


Abstract
The NF-κB is found in almost all animal cell types and is involved in a myriad of cellular responses. Aberrant expression of NF-κB has been linked to cancer, inflammatory diseases and improper development. Little is known about transcriptional regulation of the NF-κB family member gene RelA/p65. Sp1 plays a key role in the expression of the RelA/p65 gene. ZBTB2 represses transcription of the gene by inhibiting Sp1 binding to a Sp1-binding GC-box in the RelA/p65 proximal promoter (bp, −31 to −21). Moreover, recent studies revealed that RelA/p65 directly binds to the peroxisome proliferator-activated receptor-γ coactivator1α (PGC1α) to decrease transcriptional activation of the PGC1α target gene PDK4, whose gene product inhibits pyruvate dehydrogenase (PDH), a key regulator of TCA cycle flux. Accordingly, we observed that RelA/p65 repression by ZBTB2 indirectly results in increased PDK4 expression, which inhibits PDH. Consequently, in cells with ectopic ZBTB2, the concentrations of pyruvate and lactate were higher than those in normal cells, indicating changes in glucose metabolism flux favoring glycolysis over the TCA cycle. Knockdown of ZBTB2 in mouse xenografts decreased tumor growth. ZBTB2 may increase cell proliferation by reprogramming glucose metabolic pathways to favor glycolysis by upregulating PDK4 expression via repression of RelA/p65 expression.
INTRODUCTION
Among the nearly 200 different members of the POZ-domain protein family, 48 members have a C-terminal C2H2 Krüppel-type zinc finger DNA-binding domain (http://btb.uhnres.utoronto.ca). Some of the POZ-ZF proteins are characterized as important transcription factors implicated in cancer and development (1). We previously investigated the biological functions of various POK family proteins, including KR-POK, FBI-1, ZBTB5 and ZBTB2 (zinc finger and BTB domain containing 2) (2–7). While numerous studies have identified ZBTB2 as part of various protein complexes (8–10), its functional characteristics remain largely unknown. We demonstrated that ZBTB2 is a master proto-oncoprotein that controls the expression of genes in the p53 pathway (ARF-HDM2-p53-p21) and, in particular, is a potent transcriptional repressor of the cell cycle arrest gene CDKN1A through inhibition of p53 and Sp1 (7). Moreover, targeting of ZBTB2 in human gastric cancer by microRNA-149 inhibits proliferation and cell cycle progression (11), and a genome-wide association study identified ZBTB2 in a gene cluster associated with susceptibility to chronic myeloid leukemia (12). ZBTB2 was also identified as one of the proteins that belong to the ubiquitin-proteasome system that is required for mammalian DNA damage checkpoint control, particularly at the G1 cell cycle checkpoint (13). Interestingly, ZBTB2 was also found to associate with the transcription cofactor CBP (14), and ZBTB2 has also been identified as a candidate oncoprotein having an R261W polymorphism that potentiates its mitogenic activity in human colorectal cancers with microsatellite instability (15).
NF-κB family members, central mediators of the human immune (both innate and adaptive immunity) (16–18) and stress responses (19) are activated by various intra- and extra-cellular stimuli such as bacteria, viruses, cytokines, oxidative stress, growth factors and hormones (20). In particular, NF-κB regulates the expression of genes controlling apoptosis, cell adhesion, proliferation, inflammation and tissue remodeling (21,22), and dysregulation of NF-κB activity has been linked to inflammatory disorders, autoimmune and metabolic diseases, as well as cancer (23–25).
In mammalian cells, there are five NF-κB family members, RelA/p65, RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2). All NF-κB family members contain a structurally conserved N-terminal Rel-homology domain, composed of 300 amino acids, that mediates DNA binding and subunit dimerization. Although all NF-κB family members bind DNA, only RelA/65, c-Rel and RelB have a transactivation domain in their C-termini. While RelB and c-Rel show tissue-specific expression, RelA/p65 and p50/NF-κB1 exist in a variety of cell types, and in the nucleus, RelA/p65 induces numerous genes that regulate the cellular processes mentioned above (26). Moreover, activity of the RelA/p65 subunit of the NF-κB complex can be regulated by post-translational modifications such as phosphorylation, acetylation and methylation (27–29).
In addition to the translocation and post-translational modifications of NF-κB in response to various stimuli, the precise regulation of each subunit of NF-κB is critical. In particular, the expression of RelA/p65 is significant to NF-κB activity because RelA/p65 is a primary subunit of the NF-κB complex expressed in almost all tissues. Although post-translational modifications of RelA/p65 affecting NF-κB activity have been studied extensively, the molecular mechanism on how RelA/p65 gene transcription is regulated remains elusive. Although the RelA/p65 promoter region is highly GC-rich and has many GC boxes to which Sp1 may bind (30), little is known about how these sites may regulate RelA/p65 expression. Previously, it was shown that human cytomegalovirus infection increased Sp1 mRNA and protein which, in turn, may potently activate RelA/p65 transcription through its promoter Sp1-binding sites (31,32); these sites may also bind MDM2 to induce RelA/p65 (33).
A key regulator of TCA cycle flux, the pyruvate dehydrogenase (PDH) complex that converts pyruvate into acetyl coenzyme A (Ac-CoA), is regulated by four PDH kinase isozymes (PDK1-4) (34). These kinases are located in the matrix of the mitochondria and phosphorylate and inhibit PDH complex subunits. Additionally, it was recently shown that PGC1α (peroxisome proliferator–activated receptor-γ coactivator 1α), a key regulator of lipid and glucose oxidative metabolism, increases PDK4 gene expression by binding to a putative nuclear receptor (NR) binding site in the PDK4 promoter (35). Additionally, in the myocardium, TNFα-activated RelA/p65 binds to and inhibits PGC1α transactivation activity, resulting in downregulation of PDK4 (36).
Here, we investigated the functional significance of whether ZBTB2 regulates RelA/p65 to elicit a shift in metabolism in highly proliferating or cancer cells through enhanced PGC1α transcriptional activation of PDK4 and inhibition of PDH. We demonstrate that ZBTB2 may be a proto-oncoprotein that plays important roles in cell proliferation by reprogramming metabolic pathways via transcriptional regulation of RelA/p65 and indirect upregulation of PDK4 expression.
MATERIALS AND METHODS
Plasmids, antibodies and reagents
To express ZBTB2 protein with a FLAG tag, a full-length human ZBTB2 cDNA fragment was polymerase chain reaction (PCR)-amplified from a cDNA library and cloned into pcDNA3 vector (Invitrogen). The pcDNA3-ΔPOZ-ZBTB2 expression vector was prepared by cloning the ZBTB2 cDNA fragment lacking the POZ domain and reported elsewhere (7). To prepare recombinant GST-POZ-ZBTB2 and GST-ZF-ZBTB2 proteins, cDNA fragments encoding the POZ domain (a.a. 24–117) and zinc fingers (a.a. 254–468) were cloned into pGEX4T3 (Amersham Biosciences). Recombinant His-ZBTB2 full-length protein was produced by cloning ZBTB2 cDNA into pET-21(a) expression vector (EMD Millipore) and induced expression in Escherichia coli DH5α cells. To prepare the pGL2-RelA/p65-Luc plasmid, the human RelA/p65 promoter region (bp, −401 to +92) was PCR-amplified from human genomic DNA isolated from human embryonic kidney HEK293 cells and cloned into the XhoI and HindIII sites of the pGL2-basic vector. Other pGL2-RelA/p65-Luc constructs with shorter promoter sequences were cloned in the same way. The pGL2-RelA/p65-Luc −47-bp construct with mutation at GC-box #3 (bp, −30 to −20) was prepared by site-directed mutagenesis. The pNF-κB-Luc plasmid was purchased from Clontech. All plasmid constructs were verified by sequencing. Antibodies against RelA/p65, Sp1 and PGC1α were purchased from Santa Cruz Biotechnology. Antibodies recognizing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and PDK4 were purchased from Millipore and Abcam, respectively. Most of the chemical reagents were purchased from Sigma.
Cell culture and transcription analysis
Human HEK293 and U343 cells were cultured in Dulbecco's Modified Eagle Medium (Life Technologies) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). To analyze transcriptional regulation of the RelA/p65 promoter or pNF-κB-Luc by ZBTB2, various promoter–reporter construct plasmids, in various combinations with pcDNA3-ZBTB2 or empty pcDNA3 vectors, were transiently transfected into HEK293 cells using Lipofectamine Plus reagent (Invitrogen). The transfected cells were further cultured for 48 h, harvested and analyzed for luciferase activity. Reporter activity was normalized to the total protein concentration of the cell lysates.
RNA isolation and real-time PCR quantification of mRNA
Total RNA was isolated from HEK293 and U343 cells using TRIzol reagent (Invitrogen). A cDNA pool was then synthesized using 5 μg of total RNA, oligo-dT primers (10 pmol) and reverse transcriptase II (200 units) in a 20-μl reaction volume, using a reverse transcription kit (Promega). RT-qPCR was then performed using SYBR Green Master Mix (Applied Biosystems) in a StepOnePlus real-time PCR instrument (Applied Biosystems). The following oligonucleotide PCR primer sets were used for qPCR quantification of ZBTB2, RelA/p65, Sp1 and 18S RNA: ZBTB2 forward, 5′-GATCGGATCCGATTTGGCCAACCATGGA-3′, reverse, 5′-GATCCTCGAGAGAAAAGGCTCCCTGGCT-3′; RelA/p65 forward, 5′-CTGCAGTTTGATGATGAAGA-3′, reverse, 5′-TAGGCGAGTTATAGCCTCAG-3′; Sp1 forward, 5′-GGAGAGCAAAACCAGCAGAC-3′, reverse, 5′-CAATGGGTGTCAGAGTGGTG-3′; 18S RNA forward, 5′- AGTCCCTGCCCTTTGTACACA-3′, reverse, 5′-GATCCGAGGGCCTCACTAAAC-3′; IL-1α forward, 5′-ATCAGTACCTCACGGCTGCT-3′, reverse, 5′-TGGGTATCTCAGGCATCTCC-3′; IL-6 forward, 5′-CCACACAGACAGCCACTCACC-3′, reverse, 5′-CTACATTTGCCGAAGAGCCCTC-3′; IL-8 forward, 5′-ATGACTTCCAAGCTGGCCGTGGCT-3′, reverse, 5′-TCTCAGCCCTCTTCAAAAACTTCTC-3′.
Western blot analyses
HEK293 and U343 cells were harvested 48 h after transfection and lysed in RIPA buffer (50-mM Tris-HCl pH 8.0, 150-mM NaCl, 2-mM ethylenediaminetetraacetic acid (EDTA) pH 8.0, 1% Nonidet P-40, 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulphate (SDS)) for 30 min on ice and pelleted. Protein concentrations were determined using the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad). Proteins (40–60 μg) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) (8–10%) and transferred onto Immun-BlotTM polyvinylidene difluoride membranes (Bio-Rad), and blocked with 3% bovine serum albumin (BSA) (Sigma-Aldrich). The membranes were then incubated overnight at 4°C with primary antibody, and protein bands were detected using horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Thermo Fisher Scientific) and visualization using ECL solution (Thermo Fisher Scientific).
Knockdown of endogenous ZBTB2 and RelA/p65 mRNA by siRNA
Three siRNAs against ZBTB2 mRNA were designed and purchased from Bioneer (Seoul, Korea): siZBTB2–1 5′-GAUUAGAACAGGGCAUCAA(dTdT)-3′, 5′-UUGAUGCCCUGUUCUAAUC(dTdT)-3′; siZBTB2–2 5′-CUCCACUCCCAAUGAGGUU(dTdT)-3′, 5′-AACCUCAUUGGGAGUGGAG(dTdT)-3′ and siZBTB2–3 5′-CUCUUACACUUGAUGUACA(dTdT)-3′, 5′-UGUACAUCAAGUGUAAGAG(dTdT)-3′. The siRNAs against RelA/p65 mRNA were purchased from Bioneer: siRelA/p65 5′-CCUGAGCACCAUCAACUAU(dTdT)-3′, 5′-AUAGUUGAUGGUGCUCAGG(dTdT)-3′. siRNAs were transfected into HEK293 and U343 cells using the Lipofectamine RNAiMAX Reagent (Invitrogen).
Purification of GST fusion proteins and GST fusion protein pull-down assays
pGEX4T3-POZ-ZBTB2 and pGEX4T3-ZFDBD-ZBTB2 expression plasmids, encoding the ZBTB2 POZ and zinc finger domains, respectively, were prepared as described in the plasmid section. The pGEX4T3 (Control), pGEX4T3-POZ-ZBTB2 and pGEX4T3-ZFDBD-ZBTB2 plasmids were transformed into E. coli BL21 (DE3) cells and grown for 4 h at 37°C in standard media. After 4 h, the bacteria were further cultured for 14 h at 18°C with 0.2-mM isopropyl 1-thio-β-D-galactopyranoside (IPTG, Calbiochem) to induce protein expression. E. coli was then lysed in E. coli lysis buffer (1-mM PMSF, 2-mM EDTA, 0.2-mg/ml lysozyme up to 1x phosphate buffered saline (PBS)) and recombinant proteins purified by glutathione sepharose bead affinity chromatography (GE Healthcare). The purified proteins were resolved by SDS-PAGE (12%) to quantitate and assess purity. The Sp1 polypeptide was prepared using TNT Quick-coupled Transcription/Translation Extract (Promega) containing 40 μl of TNT Quick Master Mix and 2 μl of [35S]-methionine for 90 min at 30°C. GST or GST fusion proteins bound to glutathione sepharose beads were washed three times with cold HEMG buffer (40-mM HEPES pH 7.9, 100-mM KCl, 0.2-mM EDTA pH 8.0, 5-mM MgCl2, 0.1% NP-40, 10% glycerol and 1.5-mM DTT) and collected by centrifugation at 3000 rpm at 4°C for 3 min. GST beads or GST fusion protein beads were incubated with [35S]-methionine-labeled synthetic Sp1 protein in HEMG buffer overnight at 4°C with shaking. The reaction mixtures were then centrifuged and the pelleted precipitates washed three times with 1-ml cold HEMG buffer. The bound proteins were resolved by SDS-PAGE (8%) and the gels then dried and exposed to X-ray film with an image-intensifying screen (Agfa-Gevaert).
Immunoprecipitation assays
HEK293 cells were harvested 48 h after transfection with ZBTB2- and Sp1-overexpressing vectors and lysed in RIPA buffer (50-mM Tris-HCl pH 8.0, 150-mM NaCl, 2-mM EDTA pH 8.0, 1% Nonidet P-40, 0.5% Na-deoxycholate, 0.1% SDS). Cell lysates were pre-cleared, and the supernatants were incubated with anti-ZBTB2 or anti-Sp1 antibodies on a rotating platform at 4°C for 15 h. After incubation, the cell lysates and antibody complexes were incubated with protein A-sepharose fast flow beads for 4 h at 4°C. The beads were then washed three times with PBS, collected and suspended in equal volumes of 5x SDS loading buffer. Immunoprecipitated proteins were separated by 8% SDS-PAGE, transferred to membranes, and western blot, using the appropriate antibodies, was performed as described above.
Oligonucleotide pull-down assays
HEK293 cells were transfected with increasing amounts of pcDNA3-ZBTB2, cultured for 48 h and lysed in HKMG buffer (10-mM HEPES pH 7.9, 100-mM KCl, 5-mM MgCl2, 10% glycerol, 1-mM dithiothreitol, 0.5% NP-40). Cell lysates were then incubated with 1 μg of biotinylated double-stranded oligonucleotide (GC-box #3, GC-box #7, GC-box #12) at 4°C overnight. The sequences of the oligonucleotides were as follows (only sense strands are shown): GC-box #3, 5′-CCCTGCGCGGGGCGGGCGGCCGCAG-3′; GC-box #7, 5′-CTCCAGGCGGGGCCGGGACCCGGGA-3′; GC-box #12, 5′-CACTTGCTCCCGGCCCCTGCGCCGG-3′. Both forward and reverse oligonucleotides were incubated with 10x reaction buffer (3) (500-mM Tris-HCl pH 8.0, 100-mM MgCl2, 1000-mM NaCl) at 95°C for 5 min. After incubation, the temperature was gradually reduced to room temperature. To collect DNA-bound proteins, the mixtures were incubated overnight with streptavidin agarose beads (Thermo Scientific), washed with PBS buffer and precipitated by centrifugation. The precipitate was then analyzed by western blot as described above, using anti-SP1 or anti-ZBTB2 antibodies.
Quantitative chromatin immunoprecipitation (qChIP) PCR and ChIP-reChIP assays
HEK293 cells were transfected with pcDNA3 or pcDNA3-ZBTB2, grown for an additional 48 h and treated with formaldehyde (final concentration 1%) to cross-link proteins to DNA, followed by cell lysis in RIPA buffer. Chromatin was sheared three times (amplitude: 50%; cycle: 0.5; sec: 30 s) by sonication (Bioruptor, Diagenode) and the supernatant then incubated with specific antibodies at 4°C overnight. The next day, the mixture was incubated with salmon sperm DNA/protein-A agarose beads at 4°C for 3 h. The beads were then collected, washed with PBS and eluted with elution buffer (1% SDS, 0.1-M NaHCO3). Input and immunoprecipitated chromatin were then incubated with 5-M NaCl at 65°C for 4 h to reverse crosslinks. After incubation, the temperature was gradually reduced to room temperature. To precipitate the DNA, phenol/chloroform/isoamyl alcohol (Calbiochem) was added and the DNA pellet obtained by centrifugation. As a negative control for the qChIP assays, IgG was used. To amplify the promoter regions, PCR reactions of the immunoprecipitated DNA were carried out using the following sets of oligonucleotide primers: GC-box #3 region within the RelA/p65 promoter, forward, 5′-CGAGGCGCGCACTTGGCCCCGAC-3′, reverse, 5′-CCGTCGCGTCACTGCCCGGAATC-3′; NR binding site within the PDK4 promoter, forward, 5′-AATGAGCACGCGGAGTCCAAACTCTT-3′, reverse, 5′-CAAGCTGGGCTTAAGATTAGCCTCTT-3′. PCR was performed using the following cycling conditions: denaturation at 94°C for 5 min, followed by 40 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 35 s and a final extension reaction at 72°C for 5 min, using a GeneAmp PCR system 9700 (Applied Biosystems).
For ChIP-reChIP assays of Sp1 binding to the RelA/p65 proximal promoter, the first chromatin immunoprecipitates by an anti-ZBTB2 antibody were diluted 10 times with buffer (15-mM Tris–HCl pH 8.1, 1% Triton X-100, 1-mM EDTA,150-mM NaCl) and re-immunoprecipitated using an anti-Sp1 antibody. Negative control of ChIP assays was carried out using a rabbit IgG antibody.
Electrophoretic mobility shift assays
The sequences of the three 32P-labeled GC-box probes of the RelA/p65 promoter are described below. Electrophoretic mobility shift assay (EMSA) was performed by incubating with recombinant GST-ZFDBD-ZBTB2 protein with the following probes (only the sequences of the top strands are shown). GC-box #3, 5′-GATCTTTCCCTGCGCGGGGCGGGCGGCCGCAGCCC-3′; GC-box #7, 5′-GATCACCCTCCAGGCGGGGCCGGGACCCGGGAGCT-3′; GC-box #12, 5′-GATCGTGCACTTGCTCCCGGCCCCTGCGCCGGGCG-3′. The probes were labeled by the addition of Klenow enzyme and 32P-α-[dATP], followed by purification using Sephadex G-25 columns. Binding reactions were performed at room temperature for 30 min in 5x binding buffer (50-mM HEPES pH 7.9, 300-mM KCl, 50% glycerol, 10-mM DTT and 1% BSA) containing recombinant GST-ZFDBD-ZBTB2 protein or recombinant Sp1 protein (Enzo Life Sciences) and labeled probe (50 000 cpm). Where indicated, an antibody against Sp1 was also added. To demonstrate the specificity of the protein–DNA complexes, a large excess of an unlabeled (‘cold’) oligonucleotide containing the Sp1 binding GC-boxes #3, #7 and #12 was added before the labeled probes to compete for Sp1 binding. To investigate competitive binding between Sp1 and ZBTB2, the probe was incubated with recombinant Sp1 and increasing amounts of GST-ZFDBD-ZBTB2 protein.
DNase I footprinting assays
To identify Sp1- or ZBTB2-binding sites, the RelA/p65 proximal promoter (246 bp) was cloned into the pcDNA3 vector, digested with EcoRI and labeled with Klenow fragment and 32P-α-[dATP] (PerkinElmer). The labeled plasmid was then digested with a second restriction enzyme, BamHI, separated by 4.5% PAGE, and the labeled promoter fragment then isolated by electro-elution using a Gebaflex electro-elution tube (GENTAUR). Sp1 (100, 300, 900 ng) or ZBTB2 (1, 3, 9 μg) was mixed in binding buffer (50-mM HEPES pH 7.9, 300-mM KCl, 50% glycerol, 10-mM ZnCl2, 10-mM DTT) and incubated at room temperature for 10 min. The 32P-labeled RelA/p65 promoter fragment was mixed with a pre-incubated mixture containing recombinant Sp1 or ZBTB2 and incubated for an additional 20 min at room temperature. A 20-μl aliquot of diluted DNase I (1:500) was then added, incubated for 2 min, and the DNase I digestion reaction stopped by adding 320 μl of stop solution (20-mM Tris-Cl pH 8.0, 20-mM EDTA pH 8.0, 250-mM NaCl, 0.5% SDS, 10-mg/ml yeast tRNA, 20-mg/ml proteinase K). The mixture was extracted with phenol/CHCl3 and precipitated by the addition of 0.1 volumes 3-M sodium acetate and 2.5 volumes ethanol. Centrifuged pellets were resuspended in 10 μl of distilled water and 10 μl of formamide loading buffer (Fisher Scientific). The digested DNA (20 000 cpm) mixtures were separated by 6% polyacrylamide gels containing 7-M urea (Duchefa Biochemie). An autoradiogram was obtained by exposing the dried gel to X-ray film (FUJIFILM) overnight at −70°C with an image enhancing screen (Kodak).
Cytosolic and nuclear protein fractionation
HEK293 cells were transfected with pcDNA3-ZBTB2 and/or pcDNA3-Sp1 expression vectors and grown for an additional 48 h. Cells were lysed by buffer A (10-mM HEPES pH 7.9, 1.5-mM MgCl2, 10-mM KCl, 1-mM DTT, 0.2-mM PMSF, 0.1% NP-40) and incubated on ice for 15 min. After centrifugation at 850 x g for 10 min, the supernatants of cell lysates were collected (cytoplasmic fractions) and pellets were resuspended in buffer A* (10-mM HEPES pH 7.9, 1.5-mM MgCl2, 10-mM KCl, 1-mM DTT, 0.2-mM PMSF). After centrifugation at 1500 x g for 10 min, the pellets were resuspended with buffer B (20-mM HEPES pH 7.9, 1.5-mM MgCl2, 25% glycerol, 0.42-M NaCl, 0.2-mM EDTA, 1-mM DTT, 0.2-mM PMSF) and incubated on ice for 30 min. The nuclear fractions were then collected by centrifugation at 13 000 rpm for 10 min. Final pellets containing nuclei were resuspended for western blot.
PDH activity assays
HEK293 and U343 cells were transfected with pcDNA3 or pcDNA3-ZBTB2 expression plasmid, harvested 48 h after transfection and resuspended in PBS. Cells were lysed by sonication (Bioruptor) (amplitude: 50%, cycle: 0.5, 5 times). Twenty microliter of the cell lysates was then placed in 96-well plates followed by the addition of 25-mM oxaloacetate and DTNB (5–5′-dithiobis(2-nitrobenzoic acid)) and 165-μl reaction assay mixture (0.25-M Tris-HCl pH 8.0, 40-mM β-NAD+, 32.5-mM CoA-SH, 20-mM TPP, 56-mM pyruvate, 1-M MgCl2, 100-mM DTT, 1-μl citrate synthase (0.25 unit)). After 10-min incubation, the reaction mixtures were analyzed using a VERSAMAX microplate reader (Molecular Devices) (412 nm, run time: 20 min, interval: 15 s).
Analysis of lactate concentration
HEK293 and U343 cells were transfected with pcDNA3 or pcDNA3-ZBTB2 expression plasmid and harvested 48 h after transfection. Lactate concentration was determined by measuring absorbance at 570 nm using a colorimetric assay kit (Sigma).
Preparation of recombinant expressing adenovirus siRNA against ZBTB2 mRNA
To prepare recombinant adenovirus expressing siRNA against ZBTB2 mRNA, annealed shRNA (sense: 5′-GATC-CAGGTGAATCGGACAAATATT-TTCAAGAGA(loop)-TATTTGTCCGATTCACCTGTT-TTTTTGGAA(loop)-A-3′), antisense: 5′-GATC-TTCCAAAAA(loop)-AACAGGTGAATCGGACAAATA-TCTCTTGAA(loop)-AATATTTGTCCGATTCACCTG-G-3′) was cloned into pSilencer 2.0-U6 (Ambion) and subcloned into the pΔE1sp1A vector. pΔE1sp1A-U6-shZBTB2 vector and the adenovirus vector vmdl324Bst were linearized by restriction enzyme digestion. Linearized pΔE1sp1A-U6-shZBTB2 was co-transformed into E. coli BJ518 with vmdl324Bst for homologous recombination. Homologous recombinant adenoviral plasmid was digested with PacI and transfected into HEK293 cells to generate the adenovirus expressing siRNA against ZBTB2 mRNA(dl324-shZBTB2).
Effect of ZBTB2 on tumor growth in vivo
U343 glioblastoma tumor cells were implanted under the abdominal skin of female BALB/c mice. Once tumors had reached 100–120 mm3 in volume, mice were injected intra-tumorally five times at 2-day intervals with either control dl324 or dl324-shRNA ZBTB2 viruses (2 × 108 PFU). Tumor growth was monitored by measuring the length and width of the tumor three times a week using a caliper. Tumor volume = 0.523Lw2, where L is length and w is width in mm.
Immunohistochemical analyses of human glioblastoma tissues
Formaldehyde-fixed, paraffin-embedded glioblastoma tissue sections were obtained from Dr Se-Hoon Kim of the Department of Pathology, Yonsei University School of Medicine. DAB immunohistochemical (IHC) staining was done according to the manufacturer's instructions (VECTASTAIN ABC Kit, Vector Laboratories) after incubating with anti-ZBTB2, anti-RelA/p65 and anti-PDK4 antibodies overnight at 4°C.
Statistical analysis
Student's t-test was used for the statistical analyses. P-values of <0.05 were considered statistically significant.
RESULTS
The RelA/p65 promoter is activated by Sp1 and a specific proximal GC-rich element (bp, −30 to −1) is important for transcriptional initiation/activation of the RelA/p65 gene by Sp1
Promoter analysis of the RelA/p65 gene for potential transcription factor binding sites using the MacVector program revealed that the promoter lacks a TATA consensus sequence and is GC-rich (71%) in a 445-bp region (bp, +44 to −401) from the transcription start point. The promoter region contains more than 14 potential Sp1-binding GC-boxes (bp, +25 to +34, +1 to +17, −1 to −30, −39 to −51, −82 to −95, −108 to −115, −177 to −192, −200 to −218, −240 to −262, −279 to −289, −300 to −309, −322 to −336, −361 to −378, −385 to −394) (Supplementary Figure S1). Sp1-binding GC-boxes cover the RelA/p65 promoter region extensively, suggesting that Sp1 might be a critical transcription regulator of RelA/p65 transcription. Accordingly, we investigated whether the RelA/p65 gene is regulated by Sp1 at the transcriptional level. Knockdown of Sp1 mRNA potently decreased RelA/p65 expression both at the mRNA and protein levels (Figure 1A and B).
Figure 1.
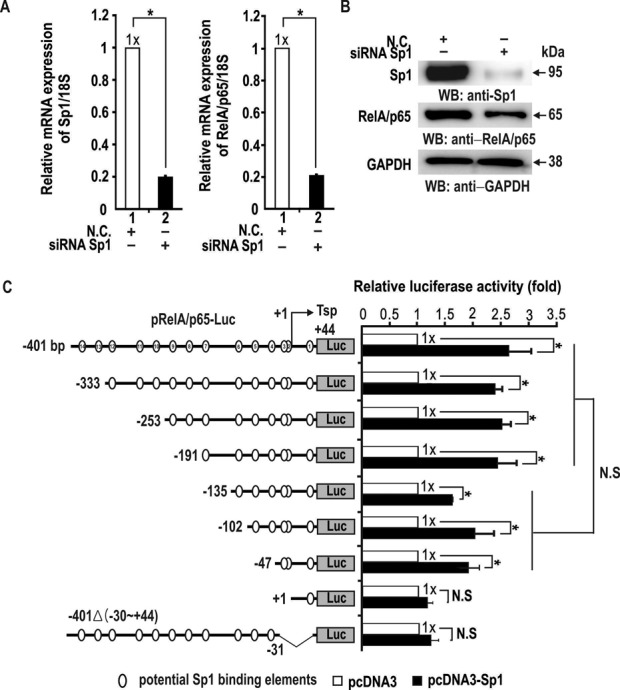
Sp1 activates transcriptional initiation/activation of RelA/p65 via a proximal promoter GC-rich element (bp, −30 to −1). (A, B) RT-qPCR and western blot analysis of RelA/p65 expression. Knockdown of Sp1 mRNA resulted in a potent decrease in RelA/p65 expression at both the mRNA and protein levels. *P < 0.05. (C) Transient transfection and transcription assays, and identification of a cis-regulatory element critical in transcriptional initiation/activation of RelA/p65 by Sp1 in HEK293 cells. The structures of various pGL2-RelA/p65-Luc constructs are shown on the left. +1 (Tsp), transcription start point. Potential Sp1-binding GC-boxes are indicated (open circles, as analyzed by MacVector program). The data presented are the average of three independent assays. Luciferase activity was normalized to total protein concentration. Bars indicate standard deviation. *P < 0.05, statistically significant.
To identify the RelA/p65 promoter regulatory elements that mediate transcriptional activation by Sp1, HEK293 cells were co-transfected with Sp1 expression vector and various RelA/p65 promoter reporter gene fusion constructs differing in the length of the 5′ upstream regulatory sequence (Figure 1C). The pRelA/p65-Luc −401Δ(bp, −30 to +44) reporter construct that retained almost all of the upstream regulatory sequence (except the promoter GC-box #3) was also tested. Transient transfection and transcription assays of various pRelA/p65-Luc plasmids showed that Sp1 could induce RelA/p65 primarily by acting on its proximal minimal promoter. Deletion of the promoter region down to −1 and deletion of a −30 to +44 region abolished transcriptional activation by Sp1. The pRelA/p65-Luc −135, −102 and −47 constructs showed nearly identical transcriptional activation by Sp1, suggesting that the Sp1 GC-boxes at bp −30 to −1 may be important for transcriptional initiation and/or activation. The pRelA/p65-Luc −401, −333, −253 and −191 constructs showed similar transcriptional activation by Sp1. Although overall transcription levels of these constructs appeared higher than those of the shorter constructs −135, −102, and −47, the differences were not statistically significant (Figure 1C). Alternatively, knockdown of Sp1 expression potently decreased transcription of the RelA/p65 promoter in most of the constructs, but not in the pRelA/p65-Luc +1 construct (Supplementary Figure S2B). These data suggest that Sp1 can activate RelA/p65 gene transcription primarily by acting on the proximal promoter element containing GC-box #3 (bp, −30 to −1).
ZBTB2 interacts directly with transcription factor Sp1 and represses the RelA/p65 promoter by acting on the proximal GC-rich element (bp, −30 to −1)
Previously, we showed that ZBTB2 interacts with Sp1 directly (7). Because the RelA/p65 gene is activated by Sp1, we tested whether the interaction between ZBTB2 and Sp1 is important for transcriptional regulation of RelA/p65. To that end, we found that the pGL2-RelA/p65-Luc (−47 bp) construct was activated by Sp1; deletion of further downstream regions resulted in no transcriptional activation by Sp1. Using the same constructs, we next investigated possible regulatory relationships between Sp1 and ZBTB2. As expected, Sp1 activated the RelA/p65 promoter by 1.8-fold and transcriptional activation by Sp1 was potently repressed by ZBTB2 in the absence or presence of Sp1 co-expression (70–80%) (Figure 2A). In contrast, the pGL2-RelA/p65-Luc (−401Δ(−30 to +44)) construct, with a deletion of the proximal promoter, was neither activated by Sp1 nor repressed by ZBTB2, suggesting that the minimal promoter region (containing GC-box #3, −30 to −1 bp) is important for transcription initiation and activation of the RelA/p65 gene by Sp1, and repression by ZBTB2. Consequently, the GC-box #3 element residing in the region from −30 to −1 bp may be the target of transcriptional repression by ZBTB2.
Figure 2.
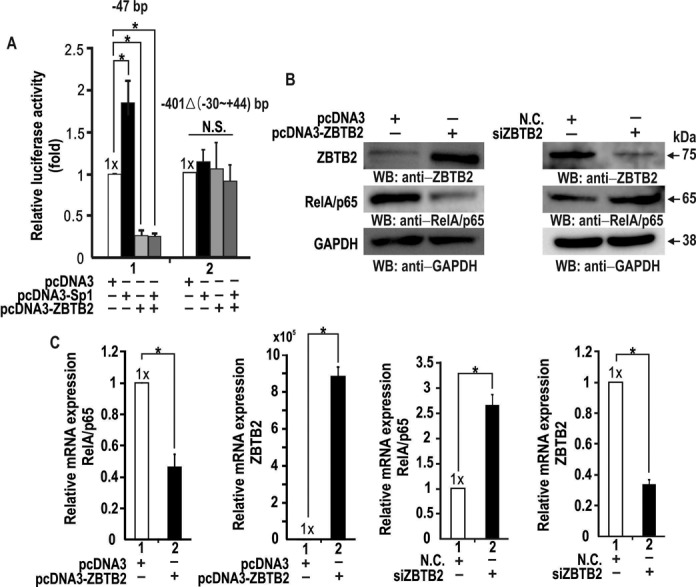
ZBTB2 potently represses endogenous RelA/p65 expression in HEK293 cells by acting on the proximal GC-rich promoter element (bp, −30 to −1). (A) Transient transfection and transcription assays in HEK293 cells. HEK293 cells were co-transfected with the pGL2-RelA/p65-Luc reporter constructs indicated (−47; −401Δ(−30 to +44)) and Sp1 and/or ZBTB2 expression vectors. Luciferase activity was measured 48 h after transfection and normalized to total protein concentration. Bars represent standard deviations. *P < 0.05. (B) Western blot analysis of regulation of endogenous RelA/p65 expression by ZBTB2. HEK293 cells were transfected with pcDNA3 or pcDNA3-ZBTB2 expression plasmid or ZBTB2 siRNA. Whole cell extracts were separated by SDS-PAGE and analyzed using the antibodies indicated. GAPDH, control. (C) RT-qPCR analysis of regulation of endogenous RelA/p65 mRNA expression by ZBTB2. Cells were treated as in (B). Total mRNA was isolated from cells and the levels of ZBTB2 and RelA/p65 mRNA determined by RT-qPCR and normalized to 18S ribosomal RNA. *P < 0.05.
ZBTB2 represses transcription of endogenous RelA/p65 gene expression in human HEK293 cells
Using RT-qPCR and western blot analysis, we investigated whether ectopic ZBTB2 overexpression or knockdown affected transcription of the endogenous RelA/p65 gene. In human HEK293 cells, ectopic ZBTB2 significantly downregulated RelA/p65, while ZBTB2 knockdown enhanced RelA/p65 expression at the protein level (Figure 2B). Ectopic ZBTB2 expression repressed RelA/p65 transcription by 0.4- to 0.5-fold, and knockdown of ZBTB2 increased transcription of the RelA/p65 gene by 2.5-fold (Figure 2C). These data further suggest that ZBTB2 represses RelA/p65 transcription by acting on its proximal promoter GC-box element (bp, −30 to −1).
ZBTB2 and Sp1 directly bind GC-boxes, including GC-box #3 (bp, −32 to −21) in the proximal RelA/p65 promoter, and the two proteins show identical DNase I footprinting patterns
Because Sp1 activates transcription of the RelA/p65 gene and ZBTB2 represses transcriptional activation by Sp1, we used DNase I footprinting to investigate DNA binding of Sp1 and ZBTB2 to five Sp1 footprinting regions (bp, +58 to +68; bp, +21 to +24; bp, −32 to −21; bp, −63 to −49; bp, −93 to −83) in the RelA/p65 proximal promoter. Intriguingly, although comparison of amino acid sequences of the zinc finger DNA-binding domains of ZBTB2 and Sp1 showed no apparent sequence homology, ZBTB2 bound to the exactly same regions recognized by Sp1 (Figure 3A). Interestingly, the pGL2-RelA/p65-Luc (−47 bp) construct, which showed transcriptional activation by Sp1 and repression by ZBTB2, contains two 11 base pair and one short four base pair DNase I footprinting regions (Figure 3B). Considering that there was no transcriptional regulation of pGL2-RelA/p65-Luc (401Δ(−30 to +44)) lacking DNase I footprinting region 3 (containing GC box #3) by Sp1 and ZBTB2 (Figure 2A), this region might be a key regulatory element important in transcription initiation and activation by Sp1 and also repression by ZBTB2 (Figure 3B).
Figure 3.

DNase I footprinting assays. ZBTB2 and Sp1 identically bind to five GC-box elements including GC-box #3 (bp, −32 to −21) of the RelA/p65 proximal promoter. (A) DNase I footprinting of the RelA/p65 promoter with recombinant Sp1 (100, 300, 900 ng) and His-ZBTB2 (1, 3, 9 μg). Left, sense strand short run; right, sense strand long run. G and G+A indicate Maxam and Gilbert sequencing reactions of the 32P-labeled promoter DNA fragment (246 bp). N, control reaction without Sp1 and ZBTB2. (B) Summary of DNase I footprinting sequences. Footprinting regions (1–5) are indicated in black. The transcription start point (+1, Tsp) is marked by an arrow.
ZBTB2 and Sp1 directly bind the GC-box #3 of the RelA/p65 promoter, and ZBTB2 blocks Sp1 binding
We next analyzed the molecular events involving DNA interactions by Sp1 and ZBTB2 at the RelA/p65 proximal minimal promoter because the region contains the key regulatory footprinting element GC-box #3 (bp, −32 to −21), which may be essential for transcriptional initiation and activation of the RelA/p65 gene by Sp1, and repression by ZBTB2. Using electrophoretic mobility shift assays (EMSAs), we further investigated whether recombinant GST-ZFDBD-ZBTB2 and Sp1 protein could bind the GC-box #3 probe flanking footprinting region 3 (bp, −32 to −21), showing that ZBTB2 and Sp1 directly interact with each other (Supplementary Figure S3), and both ZBTB2 and Sp1 bind to the GC-box #3 of the RelA/p65 promoter (Figure 4A). Oligonucleotide pull-down assays of whole cell lysates transfected with empty pcDNA3 or ZBTB2 expression vectors also showed that Sp1 and ZBTB2 bound to GC-box #3, where Sp1 binding was significantly decreased by the presence of ZBTB2, but not at GC-box #7 (bp, −177 to −192) or GC-box #12 (bp, −322 to −336) (Figure 4B). These data indicate that ZBTB2 might directly block Sp1 binding to GC-box #3 by binding competition.
Figure 4.
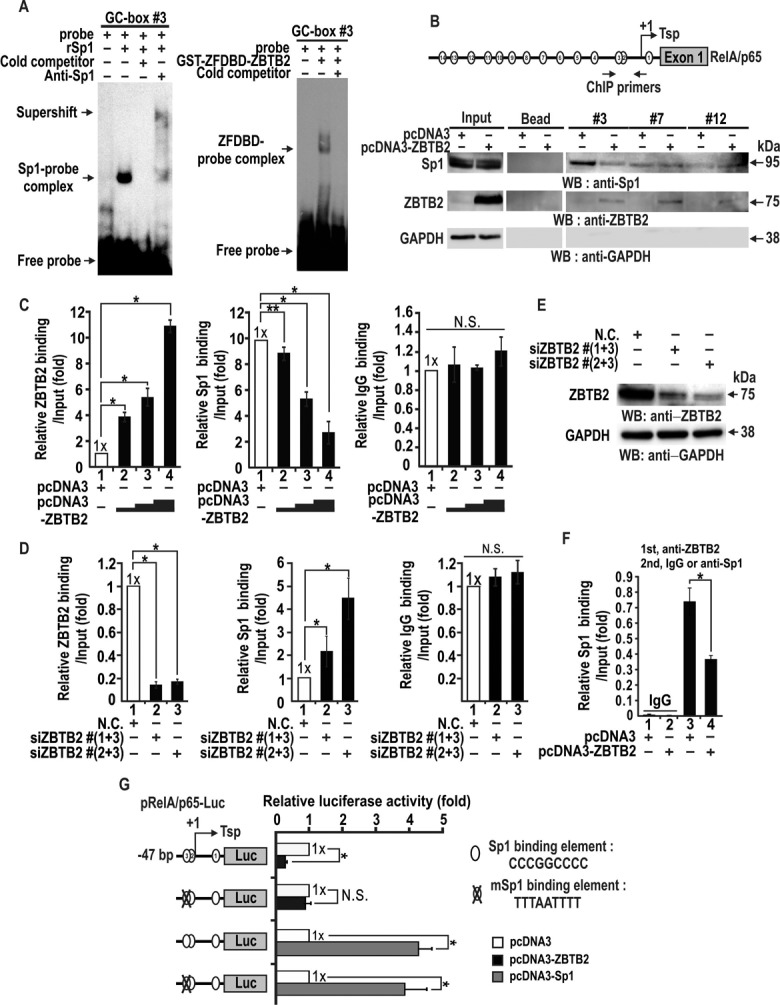
ZBTB2 and Sp1 directly bind to GC-box #3 in the proximal RelA/p65 promoter and ZBTB2 decreases Sp1 binding at the element in vitro and in vivo. (A) Electrophoretic mobility shift assays (EMSAs). The 32P-labeled GC-box #3 probe was incubated with recombinant Sp1 (100 ng) or GST-ZFDBD-ZBTB2 (1 μg), in the presence or absence of cold competitor probe (100x) or anti-Sp1 or anti-ZBTB2 antibodies, and the mixtures separated by 4% non-denaturing PAGE and analyzed by autoradiography. (B) Oligonucleotide pull-down assays of ZBTB2 and Sp1 binding. Structure of the RelA/p65 promoter with GC-box #3 and locations of ChIP oligonucleotide primers spanning the GC-box #3 are indicated above. GC-box #7 and #12 elements were used as controls. The transcription start point (Tsp, +1) is indicated by an arrow. HEK293 cells were transfected with pcDNA3 or pcDNA3-ZBTB2 plasmids and harvested after 48 h. Biotinylated oligonucleotide probes including GC-box #3 linked to streptavidin agarose beads were incubated with the cell lysates. The precipitates were then analyzed by western blotting using the antibodies indicated. GAPDH, control. (C, D) ChIP assays of ZBTB2 and Sp1 binding at the proximal promoter region flanking GC-box #3. HEK293 cells were transfected with increasing amounts of ZBTB2 expression vector or combinations of three distinct anti-ZBTB2 siRNAs. Chromatin was immunoprecipitated with the antibodies indicated and analyzed by qRT-PCR. IgG, negative control. *P < 0.05. **P = 0.06. (E) Knockdown of ZBTB2 expression and western blot analysis. HEK293 cells were transfected with an siRNA mixture. GAPDH, control. N.C., negative control scrambled siRNA. (F) ChIP-reChIP assay of ZBTB2 and Sp1 binding to the RelA/p65 promoter. HEK293 cells were transfected with pcDNA3-ZBTB2 and immunoprecipitated with ZBTB2 (first) or IgG and Sp1 antibodies (second). (G) Transient transfection and transcription assays. pRelA/p65-Luc −47-bp wild-type or mutant reporter constructs were co-transfected with control or ZBTB2 or Sp1 expression vector into HEK293 cells. Luciferase activity was measured 48 h after transfection. The data presented are the average of three independent assays. Luciferase activity was normalized to total protein concentration. *P < 0.05.
We also investigated whether ZBTB2 could block Sp1 binding at GC-box #3 by ChIP of HEK293 cells transfected with Sp1- and increasing amounts of ZBTB2-expression vectors. When ectopic ZBTB2 binding increased, Sp1 binding to the region decreased in a dose-dependent manner (Figure 4C). Alternatively, we also used ChIP to investigate whether knockdown of ZBTB2 expression could increase Sp1 binding to the region flanking GC-box #3 (Figure 4D and E). ChIP-re-ChIP assays showed that ectopic ZBTB2 decreased Sp1 binding to the element (Figure 4F).
Furthermore, to even more firmly establish the importance of GC-box #3 in RelA/p65 transcriptional initiation and activation by Sp1 and repression by ZBTB2, we made a reporter construct with a mutation introduced at the core of GC-box #3 (CCCGGCCCC to TTTAATTTT). Transient transcription assays showed that ZBTB2 potently downregulates RelA/p65 promoter having the wild-type, but not mutant, promoter GC-box #3. The mutation had no effect, however, on RelA/p65 transcriptional activation by Sp1, suggesting that GC-box #2 and/or #1 might also be important in transcription initiation and activation by Sp1 (Figure 4G).
ZBTB2 bound to the RelA/p65 proximal promoter GC-box #3 recruits the NCoR–HDAC complex and deacetylates histone H3 and H4 to repress RelA/p65 transcription
As shown Figure 4, ZBTB2 decreased Sp1 binding at the RelA/p65 proximal promoter. Using qChIP-PCR assays, we examined one possible mechanism of ZBTB2 repression, recruitment of the co-repressor NCoR-HDAC complex to the RelA/p65 proximal promoter GC-box #3 element and deacetylation of histones around its transcription initiation point. HEK293 cells were co-transfected with increasing amounts of either pcDNA3-ZBTB2 expression vector or ZBTB2 siRNA. These studies showed that ZBTB2 increased the recruitment of NCoR to the proximal promoter region to deacetylate histones H3 and H4 in a dose-dependent manner (Figure 5A). Additionally, knockdown of ZBTB2 strongly decreased NCoR recruitment to the region, resulting in increased acetylation of histones H3 and H4 (Figure 5B). These changes in epigenetic markers of histones around the transcription initiation sites suggest that ZBTB2 represses RelA/p65 transcription by nucleosome compaction around the promoter region critical in transcription.
Figure 5.
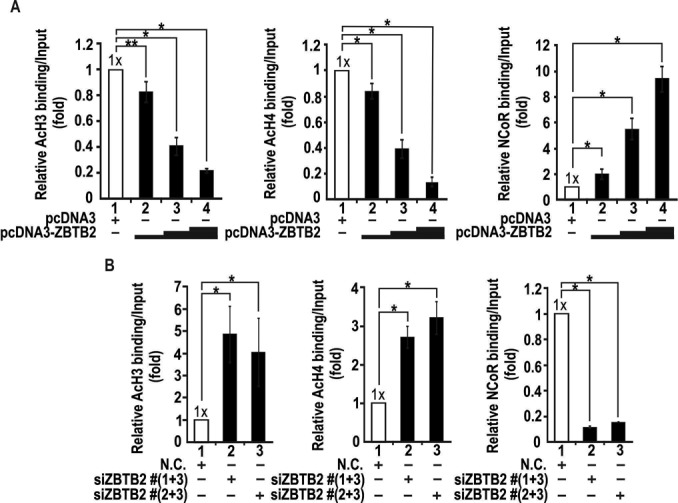
ZBTB2 bound at the proximal GC-box #3 of endogenous RelA/p65 recruits the NCoR–HDAC complex and deacetylates histones H3 and H4. ChIP assay of acetylated histones H3 and H4 and corepressor NCoR binding at the proximal promoter flanking GC-box #3 of the endogenous RelA/p65 gene. HEK293 cells were transfected with increasing amounts of ZBTB2 expression vector (A) or ZBTB2 siRNA (B). *P < 0.05. **P = 0.06. Chromatin was immunoprecipitated with the antibodies indicated and analyzed by qRT-PCR. *P < 0.05.
ZBTB2 represses transcriptional activation of the NF-κB responsive reporter plasmid pNF-κB-Luc by TNFα
Because RelA/p65 is crucial for NF-κB-mediated transcription, we examined whether ZBTB2 might affect reporter expression of pNF-κB-Luc in cells stimulated by TNFα (Figure 6A). ZBTB2 alone repressed transcription of pNF-κB-Luc, while TNFα treatment increased luciferase expression 3-fold, which was repressed by ZBTB2 to the levels of the control (Figure 6B). Knockdown of ZBTB2 derepressed transcription by 5-fold. TNFα treatment potently activated transcription of the reporter gene 43-fold (Figure 6C). These results suggest that ZBTB2 potentially plays a major role in suppressing the NF-κB target gene expression. Accordingly, we tested whether expression of endogenous NF-κB target genes such as IL-1α, -6 and -8 was affected by ZBTB2. Ectopic ZBTB2 expression repressed interleukin genes, while knockdown of ZBTB2 derepressed interleukin genes, particularly IL-1α (Supplementary Figure S4A and B).
Figure 6.
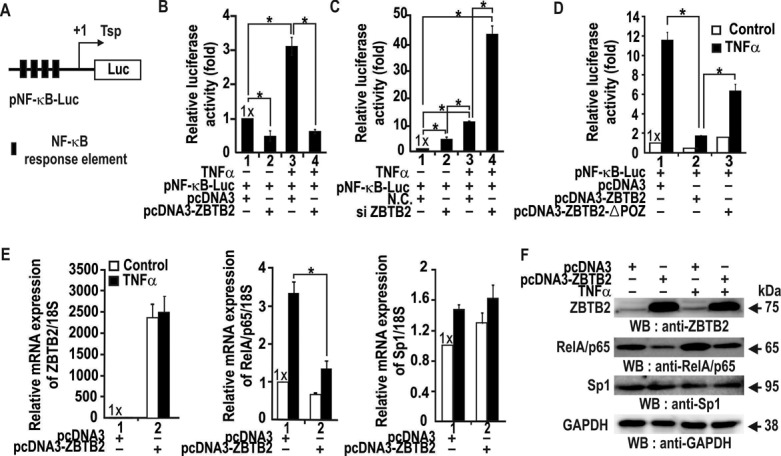
ZBTB2 represses transcriptional activation of pNF-κB-Luc and the RelA/p65 gene by TNFα in HEK293 cells. (A) Structure of pNF-κB-Luc. Filled black box, NF-κB responsive element; Tsp, transcription start point (+1). (B, C) Transient transcription assays in HEK293 cells transfected with pNF-κB-Luc and pcDNA3 or pcDNA3-ZBTB2 plasmids or ZBTB2 siRNA. Cells were treated with TNFα (20 ng/ml) for 12 h. Error bars, standard deviations; N.C., negative control (scrambled siRNA). (D) Transient transcription assays. HEK293 cells were co-transfected with pNF-κB-Luc and pcDNA3-ZBTB2 or pcDNA3-ZBTB2ΔPOZ. TNFα treatment was as described above. Luciferase activity was analyzed as in (B) and (C). (E) RT-qPCR analysis of RelA/p65 and Sp1 mRNA in TNFα-treated HEK293 cells transfected with ZBTB2 expression vector. Cells were treated with TNFα for 12 h. The data were normalized to 18S ribosomal RNA. (F) Western blot analysis of RelA/p65 and Sp1 expression in HEK293 cells treated as in (E). ZBTB2, RelA/p65, Sp1 and GAPDH protein in total cell lysates were analyzed using the antibodies indicated. GAPDH, control.
Because the POZ domain of ZBTB2 is important in transcriptional repression (7), we next investigated if the domain is also important in transcription of pNF-κB-Luc in HEK293 cells. While ZBTB2 potently repressed reporter expression, ZBTB2ΔPOZ (lacking the POZ domain) very weakly repressed reporter expression, suggesting the importance of the POZ domain in transcriptional repression (Figure 6D).
Furthermore, we investigated whether ZBTB2 influenced transcription of the endogenous RelA/p65 gene in HEK293 cells treated with TNFα. While the mRNA levels of ZBTB2 and Sp1 remained the same or slightly increased, TNFα treatment increased RelA/p65 mRNA expression. By contrast, endogenous RelA/p65 expression was potently repressed by ZBTB2 both in the absence or presence of TNFα (Figure 6E and F). These data demonstrate that ZBTB2 is a transcriptional repressor of the endogenous RelA/p65 gene in quiescent or TNFα-stimulated cells.
Previous studies show that translocation of NF-κB from cytoplasm into nucleus is important in NF-κB signaling cascade. To determine the effect of ZBTB2 and Sp1 on translocation of RelA/p65, we investigated amount of nuclear RelA/p65 protein by western blot analysis of cytosol and nuclear cell fractions. While ectopic Sp1 increases cytoplasmic and nuclear RelA/p65, ectopic ZBTB2 decreases cytoplasmic and nuclear RelA/p65. Moreover, we tested whether ectopic ZBTB2 changes RelA/p65 associated with IκB-α by co-immunoprecipitation with an anti-RelA/p65 antibody and western blot analysis with an anti-IκB-α antibody. When ZBTB2 was overexpressed, more IκB-α interacted with RelA/p65, suggesting that ZBTB2 may decrease nuclear RelA/p65 by increasing cytosolic RelA/p65-IκB-α interactions (Supplementary Figure S5A and B).
We also analyzed the temporal changes in expression of RelA/p65 and ZBTB2 in the HEK293 cells treated with TNFα. Expression of RelA/p65 was increased over 60 min and interestingly, ZBTB2 expression was decreased in a manner negatively correlates with RelA/p65 expression. It appears that TNFα treatment activates NF-κB response gene transcription by de-repressing RelA/p65 expression through decreasing ZBTB2 expression over a 60-min period. Once, NF-κB responsive gene expression is accomplished, ZBTB2 expression may be restored to repress RelA/p65 expression, as shown in Figure 6E and F, at 12 h post TNFα treatment (Supplementary Figure S6).
ZBTB2 decreases PDH activity by increasing PDK4 expression
PGC1α activates PDK4 expression by binding an NR binding site within the PDK4 promoter. RelA/p65 directly interacts with PGC1α and inhibits PDK4 transcriptional activation by PGC1α (36). Because ZBTB2 represses RelA/p65 transcription, ZBTB2 may increase PDK4 expression indirectly by increasing unbound PGC1α. Consequently, we investigated whether ZBTB2 regulates endogenous PDK4 and PGC1α expression using western blot analysis. In human HEK293 cells transfected with ZBTB2, PDK4 expression increased, with no change in PGC1α expression (Figure 7A). Because more free PGC1α is available by downregulation of its binding partner RelA/p65 by ZBTB2, PGC1α may increasingly bind to the PDK4 promoter and activate PDK4 transcription. Indeed, ChIP assays showed increased PGC1α binding to the PDK4 promoter region-flanking NR site in HEK293 cells transfected with pcDNA3-ZBTB2 expression vector, compared to the control (Figure 7B).
Figure 7.
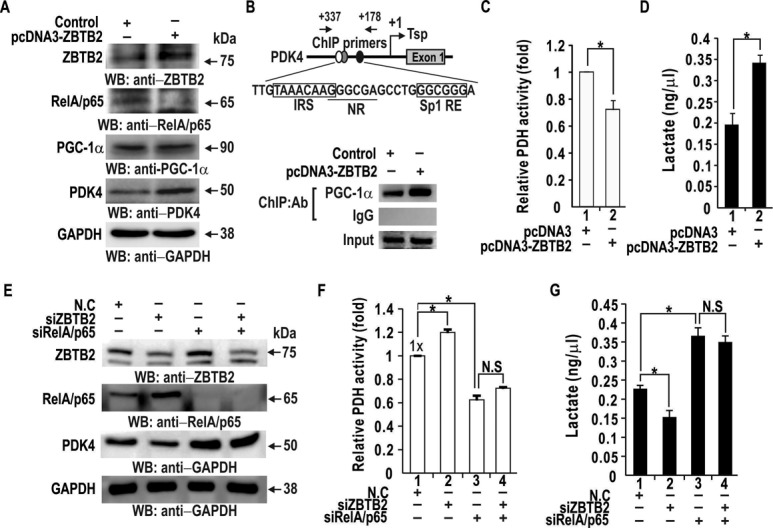
ZBTB2 decreases PDH activity by increasing PDK4 expression (A) Western blot analysis of regulation of endogenous PDK4 expression by ZBTB2. HEK293 cells were transfected with pcDNA3 or pcDNA3-ZBTB2 expression plasmid. Whole cell extracts were separated by SDS-PAGE and analyzed using the antibodies indicated. GAPDH, control. (B) ChIP assays of PGC1α binding to the PDK4 proximal promoter region flanking a nuclear receptor binding site (NR). HEK293 cells were transfected with ZBTB2 expression vector. Chromatin was immunoprecipitated with the antibodies indicated and analyzed by PCR. IgG, negative control. IRS, FOXO1-binding element; NR, nuclear receptor binding element; Sp1RE, Sp1 binding element. (C) PDH activity in HEK293 cells was measured using a coupled enzyme assay of the cell lysates. *P < 0.05. (D) Concentration of lactate produced by HEK293 cells transfected with control or ZBTB2 expression vector was measured using lactate assay kits. Data were from two independent experiments. *P < 0.05. (E) Western blot analysis of regulation of endogenous PDK4 expression by ZBTB2. HEK293 cells were transfected with siZBTB2 and/or siRelA/p65 RNA. Whole cell extracts were separated by SDS-PAGE and analyzed using the antibodies indicated. GAPDH, control. (F, G) PDH activity and lactate concentration in HEK293 cell lysates were measured using a coupled enzyme assay and lactate assay kit, respectively. HEK293 cells transfected with siZBTB2 and/or siRelA/p65 RNA. Data were from three independent experiments. *P < 0.05.
The PDH complex, a critical enzyme that converts pyruvate to acetyl coenzyme A (Ac-CoA), is a major regulator of glucose metabolism. Because PDK4 inhibits PDH activity by phosphorylation and ZBTB2 increase expression of PDK4 via PGC1α, we examined whether ZBTB2 could regulate PDH activity. Ectopic expression of ZBTB2 in HEK293 cells decreased PDH activity (Figure 7C). Furthermore, we investigated whether decreased PDH activity in cells with ectopic ZBTB2 expression affected lactate production. Lactate assays showed that the concentration of lactate was increased by ectopic ZBTB2 (Figure 7D).
We also examined whether PDK4 upregulation by ZBTB2 was through repression of RelA/p65 expression by a ‘loss of function’ approach in which ZBTB2 siRNA and/or RelA/p65 siRNA were transfected into HEK293 cells. Those results showed PDK4 expression decreased by ZBTB2 knockdown and increased by RelA/p65 knockdown (Figure 7E). Moreover, ZBTB2 knockdown could not decrease PDK4 expression in the absence of RelA/p65, eventually resulting in increased PDH activity and decreased lactate production, which was not observed in the cells with RelA/p65 knockdown (Figure 7F and G).
We also investigated whether ZBTB2 regulates RelA/p65 and PDK4 expression in other cell types. It has been previously shown that microRNA-149 downregulates ZBTB2 expression. In glioblastoma, ZBTB2 expression may be rather high because microRNA-149 expression is low. Accordingly, we selected the U343 cell line, derived from a glioblastoma, to investigate whether ZBTB2 regulates RelA/p65 and PDK4 expression. In U343 cells, ZBTB2 repressed RelA/p65 transcription and eventually also increased PDK4 expression similar to HEK293 cells. These changes resulted in decreased PDH activity and increased lactate concentration. We also confirmed that ZBTB2 increase PDK4 expression via RelA/p65 inhibition, suggesting that ZBTB2 may also act as a proto-oncogene in glioblastoma (Supplementary Figure S7).
Cancer cells or highly proliferative cells show low PDH activity and thereby metabolic contribution of the TCA cycle is compromised and glycolytic pathways supplement their metabolic needs. Because ZBTB2 can reprogram cellular glucose metabolism into the metabolic pattern similar to that of cancer cells or highly proliferative cells, we examined whether ectopic ZBTB2 could stimulate tumor cell proliferation in a mouse xenograft assay.
Those studies showed that the volume of tumors derived from xenografted U343 glioblastoma cells was significantly (47%) decreased by intra-tumoral injection of recombinant adenovirus overexpressing ZBTB2 shRNA over a 31-day period (Supplementary Figure S8A). These data suggest that ZBTB2 may increase cell proliferation by shifting glucose metabolic pathways to those used by cancer cells or highly proliferative cells.
Other reported that ZBTB2 expression may be high in glioblastoma tissues that express low levels of microRNA-149 against ZBTB2 (11). Because ZBTB2 affects pathways from RelA/p65 down to lactate production, we tested whether changes in RelA/p65 and PDK4 expression caused by high ZBTB2 expression were also observed in glioblastoma by immunocytochemistry. Indeed, ZBTB2 and PDK4 expression in glioblastoma tissues was high, while RelA/p65 expression was low, compared to that of the control (Supplementary Figure S8B). These changes suggest that a metabolic shift to glycolysis in cells with ectopic ZBTB2 may also be applicable to glioblastoma.
DISCUSSION
NF-κB is involved in a variety of cellular stress responses and is also a key regulator of immune responses. Although regulation of the activity of the RelA/p65 protein, a key subunit of NF-κB, is well known (16), the mechanisms of RelA/p65 regulation at the transcriptional level, which may be important for sustained NF-κB responses, remain largely unknown. The RelA/p65 promoter is GC-rich (>71%) and has many potential Sp1-binding sites, of which three have been proposed to be important for increasing promoter activity of the RelA/p65 gene (30). Here, we found that Sp1 activates transcription of the RelA/p65 gene (Figure 1).
ZBTB2 potently repressed transcriptional activation of RelA/p65 by Sp1. The presence of ZBTB2 potently blocks the DNA-binding activity of Sp1 and also nearly completely blocks transcription of a RelA/p65 reporter by Sp1 through the key regulatory element, GC-box #3 (Figures 2–4). We showed that molecular interactions between Sp1, ZBTB2 and the GC-box regulated the expression levels of RelA/p65. Specifically, ZBTB2 bound to the RelA/p65 promoter and increased the recruitment of the NCoR–HDAC complex to the RelA/p65 promoter region, which deacetylated histone H3 and H4 in a dose-dependent manner (Figure 5). Thus, ZBTB2 may repress RelA/p65 by inhibition of Sp1 binding and increased nucleosome compaction of the RelA/p65 proximal promoter.
RelA/p65 not only acts as an NF-κB subunit but also plays a direct role in various biological processes. Overexpression of RelA/p65 has been shown to activate curcumin-induced apoptosis in HCT116 human colon cancer cells (37), and to maintain cellular senescence by promoting DNA repair and genomic stability in pre-neoplastic cells (38). Also, RelA/p65 can interact with histone deacetylase/co-repressors (including HDAC1 and HDAC2), resulting in gene repression (39). Thus, RelA/p65 may act either as a tumor suppressor or an oncoprotein, depending on cellular contexts.
Because ZBTB2 is a transcriptional repressor of the RelA/p65 gene, we investigated the effect of ZBTB2 on NF-κB responsive reporter gene expression activated by TNFα. ZBTB2 significantly decreased reporter expression of the pNF-κB-Luc. Interestingly, transcription of RelA/p65 increased 3.5-fold by TNFα treatment (Figure 6). It is well known that TNFα induces phosphorylation of RelA/p65, but TNFα effect on transcriptional activation of the RelA/p65 gene is relatively unknown. There are a few reports indicating that the mRNA level of the RelA/p65 gene increases in response to TNFα, but the mechanism remains unknown (40,41).
In various cellular stress responses, the precise activation and termination of NF-κB activity and expression are important. The well-accepted mechanism for termination of the NF-κB response involves the resynthesis of IκB proteins induced by activated NF-κB (28). Newly synthesized IκBα enters the nucleus, releases NF-κB from the DNA and relocalizes NF-κB to the cytosol (29). Additionally, acetylation of RelA/p65 has been demonstrated to decrease its DNA-binding affinity (28), and post-translational modifications of RelA/p65 by HATs or HDACs influence termination of the NF-κB response (39). Our data showed that ZBTB2 potently decreases NF-κB expression both in the presence or absence of TNFα. ZBTB2, as a transcriptional repressor, keeps the RelA/p65 gene silent. This is the first report showing how the expression of the RelA/p65 gene, a critical component of NF-κB, is repressed by ZBTB2 at the transcriptional level, and how de novo repression of RelA/p65 gene by ZBTB2 is deregulated by NF-κB-activating signals such as TNFα (Figure 6). ZBTB2, as a repressor of transcription, may regulate various biological processes controlled by NF-κB such as apoptosis, cell adhesion, immunity and inflammation.
The PDH complex converts pyruvate into acetyl-CoA, the first substrate used in the TCA cycle. Regulation of PDH expression and activity is important for metabolism of the pyruvate that derives from glycolysis and accordingly, for glucose metabolism (42). Expression of PDK4, a negative regulator of the PDH complex, is regulated by diverse factors such as ERRγ, C/EBPβ, FOXO1 and PGC1α, both transcriptionally and post-transcriptionally (35,43–44). PDK4 is highly expressed in the liver, heart and skeletal muscle, and is also upregulated in cancer cells.
Interestingly, cells overexpressing the oncoprotein ZBTB2 proliferate more rapidly (7). Consequently, we hypothesize that ZBTB2 may reprogram cellular metabolic pathways to favor rapid cell proliferation, similar to cancer cell metabolic pathways. PGC1α, an activator of PDK4 transcription, directly interacts with RelA/p65; that interaction limits the amount of PGC1α available to bind the promoter of PDK4. Accordingly, by transcriptional repression of RelA/p65, ZBTB2 indirectly increases PDK4 expression (via increased unbound PGC1α), leading to inhibition of PDH and attenuated glucose metabolic processing via the TCA cycle. The molecular events triggered by overexpressed ZBTB2 may cause metabolic switching to favor glycolysis over the TCA cycle, a metabolic feature of cancer cells (45,46). This conjecture was supported by our finding a negative correlation between PDH activity and PDK4 expression and lactate (Figure 7 and Supplementary Figure S7). Thus, metabolic reprogramming may be one of the features important for rapid proliferation of cells with ectopic ZBTB2 (Figure 8).
Figure 8.
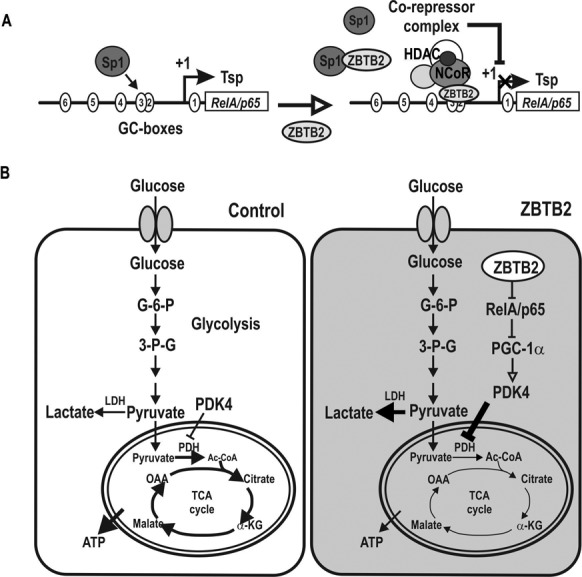
Hypothetical model of transcriptional repression of RelA/p65 by ZBTB2, resulting in a glucose metabolism switch. (A) RelA/p65 proximal promoter (up to −401 bp) is highly GC-rich and has many potential Sp1 binding GC-boxes (as many as 14). Among the potential Sp1-binding GC-boxes, GC-box #3 is particularly important. The element is bound by Sp1 relatively strongly and plays a critical role in transcriptional initiation and activation by Sp1. ZBTB2 interacts with Sp1 and affects Sp1 binding activity or competes with Sp1 to bind GC-box #3. ZBTB2 recruits the NCoR co-repressor complex to repress transcription by histone compaction. (B) ZBTB2 switches glucose metabolism to favor glycolysis over the TCA cycle by indirectly increasing PDK4 expression. Repression of RelA/p65 expression by ZBTB2 frees PGC1α to bind and activate the PDK4 promoter to eventually increase PDK4 expression. PDK4 inhibits PDH activity, attenuating glucose metabolism through the TCA cycle. ZBTB2 also indirectly increases pyruvate and lactate production, indicating activation of glycolysis. Thus, ZBTB2 may reprogram glucose metabolic pathways similar to those in cancer cells or highly proliferative cells.
Proliferating cells use aerobic glycolysis to meet their metabolic needs other than only ATP (45). ZBTB2 not only controls the genes of the p53 pathway such as CDKN1A to increase cell proliferation (7), but also to reprogram metabolic pathways favoring glycolysis to provide resources including ATP, ribose and glycerol for nucleotide, amino acid and lipid biosynethesis, and NADPH for rapid cell proliferation, at the cost of massive glucose intake (45,46). Overall, our results suggest that the proto-oncoprotein ZBTB2 may stimulate cell proliferation through indirect regulation of PDK4 expression via transcriptional repression of RelA/p65 (41).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Medical Research Center Grant [2011-0030086 to M.-W.H.]; DOYAK, Korea Research Foundation of the Korea Ministry of Education, Science and Technology [2011-0028817 to M.-W.H.]. Funding for open access charge: Medical Research Center Grant [2011-0030086 to M.-W.H.]; DOYAK, Korea Research Foundation of the Korea Ministry of Education, Science and Technology [2011-0028817 to M.-W.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Kelly K.F., Daniel J.M. POZ for effect–POZ-ZF transcription factors in cancer and development. Trends Cell Biol. 2006;16:578–587. doi: 10.1016/j.tcb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Jeon B.N., Kim M.K., Choi W.I., Koh D.I., Hong S.Y., Kim K.S., Kim M., Yun C.O., Yoon J., Choi K.Y., et al. KR-POK interacts with p53 and represses CDKN1A transcription activation by p53. Cancer Res. 2012;72:1137–1148. doi: 10.1158/0008-5472.CAN-11-2433. [DOI] [PubMed] [Google Scholar]
- 3.Choi W.I., Jeon B.N., Park H., Yoo J.Y., Kim Y.S., Koh D.I., Kim M.H., Kim Y.R., Lee C.E., Kim K.S., et al. Proto-oncogene FBI-1 (Pokemon) and SREBP-1 synergistically activate transcription of fatty-acid synthase gene (FASN) J. Biol. Chem. 2008;283:29341–29354. doi: 10.1074/jbc.M802477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeon B.N., Yoo J.Y., Choi W.I., Lee C.E., Yoon H.G., Hur M.W. Proto-oncogene FBI-1 (Pokemon/ZBTB7A) represses transcription of the tumor suppressor Rb gene via binding competition with Sp1 and recruitment of co-repressors. J. Biol. Chem. 2008;283:33199–33210. doi: 10.1074/jbc.M802935200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi W.I., Jeon B.N., Yun C.O., Kim P.H., Kim S.E., Choi K.Y., Kim S.H., Hur M.W. Proto-oncogene FBI-1 represses transcription of p21CIP1 by inhibition of transcription activation by p53 and Sp1. J. Biol. Chem. 2009;284:12633–12644. doi: 10.1074/jbc.M809794200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koh D.I., Choi W.I., Jeon B.N., Lee C.E., Yun C.O., Hur M.W. A novel POK family transcription factor, ZBTB5, represses transcription of p21CIP1 gene. J. Biol. Chem. 2009;284:19856–19866. doi: 10.1074/jbc.M109.025817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeon B.N., Choi W.I., Yu M.Y., Yoon A.R., Kim M.H., Yun C.O., Hur M.W. ZBTB2, a novel master regulator of the p53 pathway. J. Biol. Chem. 2009;284:17935–17946. doi: 10.1074/jbc.M809559200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardo M., Lang B., Yu L., Prosser H., Bradley A., Babu M.M., Choudhary J. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell. 2010;6:382–395. doi: 10.1016/j.stem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 10.van Nuland R., Smits A.H., Pallaki P., Jansen P.W., Vermeulen M., Timmers H.T. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol. Cell. Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Zheng X., Zhang Z., Zhou J., Zhao G., Yang J., Xia L., Wang R., Cai X., Hu H., et al. MicroRNA-149 inhibits proliferation and cell cycle progression through the targeting of ZBTB2 in human gastric cancer. PLoS ONE. 2012;7:e41693. doi: 10.1371/journal.pone.0041693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D.H., Lee S.T., Won H.H., Kim S., Kim M.J., Kim H.J., Kim S.H., Kim J.W., Kim H.J., Kim Y.K., et al. A genome-wide association study identifies novel loci associated with susceptibility to chronic myeloid leukemia. Blood. 2011;117:6906–6911. doi: 10.1182/blood-2011-01-329797. [DOI] [PubMed] [Google Scholar]
- 13.Mu J.J., Wang Y., Luo H., Leng M., Zhang J., Yang T., Besusso D., Jung S.Y., Qin J. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 2007;282:17330–17334. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 14.Jung S.Y., Malovannaya A., Wei J., O'Malley B.W., Qin J. Proteomic analysis of steady-state nuclear hormone receptor coactivator complexes. Mol. Endocrinol. 2005;19:2451–2465. doi: 10.1210/me.2004-0476. [DOI] [PubMed] [Google Scholar]
- 15.Gylfe A.E., Kondelin J., Turunen M., Ristolainen H., Katainen R., Pitkänen E., Kaasinen E., Rantanen V., Tanskanen T., Varjosalo M., et al. Identification of candidate oncogenes in human colorectal cancers with microsatellite instability. Gastroenterology. 2013;145:540–543. doi: 10.1053/j.gastro.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Hayden M.S., West A.P., Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 17.Li Q., Verma I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 18.Bonizzi G., Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Wong H.R., Ryan M., Wispé J.R. Stress response decreases NF-kappaB nuclear translocation and increases I-kappaBalpha expression in A549 cells. J. Clin. Invest. 1997;99:2423–2428. doi: 10.1172/JCI119425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pahl H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 21.Hayden M.S., Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 22.Gerondakis S., Grossmann M., Nakamura Y., Pohl T., Grumont R. Genetic approaches in mice to understand Rel/NF-kappaB and IkappaB function: transgenics and knockouts. Oncogene. 1999;18:6888–6895. doi: 10.1038/sj.onc.1203236. [DOI] [PubMed] [Google Scholar]
- 23.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tak P.P., Firestein G.S. NF-κB: a key role in inflammatory diseases. J. Clin. Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker R.G., Hayden M.S., Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayden M.S., Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong H., Voll R.E., Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 28.Rothgiesser K.M., Valovka T., Owen H.R., Rehrauer H., Fey M. Functional relevance of novel p300-mediated lysine 314 and 315 acetylation of RelA/p65. Nucleic Acids Res. 2008;36:1665–1680. doi: 10.1093/nar/gkn003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ea C.K., Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueberla K., Lu Y., Chung E., Haseltine W.A. The NF-kappa B p65 promoter. J. Acquir. Immune Defic. Syndr. 1993;6:227–230. [PubMed] [Google Scholar]
- 31.Yurochko A.D., Kowalik T.F., Huong S.M., Huang E.S. Human cytomegalovirus upregulates NF-kappa B activity by transactivating the NF-kappa B p105/p50 and p65 promoters. J. Virol. 1995;69:5391–5400. doi: 10.1128/jvi.69.9.5391-5400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yurochko A.D., Mayo M.W., Poma E.E., Baldwin A.S. Jr, Huang E.S. Induction of the transcription factor Sp1 during human cytomegalovirus infection mediates upregulation of the p65 and p105/p50 NF-kappaB promoters. J. Virol. 1997;71:4638–4648. doi: 10.1128/jvi.71.6.4638-4648.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu L., Findley H.W., Zhou M. MDM2 induces NF-kappaB/p65 expression transcriptionally through Sp1-binding sites: a novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood. 2002;9:3367–3375. doi: 10.1182/blood.v99.9.3367. [DOI] [PubMed] [Google Scholar]
- 34.Sutendra G., Michelakis E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013;3:38. doi: 10.3389/fonc.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wende A.R., Huss J.M., Schaeffer P.J., Giguère V., Kelly D.P. PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol. 2005;25:10684–10694. doi: 10.1128/MCB.25.24.10684-10694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez-Guardia D., Palomer X., Coll T., Davidson M.M., Chan T.O., Feldman A.M., Laguna J.C., Vázquez-Carrera M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 2010;87:449–458. doi: 10.1093/cvr/cvq080. [DOI] [PubMed] [Google Scholar]
- 37.Collett G.P., Campbell F.C. Overexpression of p65/RelA potentiates curcumin-induced apoptosis in HCT116 human colon cancer cells. Carcinogenesis. 2006;27:1285–1291. doi: 10.1093/carcin/bgi368. [DOI] [PubMed] [Google Scholar]
- 38.Wang J., Jacob N.K., Ladner K.J., Beg A., Perko J.D., Tanner S.M., Liyanarachchi S., Fishel R., Guttridge D.C. RelA/p65 functions to maintain cellular senescence by regulating genomic stability and DNA repair. EMBO Rep. 2009;10:1272–1278. doi: 10.1038/embor.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashburner B.P., Westerheide S.D., Baldwin A.S., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurokouchi K., Kambe F., Yasukawa K., Izumi R., Ishiguro N., Iwata H., Seo H. TNF-alpha increases expression of IL-6 and ICAM-1 genes through activation of NF-kappaB in osteoblast-like ROS17/2.8 cells. J. Bone Miner. Res. 1998;13:1290–1299. doi: 10.1359/jbmr.1998.13.8.1290. [DOI] [PubMed] [Google Scholar]
- 41.Higai K., Ishihara S., Matsumoto K. NFkappaB-p65 dependent transcriptional regulation of glycosyltransferases in human colon adenocarcinoma HT-29 by stimulation with tumor necrosis factor alpha. Biol. Pharm. Bull. 2006;29:2372–2377. doi: 10.1248/bpb.29.2372. [DOI] [PubMed] [Google Scholar]
- 42.McFate T., Mohyeldin A., Lu H., Thakar J., Henriques J., Halim N.D., Wu H., Schell M.J., Tsang T.M., Teahan O., et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Attia R.R., Sharma P., Janssen R.C., Friedman J.E., Deng X., Lee J.S., Elam M.B., Cook G.A., Park E.A. Regulation of pyruvate dehydrogenase kinase 4 (PDK4) by CCAAT/enhancer-binding protein beta (C/EBPbeta) J. Biol. Chem. 2011;286:23799–23807. doi: 10.1074/jbc.M111.246389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piao L., Sidhu V.K., Fang Y.H., Ryan J.J., Parikh K.S., Hong Z., Toth P.T., Morrow E., Kutty S., Lopaschuk G.D., et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J. Mol. Med. 2013;91:333–346. doi: 10.1007/s00109-012-0982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vander Heiden M.G., Locasale J.W., Swanson K.D., Sharfi H., Heffron G.J., Amador-Noguez D., Christofk H.R., Wagner G., Rabinowitz J.D., Asara J.M., et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.