


Abstract
Overexpression of Oct4, a stemness gene encoding a transcription factor, has been reported in several cancers. However, the mechanism by which Oct4 directs transcriptional program that leads to somatic cancer progression remains unclear. In this study, we provide mechanistic insight into Oct4-driven transcriptional network promoting drug-resistance and metastasis in lung cancer cell, animal and clinical studies. Through an integrative approach combining our Oct4 chromatin-immunoprecipitation sequencing and ENCODE datasets, we identified the genome-wide binding regions of Oct4 in lung cancer at promoter and enhancer of numerous genes involved in critical pathways which promote tumorigenesis. Notably, PTEN and TNC were previously undefined targets of Oct4. In addition, novel Oct4-binding motifs were found to overlap with DNA elements for Sp1 transcription factor. We provided evidence that Oct4 suppressed PTEN in an Sp1-dependent manner by recruitment of HDAC1/2, leading to activation of AKT signaling and drug-resistance. In contrast, Oct4 transactivated TNC independent of Sp1 and resulted in cancer metastasis. Clinically, lung cancer patients with Oct4 high, PTEN low and TNC high expression profile significantly correlated with poor disease-free survival. Our study reveals a critical Oct4-driven transcriptional program that promotes lung cancer progression, illustrating the therapeutic potential of targeting Oc4 transcriptionally regulated genes.
INTRODUCTION
Lung cancer is the leading cause of cancer deaths (1). The 5-year survival of non-small cell lung cancer (NSCLC) is <16%, partly due to metastasis and relapse from current therapeutic strategies (1). Traditionally, platinum-based chemotherapy is considered the first line treatment but it shows limited effectiveness (2). Recently, new encouraging targeted therapies for sub-populations of NSCLC patients harboring epidermal growth factor receptor or anaplastic lymphoma kinase mutations have been developed (3,4). Therefore, further understanding of the molecular mechanisms that drive NSCLC will be useful for development of innovative therapeutic interventions.
Tumor-initiating cells (TICs) are defined as a sub-population of tumors that have the ability to self-renew and differentiate into nontumorigenic cells, thereby contributing to the functional and phenotypic heterogeneity in diverse cancer types (5). TICs have been shown to be more resistant to chemotherapy and radiotherapy and possess metastatic potential (5). Previous study demonstrated that regional pulmonary stem cell population, termed bronchioalveolar stem cells (BASCs), exhibit properties of self-renewal and multipotency. Oncogenically transformed BASCs give rise to lung adenocarcinoma, suggesting the existence of lung TICs (6). Primary lung tumors contain sub-populations of cells, which are characterized by CD133+ surface marker, showing stem-like features with increased tumorigenicity and drug-resistance (7–9). The CD133+ tumorigenic cancer-initiating cells are maintained by highly expressed Oct4 transcription factor (8). Additionally, ectopically co-expression of Oct4 and Nanog in lung cancer cell lines induced TICs properties and promoted epithelial-mesenchymal transition (10). These findings suggest that Oct4-driven lung TICs may be effectively targeted to benefit therapeutic response. Recently, accumulative evidences demonstrate that Oct4 is overexpressed in various solid tumors, including breast cancer (11), bladder cancer (12) and lung cancer (10,13). Targeting of Oct4 promotes cell death in breast and lung cancer cells (14,15), and sensitizes drug-resistant hepatocellular carcinoma to chemotherapy (16). However, the mechanism by which Oct4 influences transcriptional reprogramming that leads to somatic cancer progression remains largely unclear. The target gene networks of Oct4 in somatic cancer are also undiscovered.
Oct4, encoded by POU5F1, belongs to a member of the POU (Pit-1, Oct1/2 and UNC-86) family of transcription factors. Oct4 is a key regulator for maintaining pluripotency of embryonic stem cells (ESCs) by regulating transcriptional programs in coordination with various transcription factors, such as Sox2, Nanog and Klf4 (17–19), or with transcriptional repression complexes, such as histone deacetlyases (HDACs)-containing complex (20). The genome-wide binding profile of Oct4 in ESCs has been revealed in independent studies using different genome-wide approaches (17,21–23). The functions of Oct4 in ESCs are well defined, however, little is known about those in somatic cancer cells or TICs. Investigation of Oct4-centered transcriptional networks in somatic cancers will not only uncover how Oct4 cooperates with other transcriptional factors to regulate downstream genes, but also reveal the cellular mechanisms of Oct4-induced stem-like characteristics, such as drug resistance and metastasis.
Here, we demonstrate the first genome-wide binding profile of Oct4 in lung cancer cells using chromatin-immunoprecipitaion followed by deep sequencing (ChIP-seq). We identified that Oct4 binds to thousands genomic regions in lung cancer cells. De novo motif and sequence similarity analyses showed that some novel Oct4-binding motifs contained the DNA elements for transcription factors such as Sp1, Klf4, ZNF219 and Stat3. Pathway analyses showed that Oct4 downstream target genes play key roles in several tumorigenesis events and important signaling pathways, such as phosphatase and tensin homolog (PTEN) signaling. Mechanistically, Oct4 suppressed tumor suppressor genes, such as PTEN, whereas Oct4 activated oncogenes, such as Tenascin-C (TNC) via differential interplay with the Sp1 transcription factor. Oct4-mediated downregulation of PTEN contributed to drug resistance, while TNC induction was required for Oct4-elicited cell invasion and metastasis in cell and animal models. Lung cancer patients with advanced disease showed high Oct4 protein expression coinciding with low PTEN and high TNC in tumors.
MATERIALS AND METHODS
Cell lines and culture conditions
Human lung adenocarcinoma cell lines A549 were purchased from American Tissue Culture Company (ATCC). Human lung adenocarcinoma cell line CL1–0 and CL1–5 was obtained from Dr Pan-Chyr Yang (Department of Internal Medicine, Medical College, National Taiwan University, Taiwan). All media were supplemented with 10% Fetal Bovine Serum (Gibco, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco). Stable cell line expressing Oct4 (stable Oct4 expressing cells) or vector was established by ectopic transfection of Flag-Oct4 or empty vector plasmid into A549 cells with puromycin selection.
Plasmid, RNAi and transfection
The plasmids and interference RNA (RNAi) used in the study are listed in Supplementary Tables S1 and S2. Plasmids and RNAi transfections were carried out with lipofetamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols.
Tumor-sphere formation assay
Cells were expanded as spheres in a 10-cm ultra-low adhesion culture dish (Corning Inc., Corning, New York, NY, USA) containing DMEM/F-12 with N2 supplement (Invitrogen), 20 ng/ml epithelial growth factor and 20 ng/ml basic fibroblast growth factor (PeproTech Inc., Rocky Hill, NJ, USA), referred to as stem cell medium. Tumor spheres consisting of >30 cells were counted and expressed as the means ± SEM of triplicate within the same experiment.
Tumor formation assay
The 5–6-week-old BALB/c nude female mice were subcutaneously implanted with varying number (1 × 102, 1 × 103 or 5 × 103) of vector control (vector) or Oct4-stably expressing A549 cells (Oct4#1). Vector and Oct4#1 cells in 50 μl Hanks’ balanced salt solution (HBSS) were mixed with 50 μl matrigel (2.5 mg/ml, Sigma-Aldrich, St Louis, MO, USA) and then subcutaneously injected into each flank of a mouse. The incidence of tumor formation was monitored within 8 weeks after implantation.
Anchorage-independent growth assay
Anchorage-independent colony formation assay was carried out by growing 3 × 103 cells in 0.4% bactoagar on a bottom layer of solidified 0.6% bactoagar in 6-well plates. After 12–17 days, colonies consisting of >30 cells were counted and expressed as the means ± SEM of triplicate within the same experiment.
Transwell migration and invasion assay
The transwell insert with millipore membrane (pore size of 8 μm, Falcon, BD, Franklin Lakes, NJ, USA) was used. For transwell migration assay, 2 × 105 cells were seeded onto the upper chamber with 1 ml serum-free medium. For invasion assay, the transwell insert membrane was pre-coated with Matrigel (2.5 mg/ml, Sigma-Aldrich) one day before cells were seeded. Complete medium containing 20% FBS was added to the lower chamber as chemoattractants and cells were incubated for 24 h. The cells attached on the reverse side of the membrane were then fixed and stained with 1% crystal violet/MeOH for 15 min at room temperature. Seven random views were photographed and quantified under an upright microscope (Nikon E400, Yurakucho, Tokyo, Japan).
ChIP-seq assay
Stable Oct4 expressing cells or empty vector control (1 × 107 cells) were cross-linked with 1% formaldehyde, followed by preparation of nuclear lysates using Magna ChIP™ protein G Kit (Millipore Co., Billerica, MA, USA) according to the manufacturer's protocols. Nuclear lysates were sonicated to shear crosslinked DNA, followed by immunoprecipitation with anti-Oct4 antibody using the conditions described in Supplementary Table S3. Purified ChIP DNA was used for the preparation of fragment libraries, followed by high-throughput sequencing using a SOLiDTM 5500xl sequencer (Applied Biosystems, Foster City, CA, USA). We obtained about 24–29 million raw reads from vector and stable Oct4 cell line. The raw reads were further analyzed using LifeScopeTM Genomic Analysis Software (version 2.5) and mapped to human genome (hg19) released from UCSC database. To find the significant peak, the mapped profiles were analyzed using the ChIP-seq tool in CLC Genomics Workbench (version 4.9). Window size and false discovery rate (FDR) were set to 200 bp and 5%, respectively. To determine the high confidence Oct4 binding loci, the ChIP-region was identified by scanning the peaks with significantly higher read count in stable cells expressing Oct4 compared to those in the vector control cells. Oct4 ChIP-seq data can be viewed online under GEO accession number GSE58462.
Quantitative ChIP-PCR assay
ChIP assays were performed as described above using anti-Oct4, anti-Sp1, anti-HDAC1, anti-HDAC2 or anti-Rabbit IgG antibody in vector control and A549 cells stably-expressing Oct4. Quantitative polymerase chain reaction (PCR) analysis was performed using Fast SYBR Green Master Mix and StepOnePlus™ System (Applied Biosystems). The primer sequences are listed in Supplementary Table S4.
DNA sequence motif analysis
For de novo motif discovery algorithm, the top 150 ranked Oct4 ChIP-seq peaks (±100 bp from center of the ChIP-seq peaks) were analyzed by MEME software (24) using the recommended default settings. To identify specific transcription factors that potentially interact with Oct4-targeting motifs, the STAMP software (25) (using the recommended default settings), which compares the motifs discovered by MEME with two existing databases of known motifs, TRANSFAC (v11.3) and JASPAR (v3), was used.
ENCODE datasets analysis
To identify functional DNA elements targeted by Oct4, we correlated our Oct4 ChIP-seq results with datasets from ENCODE project (26) that were deposited to UCSC website. The genomic regions of DNase I hypersensitive sites (DHS), H3K4me3 and H3K4me1 obtained from A549 lung cancer cells were downloaded and analyzed. We used a 1-bp minimum cutoff for the overlap between regions to define common genomic targets across all datasets. The Oct4-targeted functional promoters were obtained by integration of four datasets, including Oct4 ChIP-seq, DHS, H3K4me3, and gene promoter regions (within 2 kb upstream from transcriptional start site) annotated by ENCODE (release v17). The Oct4-targeted enhancers were revealed by integration of three datasets, including Oct4 ChIP-seq, DHS, and H3K4me1. To investigate the coordinates of Oct4 and Sp1 binding regions in A549 lung cancer cells, we integrated Oct4 ChIP-seq and Sp1 ChIP-seq, which is obtained from ENCODE project, using a 1-bp minimum cutoff as described above.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was conducted to determine whether Oct4-target gene set was highly enriched in the previously reported gene signatures of aggressive lung adenocarcinoma (27) or lung TICs (28). GSEA was performed using Oct4-target gene set comprising Oct4-bound top 150, promoter-associated and enhancer-associated genes and using default settings (29). The genes with 1.5-fold increased or decreased change on the microarrays from two previous studies (27,28) were ranked according to their differential expression levels across the two distinct phenotypes using a t-Test metric. Gene sets were considered to be highly enriched if FDR q < 0.25. The P-values were determined by a random permutation test.
RNA extraction and qRT-PCR assays
The primers used for quantitative RT-PCR analyses are described in Supplementary Table S4.
Western blot analysis
Samples containing equal amounts of protein (50 μg) were separated on a 10% sodium dodecylsulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto Immobilon-P membranes (Millipore). Immunoblotting was performed using the conditions described in Supplementary Table S3.
Immunoprecipitation assay
For immunoprecipitation, 600 μg nuclear lysates were incubated with 4 μg of the appropriate antibody, including Oct4, Sp1, Rabbit-IgG and Mouse-IgG, then 1× wash buffer was added to a final volume of 480 μl. After incubation at 4°C for 3 h, 20 μl Protein G/Protein A agarose beads (Calbiochem Co., Darmstadt, Germany) were added. After 1 h of incubation at 4°C, complexes were washed 3× with 1× immonoprecepitation buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 20 mM α-glycerol-phosphate, 1% NP-40, 5 mM ethylenediaminetetraacetic acid (EDTA)). Proteins were eluted by boiling in 2× SDS loading buffer, separated by 8% SDS-PAGE, then blotted with Oct4, Sp1, HDAC1 or HDAC2 antibody.
Cell cytotoxicity analyses
Cells were incubated with solvent control or various concentrations of cisplatin, Suberoylanilide hydroxamic acid (SAHA), MS275 or LY294002 compound for indicated times. Cell cytotoxicity was assayed with 3-(4.5-dimethylthiazol-2-ly)-2,5-diphenyl tetrazolium bromide (MTT, Sigma-Aldrich) according to the manufacturer's instructions.
Site-directed mutagenesis
Mutations of Sp1 binding sites within PTEN-1 and/or PTEN-2 regions were generated by site-directed mutagenesis using wild-type (WT) PTEN promoter vector and specific primers listed in Supplementary Table S4. The Oct4 binding site or Sp1 binding site was mutated by site-directed mutagenesis using WT TNC promoter vector and specific primers listed in Supplementary Table S4.
Dual luciferase promoter assay
Cells were plated in 12-well plates the day before transfection. The pGL4-Renilla construct was included as an internal control. After 16 h co-transfection with empty vector or gene promoter vector and pGL4-Renilla, the dual luciferase reporter assay kit (Promega, Madison, WI, USA) was used to determine gene promoter activity according to the protocols provided by the manufacturer. The data were represented as the means of ratio of firefly luciferase to Renilla luciferase activity by triplicate experiments.
DNA affinity precipitation assay (DAPA)
Biotin-labeled DNA probes containing Oct4 and Sp1 binding sites on PTEN and TNC promoter regions were used in DAPA assay. The Oct4 and Sp1 binding motifs within these probes were either WT or mutated (Mut), and the sequences are shown in Supplementary Table S5. Nuclear lysates of A549 cells transfected with empty vector or Oct4 expression vector were incubated with 10 μl streptavidin agarose beads (Thermo Fisher Scientific Inc., Waltham, MA, USA) with wash buffer (20 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonicacid, 0.1 mM KCl, 2 mM MgCl2, 0.2 mM EDTA. 1 mM DTT, 10% glycerol, 0.01% NP-40 (pH 7.9)) at room temperature for 20 min. Nuclear lysates were then incubated with 2 μg biotin-probes for 30 min on ice, followed by rotation at room temperature for 1 h with streptavidin agarose beads. The DNA–protein complexes were washed 3× with wash buffer (with additional 0.25% Triton X-100). Proteins were eluted by boiling in 2× SDS loading buffer, separated by 8% SDS-PAGE, then blotted with Oct4, Sp1, HDAC1 or HDAC2 antibody.
In vivo tumor growth assay
For subcutaneous xenograft model, 5–6-week-old BALB/c nude female mice were subcutaneously implanted with vector control cells or A549 cells stably expressing Oct4 (1 × 107 cells per mouse). When tumor xenograft volume reached 50 mm3, animals were treated intraperitoneally with LY294002 (25 mg/kg) and/or SAHA (30 mg/kg) on days 1, 3 and 5 for three weeks. The volume of the xenograft were measured and calculated as (length × width square)/2 in mm3.
In vivo tumor metastasis assay
Parental A549 cells were transfected with control or Oct4 siRNAs for 24 h, followed by ectopic expression of TNC for another 24 h. Cells (1 × 106 cells/200 μl) were injected intravenously into NOD/SCID mice via the tail vein. Mice were sacrificed at 9 weeks after tail vein injection. The lungs with colonized tumor nodules were dissected and stained for further confirmation.
Study population
We recruited 91 lung cancer patients from Veterans General Hospital, Taipei, Taiwan and 42 lung cancer patients from National Cheng Kung University Hospital after obtaining appropriate institutional review board permission and informed consent from the patients. Paraffin blocks of tumors were collected. The detailed clinicopathological characteristics of the enrolled patients are listed in Supplementary Table S6.
Immunohistochemistry assay
Antibodies used and their experimental conditions are listed in Supplementary Table S3. Staining was scored as +++ if >75% tumor cells were immunostaining-positive; ++ for 50–75%; + for 25–50%; +/− for 10–25% cells and − if <10% were positive. For Oct4 or TNC protein expression level, the stains were graded as ‘overexpression’ if the score were ++ and +++. For PTEN protein expression level, the stains were graded as ‘low expression’ if the score was equal or less than +.
Statistical analysis
Pearson's χ2 t-test was used to determine the significance of difference in cell and animal model experiments. Chi-square test was conducted to examine the association between Oct4, PTEN and TNC protein expression levels and clinical pathological parameters. Overall and progression-free survival curves were calculated according to the Kaplan-Meier method using the log-rank test. P<0.05 was considered to be statistically significant.
RESULTS
Identification of global target regions of Oct4 by ChIP-seq in lung cancer cells
Oct4 is a master regulator of pluripotent ESCs and maintains stem cell-like properties of TICs. The genomic binding pattern of Oct4 in somatic cancer is still unknown and uncovering it could reveal how Oct4 drives somatic cancer into TIC-like states. To this end, we established stable Oct4-expressing cell line using A549 lung adenocarcinoma cells. Interestingly, ectopic expression of Oct4 alone promoted dedifferentiation of A549 cells into TIC-like cells, which possessed increased in vitro self-renewal capacity and increased expression of stemness-related genes, such as SOX2 and NOTCH (Supplementary Figure S1A and SB). Limited numbers of stable Oct4 expressing cells formed tumors in immunodeficiency mice compared to vector control cells (Supplementary Figure S1C). Enhanced cell proliferation, cell motility, as well as drug-resistance to chemotherapy reagent (cisplatin) and HDAC inhibitors (SAHA and MS275) were observed in stable Oct4 expressing cells (Supplementary Figure S2). In contrast, knockdown of Oct4 diminished anchorage-independent growth and cell invasion/migration ability in parental A549 cells, while restoration of Oct4 expression in Oct4-depleted A549 cells completely reversed the effect of Oct4 siRNA on such phenotypes (Supplementary Figure S3). These data suggest that enforced Oct4 expression alone in A549 cells induces TIC-like states.
Since TICs account for less than 1% in somatic cancer cell lines, it is difficult to address the genome-wide binding regions of Oct4 using A549 lung cancer cell lines. Therefore, we chose the stable Oct4 expressing A549 cells, which showed TIC-like properties (Supplementary Figures S1 and S2), to interrogate the Oct4 occupancy using ChIP-seq approach. Based on two biological replicates, a total of 5,380 ChIP regions corresponding to 3,350 unique RefSeq genes were identified (Supplementary Table S7). A large proportion of Oct4 occupancy located at gene body and, to a lesser extent, distal region (Figure 1A). With respect to gene targeting, our ChIP-seq data showed that Oct4 heavily occupied its own promoter and gene body regions (POU5F1) (Figure 1B) as reported in ESC. Our ChIP-seq data revealed new Oct4-targeted sites in many cancer-related genes (Supplementary Table S7) in addition to several Oct4 binding loci previously identified in ESCs, including ESRRB, JARID2, TCF3 (21), ZFP42, SIRPA, MYBL2, PCSK6 (30), EOMES, and PAX6 (31). For example, newly identified Oct4-targeted sites were found at promoter region of oncogenic gene GSK3A and intragenic region of tumor suppressor gene STAG3 (Figure 1B).
Figure 1.
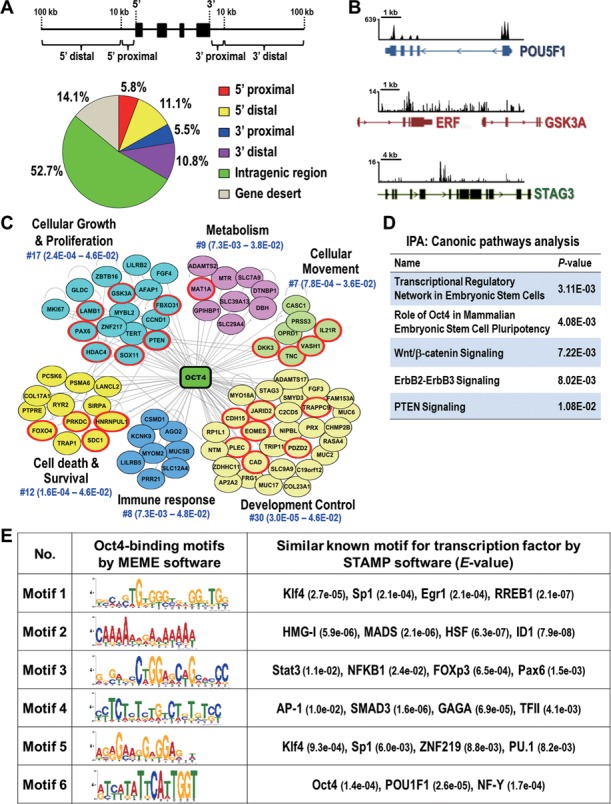
Identification of global binding sites and potential interacting partners of Oct4 in lung cancer cells. (A) Distribution of Oct4 binding sites from ChIP-seq. Schematic diagram illustrates the definition of the location of a binding site in relation to a transcription unit (upper). Pie diagram shows the location of Oct4 binding sites relative to the nearest transcription unit (lower). Gene desert was defined as loci location >100 kb away from the nearest gene. (B) Snapshots of the ChIP-seq binding profiles of Oct4 at POU5F1, GSK3A and STAG3 genes from Integrative Genomics Viewer. (C) Oct4-centered transcriptional network analyzed by Ingenuity-IPA software is shown. Genes validated by qChIP-PCR and qRT-PCR in Figure 2 are in red circle. (D) Top five canonical signaling pathways analyzed by Ingenuity-IPA using gene set from (C). (E) Potential interaction partners of Oct4. Oct4 binding motifs were obtained by MEME software. The potential transcription factor binding sites related to Oct4 targeting motifs were analyzed by STAMP software. The E-value for specific transcription factor is shown.
Oct4-centered transcriptional network contributes to tumorigenesis
To delineate the importance of Oct4-driven transcriptional network in cancer progression, we conducted Ingenuity Pathway Analysis (IPA) for network and canonic pathway analyses. As shown in Figure 1C, Oct4 bound genes in the top 150 binding sites were enriched in several cellular functions related to tumorigenesis events, such as cellular growth and proliferation, cell death and survival, and cellular movement (P<0.05) in addition to development control. Importantly, these genes are directly involved in several critical pathways, including Wnt/β-catenin, ErbB2-ErbB3 and PTEN signaling (P<0.05) (Figure 1D).
To further verify the effects of Oct4 occupancy on gene expression relevant to tumorigenesis, we next classified these top Oct4 bound cancer-related genes into two groups, oncogenic-like and tumor suppressor-like genes, based on literature survey. qChIP-PCR confirmed the enriched occupancy of endogenous Oct4 at these target genes in parental A549 cells (Figure 2A). Interestingly, among 29 genes validated, Oct4 was found to regulate many genes, most of which were reported for the first time, such as transactivation of oncogenes TNC, HDAC4, LAMB1 and GSK3A (Figure 2B, left), or transcriptional suppression of tumor suppressor-like genes, including PTEN, DKK3, FBXO31 and RAB37 (Figure 2B, right). Knockdown of Oct4 by two independent Oct4 siRNAs in A549 cells showed opposite effects on gene expression compared to overexpression of Oct4 (Figure 2C), thus confirming differential modulation of target genes by Oct4. Collectively, these data showed high confidence of our ChIP-seq results and suggested the important roles of Oct4 in transcriptional regulation of oncogenic-like and tumor suppressor-like genes in lung cancer cells.
Figure 2.
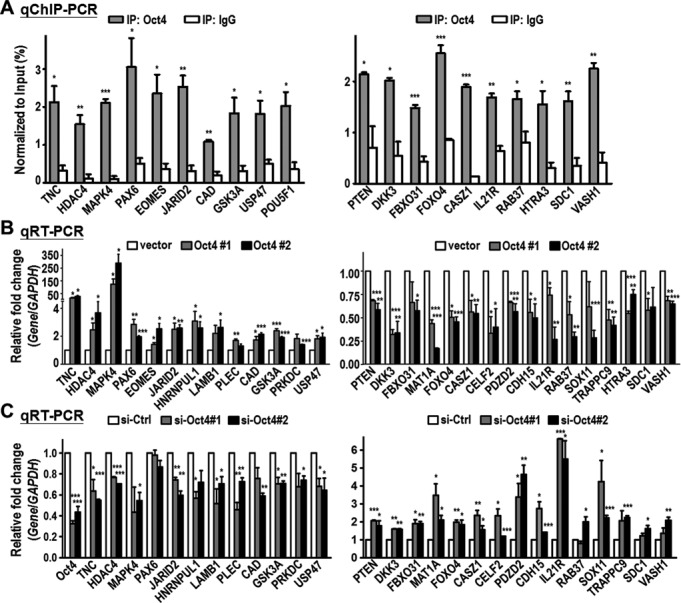
Oct4 induces expression of oncogenic genes while represses tumor suppressor-like genes. Oct4 target genes were classified into two groups: oncogenic genes (left panel) and tumor suppressor-like genes (right panel). (A) qChIP-PCR analysis confirmed the endogenous Oct4 occupancy at the binding sites of genes obtained from Oct4 ChIP-seq. Oct4-bound chromatin was immunoprecipitated (IP) in parental A549 lung cancer cells using anti-Oct4 antibody. Normal IgG served as negative control. (B) qRT-PCR analysis of genes expression in A549 stable clones (vector, Oct4#1 and Oct4#2). GAPDH gene served as internal control. (C) qRT-PCR analysis of genes expression in parental A549 cells after knockdown of Oct4 (si-Oct4#1 and si-Oct4#2) or control (si-Ctrl). For all graphs, data are mean ± SEM. (n = 3). P-values determined using two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
Oct4 occupies promoter and enhancer regions of genes that play key roles in critical signaling pathways involved in cancer progression
Since DNA regulatory elements (i.e. promoter and enhancer) comprise important information that determines gene expression, we correlated Oct4 binding profiles with datasets from the ENCODE project (26), which deposit informative ChIP-seq of histone modifications and chromatin accessibility characterized by DHS of numerous cell lines, including A549 lung cancer cells. We found that Oct4 occupied 258 functional promoters, including PTEN gene, which are characterized by simultaneous DHS and histone H3 lysine 4 tri-methylation (H3K4me3) (Figure 3A). In addition, Oct4 binding sites were well associated with 431 enhancers, characterized by simultaneous DHS and histone H3 lysine 4 mono-methylation (H3K4me1) (Figure 3B). IPA analysis revealed that promoter-associated and enhancer-associated genes were both significantly associated with cancer disease (P<0.05) and important signaling pathways, such as PTEN signaling (P<0.001) and integrin signaling (P<0.01) (Figure 3C and D). Moreover, we conducted GSEA using a gene set comprising Oct4-bound top 150, promoter- and enhancer-associated genes to compare with those aggressive gene signatures of lung cancer published previously. GSEA demonstrated that our Oct4-target gene set correlated with an aggressive gene signature of lung adenocarcinoma (27) and a gene signature of lung TICs (28) (Supplementary Figure S4). These data suggested that Oct4 occupies functional regulatory elements of critical genes and is involved in tumorigenesis.
Figure 3.
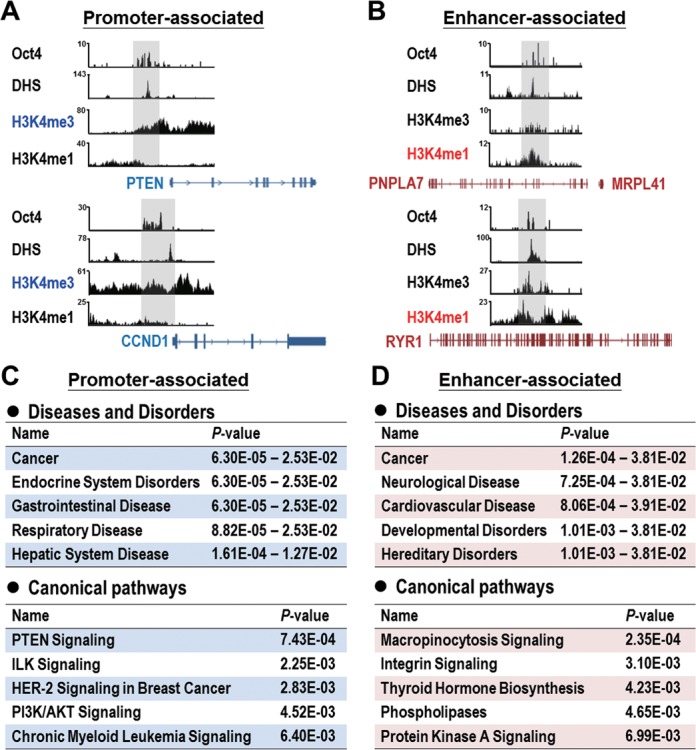
Involvement of Oct4-targeted promoter- and enhancer-associated genes in critical signaling pathways relevant to cancer progression. (A and B) Configurations of Oct4 binding (data from Oct4 ChIP-seq), DHS, H3K4me3 and H3K4me1 profiles (data from ENCODE datasets) at promoter-associated (A) and enhancer-associated (B) genes. Snapshots of the binding profiles from Integrative Genomics Viewer. (C and D) Diseases and disorders (upper) and canonical pathways analyses (lower) of promoter-associated (C) and enhancer-associated (D) genes targeted by Oct4 conducted by Ingenuity-IPA.
Oct4 co-occupies distinct binding motifs in lung cancer with transcription factors
The interacting partners of Oct4 have not been defined clearly in somatic cancer. Thus, we adopted MEME software (24) for de novo motif analysis followed by STAMP software (25) to explore the Oct4 binding DNA sequences and potential Oct4-interacting transcription factors that may mediate the preferential recruitment of Oct4. We analyzed the top 150 Oct4 binding sites and identified not only the known Oct4 consensus binding motif in ESCs (Motif 6, E-value: 1.4e-04) but also several novel binding motifs (Motifs 1–5) containing DNA binding sequence of known transcription factors such as Klf4, Sp1, Stat3 and ZNF219 (Figure 1E). Klf4 has been shown experimentally to interact with Oct4 (19) while Sp1 and ZNF219 have been reported as candidate partners of Oct4 in ESCs using mass spectrometry without further validation (30,32). These data supported our predicted results and suggested that combinatorial action of Oct4 with other co-regulators is also prevalent in lung cancer.
Differential interplay of Oct4 and Sp1 leads to downregulation of PTEN and upregulation of TNC
We identified that important tumor suppressor PTEN is a novel Oct4-repressed gene and PTEN signaling was a significantly enriched pathway by bioinformatics analyses using different subsets of Oct4 bound genes (Figures 1D and 3C). Therefore, we investigated the mechanism by which Oct4 transcriptionally downregulated PTEN. PTEN promoter contained two Oct4 ChIP-seq binding regions (indicated by PTEN-1 and PTEN-2, Figure 4A) belonged to ‘Motif 1’, which contained Sp1 transcription factor binding sites (Figure 1E, E-value: 2.1e-04). Indeed, we confirmed that Oct4 interacted with Sp1 in the nuclear fractions of A549 cells by IP-western assay (Supplementary Figure S5A). To gain insight into the effect of interplay between Oct4 and Sp1 on target genes expression, we included another novel Oct4-transactivated oncogene TNC, which contains a separate Sp1 site from the Oct4 consensus binding site (Motif 6) in its promoter region (Figure 4B), serving as a comparison in the following transcriptional regulation assays.
Figure 4.
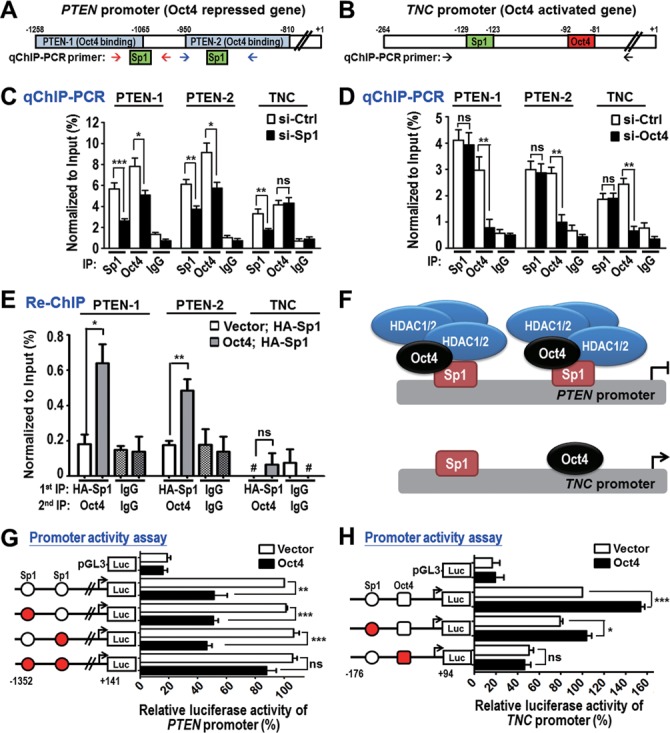
Differential targeting and regulation of Oct4 and Sp1 on PTEN and TNC promoters. (A and B) Promoter regions of PTEN (A) and TNC (B) were analyzed. Oct4 ChIP-seq regions (blue; PTEN-1 and PTEN-2), Sp1 binding site (green) and Oct4 binding site (red) are shown. qChIP-PCR primers for amplification of PTEN-1, PTEN-2 and TNC promoters are indicated by arrows. (C and D) qChIP-PCR analysis of Oct4 and Sp1 occupancies at PTEN and TNC promoters in A549 cells stably expressing Oct4 after knockdown of Sp1 (si-Sp1) (C) or Oct4 (si-Oct4) (D) compared to si-control (si-Ctrl). (E) Re-ChIP assay was performed in control (vector) and A549 cells stably expressing Oct4 (Oct4) with overexpression of HA-tagged Sp1 (HA-Sp1). Sequential IP was performed using anti-HA antibody, followed by anti-Oct4 antibody. #, no detectable signal; ns, non-significant. (F) Working models of differential targeting of Oct4, Sp1 and HDAC1/2 complex at PTEN and TNC promoters. (G and H) Dual luciferase activity assays were performed using PTEN promoter (G) or TNC promoter (H) containing WT (white circle), mutant Sp1 binding site (red circle) and/or mutant Oct4 binding site (red square) in A549 cells overexpressing vector control or Oct4. Data are mean ± SEM. (n = 3). P-values determined using two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
The cell-based qChIP-PCR assays were performed using Oct4 and Sp1 antibodies in cells with Sp1 knockdown (Figure 4C) or Oct4 knockdown (Figure 4D). In the context of PTEN promoter, knockdown of Sp1 led to loss of Oct4 binding (Figure 4C, PTEN-1 and PTEN-2 panels), whereas knockdown of Oct4 did not affect Sp1 binding (Figure 4D, PTEN-1 and PTEN-2 panels), suggesting that Sp1 protein binding is essential for Oct4 binding at PTEN promoter. Re-ChIP results further supported the co-occupancy of Oct4 and Sp1 at PTEN promoter (Figure 4E). Notably, it is reported that HDAC1/2-containing repressive complex participates in Oct4- or Sp1-mediated gene suppression (20,33). Indeed, we found that Oct4 facilitated formation of protein complex composed of Sp1 and HDAC1/2 (Supplementary Figure S5A), thereby recruiting HDAC1/2 to PTEN promoter as evident in the loss of HDAC1/2 binding in si-Oct4 cells (Supplementary Figure S5B and SC, PTEN-1 and PTEN-2 panels).
In contrast, at the TNC promoter, knockdown of Sp1 did not affect Oct4 binding (Figure 4C, TNC panel), and vice versa (Figure 4D, TNC panel), suggesting that Oct4 could recognize and bind to its own consensus binding site independent of Sp1. Consistently, we did not find co-occupancy of Oct4 and Sp1 at TNC promoter by Re-ChIP assay (Figure 4E). Moreover, Sp1, but not Oct4, recruited the HDAC1/2 complex to TNC promoter (Supplementary Figure S5B and SC, TNC panel), which was in agreement with positive regulation of TNC by Oct4. Similar results were observed by in vitro DNA affinity precipitation assay (DAPA) (Supplementary Figure S6A-D). Interestingly, integration of our Oct4 ChIP-seq data with Sp1 ChIP-seq in A549 cells from ENCODE dataset further confirmed the co-occupancy of Oct4 and Sp1 at PTEN promoter, but not at TNC promoter (Supplementary Figure S7). Together, these results indicated that, in the context of PTEN promoter, Sp1 serves as a platform for Oct4 binding, then Oct4 recruits HDAC1/2 complex to the PTEN promoter. On the other hand, Oct4 binds to TNC promoter independent of Sp1 (Figure 4F).
To investigate the differential effects of Oct4 and Sp1 on promoter activity of PTEN and TNC, dual luciferase activity assays were conducted. As shown in Figure 4G, Oct4 suppressed the activity of PTEN promoters containing either one of WT Sp1 binding sites. However, Oct4 did not affect the activity of PTEN promoter with double mutant Sp1 binding sites (Figure 4G). Consistently, the repressive effect of Oct4 relied on the HDAC1/2 complex as evident in restored PTEN expression and promoter activity upon knockdown of HDAC1/2 (Supplementary Figure S8). In addition, Oct4 significantly induced activity of TNC promoter in the presence of WT Oct4 binding site, but not the TNC promoter containing mutant Oct4 binding site (Figure 4H). Collectively, these data further supported our model that Oct4 cooperates with Sp1 and HDAC1/2 complex to suppress PTEN expression while Oct4 can transactivate TNC expression independent of Sp1.
Oct4 transcriptionally represses PTEN, leading to AKT signaling activation and drug-resistance
Dysregulation of PTEN/AKT pathway has been implicated in gain of drug-resistance in cancers (34). Therefore, we speculated that Oct4 could modulate drug-resistance (Supplementary Figure S2C) through PTEN. We confirmed that overexpression of Oct4 decreased PTEN protein expression and subsequently induced phosphorylation of AKT in A549 and CL1–0 lung cancer cell lines (Supplementary Figure S9A), while knockdown of Oct4 by two independent siRNAs showed opposite effects on PTEN/AKT signaling (Figure 5A). In contrast, expression of Oct4 did not affect phospho-AKT levels in CL1–5 cells (Figure 5A; Supplementary Figure S9B), which are PTEN deleted cells (35), suggesting that Oct4 up-regulated AKT signaling via suppression of PTEN expression.
Figure 5.
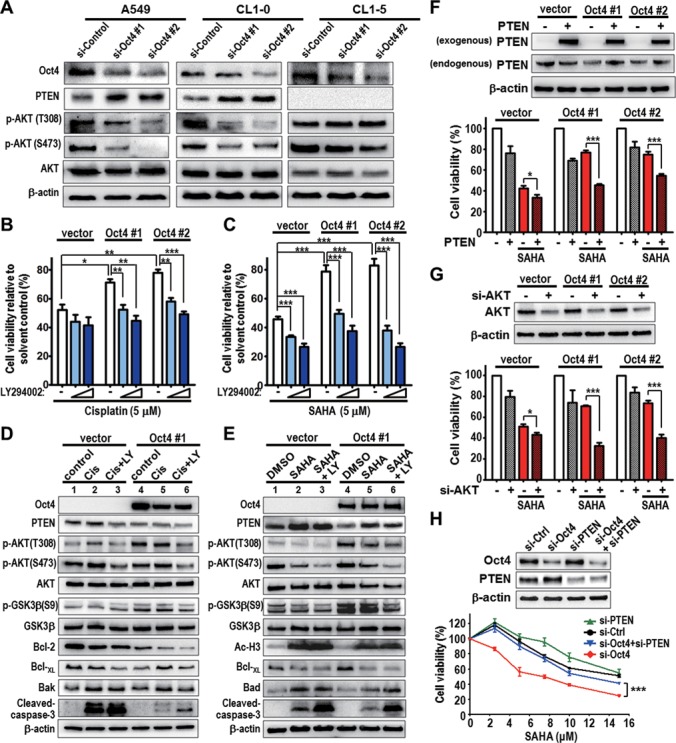
Oct4 induces drug-resistance via dysregulation of PTEN/AKT signaling. (A) A549, CL1–0 and CL1–5 lung cancer cells were transiently transfected with control (si-Control) and Oct4 (si-Oct4#1 and si-Oct4#2) siRNAs for 48 h and then subjected to western blot analysis of PTEN, AKT and AKT phosphorylation levels. β-actin was used as internal control. (B and C) Cell viability in vector control (vector) or A549 cells stably expressing Oct4 (Oct4#1 and Oct4#2) after treatment with solvent control, LY294002 (LY, 5 or 10 μM) and (B) cisplatin (5 μM), or (C) SAHA (5 μM) for 48 h. Cell viability was normalized to that of solvent control. (D and E) Western blot analysis of AKT signaling and apoptosis-related proteins in vector and Oct4#1 cells after treatment with LY294002 (LY) and (D) cisplatin (Cis), or (E) SAHA for 24 h. (F and G) A549 stable clone cells were transfected with PTEN expression vector (PTEN, F) or with AKT siRNA (si-AKT, G), followed by treatment with SAHA for 48 h. Western blot shows the efficiency of PTEN overexpression or AKT knockdown (upper). Cell viability was analyzed after drug treatment (lower). (H) A549 cells stably expressing Oct4 cells (Oct4#2) were transfected with Oct4 (si-Oct4#1) and/or PTEN siRNA (si-PTEN), followed by treatment with different concentration of SAHA for 48 h. Western blots show the efficiency of Oct4 and/or PTEN knockdown (upper). Cell viability was analyzed after drug treatment (lower). For all graphs, data are mean ± SEM. (n = 3). P-values determined using two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
To address the involvement of PTEN/AKT signaling in Oct4-mediated drug-resistance, we performed cytotoxicity and western blot assays in A549 cells stably expressing Oct4 treated with PI3K/AKT inhibitor LY294002 in combination with cisplatin (Figure 5B) or SAHA (Figure 5C). The results showed that combined treatment significantly abolished drug resistance in stable Oct4 expressing cells (Oct4#1 and Oct4#2). Inhibition of AKT/GSK3β signaling by combinatorial treatment further triggered apoptosis signaling compared to single drug treatment (Figure 5D and E, lane 5 versus lane 6). Importantly, re-enforced PTEN expression (Figure 5F) or knockdown of AKT (Figure 5G) sensitized stable Oct4 expressing drug-resistant cells to SAHA treatment. Knockdown of Oct4 sensitized stable Oct4 expressing drug-resistant cells to SAHA treatment, whereas simultaneous knockdown of PTEN rescued the cell viability of si-Oct4 cells upon SAHA treatment (Figure 5H).
To further characterize Oct4-mediated drug-resistance in vivo, Balb/c nude mice bearing vector or stable Oct4 expressing (Oct4#1) xenograft were established. Low dose of LY294002 or SAHA single treatment significantly suppressed tumor growth of vector xenograft (Figure 6A, left), but had no effects on Oct4#1 xenograft (Figure 6A, right). Notably, anti-tumor activity was significantly increased by combinatorial therapeutics in Oct4#1 xenograft as well as vector xenograft (Figure 6A and B). In addition, immunohistochemistry (IHC) analyses confirmed that Oct4 expression was inversely correlated with PTEN expression in xenograft tissues (Figure 6C). Combined LY294002 and SAHA treatment effectively increased apoptosis in vivo, indicated by Caspase-3 activation (Figure 6D). These results collectively suggested that Oct4 induces drug-resistance, in part, by triggering loss of PTEN and activation of AKT signaling.
Figure 6.
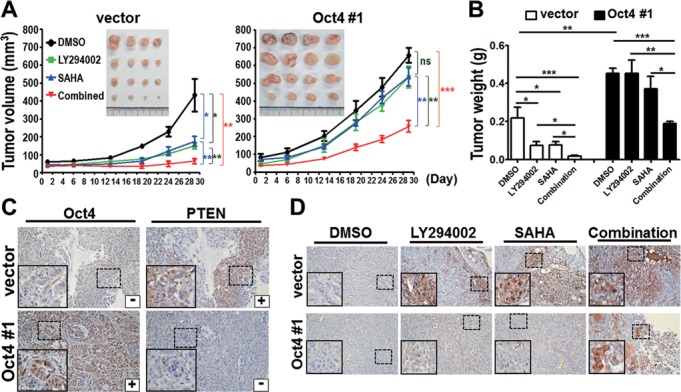
Combinatorial treatment of LY294002 and SAHA overcomes Oct4-mediated drug resistance in vivo. (A) Nude mice bearing xenografts of A549 stable clone, vector (left) or Oct4#1 (right), were treated with DMSO, LY294002 (25 mg/kg) and/or SAHA (30 mg/kg). Tumor volume and image of xenografts are shown. (B) Tumor weight of vector and Oct4#1 xenografts. (C) IHC analyses of Oct4 and PTEN proteins. (D) IHC analysis of cleaved Caspase-3 proteins. For all IHC images, the insets are a higher magnification of the boxed areas. Original magnification × 200. For all graphs, data are mean ± SEM. (n = 5 mice per group). P-values determined using two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001.
TNC is required for Oct4-mediated cancer cell invasiveness
Previous studies showed that overexpression of Oct4 increased cell motility in cancer cells (12), however, little is known about the underlying mechanism. TNC has been reported to be involved in lung metastasis of breast cancer (36). We found that Oct4 increased TNC protein level in multiple lung cancer cell lines (Supplementary Figure S9), this prompted us to investigate whether Oct4-induced TNC is required for cancer invasion and metastasis. We first performed in vitro transwell invasion/migration assay in vector control (vector) and stable Oct4 expressing cells (Oct4#1 and Oct4#2). Knockdown of TNC significantly abrogated Oct4-mediated cell invasion and migration (Figure 7A–C). Conversely, knockdown of Oct4 led to decrease in cell motility, which could be recovered by reconstitution of TNC expression in A549 cells (Figure 7D–F). The requirement of TNC expression for Oct4-induced cell invasion and migration was confirmed in CL1–5 cells (Supplementary Figure S10). Notably, we performed in vivo extravasation assay via tail vein injection model and verified that ectopically expressed TNC reestablished lung colonization of Oct4 knocked down A549 cells in immunodeficiency mice (Figure 7G). These data suggested that Oct4 transactivates TNC, in part, leading to increased invasiveness and extravasation of lung cancer.
Figure 7.
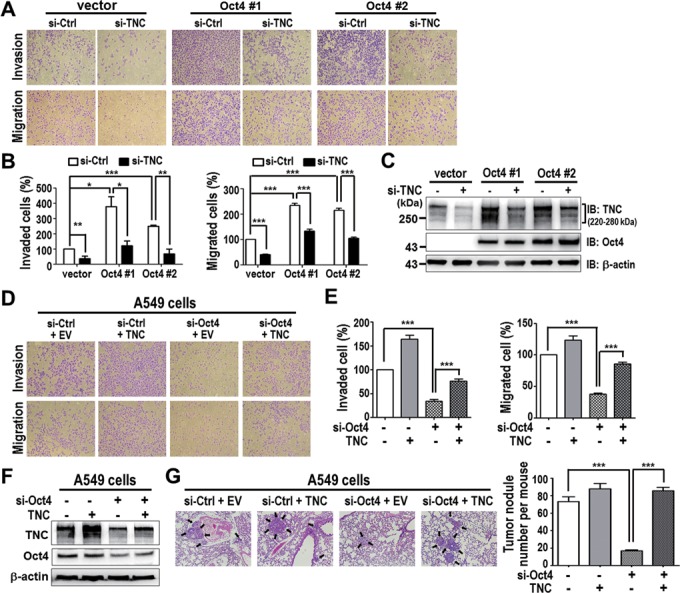
TNC expression is required for Oct4-mediated cancer invasiveness in vitro and in vivo. (A and B) Transwell invasion/migration assay was performed in A549 stable clones (Oct4#1 and Oct4#2) after TNC knockdown (si-TNC) or control siRNA (si-Ctrl). Representative image (A) and quantitation (B) are shown. Data are mean ± SEM. (n = 3). (C) Western blot analysis of TNC and Oct4 protein levels in cells from (A and B). (D and E) Transwell invasion/migration assay was performed in si-Ctrl, TNC, si-Oct4, si-Oct4/TNC cells. Representative image (D) and quantitation (E) are shown. Data are mean ± SEM. (n = 3). (F) Western blot analysis of TNC and Oct4 protein levels in cells from (D and E). (G) Representative lung images of mice intravenously injected via tail vein with si-Ctrl, TNC, si-Oct4, si-Oct4/TNC A549 cells (left). Tumor nodules were indicated by arrows. Number of tumor nodules in lungs per mice is shown (right). Data are mean ± SEM. (n = 8 mice per group). P-values determined using two-tailed Student's t-test. *P<0.05; **P<0.01; ***P<0.001. Original magnification × 100 for all microscopic images.
Lung cancer patients with high Oct4, low PTEN and high TNC expression profiles correlate with poor prognosis
We have demonstrated above that Oct4 transcriptionally repressed PTEN to induce drug resistance and activated TNC to promote tumor invasiveness. Since the relationship between Oct4, PTEN and TNC protein expression has never been examined in human cancer patient, we investigated whether high Oct4 protein expression correlates with low PTEN and high TNC levels in lung cancer patients with poor prognosis or cancer metastasis. For this purpose, we examined the expression profiles of Oct4, PTEN and TNC proteins in lung tumor specimens from 133 lung cancer patients by IHC analyses (Figure 8A; Supplementary Table S6). Importantly, an inverse correlation was found between Oct4 and PTEN protein expressions in lung cancer patients (P=0.022, Figure 8B; Supplementary Table S8), while patients with high Oct4 showed concordantly increased TNC (P<0.0001, Figure 8C; Supplementary Table S8). In addition, 69.4% (43/62) patients with high Oct4 and low PTEN expression levels also demonstrated high TNC expression levels. Consistently, 72.7% (16/22) patients with normal Oct4 and PTEN expression levels demonstrated normal TNC expression (P=0.003, Supplementary Table S9). These data further supported our finding that Oct4 negatively regulates PTEN and positively regulates TNC expression in lung cancer.
Figure 8.
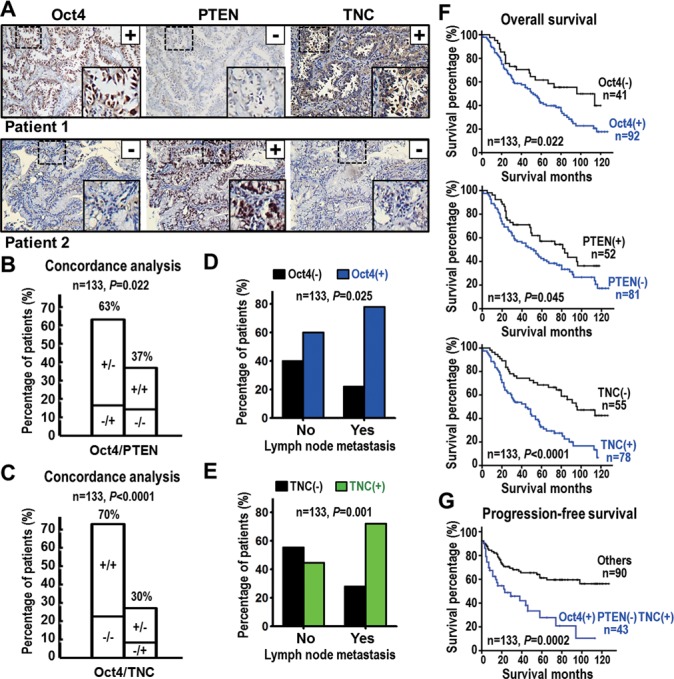
High Oct4 protein coincides with low PTEN and high TNC levels and poor survival of lung cancer patients. (A) Representative IHC images of Oct4, PTEN and TNC proteins in tumor specimen of lung cancer patients. Oct4 positive immunoreactivity (+), PTEN negative immunoreactivity (–) and TNC positive immunoreactivity (+) were found in patient 1, whereas patient 2 shows a reverse pattern. Original magnification × 200. The insets are a higher magnification of the boxed areas. (B and C) Concordance analysis between Oct4 and PTEN (B) or TNC (C) proteins expression (+, positive immunoreactivity; –, negative immunoreactivity) according to the four molecular subtypes. The percentage of the concordant group (left columns) and discordant group (right columns) is indicated above. (D and E) Histogram showing frequency of high Oct4 (D) or high TNC (E) protein expression in patients with different lymph node metastasis status. (F) Overall survival curves of Kaplan–Meier method indicated that patients with high Oct4, low PTEN or high TNC expression had significantly poorer survival than patients with normal expression of the corresponding protein. (G) Progression-free survival analyses indicated that patients with high Oct4 combined with both low PTEN and high TNC expression had significantly poorer survival than other patients. P-values for correlation analyses (B–E) were determined using Pearson χ2 test; and for survival analyses (F and G) using log-rank test.
Of note, patients with high Oct4 expression (P=0.025, Pearson χ2 test) or high TNC expression (P=0.001, Pearson χ2 test) were significantly associated with lymph node metastasis (Figure 8D and E; Supplementary Table S6). Kaplan–Meier analysis showed high expression of Oct4 (P=0.022), low expression of PTEN (P=0.045) or high expression of TNC (P<0.0001) in lung cancer patients correlated with poor overall survival (Figure 8F). Patients with high Oct4, low PTEN and high TNC levels showed worse progression-free survival compared to patients with normal expression of the corresponding proteins (P=0.0002, Figure 8G). To further define the relative risk of death of Oct4, TNC and PTEN expression, we performed the multivariate Cox-regression analysis and the data indicated that patients with Oct4 overexpression had poor outcome (P=0.052, hazard ratio [HR] = 1.83, 95% confidence interval = 1.00–3.35) after adjusting for all the clinicopathological characteristics (Table 1, left panel). Notably, the patients with aberrant expression of all three proteins Oct4, PTEN and TNC showed an HR of 3.24 (P<0.001) (Table 1, right panel). Altogether, these clinical data demonstrated that high Oct4 protein coincides with low PTEN and high TNC levels in surgical tumor tissues, which could be prognostic biomarkers for lung cancer.
Table 1. Multivariate Cox regression analysis of risk factors for cancer-related death in lung cancer patients.
| Characteristics | Multivariate analysis | Characteristics | Multivariate analysis | ||
|---|---|---|---|---|---|
| HRa (95% CIb) | P-valuec | HRa (95% CIb) | P-valuec | ||
| Oct4 protein expression | Oct4/PTEN/TNC protein expression | ||||
| Normal expression | 1.00 | Others | 1.00 | ||
| Overexpression | 1.83 (1.00–3.35) | 0.052 | Aberrant expression | 3.24 (1.77–5.94) | <0.001 |
| Age | Age | ||||
| <60 | 1.00 | <60 | 1.00 | ||
| ≥60 | 1.44 (0.75–2.75) | 0.273 | ≥60 | 1.14 (0.59–2.22) | 0.700 |
| Gender | Gender | ||||
| Female | 1.00 | Female | 1.00 | ||
| Male | 1.20 (0.48–3.01) | 0.693 | Male | 1.12 (0.44–2.87) | 0.816 |
| Smoking habit | Smoking habit | ||||
| Non-smoker | 1.00 | Non-smoker | 1.00 | ||
| Smoker | 1.79 (0.80–4.00) | 0.154 | Smoker | 1.67 (0.72–3.84) | 0.230 |
| Tumor typed | Tumor typed | ||||
| SCC | 1.00 | SCC | 1.00 | ||
| ADC | 2.83 (1.34–5.98) | 0.006 | ADC | 1.75 (0.81–3.79) | 0.156 |
| Stage | Stage | ||||
| Stage I-II | 1.00 | Stage I-II | 1.00 | ||
| Stage III-IV | 1.29 (0.55–3.03) | 0.561 | Stage III-IV | 1.95 (0.80–4.77) | 0.144 |
| T stage | T stage | ||||
| Stage 1–2 | 1.00 | Stage 1–2 | 1.00 | ||
| Stage 3–4 | 2.99 (1.49–5.98) | 0.002 | Stage 3–4 | 2.30 (1.15–4.58) | 0.018 |
| N stage | N stage | ||||
| N0 | 1.00 | N0 | 1.00 | ||
| ≥N1 | 3.11 (1.31–7.40) | 0.010 | ≥N1 | 2.17 (0.88–5.37) | 0.093 |
| M stage | M stage | ||||
| M0 | 1.00 | M0 | 1.00 | ||
| ≥M1 | 0.75 (0.26–2.16) | 0.591 | ≥M1 | 0.46 (0.15–1.36) | 0.159 |
aHR, Hazard ratio.
bCI, Confidence interval.
cBold values indicatestatisticalsignificance (P<0.05).
dADC, Adenocarcinoma; SCC, Squamous cell carcinoma.
DISCUSSION
Here, we provide compelling evidence from lung cells, animal and clinical studies that Oct4-driven transcriptional program promotes lung cancer progression. Through Oct4 ChIP-seq and cross-validation of ENCODE datasets, we identify important Oct4 target genes involved in tumorigenesis and critical signaling pathways, especially PTEN signaling. We further provide the mechanistic insight into the roles of Oct4 in modulating downstream target genes. Oct4 suppresses PTEN and activates TNC expression via differential interplay with Sp1 transcription factor, resulting in drug-resistance and cancer metastasis, respectively. Clinically, lung cancer patients with Oct4 high, PTEN low and TNC high expression profile correlate with cancer progression and poor disease-free survival (see schematic in Supplementary Figure S6E and SF).
Recent study addressed the upstream signaling (i.e. IGF signaling) that initiates stem-like features in lung cancer for which Oct4 is an indispensable effector (13,28), but the relevant functions of Oct4 are not defined. In this study, we present the first genome-wide Oct4 binding profile in somatic cancer using ChIP-seq approach and strengthen the critical role of Oct4 in lung cancer progression. Interestingly, we found high Oct4 occupancy at gene body and, to a lesser extent, distal region in lung cancer cells (Figure 1A). Such distribution pattern was different from that in human ESCs (37), in which Oct4 mainly occupied distal region. In addition, only 75 common genes out of 3350 genes in our ChIP-seq have been identified in human ESCs dataset (17) (Supplementary Table S7), suggesting cell-type specificity of Oct4 binding profile. Although such discrepancy could be explained, in part, by the different platforms and analysis tools employed in our and ESC studies, our GSEA analysis showed that Oct4-target gene set correlated with previously published gene signature of lung TICs (Supplementary Figure S4B), supporting that our Oct4 target genes were specific to lung cancer cells. In addition, we identified that many cancer-related genes were previously undefined direct targets of Oct4 in lung cancer (Figure 2). The validation rate of Oct4 occupancy at cancer-related genes was 100% (20/20) by qChIP-PCR assay, while the validation rate was 58% (29/50) by qRT-PCR assay demonstrating the differential regulation between oncogene and tumor suppressor-like genes by Oct4 (Figure 2). For example, oncogenic HDAC4 is able to activate Stat1 and promote drug-resistance in ovarian cancer (38), and is involved in bone-metastasis of lung cancer (39). Enhanced translation of LABM1 is required for epithelial to mesenchymal transition in hepatocellular carcinoma (40). In terms of Oct4-suppressed genes, DKK3, a well-known Wnt antagonist, exhibits low expression level in many cancer types and serves as a poor prognostic marker (41). RAB37 is a novel metastasis suppressor that regulates exocytosis machinery in lung cancer (42,43). In addition, we confirmed the roles of Oct4 in transcriptional regulation of the tumor suppressor gene PTEN and oncogene TNC that lead to drug-resistance and metastasis. Together, we propose that these cancer-related genes comprise the Oct4-centered transcriptional network that collectively drives lung cancer progression.
PTEN is a bona fide tumor suppressor gene, which acts as negative regulator of PI3K/AKT signaling pathway (34). However, somatic PTEN mutations occur at a low frequency in lung cancer compared to other cancer types (34). Here, we show a new dysregulation mechanism of PTEN in cancer by discovering that Oct4 transcriptionally suppressed PTEN via cooperation with Sp1 and HDAC1/2 in lung cancer cells (Figure 4; Supplementary Figures S5–S8). Previous study showed that mice with lung-specific homozygous deletion of Pten in alveolar type II cells develop lung adenocarcinoma (44), suggesting Pten is critical for prevention of onset of lung tumorigenesis. However, there was no evidence of the mechanism leading to PTEN loss in lung TICs. Our finding and previous study indicate that Oct4-mediated transcriptional repression of PTEN may account for expansion of lung TICs and gain of drug-resistant capacity. Whether lung cancer patients with both high Oct4 and low PTEN expressions show poor response to current chemotherapy needs further interrogation.
TNC is a secreted extracellular matrix molecule and is highly expressed in the microenvironment of several solid tumors, including breast and lung cancers (45). TNC secreted by breast cancer cells acts as a metastatic niche component for colonization to lungs (36). TNC downregulates Dickkopf-1 expression by blocking actin stress fiber formation, thereby activating Wnt signaling in neuroendocrine tumor cells (46). However, little is known about the mechanisms or stimulations leading to TNC overexpression in cancers. In this study, we provide evidences that Oct4 transcriptionally activated TNC expression (Figure 4), which was required for cancer invasion and metastasis (Figures 7 and 8). It has been shown that TNC binds to and activates integrin αvβ3, a cell surface adhesion molecule (47). Recently, Seguin et al. demonstrated that activation of integrin αvβ3-KRAS-RalB-NFκB pathway drives lung tumor stemness (48). Therefore, whether Oct4-induced TNC triggers integrin αvβ3-mediated signaling, which in turn coordinates with Oct4-driven transcriptional program to promote stem-like features in lung cancer is worthy of further investigation.
Our de novo motif and sequence similarity analyses showed that some novel Oct4-binding motifs containing DNA elements for known transcription factors, Sp1, Klf4 and ZNF219 (Figure 1E), which also appeared in the interactome databases of Oct4 from ESC studies (30,32). Mechanistically, we identified that Sp1 served as a platform for Oct4 binding at PTEN promoter, and the binding subsequently facilitated HDAC1/2 recruitment. In contrast, Sp1 and Oct4 could bind to TNC promoter independently (Figure 4; Supplementary Figures S5 and S6). However, question as to what determines the differential interplay between Oct4 and Sp1 in the context of PTEN or TNC gene remains. It is possible that the differential binding affinity of Oct4 to its cis-elements may diverge in the configuration of PTEN or TNC gene promoters as suggested by different Oct4-binding motifs that they possessed. Notably, there was only 4% overlap between Oct4 and Sp1 binding regions in A549 cells by integrating our Oct4 ChIP-seq data and Sp1 ChIP-seq from ENCODE database, suggesting the collaboration of Oct4 and Sp1 in gene regulation may be in the context- or gene-specific manner. Alternatively, phosphorylation of Oct4 and Sp1 is involved in such a differential regulation. Previous studies demonstrated that T739 phosphorylation of Sp1 was required for its DNA binding activity and promoted interaction with other transcription factors (33). In addition, T235 phosphorylation of Oct4 increases its interaction with Sox2, and promotes Oct4-Sox2 targeting to specific genes in embryonic carcinoma cells (49). Therefore, whether Oct4 undergoes phosphorylation that alters its transcriptional activity and DNA binding affinity to targeted promoters in promoting lung tumorigenesis needs further investigations.
From a clinical point of view, expression level of Oct4 alone or together with that of PTEN or TNC in resected lung tumor specimens could be an effective prognosis biomarker. The present results provide compelling evidences that Oct4-driven transcriptional program accelerates lung cancer progression. Therapeutic strategies such as efficient delivery of Oct4 interference RNAs (14) or combinatorial therapeutic strategies targeting PI3K/AKT, and targeted inhibitors of Oct4 downstream genes could facilitate the development of cancer therapy by targeting TICs.
ACCESSION NUMBER
Gene Expression Omnibus (GEO) accession number: GSE58462.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the Human Biobank, Research Center of Clinical Medicine, National Cheng Kung University Hospital for providing the clinical specimens and Ingenuity Pathway Analysis and Taiwan Bioinformatics Core at the National Cheng Kung University, supported by the Taiwan Ministryof Science for assisting with bioinformatics analyses.
FUNDING
Taiwan Ministry of Science and Technology Grant [MOST103-2320-B-006-045-MY3]; Taiwan Ministry of Health and Welfare Grant [MOHW103-TD-B-111-06]; Aim for the Top University Project Grant [D103-35A02]. Funding for open access charge: Taiwan Ministry of Science and Technology Grant [MOST103-2320-B-006-045-MY3].
Conflict of interest statement. None declared.
REFERENCES
- 1.Siegel R., Naishadham D., Jemal A. Cancer statistics, 2013. CA Cancer J. Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Keith R.L., Miller Y.E. Lung cancer chemoprevention: current status and future prospects. Nat. Rev. Clin. Oncol. 2013;10:334–343. doi: 10.1038/nrclinonc.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Shaw A.T., Kim D.W., Nakagawa K., Seto T., Crino L., Ahn M.J., De Pas T., Besse B., Solomon B.J., Blackhall F., et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 5.Visvader J.E., Lindeman G.J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Kim C.F., Jackson E.L., Woolfenden A.E., Lawrence S., Babar I., Vogel S., Crowley D., Bronson R.T., Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 7.Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S., et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y.C., Hsu H.S., Chen Y.W., Tsai T.H., How C.K., Wang C.Y., Hung S.C., Chang Y.L., Tsai M.L., Lee Y.Y., et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One. 2008;3:2637. doi: 10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eramo A., Lotti F., Sette G., Pilozzi E., Biffoni M., Di Virgilio A., Conticello C., Ruco L., Peschle C., Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 10.Chiou S.H., Wang M.L., Chou Y.T., Chen C.J., Hong C.F., Hsieh W.J., Chang H.T., Chen Y.S., Lin T.W., Hsu H.S., et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433–10444. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- 11.Ezeh U.I., Turek P.J., Reijo R.A., Clark A.T. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104:2255–2265. doi: 10.1002/cncr.21432. [DOI] [PubMed] [Google Scholar]
- 12.Chang C.C., Shieh G.S., Wu P., Lin C.C., Shiau A.L., Wu C.L. Oct-3/4 expression reflects tumor progression and regulates motility of bladder cancer cells. Cancer Res. 2008;68:6281–6291. doi: 10.1158/0008-5472.CAN-08-0094. [DOI] [PubMed] [Google Scholar]
- 13.Xu C., Xie D., Yu S.C., Yang X.J., He L.R., Yang J., Ping Y.F., Wang B., Yang L., Xu S.L., et al. beta-Catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res. 2013;73:3181–3189. doi: 10.1158/0008-5472.CAN-12-4403. [DOI] [PubMed] [Google Scholar]
- 14.Chiou G.Y., Cherng J.Y., Hsu H.S., Wang M.L., Tsai C.M., Lu K.H., Chien Y., Hung S.C., Chen Y.W., Wong C.I., et al. Cationic polyurethanes-short branch PEI-mediated delivery of Mir145 inhibited epithelial-mesenchymal transdifferentiation and cancer stem-like properties and in lung adenocarcinoma. J. Control Release. 2012;159:240–250. doi: 10.1016/j.jconrel.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Hu T., Liu S., Breiter D.R., Wang F., Tang Y., Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008;68:6533–6540. doi: 10.1158/0008-5472.CAN-07-6642. [DOI] [PubMed] [Google Scholar]
- 16.Wang X.Q., Ongkeko W.M., Chen L., Yang Z.F., Lu P., Chen K.K., Lopez J.P., Poon R.T., Fan S.T. Octamer 4 (Oct4) mediates chemotherapeutic drug resistance in liver cancer cells through a potential Oct4-AKT-ATP-binding cassette G2 pathway. Hepatology. 2010;52:528–539. doi: 10.1002/hep.23692. [DOI] [PubMed] [Google Scholar]
- 17.Boyer L.A., Lee T.I., Cole M.F., Johnstone S.E., Levine S.S., Zucker J.P., Guenther M.G., Kumar R.M., Murray H.L., Jenner R.G., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols J., Zevnik B., Anastassiadis K., Niwa H., Klewe-Nebenius D., Chambers I., Scholer H., Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 19.Wei Z., Yang Y., Zhang P., Andrianakos R., Hasegawa K., Lyu J., Chen X., Bai G., Liu C., Pera M., et al. Klf4 interacts directly with Oct4 and Sox2 to promote reprogramming. Stem Cells. 2009;27:2969–2978. doi: 10.1002/stem.231. [DOI] [PubMed] [Google Scholar]
- 20.Liang J., Wan M., Zhang Y., Gu P., Xin H., Jung S.Y., Qin J., Wong J., Cooney A.J., Liu D., et al. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat. Cell Biol. 2008;10:731–739. doi: 10.1038/ncb1736. [DOI] [PubMed] [Google Scholar]
- 21.Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J., et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 22.Soufi A., Donahue G., Zaret K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bailey T.L., Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- 25.Mahony S., Benos P.V. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B., et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung W.K., Zhao M., Liu Z., Stevens L.E., Cao P.D., Fang J.E., Westbrook T.F., Nguyen D.X. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell. 2013;23:725–738. doi: 10.1016/j.ccr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W.J., Ho C.C., Chang Y.L., Chen H.Y., Lin C.A., Ling T.Y., Yu S.L., Yuan S.S., Chen Y.J., Lin C.Y., et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014;5:3472. doi: 10.1038/ncomms4472. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Berg D.L., Snoek T., Mullin N.P., Yates A., Bezstarosti K., Demmers J., Chambers I., Poot R.A. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chavez L., Bais A.S., Vingron M., Lehrach H., Adjaye J., Herwig R. In silico identification of a core regulatory network of OCT4 in human embryonic stem cells using an integrated approach. BMC Genomics. 2009;10:314. doi: 10.1186/1471-2164-10-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pardo M., Lang B., Yu L., Prosser H., Bradley A., Babu M.M., Choudhary J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell. 2010;6:382–395. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang W.C., Hung J.J. Functional role of post-translational modifications of Sp1 in tumorigenesis. J. Biomed. Sci. 2012;19:94. doi: 10.1186/1423-0127-19-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hollander M.C., Blumenthal G.M., Dennis P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong T.M., Yang P.C., Peck K., Chen J.J., Yang S.C., Chen Y.C., Wu C.W. Profiling the downstream genes of tumor suppressor PTEN in lung cancer cells by complementary DNA microarray. Am. J. Respir. Cell Mol. Biol. 2000;23:355–363. doi: 10.1165/ajrcmb.23.3.4002. [DOI] [PubMed] [Google Scholar]
- 36.Oskarsson T., Acharyya S., Zhang X.H., Vanharanta S., Tavazoie S.F., Morris P.G., Downey R.J., Manova-Todorova K., Brogi E., Massague J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.M., et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stronach E.A., Alfraidi A., Rama N., Datler C., Studd J.B., Agarwal R., Guney T.G., Gourley C., Hennessy B.T., Mills G.B., et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011;71:4412–4422. doi: 10.1158/0008-5472.CAN-10-4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luis-Ravelo D., Anton I., Zandueta C., Valencia K., Ormazabal C., Martinez-Canarias S., Guruceaga E., Perurena N., Vicent S., De Las Rivas J., et al. A gene signature of bone metastatic colonization sensitizes for tumor-induced osteolysis and predicts survival in lung cancer. Oncogene. 2013;33:5090–5099. doi: 10.1038/onc.2013.440. [DOI] [PubMed] [Google Scholar]
- 40.Petz M., Them N.C., Huber H., Mikulits W. PDGF enhances IRES-mediated translation of Laminin B1 by cytoplasmic accumulation of La during epithelial to mesenchymal transition. Nucleic Acids Res. 2012;40:9738–9749. doi: 10.1093/nar/gks760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veeck J., Dahl E. Targeting the Wnt pathway in cancer: the emerging role of Dickkopf-3. Biochim. Biophys. Acta. 2012;1825:18–28. doi: 10.1016/j.bbcan.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 42.Tsai C.H., Cheng H.C., Wang Y.S., Lin P., Jen J., Kuo I.Y., Chang Y.H., Liao P.C., Chen R.H., Yuan W.C., et al. Small GTPase Rab37 targets tissue inhibitor of metalloproteinase 1 for exocytosis and thus suppresses tumour metastasis. Nat. Commun. 2014;5:4804. doi: 10.1038/ncomms5804. [DOI] [PubMed] [Google Scholar]
- 43.Wu C.Y., Tseng R.C., Hsu H.S., Wang Y.C., Hsu M.T. Frequent down-regulation of hRAB37 in metastatic tumor by genetic and epigenetic mechanisms in lung cancer. Lung Cancer. 2009;63:360–367. doi: 10.1016/j.lungcan.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 44.Yanagi S., Kishimoto H., Kawahara K., Sasaki T., Sasaki M., Nishio M., Yajima N., Hamada K., Horie Y., Kubo H., et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J. Clin. Invest. 2007;117:2929–2940. doi: 10.1172/JCI31854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orend G., Chiquet-Ehrismann R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006;244:143–163. doi: 10.1016/j.canlet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 46.Saupe F., Schwenzer A., Jia Y., Gasser I., Spenle C., Langlois B., Kammerer M., Lefebvre O., Hlushchuk R., Rupp T., et al. Tenascin-C downregulates wnt inhibitor dickkopf-1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep. 2013;5:482–492. doi: 10.1016/j.celrep.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 47.Ishigaki T., Imanaka-Yoshida K., Shimojo N., Matsushima S., Taki W., Yoshida T. Tenascin-C enhances crosstalk signaling of integrin alphavbeta3/PDGFR-beta complex by SRC recruitment promoting PDGF-induced proliferation and migration in smooth muscle cells. J. Cell Physiol. 2011;226:2617–2624. doi: 10.1002/jcp.22614. [DOI] [PubMed] [Google Scholar]
- 48.Seguin L., Kato S., Franovic A., Camargo M.F., Lesperance J., Elliott K.C., Yebra M., Mielgo A., Lowy A.M., Husain H., et al. An integrin beta3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014;16:457–468. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin Y., Yang Y., Li W., Chen Q., Li J., Pan X., Zhou L., Liu C., Chen C., He J., et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell. 2012;48:627–640. doi: 10.1016/j.molcel.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.