure 6A. One of the most significantly enriched molecular functions is ‘RNA polymerase III activity’ (adjusted P = 3.59 × 10−4) which indicates that fungal cells strongly and quickly adapt to the new environment. This is further supported by several enriched GO categories dealing with RNA processing steps as well as translation. The biological process enriched with the most DEGs is ‘oxidation–reduction process’ (adjusted P = 8.59 × 10−6) which clearly indicates that fungal cells are confronted with stress. Compared to S. cerevisiae (55), both pathogenic yeast regulate a substantially larger number of genes in response to nitrosative stress underlining the importance of stress response during interaction with the host (Figure 6B). There are 784 genes exclusively regulated in C. albicans (33) and C. glabrata, while only 58 are shared by all fungi. The expression pattern of the shared genes is different for each of the three fungal species (Figure 6C). Only the two genes YHB1 and INO1 were upregulated in all three species. C. albicans YHB1 encodes a flavohemoglobin involved in the detoxification of nitric oxide (88). Its C. glabrata homolog, CAGL0L06666g, has not yet been functionally characterized, but is strongly upregulated in GNSO-containing media which indicates that CAGL0L06666g may be an important nitrosative stress gene for C. glabrata as well. Six genes are more similarly regulated by the more closely related species C. glabrata and S. cerevisiae. Four of them are upregulated in C. glabrata and S. cerevisiae and downregulated in C. albicans, while for FBP1 and SSA4 the opposite is true. Interestingly, five genes are specifically upregulated in C. glabrata, but not in the two other fungi indicating a specific importance for C. glabrata. These include four genes involved in RNA interaction: NOP13, NOP58, CBF5, NSA2 as ENP2 (Figure 6C).
Figure 6.
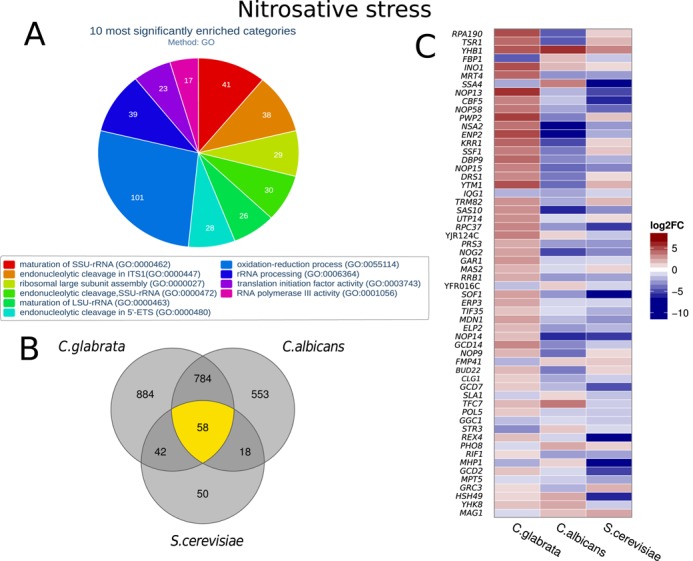
Gene expression of C. glabrata during nitrosative stress. (A) Systematic analysis of DEGs with the help of enriched Gene Ontologies categories calculated with FungiFun2. The top significantly enriched molecular functions ore biological processes are shown. (B) Number of differentially expressed genes in response to nitrosative stress in C. glabrata compared to C. albicans and S. cerevisiae. Only homologous genes (defined by CGDB) are shown. Pathogenic yeasts regulate more genes in response to nitrosative stress than non-pathogenic. (C) Heatmap comparing all homologous DEGs shared between C. glabrata and C. albicans and S. cerevisiae (yellow in (B)). Names of S. cerevisiae genes are shown; see Supplementary Table S1 for IDs and names of the other homologs. Similarly regulated genes include the marker gene for nitrosative stress YHB1. On the other hand, RNA processing and ribosome biogenesis genes like HAS1, TSR1 and RPA190 are strongly upregulated in C. glabrata, but not in C. albicans.
DISCUSSION
Understanding microbial virulence is a prerequisite for the identification of novel diagnostic approaches and innovative therapeutic tools. With modern tools, systems biology based analyses of host–pathogen interactions have gained considerably in importance and impact. A major qualification for this is exact and complete knowledge of the genomic structure. This is especially important as growing information suggests that beside protein-coding genes, non-coding transcripts have a major impact on the biology of microorganisms. For this reason the transcriptional landscapes of several fungal species, including pathogens, have previously been studied. However, in most of these studies TARs were identified as genomic regions, with so far unknown genes, to which some of the sequenced reads could be mapped. It remained unclear if mapped reads represented noise, were mapped erroneously, coded for proteins or coded for ncRNA species. For example, previously solely the length of the identified TARs was used to decide whether or not they might code for proteins (33). In this study, we have advanced this approach to not only identify novel TARs but also reliably predict which TARs code for proteins or for non-protein-coding RNA species using state-of-the-art bioinformatics tools. For this we used deep sequencing data for analysis of the transcriptional landscape of the human fungal pathogen C. glabrata. This pathogenic yeast is the second most common Candida spp. and accounts for a growing number of invasive infections associated with high mortality (17). Genome wide transcription was analyzed under standard growth conditions as well as under pH and nitrosative stress, which represent major characteristics of niches encountered during colonization or infection of the human host (89,90).
Although time-course analyses would be helpful for a more in-depth understanding of temporal expression changed during adaptation to environmental stresses (91,92), our data suggest that the transcriptional response of C. glabrata to pH is similar to C. albicans (Figure 5B) and S. cerevisiae. Additionally, C. glabrata seems to react specifically to pH changes. Here, we report four genes (CAR1, CAR2, RHR2, PHO84), which were upregulated during pH shift in C. albicans, but downregulated in C. glabrata. Differences in the pH responses of the two pathogenic yeasts have been reported on protein level (93). Despite this overall pattern, we could identify 15 genes that are differentially regulated by the two pathogenic species but not by S. cerevisiae. These may relate to host-specific adaptation of C. glabrata and C. albicans which is not present in S. cerevisiae. Interestingly, the response of the three fungal species to nitrosative stress is much more divergent and differs largely between the pathogenic species and non-pathogenic S. cerevisiae. A remarkable fraction of the C. glabrata genome is differentially expressed during the response to nitrosative stress (48% of transcripts are DEGs; 2598 of the total number of 5378). Our data, together with the findings of Causton et al. (94), provide further evidence that the proportion of yeast genome regulated in any given condition can in fact be the majority of the genome. This highlights the efforts required by the organism to respond to changing environments. Our data indicate that the response of C. glabrata to nitrosative stress is different to the responses of C. albicans and S. cerevisiae (Figure 6C). After phagocytosis macrophages exert nitrosative stress on fungal cells. Since C. glabrata is the only yeast under consideration which can replicate within macrophages (10), it may require a distinct stress response.
In addition to new insights into fungal gene regulation under environmental conditions relevant for host–pathogen interaction, we were able to significantly refine knowledge about the genome structure of C. glabrata. Previously, transcriptional start sites were mainly unknown in this pathogen. With the help of predicted untranslated upstream regions derived from our data, future research can now more precisely address regulatory functions located in the promoter regions. Furthermore, UTR prediction will significantly improve the process of counting the number of reads per transcript during future RNA-Seq data preprocessing, thus generating counts which better represent expression levels.
In addition to UTR prediction, we also performed a homology based non-protein-coding ncRNA analysis for C. glabrata. Interestingly, within our transcriptome assembly we found human (metazoan) 7SK RNA as well as bacteria-specific tmRNA, RNAI and Spot42 (spf) RNA. Since these ncRNAs most probably do not exist in a fungal genome, the predictions likely result from low level contamination, which typically occur in deep sequencing. This is also suggested by the fact that only a very minor fraction of reads mapped outside the fungal genome (0.19% bacterial collection, 7 × 10−8% human) and indicates the high sensitivity of the GORAP tool used for identification of ncRNAs. In total, 68 C. glabrata ncRNAs were identified by GORAP, of which 58 had not been previously detected (Table 1 and Supplementary Table S1). Seven of the novel ncRNAs were located in the introns of C. glabrata genes, five new ncRNAs were located on the opposite strand of coding sequences and 23 novel ncRNAs were identified in UTRs of different ORFs. In comparison to the CGDB annotation, GORAP failed to identify three ncRNAs that we added manually: the telomerase TLC1 (CAGL0I04700r), which extends telomere ends by a repetitive sequence motif (81), the RNA component of the mitochondrial RNase and RNase P (CaglfM01) (82) and the snRNA H1 (CAGL0L08044r). With the help of the GORAP software, we were also able to identify the evolutionary-related RNase MRP that initiates mitochondrial replication and separation of 18S from 5.8S rRNA in basal eukaryotes (83). We re-annotated the RNA component in the C. glabrata spliceosome: in detail, the U1, U2, U4, U5 and U6 RNAs as well as the part of the U2-type complex major spliceosome were extended. Finally, we shortened the ncRNA part of the signal recognition particle SRP (CAGL0K01961r), which guides proteins to the endoplasmic reticulum (84). Of six rRNAs in CGDB, GORAP found three while the other three were added after manually checking their expression in the Genome browser.
Importantly, we were able to identify 49 novel protein-coding and 55 non-coding genes. With regard to previous studies, the number of new genes is comparatively low. One reason could be that C. glabrata is closely related to one of the best studied eukaryotic organisms, S. cerevisiae. Thus, a closer similarity of their genomes may have allowed a better annotation of C. glabrata in previous efforts (9). It should also be noted that the majority of the novel proteins is rather short (40 proteins with less than 100 amino acids). This indicates that these loci may have been overseen in previous annotations, stressing the advantage of using RNA-Seq for gene prediction. Among the newly annotated genes, 17 were found in gene loci which were conserved between C. glabrata and S. cerevisiae (Figure 3 and Supplementary Table S1), while others are in totally different loci or do not have homologs. This hints toward two ways in which C. glabrata might have evolved differently from S. cerevisiae. Firstly, genes without synteny of homologs may have derived from the common ancestor and been lost during adaptation of the different fungi to their respective niches. Secondly, novel genes in synteny may have been gained by C. glabrata during co-evolution with the human host and therefore may be virulence factors.
Interestingly, only six of the novel protein-coding genes have homologs in C. albicans or the other CUG-clade species, while a total number of 14 have homologs in S. cerevisiae. The fact that no newly identified gene (NP) was shared by C. glabrata and the other Candida, but not by S. cerevisiae, again relates to the phylogenetic closeness of C. glabrata and S. cerevisiae. One interesting example was CgNP1 that likely encodes the homolog of ScWIP1. The ScWip1 protein is the CENP-W homolog in S. cerevisiae (86). In humans, CENP-W forms a complex with CENP-T and its assembly is a precondition for the conversion of centromeric chromatin into a mitotic state (95). Among all Candida spp., only C. glabrata possesses homologs for CENP-T and CENP-W, indicating kinetochore assembly mechanisms similar to that of S. cerevisiae but distinct from other Candida spp.(96,97).
Despite their small sizes, many NP contain a transmembrane domain (Supplementary Table S1), which indicates potential roles for these proteins in interaction with the abiotic or biotic environment. Thus, as potential interaction partners of the human host, these proteins are attractive targets of future studies. It will be interesting to elucidate which other conditions regulate expression of these genes and, subsequently, study their function. As a proof of concept, we have shown that two NP genes (CgNP4 and CgNP38) are in fact differentially regulated upon contact with human neutrophils, which are a central component of antifungal immunity (98,99). Therefore, these genes may well be involved in mediating host–pathogen interaction. Another group of virulence associated genes that has been experiencing major refinement by our data are the C. glabrata adhesins that mediate adherence to human epithelial cells (100,101). Our gene structure prediction indicated that 14 genes that were previously annotated as pseudogenes need to be split into 32 genes without an in-frame stop codon. We manually checked the pattern of mapped reads for these genes which indicated that splitting was correct. Interestingly, most of these genes are predicted or experimentally proven adhesins (80). The re-annotation of these virulence-related proteins and their corrected protein sequence will allow more detailed studies focusing on C. glabrata adherence. Furthermore, we provide clear evidence for alternative splicing in C. glabrata also affecting adhesin genes. Alternative splicing is a process that helps organisms to create a broad range of proteins with a smaller number of genes and thus contributes to the complexity of the genome and the organism. Even though the C. glabrata genome generally contains a small number of introns (175 introns, including 38 novel introns), we were able to detect condition-specific alternative splicing events with the adhesins EPA6 (CAGL0C00110g) and EPA20 (CAGL0E0275g) expressing different isoforms in both pH shift and nitrosative stress and EPA3 (CAGL0E006688g) showing differential splicing under nitrosative stress. These data clearly suggest that C. glabrata uses alternative splicing to increase the number of possible proteins per gene in the adhesin gene family.
Taken together, the data described in this study provide important insight into virulence of C. glabrata. Furthermore, re-annotation of the genome and identification of unknown ncRNAs offers new options for studying virulence and host–pathogen interaction in C. glabrata. These studies will be a major prerequisite for further elucidating the evolution of discrete virulence traits in pathogenic ascomycete yeasts.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
These authors contributed equally to the paper as first authors.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) CRC/Transregio 124 ‘Pathogenic fungi and their human host: Networks of interaction’ subproject INF [to J.L., R.G.] subproject B3 [to K.R., M.M.]; Zeiss Stiftung [to M.M.]; DFG project MA-5082/1 [to M.M., K.R.]; Excellence Graduate School, Jena School for Microbial Communication [to S.D.]; German Ministry for Education and Science in the Program ‘Unternehmen Region’ BMBF 03Z2JN21 [to O.K., R.M., S.D., M.W, D.H.]; DFG project KU1540/3-1 [to O.K., R.M., S.D., M.W, D.H.]. Funding for open access charge: Deutsche Forschungsgemeinschaft (DFG) CRC/Transregio 124 ‘Pathogenic fungi and their human host: Networks of interaction’ subproject INF.
Conflict of interest statement. None declared.
REFERENCES
- 1.Brown G.D., Denning D.W., Levitz S.M. Tackling human fungal infections. Science. 2012;336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 2.Kronstad J.W., Attarian R., Cadieux B., Choi J., D'Souza C.A., Griffiths E.J., Geddes J.M.H., Hu G., Jung W.H., Kretschmer M., et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 2011;9:193–203. doi: 10.1038/nrmicro2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perlroth J., Choi B., Spellberg B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med. Mycol. 2007;45:321–346. doi: 10.1080/13693780701218689. [DOI] [PubMed] [Google Scholar]
- 4.Pfaller M.A., Diekema D.J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole G.T., Halawa A.A., Anaissie E.J. The role of the gastrointestinal tract in hematogenous candidiasis: from the laboratory to the bedside. Clin. Infect. Dis. 1996;22(Suppl. 2):S73–S88. doi: 10.1093/clinids/22.supplement_2.s73. [DOI] [PubMed] [Google Scholar]
- 6.Fidel P.L., Vazquez J.A., Sobel J.D. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 1999;12:80–96. doi: 10.1128/cmr.12.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L., Redding S., Dongari-Bagtzoglou A. Candida glabrata: an emerging oral opportunistic pathogen. J. Dent. Res. 2007;86:204–215. doi: 10.1177/154405910708600304. [DOI] [PubMed] [Google Scholar]
- 8.Butler G., Rasmussen M.D., Lin M.F., Santos M.A.S., Sakthikumar S., Munro C.A., Rheinbay E., Grabherr M., Forche A., Reedy J.L., et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009;459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dujon B., Sherman D., Fischer G., Durrens P., Casaregola S., Lafontaine I., Montigny J.D., Marck C., Neuvéglise C., Talla E., et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 10.Brunke S., Hube B. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell. Microbiol. 2013;15:701–708. doi: 10.1111/cmi.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaur R., Domergue R., Zupancic M.L., Cormack B.P. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 2005;8:378–384. doi: 10.1016/j.mib.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Zakikhany K., Naglik J.R., Schmidt-Westhausen A., Holland G., Schaller M., Hube B. In vivo transcript profiling of Candida albicans identifies a gene essential for interepithelial dissemination. Cell. Microbiol. 2007;9:2938–2954. doi: 10.1111/j.1462-5822.2007.01009.x. [DOI] [PubMed] [Google Scholar]
- 13.Roetzer A., Gratz N., Kovarik P., Schüller C. Autophagy supports Candida glabrata survival during phagocytosis. Cell. Microbiol. 2010;12:199–216. doi: 10.1111/j.1462-5822.2009.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seider K., Brunke S., Schild L., Jablonowski N., Wilson D., Majer O., Barz D., Haas A., Kuchler K., Schaller M., et al. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J. Immunol. 2011;187:3072–3086. doi: 10.4049/jimmunol.1003730. [DOI] [PubMed] [Google Scholar]
- 15.Jacobsen I.D., Brunke S., Seider K., Schwarzmüller T., Firon A., d’ Enfért C., Kuchler K., Hube B. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect. Immun. 2010;78:1066–1077. doi: 10.1128/IAI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobsen I.D., Grosse K., Berndt A., Hube B. Pathogenesis of Candida albicans infections in the alternative chorio-allantoic membrane chicken embryo model resembles systemic murine infections. PLoS One. 2011;6:e19741. doi: 10.1371/journal.pone.0019741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moran C., Grussemeyer C.A., Spalding J.R., Benjamin D.K., Reed S.D. Comparison of costs, length of stay, and mortality associated with Candida glabrata and Candida albicans bloodstream infections. Am. J. Infect. Control. 2010;38:78–80. doi: 10.1016/j.ajic.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horn F., Heinekamp T., Kniemeyer O., Pollmächer J., Valiante V., Brakhage A.A. Systems biology of fungal infection. Front. Microbiol. 2012;3:108. doi: 10.3389/fmicb.2012.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alves C.T., Wei X.-Q., Silva S., Azeredo J., Henriques M., Williams D.W. Candida albicans promotes invasion and colonisation of Candida glabrata in a reconstituted human vaginal epithelium. J. Infect. 2014;69:396–407. doi: 10.1016/j.jinf.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Brunke S., Seider K., Almeida R.S., Heyken A., Fleck C.B., Brock M., Barz D., Rupp S., Hube B. Candida glabrata tryptophan-based pigment production via the Ehrlich pathway. Mol. Microbiol. 2010;76:25–47. doi: 10.1111/j.1365-2958.2010.07052.x. [DOI] [PubMed] [Google Scholar]
- 21.Caudle K.E., Barker K.S., Wiederhold N.P., Xu L., Homayouni R., Rogers P.D. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell. 2011;10:373–383. doi: 10.1128/EC.00073-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai H.-F., Sammons L.R., Zhang X., Suffis S.D., Su Q., Myers T.G., Marr K.A., Bennett J.E. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob. Agents Chemother. 2010;54:3308–3317. doi: 10.1128/AAC.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda Y., Tsai H.-F., Myers T.G., Bennett J.E. Transcriptional profiling of Candida glabrata during phagocytosis by neutrophils and in the infected mouse spleen. Infect. Immun. 2013;81:1325–1333. doi: 10.1128/IAI.00851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haas B.J., Zeng Q., Pearson M.D., Cuomo C.A., Wortman J.R. Approaches to Fungal Genome Annotation. Mycology. 2011;2:118–141. doi: 10.1080/21501203.2011.606851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Besemer J., Lomsadze A., Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stanke M., Diekhans M., Baertsch R., Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 28.Mardis E.R. Next-generation DNA sequencing methods. Annu. Rev. Genomics Hum. Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 29.Hoff K. Incorporating RNA-Seq into AUGUSTUS with TOPHAT. 2014 http://bioinf.uni-greifswald.de/bioinf/wiki/pmwiki.php?n=IncorporatingRNAseq.Tophat. [Google Scholar]
- 30.Garber M., Grabherr M.G., Guttman M., Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 31.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., White O., Buell C.R., Wortman J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7.1–R7.22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruno V.M., Wang Z., Marjani S.L., Euskirchen G.M., Martin J., Sherlock G., Snyder M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010;20:1451–1458. doi: 10.1101/gr.109553.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guida A., Lindstädt C., Maguire S.L., Ding C., Higgins D.G., Corton N.J., Berriman M., Butler G. Using RNA-seq to determine the transcriptional landscape and the hypoxic response of the pathogenic yeast Candida parapsilosis. BMC Genomics. 2011;12:628. doi: 10.1186/1471-2164-12-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grumaz C., Lorenz S., Stevens P., Lindemann E., Schöck U., Retey J., Rupp S., Sohn K. Species and condition specific adaptation of the transcriptional landscapes in Candida albicans and Candida dubliniensis. BMC Genomics. 2013;14:212. doi: 10.1186/1471-2164-14-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerqueira G.C., Arnaud M.B., Inglis D.O., Skrzypek M.S., Binkley G., Simison M., Miyasato S.R., Binkley J., Orvis J., Shah P., et al. The Aspergillus genome database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res. 2013;42:D705–D710. doi: 10.1093/nar/gkt1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller S., Baldin C., Groth M., Guthke R., Kniemeyer O., Brakhage A.A., Valiante V. Comparison of transcriptome technologies in the pathogenic fungus Aspergillus fumigatus reveals novel insights into the genome and MpkA dependent gene expression. BMC Genomics. 2012;13:519. doi: 10.1186/1471-2164-13-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novodvorska M., Hayer K., Pullan S.T., Wilson R., Blythe M.J., Stam H., Stratford M., Archer D.B. Trancriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genomics. 2013;14:246. doi: 10.1186/1471-2164-14-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin J.-Q., Zhao X.-X., Zhi Q.-Q., Zhao M., He Z.-M. Transcriptomic profiling of Aspergillus flavus in response to 5-azacytidine. Fungal Genet. Biol. 2013;56:78–86. doi: 10.1016/j.fgb.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Wang B., Guo G., Wang C., Lin Y., Wang X., Zhao M., Guo Y., He M., Zhang Y., Pan L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–5087. doi: 10.1093/nar/gkq256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin R., Albrecht-Eckardt D., Brunke S., Hube B., Hünniger K., Kurzai O. A core filamentation response network in Candida albicans is restricted to eight genes. PLoS One. 2013;8:e58613. doi: 10.1371/journal.pone.0058613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ullah A., Lopes M.I., Brul S., Smits G.J. Intracellular pH homeostasis in Candida glabrata in infection-associated conditions. Microbiology. 2013;159:803–813. doi: 10.1099/mic.0.063610-0. [DOI] [PubMed] [Google Scholar]
- 43.Peñalva M.A., Arst H.N., Jr Regulation of gene expression by ambient pH in filamentous fungi and yeasts. Microbiol. Mol. Biol. Rev. 2002;66:426–446. doi: 10.1128/MMBR.66.3.426-446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seider K., Gerwien F., Kasper L., Allert S., Brunke S., Jablonowski N., Schwarzmüller T., Barz D., Rupp S., Kuchler K., et al. Immune evasion, stress resistance, and efficient nutrient acquisition are crucial for intracellular survival of Candida glabrata within macrophages. Eukaryot. Cell. 2014;13:170–183. doi: 10.1128/EC.00262-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwartze V.U., Winter S., Shelest E., Marcet-Houben M., Horn F., Wehner S., Linde J., Valiante V., Sammeth M., Riege K., et al. Gene expansion shapes genome architecture in the human pathogen Lichtheimia corymbifera: an evolutionary genomics analysis in the ancient terrestrial mucorales (Mucoromycotina) PLoS Genet. 2014;10:e1004496. doi: 10.1371/journal.pgen.1004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linde J., Schwartze V., Binder U., Lass-Fl’ørl C., Voigt K., Horn F. De novo whole-genome sequence and genome annotation of Lichtheimia ramosa. Genome Announc. 2014;2 doi: 10.1128/genomeA.00888-14. doi:10.1128/genomeA.00888-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon Anders P.T.P., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. bioRxiv. 2014 doi: 10.1093/bioinformatics/btu638. pii:btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 51.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:1–12. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Priebe S., Kreisel C., Horn F., Guthke R., Linde J. FungiFun2: a comprehensive online resource for systematic analysis of gene lists from fungal species. Bioinformatic. 2014;2014 doi: 10.1093/bioinformatics/btu627. pii:btu627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casado C., González A., Platara M., Ruiz A., Ariño J. The role of the protein kinase A pathway in the response to alkaline pH stress in yeast. Biochem. J. 2011;438:523–533. doi: 10.1042/BJ20110607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarver A., DeRisi J. Fzf1p regulates an inducible response to nitrosative stress in Saccharomyces cerevisiae. Mol. Biol. Cell. 2005;16:4781–4791. doi: 10.1091/mbc.E05-05-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen S., Park J.W., Huang J., Dittmar K.A., Lu Z., Zhou Q., Carsatens R.P., Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aschoff M., Hotz-Wagenblatt A., Glatting K.-H., Fischer M., Eils R., König R. SplicingCompass: differential splicing detection using RNA-seq data. Bioinformatics. 2013;29:1141–1148. doi: 10.1093/bioinformatics/btt101. [DOI] [PubMed] [Google Scholar]
- 58.Hu Y., Huang Y., Du Y., Orellana C.F., Singh D., Johnson A.R., Monroy A., Kuan P.-F.M., Hammond L.M., Randell S.H., et al. DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Res. 2013;41:e39. doi: 10.1093/nar/gks1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inglis D.O., Arnaud M.B., Binkley J., Shah P., Skrzypek M.S., Wymore F., Binkley G., Miyasato S.R., Simison M., Sherlock G. The Candida genome database incorporates multiple Candida species: multispecies search and analysis tools with curated gene and protein information for Candida albicans and Candida glabrata. Nucleic Acids Res. 2012;40:D667–D674. doi: 10.1093/nar/gkr945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Slater G.S.C., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haas B.J., Delcher A.L., Mount S.M., Wortman J.R., Smith R.K., Hannick L.I., Maiti R., Ronning C.M., Rusch D.B., Town C.D., et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R., et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 66.Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hornbach A., Heyken A., Schild L., Hube B., Löffler J., Kurzai O. The glycosylphosphatidylinositol-anchored protease Sap9 modulates the interaction of Candida albicans with human neutrophils. Infect. Immun. 2009;77:5216–5224. doi: 10.1128/IAI.00723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wozniok I., Hornbach A., Schmitt C., Frosch M., Einsele H., Hube B., Löffler J., Kurzai O. Induction of ERK-kinase signalling triggers morphotype-specific killing of Candida albicans filaments by human neutrophils. Cell. Microbiol. 2008;10:807–820. doi: 10.1111/j.1462-5822.2007.01086.x. [DOI] [PubMed] [Google Scholar]
- 69.Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zordan R.E., Ren Y., Pan S.-J., Rotondo G., De Las Peñas A., Iluore J., Cormack B.P. Expression plasmids for use in Candida glabrata. G3 Bethesda. 2013;3:1675–1686. doi: 10.1534/g3.113.006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 72.Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lagesen K., Hallin P., Rødland E.A., Staerfeldt H.-H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yusuf D., Marz M., Stadler P.F., Hofacker I.L. Bcheck: a wrapper tool for detecting RNase P RNA genes. BMC Genomics. 2010;11:432. doi: 10.1186/1471-2164-11-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burge S.W., Daub J., Eberhardt R., Tate J., Barquist L., Nawrocki E.P., Eddy S.R., Gardner P.P., Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bartschat S., Kehr S., Tafer H., Stadler P.F., Hertel J. snoStrip: a snoRNA annotation pipeline. Bioinformatics. 2014;30:115–116. doi: 10.1093/bioinformatics/btt604. [DOI] [PubMed] [Google Scholar]
- 78.Grützmann K., Szafranski K., Pohl M., Voigt K., Petzold A., Schuster S. Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Res. 2014;21:27–39. doi: 10.1093/dnares/dst038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eisenhaber B., Schneider G., Wildpaner M., Eisenhaber F. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J. Mol. Biol. 2004;337:243–253. doi: 10.1016/j.jmb.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 80.Weig M., Jänsch L., Gross U., Koster C.G.D., Klis F.M., Groot P.W.J.D. Systematic identification in silico of covalently bound cell wall proteins and analysis of protein-polysaccharide linkages of the human pathogen Candida glabrata. Microbiology. 2004;150:3129–3144. doi: 10.1099/mic.0.27256-0. [DOI] [PubMed] [Google Scholar]
- 81.Feng J., Funk W.D., Wang S.S., Weinrich S.L., Avilion A.A., Chiu C.P., Adams R.R., Chang E., Allsopp R.C., Yu J. The RNA component of human telomerase. Science. 1995;269:1236–1241. doi: 10.1126/science.7544491. [DOI] [PubMed] [Google Scholar]
- 82.Kachouri R., Stribinskis V., Zhu Y., Ramos K.S., Westhof E., Li Y. A surprisingly large RNase P RNA in Candida glabrata. RNA. 2005;11:1064–1072. doi: 10.1261/rna.2130705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmitt M.E., Clayton D.A. Nuclear RNase MRP is required for correct processing of pre-5.8S rRNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1993;13:7935–7941. doi: 10.1128/mcb.13.12.7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walter P., Ibrahimi I., Blobel G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981;91:545–550. doi: 10.1083/jcb.91.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heringa J. Two strategies for sequence comparison: profile-preprocessed and secondary structure-induced multiple alignment. Comput. Chem. 1999;23:341–364. doi: 10.1016/s0097-8485(99)00012-1. [DOI] [PubMed] [Google Scholar]
- 86.Schleiffer A., Maier M., Litos G., Lampert F., Hornung P., Mechtler K., Westermann S. CENP-T proteins are conserved centromere receptors of the Ndc80 complex. Nat. Cell Biol. 2012;14:604–613. doi: 10.1038/ncb2493. [DOI] [PubMed] [Google Scholar]
- 87.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ullmann B.D., Myers H., Chiranand W., Lazzell A.L., Zhao Q., Vega L.A., Lopez-Ribot J.L., Gardner P.R., Gustin M.C. Inducible defense mechanism against nitric oxide in Candida albicans. Eukaryot. Cell. 2004;3:715–723. doi: 10.1128/EC.3.3.715-723.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinz W.J., Kurzai O., Brakhage A.A., Fonzi W.A., Korting H.C., Frosch M., Mühlschlegel F.A. Molecular responses to changes in the environmental pH are conserved between the fungal pathogens Candida dubliniensis and Candida albicans. Int. J. Med. Microbiol. 2000;290:231–238. doi: 10.1016/S1438-4221(00)80120-4. [DOI] [PubMed] [Google Scholar]
- 90.Miramón P., Dunker C., Windecker H., Bohovych I.M., Brown A.J.P., Kurzai O., Hube B. Cellular responses of Candida albicans to phagocytosis and the extracellular activities of neutrophils are critical to counteract carbohydrate starvation, oxidative and nitrosative stress. PLoS One. 2012;7:e52850. doi: 10.1371/journal.pone.0052850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Linde J., Hortschansky P., Fazius E., Brakhage A.A., Guthke R., Haas H. Regulatory interactions for iron homeostasis in Aspergillus fumigatus inferred by a Systems Biology approach. BMC Syst. Biol. 2012;6:6. doi: 10.1186/1752-0509-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramachandra S., Linde J., Brock M., Guthke R., Hube B., Brunke S. Regulatory networks controlling nitrogen sensing and uptake in Candida albicans. PLoS One. 2014;9:e92734. doi: 10.1371/journal.pone.0092734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt P., Walker J., Selway L., Stead D., Yin Z., Enjalbert B., Weig M., Brown A.J.P. Proteomic analysis of the pH response in the fungal pathogen Candida glabrata. Proteomics. 2008;8:534–544. doi: 10.1002/pmic.200700845. [DOI] [PubMed] [Google Scholar]
- 94.Causton H.C., Rein B., Koh S.S., Harbison C.T., Kanin E., Jennings E.G., Lee T.I., True H.L., Lander E.S., Young R.A. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prendergast L., van Vuuren C., Kaczmarczyk A., Doering V., Hellwig D., Quinn N., Hoischen C., Diekmann S., Sullivan K.F. Premitotic assembly of human CENPs -T and -W switches centromeric chromatin to a mitotic state. PLoS Biol. 2011;9:e1001082. doi: 10.1371/journal.pbio.1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Westermann S., Schleiffer A. Family matters: structural and functional conservation of centromere-associated proteins from yeast to humans. Trends Cell Biol. 2013;23:260–269. doi: 10.1016/j.tcb.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 97.Roy B., Varshney N., Yadav V., Sanyal K. The process of kinetochore assembly in yeasts. FEMS Microbiol. Lett. 2013;338:107–117. doi: 10.1111/1574-6968.12019. [DOI] [PubMed] [Google Scholar]
- 98.Hünniger K., Lehnert T., Bieber K., Martin R., Figge M.T., Kurzai O. A virtual infection model quantifies innate effector mechanisms and candida albicans immune escape in human blood. PLoS Comput. Biol. 2014;10:e1003479. doi: 10.1371/journal.pcbi.1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cunha C., Kurzai O., Löffler J., Aversa F., Romani L., Carvalho A. Neutrophil responses to aspergillosis: new roles for old players. Mycopathologia. 2014;178:387–393. doi: 10.1007/s11046-014-9796-7. [DOI] [PubMed] [Google Scholar]
- 100.Cormack B.P., Ghori N., Falkow S. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science. 1999;285:578–582. doi: 10.1126/science.285.5427.578. [DOI] [PubMed] [Google Scholar]
- 101.de Groot P.W.J., Kraneveld E.A., Yin Q.Y., Dekker H.L., Gross U., Crielaard W., de Koster C.G., Bader O., Klis F.M., Weig M. The cell wall of the human pathogen Candida glabrata: differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell. 2008;7:1951–1964. doi: 10.1128/EC.00284-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.