

Abstract
Disorders of sexual development are conditions where sexual phenotype and genotype are discordant. Genetic sex is determined at conception as the ovum is fertilised by a spermatozoon that contains either an X or Y chromosome. A complex pathway determined by genes and hormones leads to gonadal differentiation into testis or ovary and promotes the development of internal and external genitalia. We present a case of an 18-year-old woman who presented with primary amenorrhoea. She was a virgin, and apart from hirsutism and overweight, had no complaints. Her family history was insignificant. The patient was tall and had underdeveloped breasts. Her blood results showed hypergonadotropic hypogonadism. A 46, XY genotype was detected with karyotype analysis. Ultrasound and MRI demonstrated the presence of a uterus, but no overt gonads. Laparoscopy was performed, with bilateral removal of streak ovaries.
Background
The most important gene to determine the gonadal sex of a fetus is TDF (testis-determining factor) in the SRY region (sex-determining region of the Y) on the Y chromosome.1 If SRY is present, Sertoli cells and Leydig cells develop, and the undifferentiated primordial gonads differentiate to testes. This differentiation starts from the sixth week of fetal life. If SRY is absent, the gonads develop into ovaries. The testicular Sertoli cells produce AMH (antimüllerian hormone), which triggers the degeneration of all derivatives of the müllerian duct (eg, uterus, fallopian tubes and upper vagina). The testicular Leydig cells produce testosterone and other androgens, which stabilise the wolffian ducts (mesonefric duct) and promote their differentiation into vasa deferentia, epididymis and seminal vesicles. In the peripheral tissue, testosterone is metabolised to DHT (dihydrotestosterone) by the enzyme 5α-reductase; DHT induces virilisation of the external genitalia. Without testes, there is no AMH and no DHT, so the müllerian ducts develop and the external genitalia remain female.2 A large amount of other genes are important in this cascade, respectively, SOX-9 (on the Y chromosome), WT1, DHH, FGF9, M33 and DMRT1 for the differentiation into testes, and WNT4, FOXL2, DAX1 and RSPO1 for the differentiation into ovaries. The genes SF1, LIM1, EMX2 and LHX9 are important in both. If there is a mutation in one of these genes, sexual differentiation fails and disorders of sexual development (DSD) can develop.3–8
According to their chromosomal component, DSD are divided into three groups; 46XX DSD, 46XY DSD and sex chromosomal DSD. The last group includes conditions such as Turner (45X) or Klinefelter (47XXY) syndrome. The second group, which will be discussed in this article (ie, 46 XY where the sex of rearing is female), can be subdivided into conditions with abnormal testicular development (eg, gonadal dysgenesis) and conditions with defective androgen action (eg, androgen insensitivity syndrome).1 Both subgroups contain rare syndromes with a prevalence of 1–5 of 100 000 newborns.
Because DCD are rare and their consequences can be very serious (psychological stress, infertility, malignancy), it is important to clarify the different steps that need to be undertaken in the differential diagnosis.3
Case presentation
An 18-year-old woman of Albanian descent, without facial dysmorphism, presented at the gynaecology clinic with primary amenorrhea. There was no significant medical and family history. She was of tall stature and had underdeveloped breasts. She was overweight and hirsute, with scarce pubic hair. She was a virgin; clinical investigation showed normal external genitalia and an intact hymen (figure 1). Her vagina was accessible for one finger, and had a normal length; there was a cervix.
Figure 1.
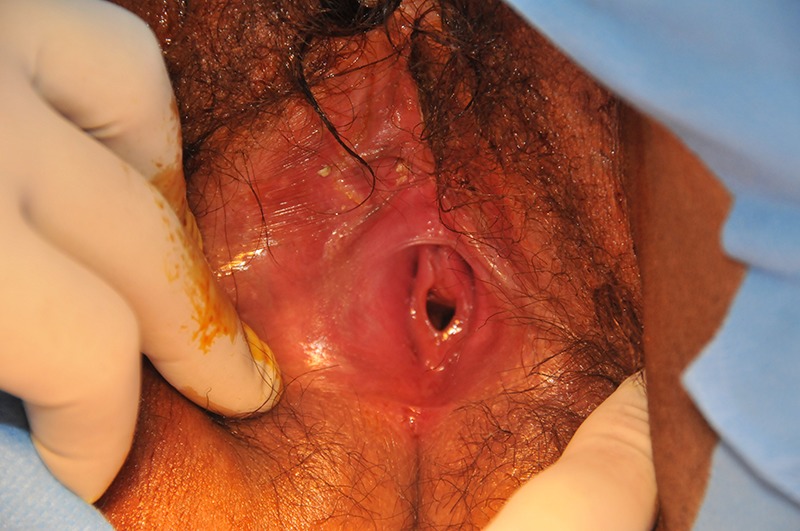
Normal external genitalia and an intact hymen in an XY female.
Investigations
Transabdominal ultrasound revealed a small uterus but no sign of overt ovaries/testes. There was no history or clinical presence of an inguinal hernia. The patient's blood results showed hypergonadotropic hypogonadism with oestradiol <5 ng/L, progesterone 0.30 µg/L, testosterone 0.3 nmol/L, FSH 52.7 U/L, LH 10.8 U/L and absence of antimüllerian hormone (<0.05 µg/L).
MRI confirmed the presence of a small uterus without overt testes or ovaria. Laparoscopic exploration revealed the presence of bilateral streak ovaries and fallopian tubes (figures 2 and 3). Such streak ovaries are not able to produce hormones.
Figure 2.
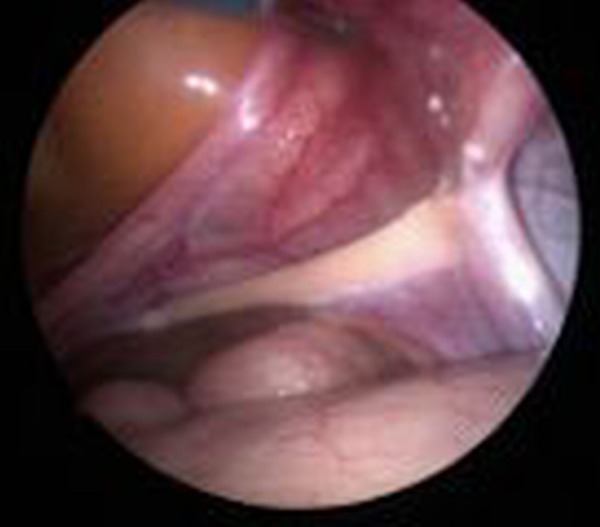
Laparoscopic exploration: streak ovary.
Figure 3.
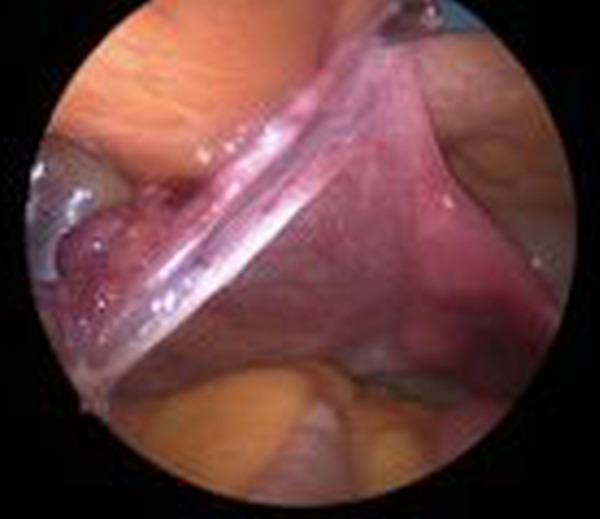
Laparoscopic exploration: streak ovary, normal fallopian tube and small uterus.
Karyotype analysis showed a 46XY genotype. A whole genome microarray showed no deletions or duplication. The literature describes a mutation of the SRY gene in 10% of patients with pure gonadal dysgenesis, but in this rare case the SRY gene revealed no microdeletions on MLPA analysis. Because of the extensiveness of possible monogenic causes, the absence of associated symptoms and the irrelevance of clinical follow-up, no further DNA analysis was performed.9
Differential diagnosis
As previously discussed, the 46 XY female (XY karyotype where the sex of rearing is female), can be subdivided into two major subdivisions: (1) conditions with abnormal testicular development (eg, gonadal dysgenesis alias Swyer syndrome) and (2) conditions with defective androgen action (eg, androgen insensitivity syndrome alias Morris syndrome).1
The androgen insensitivity syndrome (AIS) is an X linked recessive disorder in which the androgen receptor (AR) is not fully sensitive for testosterone and DHT. There are over 300 different mutations that can induce this syndrome. The degree of insensitivity determines the phenotype; this can range from complete AIS (complete female) over partial AIS (ambiguous genitalia) to minor AIS (infertile male). It is important to emphasise that these patients do not have a uterus and fallopian tubes. They have a blind vagina pouch and their gonads are testes that produce AMH. Approximately 50% present with unilateral or bilateral inguinal hernias. Since the peripheral tissues are insensitive to DHT, female external genitalia can develop. In gonadal dysgenesis, different phenotypes also exist: pure gonadal dysgenesis (as presented in this case), mixed gonadal dysgenesis (asymmetrical gonadal development, external female genitalia) and partial gonadal dysgenesis (bilateral dysgenetic testis, mix of male and female external genitalia).
Hormone analysis can make the differential diagnosis. In AIS, the androgen is up to male levels, whereas in pure gonadal dysgenesis the levels are elevated compared to female level, but they will not reach normal male levels.
Approximately 3.4% of all patients with primary amenorrhoea are XY females.3
Treatment
Because of hormone deficiency, XY females are prone to osteoporosis and cardiovascular diseases. Continuous hormonal substitution is the key treatment, especially after gonadectomy.
It is important to provide psychological help, because this diagnosis is not easy to handle for most patients and identity problems often develop.3 8
There is a larger incidence of gonadal tumours in XY females. This incidence is higher in gonadal dysgenesis compared to AIS. Patients between 30 and 50 years of age with AIS usually have a greater chance of developing seminomas. The risk at 50 years is said to be 33%. It is advised that testes be removed after puberty to maintain hormonal levels in this crucial period. In pure gonadal dysgenesis, gonadoblastomas and gonadal dysgerminomas are more frequent. The risk is estimated to be 46% or even 75%. Therefore it is advised to remove the streak ovaries early after the diagnosis. In most cases the fallopian tubes are removed as well, in order to remove the gonads with a wide margin.
Patients with pure gonadal dysgenesis can become pregnant with the use of allogenic oocytes. However, there is a higher rate of caesarean section in these individuals because of their inability to react to oxytocin and prostaglandin.3
Outcome and follow-up
Considering the higher risk of malignancy, osteoporosis and cardiovascular disease, these patients should be followed-up closely.
In our patient, continuous hormonal replacement therapy will be continued until she reaches 50 years of age.3 8
Discussion
DSD are very rare, and need to be investigated and treated in a tertiary centre. Infertility and higher risk of malignancy are important aspects, but each entity has his own particularities. Therefore good counselling, a multidisciplinary approach and regular follow-up are advised. Many published articles discuss different genetic causes for the existence of an XY female: pericentric inversion of the Y chromosome,9 SF-1 (steroidogenic factor-1) gene mutation and LH receptor gene mutation.4 It is, however, very difficult to find a monogenic cause when no other symptoms are present.
Patient's perspective.
I was very sad to hear that there is something wrong with me. It's difficult to understand for me. The doctors say I cannot get pregnant in a natural way. Now I do not have a boyfriend and I do not want one right now. My mom wants to help me by giving me an egg when I do want to have children. They gave me an appointment to talk with someone about my feelings.
Learning points.
Disorders of sexual development are conditions where the sexual phenotype and genotype are discordant.
Extensive clinical evaluation, endocrine assessment, pelvic imaging and karyotyping should be performed in women with primary amenorrhoea.
XY females can be divided into two main groups:1 conditions with abnormal testicular development (eg, gonadal dysgenesis)2 and conditions with defective androgen action (eg, androgen insensitivity syndrome). Differential diagnosis can be made by noting the androgen level and the presence of a uterus.
Footnotes
Contributors: DM and JM are responsible for the literature research, drafting of the paper and the clinical follow-up of the patient. YJ and BB critically revised and approved the final version.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Michala L, Creighton SM. The XY female. Best Pract Res Clin Obstet Gynaecol 2010;24:139–48. 10.1016/j.bpobgyn.2009.09.009 [DOI] [PubMed] [Google Scholar]
- 2.Schaaf CP, Zschocke J, Potocki L. Human genetics: from molecules to medicine. Philadelphia: Lippincott Williams & Wilkins, 2012. [Google Scholar]
- 3.Jorgensen PB, Kjartansdóttir KR, Fedder J. Care of women with XY karyotype: a clinical practice guideline. Fertil Steril 2010;94:105–13. 10.1016/j.fertnstert.2009.02.087 [DOI] [PubMed] [Google Scholar]
- 4.Philibert P, Leprieur E, Zenaty D et al. Steroidogenic factor-1 (SF-1) gene mutation as a frequent cause of primary amenorrhea in 46,XY female adolescents with low testosterone concentration. Reprod Biol Endocrinol 2010;8:28 10.1186/1477-7827-8-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helszer Z, Dmochowska A, Szemraj J et al. A novel mutation (c. 341A>G) in the SRY gene in a 46,XY female patient with gonadal dysgenesis. Gene 2013;526:467–70. 10.1016/j.gene.2013.04.027 [DOI] [PubMed] [Google Scholar]
- 6.Houk CP, Levitsky LL. Evaluation of the infant with ambiguous genitalia. UpToDate. Topic 5803 Version 11.0 2014. http://www.uptodate.com/contents/evaluation-of-the-infant-with-ambiguousgenitalia?source=searchresult&search=evaluation+infant+ambiguous&selectedTitle=1~135 (accessed 28 Feb 2013).
- 7.Online Mendelian Inheritance in Man, OMIM® Baltimore, MD: Johns Hopkins University; MIM Number: {400044 }: {12/06/2013 }. http://omim.org/ [Google Scholar]
- 8.Firth H, Hurst J. Oxford Desk Reference—Clinical Genetics. 14 July 2005.
- 9.Mitsuhashi T, Warita K, Tabuchi Y et al. Global gene profiling and comprehensive bioinformatics analysis of a 46,XY female with pericentric inversion of the Y chromosome. Congenit Anom (Kyoto) 2010;50:40–51. 10.1111/j.1741-4520.2009.00254.x [DOI] [PubMed] [Google Scholar]