

Abstract
Emotional and traumatic experiences lead to the development of particularly strong memories that can drive neuropsychiatric disorders, such as posttraumatic stress disorder (PTSD) and drug addiction. Disruption of these memories would therefore serve as a powerful treatment option, and targeting the pathologic emotional, but not declarative, component of a memory would be ideal for clinical intervention. Research reveals that after retrieval of a consolidated memory, the memory can be destabilized, and must then be reconsolidated through synaptic plasticity to allow subsequent retrieval. Disruption of reconsolidation-related plasticity would therefore impair specific, reactivated memories. Noradrenergic signaling strengthens synaptic plasticity and is essential for encoding the emotional components of memory. Consistent with this, investigations have now revealed that noradrenergic signaling is a critical mechanism for reconsolidation of emotional memories in rodent and human models. Here, we discuss these investigations and promising clinical trials indicating that disruption of noradrenergic signaling during reconsolidation may abolish the pathologic emotional, but not declarative, component of memories allowing alleviation of neuropsychiatric disorders including PTSD and drug addiction.
INTRODUCTION
Memories serve to store relevant information and were originally assumed to be static and inflexible. More recently, evidence has emerged that memories are, in fact, dynamic and modifiable. Initially, memories are acquired and consolidated into long-term representations (McGaugh, 2000; Müller and Pilzecker, 1900). Memory consolidation involves modifications in synaptic efficacy and intrinsic neuronal excitability (eg, Alkon, 1979; Bailey and Chen, 1983; Disterhoft et al, 1986; McKernan and Shinnick-Gallagher, 1997; Rogan et al, 1997; Whitlock et al, 2006; for reviews, see Kandel, 2001; Sehgal et al, 2013), and such changes allow subsequent memory retrieval. Following consolidation, memory and memory-related plasticity are dynamically regulated during and after retrieval. Retrieval can result in synaptic destabilization (Kim et al, 2010; Lee et al, 2008), and ensuing restabilization processes are required for subsequent memory expression across a variety of learning paradigms and species (eg, Kroes et al, 2014; Lee et al, 2005; Misanin et al, 1968; Nader et al, 2000; Przybyslawski et al, 1999; for a review, see Nader and Hardt, 2009; Reichelt and Lee, 2013). These restabilization processes, known as reconsolidation, involve de novo protein synthesis (Nader et al, 2000) and synaptic plasticity (Clem and Huganir, 2010). Thus, modification of reconsolidation-related plasticity allows for memory modification and even memory elimination. Reconsolidation is now being studied extensively as disruption of pathologic, emotional forms of memory could alleviate memory-related disorders, such as posttraumatic stress disorder (PTSD; Brunet et al, 2008) and drug addiction (Saladin et al, 2013; Xue et al, 2012).
Converging evidence using rodents and human subjects reveals that noradrenergic signaling is critical for memory reconsolidation. For example, inhibition of α1- and β-adrenergic receptor (AR) activity during reconsolidation leads to memory disruption within both appetitive and aversive memory paradigms (Bernardi et al, 2006, 2009; Do Monte et al, 2013; Gazarini et al, 2013; Milton et al, 2008b; Przybyslawski et al, 1999; Wouda et al, 2010). Furthermore, disruption of noradrenergic signaling during reconsolidation reduces long-term emotional memory in healthy humans (for a recent meta-analysis, see Lonergan et al, 2013), is associated with better quality of life among PTSD patients (Poundja et al, 2012), and is capable of reducing cue-induced cravings among patients with cocaine addiction (Saladin et al, 2013). Several reviews have discussed the many signaling mechanisms that regulate reconsolidation in general, such as glutamate receptor transmission, cholinergic signaling, MAP kinase activity, protein synthesis, and epigenetic modifications (for a review, see Alberini, 2011; Nader and Hardt, 2009; Reichelt and Lee, 2013; Sorg, 2012). However, none have specifically focused on the large body of literature revealing that noradrenergic signaling is critical for reconsolidation across appetitive and aversive memory paradigms. Here we discuss these encouraging findings and also highlight limitations of these studies, future research directions, and recent inconsistent results regarding the efficacy of the most commonly used amnesic, the β-AR antagonist propranolol. Finally, the ethics of reconsolidation blockade for treatment of neuropsychiatric disorders are discussed. We conclude that reconsolidation disruption, particularly by noradrenergic receptor blockade, would serve as a powerful and ethical treatment option as such memory disruption is specific to the pathologic, emotional component of a reactivated memory.
NEUROPHYSIOLOGY
Reconsolidation is regulated by noradrenergic signaling at each of its receptors, specifically α1-, α2-, and β-ARs. The effects of α1-, α2-, and β-AR activation are complex, but the general principles follow that α1- and β-AR activation increases intrinsic neuronal excitability, whereas α2-AR activation has the opposite effect. Simply stated, α1-AR activation reduces the conductance of non-gated K+ channels (K+leak), causing slow membrane depolarization (McCormick and Prince, 1988). Similarly, β-AR activation decreases the conductance of ion-gated K+ channels, reducing the slow and fast afterhyperpolarization that contribute to spike-frequency adaptation (Foehring et al, 1989; Mueller et al, 2008; Otis et al, 2013). Thus, both α1- and β-AR activation enhance intrinsic excitability, particularly by increasing the likelihood (α1) and frequency (β) of evoked action potentials. In contrast, α2-AR activation enhances inward-rectifying K+ channel conductance, leading to slow membrane hyperpolarization and reduced intrinsic excitability (Marzo et al, 2009).
In addition to the modulation of intrinsic neuronal excitability, α1-, α2-, and β-AR activation modulate synaptic transmission. Similar to the effects on intrinsic excitability, α1- and β-AR activation increase the potential for synaptic plasticity, whereas α2-AR activation has the opposite effect. Specifically, α1-AR stimulation leads to the activation of phospholipase C (PLC), which leads to the formation of the second messengers inositol triphosphate (IP3) and diacylglycerol (DAG; Graham et al, 1996; Wu et al, 1992). IP3 and DAG increase cytoplasmic Ca2+ through divergent signaling mechanisms, and such Ca2+ is critical for synaptic plasticity (Berridge, 1998). β-AR activation also enhances the potential for synaptic plasticity, by stimulation of a G protein (Gs) that is positively linked with adenylyl cyclase (Tesmer et al, 1997). Adenylyl cyclase leads to activation of the cAMP-PKA-CREB pathway, which facilitates AMPA receptor trafficking and synaptic plasticity (Nguyen and Woo, 2003). In opposition to β-ARs, α2-AR activation reduces the potential for synaptic plasticity through stimulation of an inhibitory G protein (Gi) that is negatively linked with adenylyl cyclase (Marzo et al, 2009). Moreover, α2-ARs can act as presynaptic autoreceptors, reducing total noradrenergic synaptic transmission via inhibition of presynaptic Ca2+ influx (Hirning et al, 1988; Schofield, 1990). Taken together, α1- and β-AR activation enhances intrinsic excitability and increases the potential for synaptic plasticity. In contrast, α2-AR activation can reduce intrinsic excitability and synaptic activity at both presynaptic and postsynaptic membranes. Although norepinephrine acts on multiple receptors, each are involved in reconsolidation of aversive and appetitive memories.
NORADRENERGIC REGULATION OF FEAR MEMORY RECONSOLIDATION
Reconsolidation of aversive memories has been studied extensively using fear conditioning. Fear is a powerful emotion, and the memories associated with fear-provoking events are robust (Gale et al, 2004; for a review, see Davis, 1997; Ledoux, 2000; Maren, 2001). Fear conditioning involves pairing a conditioned stimulus (CS), such as a distinct cue or environment, with an aversive unconditioned stimulus (US), such as a shock. Following conditioning, presentation of the CS in the absence of the US results in memory retrieval and conditioned fear expression. Furthermore, retrieval can induce subsequent memory destabilization that allows for modification and reconsolidation of the now-labile memory. To assess whether a particular mechanism is critical for memory reconsolidation, two basic experiments should be performed. First, manipulations (eg, pharmacological) should preferably be given after, rather than before, memory retrieval, and the long-lasting effects of these manipulations should be evaluated during a long-term memory retrieval test. The major issues with giving manipulations before retrieval, instead of after, is that such manipulations could affect memory destabilization before reconsolidation (Ben Mamou et al, 2006; Lee et al, 2008; Milton et al, 2013) or could induce memory impairments that are behaviorally similar, but mechanistically distinct from memory reconsolidation disruption (Otis et al, 2013). The importance of giving a long-term retrieval test (generally 24 h after the manipulation) is to ensure that reconsolidation is complete and that any pharmacological effects are no longer present (Dudai, 2004). The second experiment serves as a control, wherein the same manipulation is given in the absence of retrieval to ensure that observed effects require memory reconsolidation (for a review, see Nader and Hardt, 2009). Manipulations should have long-lasting effects on memory when given after, but not in the absence of, memory retrieval for investigators to conclude that reconsolidation has been affected. Finally, some investigators have also used a retrieval test briefly after the manipulation (generally 3 h), to provide further support for reconsolidation blockade (eg, Debiec and LeDoux, 2006; Nader et al, 2000). In the case that the memory is impaired long, but not briefly, after the initial retrieval test, investigators can be sure that reconsolidation has been affected.
Rodent Studies
Fear memory reconsolidation is regulated by noradrenergic signaling through both α- and β-ARs. Studies using rodent fear conditioning reveal that reconsolidation is disrupted by administration of β-AR antagonists systemically (Przybyslawski et al, 1999; Muravieva and Alberini, 2010) or directly into the amygdala (Debiec and Ledoux, 2004, 2006; Debiec et al, 2011). The effects of β-AR blockade are long lasting, as administration of the β-AR antagonist propranolol after reactivation of recent and remote fear memories prevents fear expression for weeks or longer (Abrari et al, 2008; Debiec and Ledoux, 2004). Furthermore, propranolol-induced reconsolidation disruption prevents the reinstatement of fear following presentation of a reminder shock (Abrari et al, 2008; Debiec and Ledoux, 2004). In contrast, fear memory reconsolidation can be enhanced by amygdalar infusions of β-AR agonists (Debiec et al, 2011). Taken together, rodent fear conditioning experiments reveal that β-AR activation is a critical mechanism for control of fear memory reconsolidation.
Data from the inhibitory avoidance paradigm indicate that not all aspects of fear memories are regulated by β-AR activation. Muravieva and Alberini (2010) found that systemic injections of propranolol after retrieval reduced subsequent cue-induced freezing, whereas propranolol had no effect on behavioral avoidance. In contrast to freezing, rodent avoidance learning is thought to require declarative memory (Cammarota et al, 2007). Thus, although declarative knowledge cannot be measured in rodents, these data support the idea that β-AR activation specifically regulates reconsolidation for the emotional non-declarative, but not declarative, component of fear memories. This notion is well supported by investigations examining the effectiveness of propranolol for disruption of fear memory reconsolidation in humans (eg, Kindt et al, 2009; see below).
Fear memory reconsolidation is also regulated by α-AR activity. Systemic injection of the α2-AR agonist clonidine, which reduces total noradrenergic signaling, abolishes cue-induced fear memory reconsolidation (Gamache et al, 2012). In contrast, systemic injection of the α2-AR antagonist yohimbine enhances fear memory reconsolidation leading to increased context-induced fear expression (Gazarini et al, 2013). Moreover, the effects of yohimbine are prevented by the α1-AR antagonist prazosin or β-AR antagonist propranolol (Gazarini et al, 2013). Thus, α2-AR activation impairs fear memory reconsolidation by reducing α1 and β-AR signaling. Recent data also reveal that α1-AR activity is required for fear memory reconsolidation. Systemic or intraprelimbic medial prefrontal cortex administration of the α1-AR antagonist prazosin prevents fear memory reconsolidation, an effect that reduces cue-induced fear expression for at least 3 weeks (Do Monte et al, 2013). Interestingly, these effects conflict with data revealing that prelimbic β-AR blockade does not disrupt reconsolidation of drug-associated memories (Otis et al, 2013), indicating differences between paradigms or between mechanisms required for reconsolidation processes in the prelimbic cortex. Taken together, experiments using rodents reveal that fear memory reconsolidation requires activation of α1 and β-ARs, whereas reconsolidation is hindered by α2-AR-dependent inhibition of α1 and β-AR signaling.
Human Studies
Considerable evidence reveals that noradrenergic signaling at β-ARs regulates reconsolidation of human memories. Kindt et al (2009) used human fear conditioning to determine the effects of the β-AR antagonist propranolol on reconsolidation. Propranolol was administered before memory retrieval, with the notion that the orally administered propranolol takes time to reach peak plasma concentrations (Gilman and Goodman, 1996). Propranolol had no immediate effects on conditioned fear, as measured by fear-potentiated startle (eyeblink reflex). In contrast, propranolol induced long-lasting impairments in conditioned fear expression during subsequent retrieval trials when administered before but not in the absence of retrieval. Importantly, propranolol did not disrupt expectation of the aversive US, indicating that β-AR blockade during or after retrieval disrupts the pathologic emotional, but not declarative, component of the fear memory (Kindt et al, 2009). This work has been replicated and thoroughly extended within the past several years (Sevenster et al, 2012; Soeter and Kindt, 2010, 2011, 2012a, 2012b), and these studies further reveal that propranolol administration during retrieval impairs robust fear memories in humans (Soeter and Kindt, 2012b). Moreover, propranolol-induced memory impairments provide protection against spontaneous recovery of fear 30 days following extinction (Soeter and Kindt, 2010). Crucially, further evidence now reveals that propranolol impairs human fear memories when administered after CS presentation, confirming that the observed effects are specific to reconsolidation blockade (Soeter and Kindt, 2012a, 2012b). Finally, propranolol-induced reconsolidation disruption is specific to a reactivated memory, but not to a similar non-reactivated fear memory (Soeter and Kindt, 2011), confirming that the effect is memory-specific and does not generalize to other memories. These data have been further supported by a recent meta-analysis, confirming that cue-induced fear is reduced by propranolol when given during reconsolidation (Lonergan et al, 2013). Taken together, propranolol induces long-lasting disruption of human fear memory reconsolidation in a retrieval-dependent and memory-specific manner. These data indicate that disruption of noradrenergic signaling may be effective for disruption of human fear memories outside of the laboratory, such as memories of real-life trauma.
Clinical Trials
Recent investigations have examined the necessity of β-AR activation for reconsolidation of traumatic memories in patients with PTSD. Brunet et al (2008) used script-driven memory retrieval to induce reconsolidation of real-life traumatic memories. Immediately and 2 h after memory retrieval, short- and long-acting formulations of propranolol were administered, respectively. During a follow-up reactivation test a week later, propranolol-treated patients had significantly reduced psychophysiologic responding (heart rate, skin conductance) when compared with placebo-treated patients. Moreover, these physiologic responses were reduced below threshold for PTSD diagnoses (Brunet et al, 2008). When administered in conjunction with multiple (6) memory reactivation sessions, propranolol treatment reduced the diagnosis of PTSD by more than 70% and reduced symptom severity by more than 50% (Brunet et al, 2011). Finally, propranolol treatment during memory retrieval was associated with improved quality of life and reduced depressive symptoms among patients with PTSD (Poundja et al, 2012). Although these data are promising and indicate that reconsolidation of traumatic memories may require β-AR activation, each clinical investigation lacks appropriate placebo and/or no-reactivation control groups. Future controlled investigations should fill this critical gap, permitting a better understanding and accurate control of the memory mechanisms involved in PTSD.
NORADRENERGIC REGULATION OF REWARD-RELATED MEMORY RECONSOLIDATION
Appetitive memories are associated with positive feelings, such as the rewarding effects of drugs that reinforce drug-seeking behaviors. Retrieval of these memories induces craving and drives drug seeking. Therefore, disruption of drug-associated memories would limit relapse susceptibility. Despite the differences in valence, similarities have been found between the underlying processes that regulate aversive and appetitive memory reconsolidation. Initial studies using the rodent radial arm maze revealed that systemic (Przybyslawski et al, 1999) or intracerebroventricular (Roullet and Sara, 1998) infusions of β-AR antagonists following retrieval disrupted reconsolidation of memories underlying context-driven reward-seeking behavior. Since these pioneering experiments, research has focused on the necessity of noradrenergic signaling for reconsolidation of drug-related memories using the conditioned place preference (CPP) and self-administration (SA) paradigms.
Rodent CPP Studies
The CPP paradigm is commonly used to study appetitive memories. In this paradigm, animals are conditioned to associate one chamber with a drug of abuse, and another with vehicle (control). Following conditioning, CPP retrieval tests are given during which animals are allowed full access to both chambers. An animal that spends significantly more time in the previously drug-paired context at test is said to be drug seeking, and such behavior requires context-driven memory retrieval.
Noradrenergic signaling is critical for CPP memory reconsolidation. Systemic injections of the β-AR antagonist propranolol immediately following a retrieval test (or tests) prevent subsequent expression of a cocaine CPP memory (Bernardi et al, 2006; Fricks-Gleason and Marshall, 2008). Propranolol also disrupts morphine CPP memory reconsolidation (Robinson and Franklin, 2007, 2010). Thus, memories associated with both psychostimulants and opiates are susceptible to reconsolidation blockade by propranolol. Furthermore, we and others recently demonstrated that β-AR blockade within the amygdala (Bernardi et al, 2009; Otis et al, 2013), but not the hippocampus or prelimbic cortex (Otis et al, 2013, 2014), prevents cocaine CPP memory reconsolidation. Moreover, specific blockade of amygdalar α1- or β2-ARs disrupts cocaine CPP memory reconsolidation (Bernardi et al, 2009). The effects of β-AR blockade provide long-lasting protection against drug-induced reinstatement of the CPP (Fricks-Gleason and Marshall, 2008; Robinson and Franklin, 2010), supporting the idea that β-AR blockade does not enhance extinction, but rather impairs CPP reconsolidation. Further investigation also reveals that β-AR blockade disrupts a double-conditioned morphine CPP (8 morphine pairings, instead of 4 pairings) when the CPP memory is reactivated 30 days after training (but not 1 day after training; Robinson and Franklin, 2010). These data are encouraging as they indicate that drug-associated memories can be disrupted by β-AR blockade even if these memories are acquired long before pharmacologic intervention.
Rodent SA Studies
In addition to place conditioning, appetitive memories can be studied using the SA paradigm. In this model, rodents are trained to lever press or nose poke for rewards, such as food, intravenous drug infusion, or intracranial stimulation (eg, of the lateral hypothalamus). Experiments have revealed that systemic propranolol injections administered after memory retrieval disrupt context- or cue-driven sucrose seeking (Diergaarde et al, 2006; Milton et al, 2008b), cocaine seeking (Milton et al, 2008b), and ethanol seeking (Wouda et al, 2010) within the SA paradigm (but see Milton and Everitt, 2010; Williams and Harding, 2014). However, these convincing experiments have been overshadowed by studies revealing no effect of propranolol when administered before memory retrieval (Lee and Everitt, 2008; Milton and Everitt, 2010). The latter experiments were likely performed with preretrieval infusions so that the effects could be compared with those of other pharmacological agents. However, data reveal that β-AR blockade is more effective at impairing memory during the ‘late phase' of reconsolidation. Specifically, using the rodent radial arm maze, intracebroventricular infusions of the β-AR antagonist timolol was shown to impair memory reconsolidation when administered 60 min after retrieval (Roullet and Sara, 1998). In contrast, timolol had no effect when infused 5, 30, or 300 min after retrieval. Consistent with this, we found that propranolol injections immediately before cocaine CPP memory retrieval have no effect on reconsolidation (Otis and Mueller, 2011), whereas systemic injections or amygdalar infusions of β-AR antagonists after retrieval disrupt cocaine CPP memory reconsolidation (Bernardi et al, 2006, 2009; Fricks-Gleason and Marshall, 2008; Otis et al, 2013). Thus, the lack of reconsolidation blockade could be due to metabolism of propranolol before the late phase of memory reconsolidation, which may require β-AR-dependent protein synthesis. Alternatively, preretrieval propranolol may have prevented memory destabilization such that memory reconsolidation was not necessary to maintain the memory. In support of this, β-AR activation facilitates NMDA receptor channel conductance (Huang et al, 1998; Ji et al, 2008), and NMDA receptor activation is essential for fear memory destabilization (Ben Mamou et al, 2006; Milton et al, 2013). Thus, β-AR activation may facilitate NMDA receptor signaling for memory destabilization. To assess the necessity of β-AR activation for memory destabilization, future investigations should determine if preretrieval β-AR blockade prevents the memory-impairing effects of protein synthesis inhibition during memory reconsolidation.
To determine the effects of propranolol treatment on reconsolidation of SA memories, experiments were grouped by timing of drug administration and effectiveness of propranolol (as listed in Table 1). When propranolol was administered after memory reactivation, 4/6 studies revealed a significant impairment in subsequent context- or cue-driven reward seeking. In contrast, when propranolol was administered before memory reactivation, 0/4 studies revealed significant impairments in subsequent cue-driven reward seeking. Thus, propranolol is capable of impairing reconsolidation of reward-related memories in both the CPP and SA paradigms when administered after, but not before, memory reactivation.
Table 1. Efficacy of Propranolol for Reconsolidation Disruption Within the Self Administration (SA) Paradigm.
| Timing | Effect | SA paradigm | Authors (year) |
|---|---|---|---|
| After | |||
| Impaired | Sucrose (extinction) | Diergaarde et al (2006) | |
| Impaired | Sucrose (CR) | Milton et al (2008b) | |
| Impaired | Cocaine (CR) | Milton et al (2008b) | |
| No effect | Cocaine (reinstatement) | Milton and Everitt (2010) | |
| Impaired | EtOH (extinction) | Wouda et al (2010) | |
| No effect | EtOH (extinction) | Williams and Harding (2014) | |
| Before | |||
| No effect | Sucrose (PCA) | Lee and Everitt (2008) | |
| No effect | Sucrose (PIT) | Lee and Everitt (2008) | |
| No effect | EtOH (PCA) | Milton et al (2012) | |
| No effect | EtOH (PIT) | Milton et al (2012) | |
Abbreviations: CR, conditioned reinforcement; PCA, Pavlovian conditioned approach; PIT, Pavlovian-to-instrumental transfer.
Summary of findings for research examining the effectiveness of propranolol for reconsolidation blockade within the SA paradigm.
An alternative explanation regarding the effectiveness of propranolol for reconsolidation blockade in the SA paradigm is related to the type of memory being investigated. Specifically, experiments have revealed that postretrieval injections of propranolol impair conditioned reinforcement (Milton et al, 2008b), whereas preretrieval injections of propranolol have no effect on Pavlovian conditioned approach (PCA) or Pavlovian instrumental transfer (PIT; Lee and Everitt, 2008; Milton et al, 2012). In support of the idea that preretrieval propranolol is capable of preventing reconsolidation, preretrieval propranolol disrupts fear memory reconsolidation in humans (Kindt et al, 2009; Soeter and Kindt, 2010, 2011; Zhao et al, 2011), although this may be because propranolol takes ∼90 min to reach peak plasma concentrations when orally administered in humans (Gilman and Goodman, 1996). Taken together, because propranolol was administered before memory retrieval, it is unclear whether PCA or PIT is affected by β-AR blockade during memory reconsolidation.
Human and Clinical Studies
Noradrenergic signaling is also critical for reconsolidation of human drug-related memories. Zhao et al (2011) first demonstrated that propranolol impairs memory of heroin-related, but not neutral, words among patients with heroin addiction. Participants learned a list of heroin-related positive words (eg, syringe, foil), negative words (eg, vomit, diarrhea), and neutral words (eg, table, refrigerator). The next day, propranolol was orally administered before a memory retrieval test. Although there was no effect of propranolol on retrieval during this test, 24 h later propranolol-treated patients recalled fewer heroin-related positive and negative words, but not neutral words (Zhao et al, 2011). Moreover, propranolol had no effect when administered in the absence of memory retrieval, indicating that the memory-impairing effects of propranolol required memory reactivation. Although follow-up experiments should examine the effects of postretrieval β-AR blockade on memory of heroin-related words, these data indicate that β-AR blockade selectively impaired memory reconsolidation of retrieved heroin-related words, but not neutral words, in patients with heroin addiction.
Recent data suggest that reconsolidation disruption by β-AR blockade also reduces cue-induced cocaine cravings. Saladin et al (2013) treated cocaine-dependent subjects with propranolol or placebo immediately after presentation of cocaine-related cues (video and actual cocaine). The next day, the same cues were presented to the subjects in the absence of propranolol. Propranolol-treated individuals had significantly reduced cocaine cravings during cue presentation, indicating that β-AR blockade during memory reconsolidation reduced subsequent cue-induced cocaine cravings. Despite this, follow-up experiments should replicate this study with a no-retrieval control group to confirm that the observed effects are specific to memory reconsolidation. Interestingly, patients were given another follow-up test 1 week after propranolol treatment, and although propranolol-treated individuals reported less cue-induced cocaine cravings overall, this effect did not reach the threshold for significance. However, multiple retrieval sessions followed by propranolol treatment induces more reliable and robust effects on drug-associated memory reconsolidation in rodents (Fricks-Gleason and Marshall, 2008), and is more effective at eliminating symptoms of PTSD in humans (Brunet et al, 2011). Thus, follow-up experiments should assess whether multiple cue-reactivation plus propranolol sessions will induce more pronounced and long-lasting memory impairments among patients with drug addiction.
PROPOSED MECHANISMS AND PATHWAYS
Presentation of conditioned stimuli in both aversive and appetitive conditioning paradigms induces norepinephrine release (Cassens et al, 1980; Mingote et al, 2004), which activates presynaptic (α2) and postsynaptic receptors (α1-, α2-, and β-ARs; Figure 1). Research described above provides convincing evidence that such activity controls memory reconsolidation in a bidirectional manner. Despite this, the particular mechanism by which noradrenergic signaling regulates memory reconsolidation is less clear.
Figure 1.

Cellular mechanisms of memory reconsolidation. Postsynaptic noradrenergic signaling strengthens emotional memory reconsolidation by enhancing N-methyl-D-aspartate (NMDA) receptor-dependent synaptic plasticity. α1-Adrenergic receptor (AR) activation enhances phospholipase C (PLC) signaling to activate Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII associates with the NMDA receptor subunit GluN2B, and this binding facilitates AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor phosphorylation and promotes NMDA receptor-dependent synaptic plasticity. α2-AR activation inhibits adenylyl cyclase, whereas β-AR activation stimulates adenylyl cyclase. Adenylyl cyclase activation facilitates protein synthesis and GluN1 phosphorylation via cAMP-PKA-CREB. Thus, NMDA, α1, and β receptor signaling modifies existing membrane-bound proteins and induces synthesis of new proteins for the orchestration of reconsolidation-dependent synaptic plasticity. In contrast, α2-ARs function as inhibitory presynaptic autoreceptors (not illustrated) and as postsynaptic receptors (illustrated) to reduce α1 and β-AR receptor signaling.
Cellular Mechanisms
Noradrenergic signaling likely regulates memory reconsolidation via modulation of synaptic plasticity (see Figure 1). Synaptic plasticity is a critical mechanism for reconsolidation (Clem and Huganir, 2010). Moreover, NMDA receptor activation, a well-known mechanism of synaptic plasticity and learning (Morris et al, 1986), is essential for both fear and drug-associated memory reconsolidation (Brown et al, 2008; Lee et al, 2006; Milton et al, 2008a, 2012; Suzuki et al, 2004). Noradrenergic signaling enhances NMDA receptor ion channel conductance (Huang et al, 1998; Ji et al, 2008) and NMDA receptor-dependent synaptic plasticity (Thomas et al, 1996). This likely occurs via both α1- and β-AR signaling. Specifically, PKA downstream of β-ARs promotes phosphorylation of the NMDA receptor 1 subunit (GluN1; Bird et al, 2005; Snyder et al, 1998). In contrast, CaMKII downstream of α1-ARs directly binds directly to GluN2B subunits (Leonard et al, 1999), and this binding is essential for AMPA receptor phosphorylation (Zhou et al, 2007), synaptic plasticity (Barria and Malinow, 2005; Zhou et al, 2007), and memory consolidation (Zhou et al, 2007). Interestingly, GluN2B-containing NMDA receptor neurotransmission may not be critical for restabilization of memory during reconsolidation (Ben Mamou et al, 2006; Milton et al, 2013), indicating that the GluN2B subunit may only serve as a scaffolding mechanism for reconsolidation (similar to memory consolidation, see Zhou et al, 2007). Considering that norepinephrine is released upon presentation of emotionally salient cues (Cassens et al, 1980), and reconsolidation follows presentation of those cues, norepinephrine facilitates NMDA receptor-dependent synaptic plasticity during memory reconsolidation. This is likely to allow for maintenance of cue salience, such that noradrenergic receptor blockade during reconsolidation disrupts the pathologic emotional, but not declarative, components of reactivated memories.
Neural Circuit of Reconsolidation
Much is known regarding the signaling mechanisms underlying memory reconsolidation. Despite this, the neural circuits underlying reconsolidation are less clear. Here, we describe a simplified neural circuit that likely regulates reconsolidation and expression of fear and drug-associated memories (see Figure 2).
Figure 2.
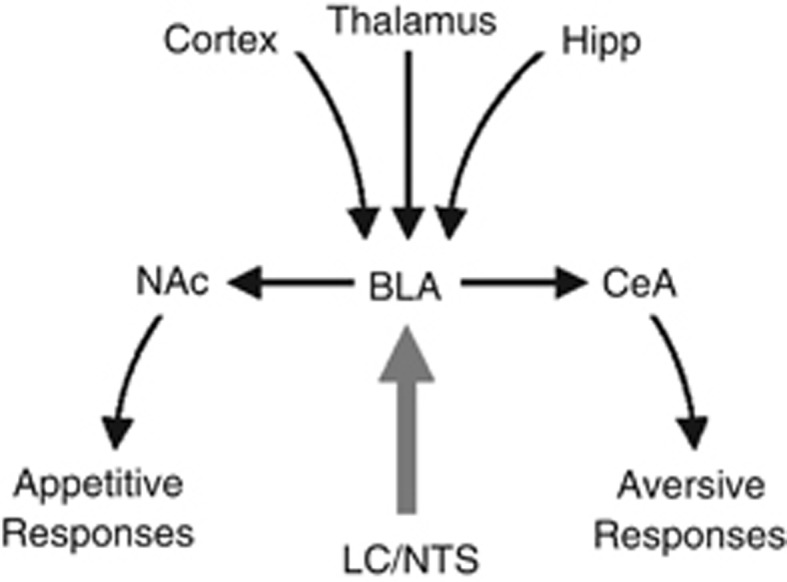
Neural circuit of emotional memory reconsolidation. Noradrenergic (thick gray arrow) and glutamatergic (black arrows) neurons converge in basolateral nucleus of the amygdala (BLA) to control N-methyl-D-aspartate (NMDA) receptor-dependent synaptic plasticity for emotional memory reconsolidation. Following reconsolidation, BLA output neurons to nucleus accumbens (NAc) and central nucleus of the amygdala (CeA) control appetitive and aversive responses, respectively, during emotional memory expression. Hipp, hippocampus; LC, locus coeruleus; NTS, nucleus tractus solitarius.
Noradrenergic and glutamatergic signaling in the BLA regulate reconsolidation of fear memories (Debiec and LeDoux, 2004; Lee et al, 2006; Milton et al, 2013) and drug-associated memories (Bernardi et al, 2009; Milton et al, 2008a; Otis et al, 2013). Thus, amygdalar noradrenergic and glutamatergic afferents must be important for reconsolidation. The amygdala receives noradrenergic input from the nucleus tractus solitarius (NTS) and locus coeruleus (LC; Fallon and Ciofi, 1992; Ricardo and Koh, 1978), which are engaged during memory consolidation (for a review, see McIntyre et al, 2012). However, whether NTS or LC is important for memory reconsolidation is unknown. The amygdala also receives glutamatergic input from thalamic, hippocampal, and cortical neurons, and these afferents underlie emotional learning and memory (for a review, see LeDoux, 2000). In fact, amygdalar glutamatergic inputs undergo synaptic potentiation following fear conditioning (Rogan et al, 1997; McKernan and Shinnick-Gallagher, 1997) and appetitive conditioning (Tye et al, 2008), suggesting that potentiation of these synapses may underlie aversive and appetitive memories. Consistent with this, the reconsolidation–extinction procedure, which induces erasure of fear memories and drug-associated memories in rodents and humans (Monfils et al, 2009; Schiller et al, 2010; Xue et al, 2012), causes synaptic depotentiation at thalamoamygdalar synapses (Clem and Huganir, 2010; also see Agren et al, 2012). Taken together, the amygdala receives both glutamatergic and noradrenergic input for memory reconsolidation. These data support the idea that noradrenergic inputs to the amygdala restabilize glutamate-dependent plasticity during reconsolidation, allowing for the persistence of emotional memories.
Although the BLA inputs underlying emotional memory reconsolidation for aversive and appetitive behaviors overlap, the outputs for these behaviors are likely distinct. BLA projection neurons innervate the central nucleus of the amygdala (CeA; Pare et al, 1995), a structure that has distinct projections for the expression of particular fear responses (LeDoux et al, 1988; for a review, see LeDoux, 2000). BLA projection neurons also innervate the nucleus accumbens (NAc; Stuber et al, 2011), a pathway that is critical for cue-induced reward seeking (Cador et al, 1989; Ambroggi et al, 2008; Stuber et al, 2011). Taken together, learning-related synaptic potentiation in BLA-CeA projection neurons promotes cue-induced fear, whereas plasticity in BLA-NAc projection neurons promotes cue-induced reward seeking. Furthermore, BLA noradrenergic signaling restabilizes this plasticity during reconsolidation for the maintenance of cue-induced behaviors.
ALTERNATIVE INTERPRETATION: FACILITATION OF EXTINCTION
Extinction is the formation of a new inhibitory memory, wherein conditioned responses to a stimulus are reduced due to repeated omission of the reinforcer (Quirk and Mueller, 2008). Thus, an alternative hypothesis to the research described above is that disruption of noradrenergic signaling facilitates extinction learning, rather than impairs reconsolidation. However, this hypothesis is inconsistent with data revealing that noradrenergic signaling enhances, rather than impedes, formation of new memories (for a review, see McGaugh, 2000). Hence, inhibition of β-ARs impairs, rather than facilitates, extinction learning in appetitive and aversive learning paradigms (eg, LaLumiere et al, 2010; Mueller et al, 2008).
ETHICAL CONSIDERATIONS FOR CLINICAL INTERVENTION
More than a decade ago, ethical opposition regarding the use of memory elimination for the treatment of neuropsychiatric disorders came from Beyond Therapy, a report published by the President's Council on Bioethics (US) (2003). This article sparked debate among ethicists regarding research and potential therapies for the treatment of neuropsychiatric disorders, particularly PTSD (Bell, 2007; Donovan, 2010; Kolber, 2007). The major argument made in Beyond Therapy is that ‘forgetting therapy' is unethical as ‘our happiness depends also on our memory, on knowing who we are in relation to who we have been' (President's Council on Bioethics (US), 2003, p 209). Despite this, research has revealed that memories are naturally dynamic, and therefore memory modification is not as artificial as it may seem. In fact, the primary therapy currently used for treatment of PTSD is extinction-based exposure therapy, which involves the formation of a new inhibitory memory that can overcome original memories (Quirk and Mueller, 2008). Although this has worked well in some patients, a more robust approach may be necessary in certain circumstances (Conklin and Tiffany, 2002). In such cases, the data described here indicates that ablation of the pathologic, emotional component of a memory by inhibition of noradrenergic signaling may be useful.
Ethical arguments made against memory disruption for the treatment of neuropsychiatric disorders (ie, ‘forgetting therapy') are outdated and in opposition to current research. First, ‘forgetting therapy' is a misnomer, particularly when describing reconsolidation blockade by noradrenergic receptor antagonism. Human research demonstrates that the declarative components of memory remain intact following propranolol-induced reconsolidation blockade, whereas the pathologic, emotional components that drive disordered behaviors are abolished (eg, Kindt et al, 2009). Thus, patients given such therapy would not forget their life experiences. Second, reconsolidation disruption by noradrenergic receptor antagonism is memory specific and does not eliminate associated or non-reactivated memories (Bernardi et al, 2006; Kindt et al, 2009; Otis et al, 2013; Przybyslawski et al, 1999; Soeter and Kindt, 2011). Third, noradrenergic receptor blockade during reconsolidation has been shown to improve quality of life in patients with PTSD (Brunet et al, 2011; Poundja et al, 2012). Finally, noradrenergic receptor blockade during reconsolidation is capable of reducing cue-induced drug cravings (Saladin et al, 2013), suggesting that such therapy could limit relapse susceptibility. Taken together, memory disruption by noradrenergic receptor antagonism could provide an ethical and powerful option for treatment of memory-related psychiatric disorders.
Although modification of memory reconsolidation via noradrenergic receptor antagonism could alleviate psychiatric disorders, clinicians should be wary of the possible deleterious effects of noradrenergic receptor antagonism when given in conjunction with prolonged memory tests, aimed to induce extinction. Specifically, investigations using rodents reveal that extinction is impaired when noradrenergic receptor antagonists are administered (LaLumiere et al, 2010; Mueller et al, 2008). Thus, when given in conjunction with prolonged retrieval trials, as opposed to brief retrieval/reconsolidation trials, noradrenergic receptor blockers could potentially promote the persistence of maladaptive behaviors associated with psychiatric dysfunction.
CONCLUDING REMARKS
Strong emotional memories contribute to the persistence of fear disorders and drug addiction, and erasure of the pathologic aspects of these memories is possible. Here we describe preclinical data that reveal that both fear and drug-associated memories are susceptible to disruption by β-AR blockade during reconsolidation. Despite this, well-controlled clinical trials must be completed to firmly conclude whether β-AR activity regulates reconsolidation of real-life human traumatic or drug-associated memories. Studies using rodents also reveal that α1-AR activity is critical for reconsolidation of fear and drug-associated memories, although such experiments have not been performed with human subjects. Considering that both β-AR and α1-AR activation are indirectly inhibited by α2-AR (autoreceptor) stimulation, future preclinical and clinical studies should determine the effectiveness of α2-AR agonists and α1-AR antagonists on disruption of human memory reconsolidation. The data demonstrating that the emotional component of a pathologic memory can be selectively disrupted leaving the declarative component intact using β-AR antagonists suggest that these antagonists effectively neutralize the affective strength of the memory without impairing the entirety of the memory. Whereas the data are generally interpreted as demonstrating reconsolidation blockade, an alternative explanation is that the memory has been modified by weakening the affective strength. Thus, β-AR antagonists could serve as targeted therapeutic agents to neutralize pathologic memories, effectively reducing their impact. Overall, the findings described here support the use of noradrenergic pharmacotherapies as efficacious adjuncts for the treatment of pathologic memory disorders, such as PTSD and addiction.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
Acknowledgments
The preparation of this manuscript was supported by NIH R01 DA038042 and a grant from the University of Wisconsin—Milwaukee Graduate School to DM, and NIH F31 DA036950 to CTW.
References
- Abrari K, Rashidy-Pour A, Semnanian S, Fathollahi Y. Administration of corticosterone after memory reactivation disrupts subsequent retrieval of a contextual conditioned fear memory: dependence upon training intensity. Neurobiol Learn Mem. 2008;89:178–184. doi: 10.1016/j.nlm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Agren T, Engman J, Frick A, Bjorkstrand J, Larsson EM, Furmark T, et al. Disruption of reconsolidation erases a fear memory trace in the human amygdala. Science. 2012;337:1550–1552. doi: 10.1126/science.1223006. [DOI] [PubMed] [Google Scholar]
- Alberini The role of reconsolidation and the dynamic process of long-term memory formation and storage. Front Behav Neurosci. 2011;5:12. doi: 10.3389/fnbeh.2011.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkon DL. Voltage-dependent calcium and potassium ion conductances: a contingency mechanism for an associative learning model. Science. 1979;205:810–816. doi: 10.1126/science.223244. [DOI] [PubMed] [Google Scholar]
- Ambroggi F, Ishikawa A, Fields HL, Nicola SM. Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron. 2008;59:648–661. doi: 10.1016/j.neuron.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Chen M. Morphological basis of long-term habituation and sensitization in aplysia. Science. 1983;220:91–93. doi: 10.1126/science.6828885. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bell JA. Preventing post-traumatic stress disorder or pathologizing bad memories. Am J Bioeth. 2007;7:29–30. doi: 10.1080/15265160701518540. [DOI] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci. 2006;9:1237–1239. doi: 10.1038/nn1778. [DOI] [PubMed] [Google Scholar]
- Bernardi RE, Lattal KM, Berger SP. Postretrieval propranolol disrupts a cocaine conditioned place preference. NeuroReport. 2006;17:1443–1447. doi: 10.1097/01.wnr.0000233098.20655.26. [DOI] [PubMed] [Google Scholar]
- Bernardi RE, Ryabinin AE, Berger SP, Lattal KM. Post-retrieval disruption of a cocaine conditioned place preference by systemic and intrabasolateral amygdala beta2- and alpha1-adrenergic antagonists. Learn Mem. 2009;16:777–789. doi: 10.1101/lm.1648509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Sorg BA. The NMDA antagonist MK-801 disrupts reconsolidation of a cocaine-associated memory for conditioned place preference but not for self-administration in rats. Learn Mem. 2008;15:857–865. doi: 10.1101/lm.1152808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Orr SP, Tremblay J, Robertson K, Nader K, Pitman RK. Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J Psychiat Res. 2008;42:503–506. doi: 10.1016/j.jpsychires.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Brunet A, Poundja J, Tremblay J, Bui E, Thomas E, Orr SP, et al. Trauma reactivation under the influence of propranolol decreases posttraumatic stress symptoms and disorder: 3 open-label trials. J Clin Psychopharmacol. 2011;31:547–550. doi: 10.1097/JCP.0b013e318222f360. [DOI] [PubMed] [Google Scholar]
- Cador M, Robbins TW, Everitt BJ. Involvement of the amygdala in stimulus–reward associations: interaction with the ventral striatum. Neuroscience. 1989;30:77–86. doi: 10.1016/0306-4522(89)90354-0. [DOI] [PubMed] [Google Scholar]
- Cammarota M, Bevilaqua LRM, Medina JH, Izquierdo I.2007Studies of short-term avoidance memoryIn: Bermúdez-Rattoni F (eds)Neural Plasticity and Memory: From Genes to Brain CRC Press: Boca Raton, FL; 193–205. [PubMed] [Google Scholar]
- Cassens G, Roffman M, Kuruc A, Orsulak PJ, Schildkraut JJ. Alterations in brain norepinephrine metabolism induced by environmental stimuli previously paired with inescapable shock. Science. 1980;209:1138–1140. doi: 10.1126/science.7403874. [DOI] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin CA, Tiffany ST. Applying extinction research and theory to cue-exposure addiction treatments. Addiction. 2002;97:155–167. doi: 10.1046/j.1360-0443.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- Davis M. Neurobiology of fear responses: the role of the amygdala. J Neuropsychiatry Clin Neurosci. 1997;9:382–402. doi: 10.1176/jnp.9.3.382. [DOI] [PubMed] [Google Scholar]
- Debiec J, Bush DE, LeDoux JE. Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats—a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety. 2011;28:186–193. doi: 10.1002/da.20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience. 2004;129:267–272. doi: 10.1016/j.neuroscience.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE. Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann NY Acad Sci. 2006;1071:521–524. doi: 10.1196/annals.1364.056. [DOI] [PubMed] [Google Scholar]
- Diergaarde L, Schoffelmeer AN, De Vries TJ. Beta-adrenoceptor mediated inhibition of long-term reward-related memory reconsolidation. Behav Brain Res. 2006;170:333–336. doi: 10.1016/j.bbr.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Coulter DA, Alkon DL. Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci USA. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Monte FH, Souza RR, Wong TT, Carobrez Ade P. Systemic or intra-prelimbic cortex infusion of prazosin impairs fear memory reconsolidation. Behav Brain Res. 2013;244:137–141. doi: 10.1016/j.bbr.2013.01.031. [DOI] [PubMed] [Google Scholar]
- Donovan E. Propranolol use in the prevention and treatment of posttraumatic stress disorder in military veterans: forgetting therapy revisited. Perspect Biol Med. 2010;53:61–74. doi: 10.1353/pbm.0.0140. [DOI] [PubMed] [Google Scholar]
- Dudai Y. The neurobiology of consolidations, or, how stable is the engram. Annu Rev Psychol. 2004;55:51–86. doi: 10.1146/annurev.psych.55.090902.142050. [DOI] [PubMed] [Google Scholar]
- Fallon JH, Ciofi P.1992Distribution of monoamines within the amygdalaIn: Aggelton JP (eds)The Amygdala Wiley-Liss: New York, NY, USA; 97–114. [Google Scholar]
- Foehring RC, Schwindt PC, Crill WE. Norepinephrine selectively reduces slow Ca2+- and Na+-mediated K+ currents in cat neocortical neurons. J Neurophysiol. 1989;61:245–256. doi: 10.1152/jn.1989.61.2.245. [DOI] [PubMed] [Google Scholar]
- Fricks-Gleason AN, Marshall JF. Post-retrieval beta-adrenergic receptor blockade: effects on extinction and reconsolidation of cocaine-cue memories. Learn Mem. 2008;15:643–648. doi: 10.1101/lm.1054608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale GD, Anagnostaras SG, Godsil BP, Mitchell S, Nozawa T, Sage JR, et al. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamache K, Pitman RK, Nader K. Preclinical evaluation of reconsolidation blockade by clonidine as a potential novel treatment for posttraumatic stress disorder. Neuropsychopharmacology. 2012;37:2789–2796. doi: 10.1038/npp.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazarini L, Stern CA, Carobrez AP, Bertoglio LJ. Enhanced noradrenergic activity potentiates fear memory consolidation and reconsolidation by differentially recruiting alpha1- and beta-adrenergic receptors. Learn Mem. 2013;20:210–219. doi: 10.1101/lm.030007.112. [DOI] [PubMed] [Google Scholar]
- Gilman AG, Goodman LS. Goodman and Gilman's the Pharmacological Basis of Therapeutics. McGraw-Hill: New York, NY; 1996. [Google Scholar]
- Graham RM, Perez DM, Hwa J, Piascik MT. Alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ Res. 1996;78:737–749. doi: 10.1161/01.res.78.5.737. [DOI] [PubMed] [Google Scholar]
- Hirning LD, Fox AP, McCleskey EW, Olivera BM, Thayer SA, Miller RJ, et al. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science. 1988;239:57–61. doi: 10.1126/science.2447647. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lin CH, Gean PW. Potentiation of N-methyl-D-aspartate currents by isoproterenol in the acutely dissociated rat amygdalar neurons. Neurosci Lett. 1998;253:9–12. doi: 10.1016/s0304-3940(98)00571-0. [DOI] [PubMed] [Google Scholar]
- Ji XH, Cao XH, Zhang CL, Feng ZJ, Zhang XH, Ma L, et al. Pre- and postsynaptic beta-adrenergic activation enhances excitatory synaptic transmission in layer V/VI pyramidal neurons of the medial prefrontal cortex of rats. Cereb Cortex. 2008;18:1506–1520. doi: 10.1093/cercor/bhm177. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kim J, Song B, Hong I, Kim J, Lee J, Park S, et al. Reactivation of fear memory renders consolidated amygdala synapses labile. J Neurosci. 2010;30:9631–9640. doi: 10.1523/JNEUROSCI.0940-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt M, Soeter M, Vervliet B. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci. 2009;12:256–258. doi: 10.1038/nn.2271. [DOI] [PubMed] [Google Scholar]
- Kolber A. Clarifying the debate over therapeutic forgetting. Am J Bioethics. 2007;7:25–27. doi: 10.1080/15265160701518508. [DOI] [PubMed] [Google Scholar]
- Kroes MC, Tendolkar I, van Wingen GA, van Waarde JA, Strange BA, Fernández G. An electroconvulsive therapy procedure impairs reconsolidation of episodic memories in humans. Nat Neurosci. 2014;17:204–206. doi: 10.1038/nn.3609. [DOI] [PubMed] [Google Scholar]
- LaLumiere RT, Niehoff KE, Kalivas PW. The infralimbic cortex regulates the consolidation of extinction after cocaine self-administration. Learn Mem. 2010;17:168–175. doi: 10.1101/lm.1576810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron. 2005;47:795–801. doi: 10.1016/j.neuron.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Lee JL, Everitt BJ. Reactivation-dependent amnesia in Pavlovian approach and instrumental transfer. Learn Mem. 2008;15:597–602. doi: 10.1101/lm.1029808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J Neurosci. 2006;26:10051–10056. doi: 10.1523/JNEUROSCI.2466-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, et al. Synaptic protein degradation underlies destabilization of retrieved fear memory. Science. 2008;319:1253–1256. doi: 10.1126/science.1150541. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci USA. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan MH, Olivera-Figueroa LA, Pitman RK, Brunet A. Propranolol's effects on the consolidation and reconsolidation of long-term emotional memory in healthy participants: a meta-analysis. J Psychiat Neurosci. 2013;38:222–231. doi: 10.1503/jpn.120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- Marzo A, Bai J, Otani S. Neuroplasticity regulation by noradrenaline in mammalian brain. Curr Neuropharm. 2009;7:286–295. doi: 10.2174/157015909790031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Noradrenergic modulation of firing pattern in guinea pig and cat thalamic neurons, in vitro. J Neurophysiol. 1988;59:978–996. doi: 10.1152/jn.1988.59.3.978. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory—a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, McGaugh JL, Williams CL. Interacting brain systems modulate memory consolidation. Neurosci Biobehav Rev. 2012;36:1750–1762. doi: 10.1016/j.neubiorev.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ. The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci. 2010;31:2308–2319. doi: 10.1111/j.1460-9568.2010.07249.x. [DOI] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Butler VJ, Gardner R, Everitt BJ. Intra-amygdala and systemic antagonism of NMDA receptors prevents the reconsolidation of drug-associated memory and impairs subsequently both novel and previously acquired drug-seeking behaviors. J Neurosci. 2008;28:8230–8237. doi: 10.1523/JNEUROSCI.1723-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Lee JL, Everitt BJ. Reconsolidation of appetitive memories for both natural and drug reinforcement is dependent on beta-adrenergic receptors. Learn Mem. 2008;15:88–92. doi: 10.1101/lm.825008. [DOI] [PubMed] [Google Scholar]
- Milton AL, Merlo E, Ratano P, Gregory BL, Dumbreck JK, Everitt BJ. Double dissociation of the requirement for GluN2B- and GluN2A-containing NMDA receptors in the destabilization and restabilization of a reconsolidating memory. J Neurosci. 2013;33:1109–1115. doi: 10.1523/JNEUROSCI.3273-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Schramm MJ, Wawrzynski JR, Gore F, Oikonomou-Mpegeti F, Wang NQ, et al. Antagonism at NMDA receptors, but not beta-adrenergic receptors, disrupts the reconsolidation of pavlovian conditioned approach and instrumental transfer for ethanol-associated conditioned stimuli. Psychopharmacology (Berl) 2012;219:751–761. doi: 10.1007/s00213-011-2399-9. [DOI] [PubMed] [Google Scholar]
- Mingote S, de Bruin JP, Feenstra MG. Noradrenaline and dopamine efflux in the prefrontal cortex in relation to appetitive classical conditioning. J Neurosci. 2004;10:2475–2480. doi: 10.1523/JNEUROSCI.4547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misanin JR, Miller RR, Lewis DJ. Retrograde amnesia produced by electroconvulsive shock after reactivation of consolidated memory trace. Science. 1968;160:554–555. doi: 10.1126/science.160.3827.554. [DOI] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction–reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller GE, Pilzecker A. Experimentelle beiträge zur lehre vom gedächtniss. Z Psychol Ergänzungsband. 1900;1:1–300. [Google Scholar]
- Muravieva EV, Alberini CM. Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem. 2010;17:306–313. doi: 10.1101/lm.1794710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nader K, Hardt O. A single standard for memory: the case for reconsolidation. Nat Rev Neurosci. 2009;10:224–234. doi: 10.1038/nrn2590. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Otis JM, Dashew KB, Mueller D. Neurobiological dissociation of retrieval and reconsolidation of cocaine-associated memory. J Neurosci. 2013;33:1271–1281a. doi: 10.1523/JNEUROSCI.3463-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Fitzgerald MK, Mueller D. Inhibition of hippocampal beta-adrenergic receptors impairs retrieval but not reconsolidation of cocaine-associated memory and prevents subsequent reinstatement. Neuropsychopharmacology. 2014;39:303–310. doi: 10.1038/npp.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Mueller D. Inhibition of beta-adrenergic receptors induces a persistent deficit in retrieval of a cocaine-associated memory providing protection against reinstatement. Neuropsychopharmacology. 2011;36:1912–1920. doi: 10.1038/npp.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Smith Y, Pare JF. Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: Phaseolus vulgaris-leucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience. 1995;69:567–583. doi: 10.1016/0306-4522(95)00272-k. [DOI] [PubMed] [Google Scholar]
- Poundja J, Sanche S, Tremblay J, Brunet A. Trauma reactivation under the influence of propranolol: an examination of clinical predictors. Eur J Psychochotraumatol. 2012;3:1–9. doi: 10.3402/ejpt.v3i0.15470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- President's Council on Bioethics (US) 2003Beyond Therapy: Biotechnology and the Pursuit of Happiness1st edn.Regan Books: New York, NY [Google Scholar]
- Przybyslawski J, Roullet P, Sara SJ. Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J Neurosci. 1999;19:6623–6628. doi: 10.1523/JNEUROSCI.19-15-06623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt AC, Lee JL. Memory reconsolidation in aversive and appetitive settings. Front Behav Neurosci. 2013;7:118. doi: 10.3389/fnbeh.2013.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricardo JA, Koh ET. Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res. 1978;153:1–26. doi: 10.1016/0006-8993(78)91125-3. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Franklin KB. Central but not peripheral beta-adrenergic antagonism blocks reconsolidation for a morphine place preference. Behav Brain Res. 2007;182:129–134. doi: 10.1016/j.bbr.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Franklin KB. Reconsolidation of a morphine place preference: impact of the strength and age of memory on disruption by propranolol and midazolam. Behav Brain Res. 2010;213:201–207. doi: 10.1016/j.bbr.2010.04.056. [DOI] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- Roullet P, Sara S. Consolidation of memory after its reactivation: involvement of beta noradrenergic receptors in the late phase. Neural Plast. 1998;6:63–68. doi: 10.1155/NP.1998.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladin ME, Gray KM, McRae-Clark AL, Larowe SD, Yeatts SD, Baker NL, et al. A double blind, placebo-controlled study of the effects of post-retrieval propranolol on reconsolidation of memory for craving and cue reactivity in cocaine dependent humans. Psychopharmacology (Berl) 2013;226:721–737. doi: 10.1007/s00213-013-3039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Monfils MH, Raio CM, Johnson DC, LeDoux JE, Phelps EA. Preventing the return of fear in humans using reconsolidation update mechanisms. Nature. 2010;463:49–53. doi: 10.1038/nature08637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine blocks a calcium current of adult rat sympathetic neurons via an alpha 2-adrenoceptor. Eur J Pharmacol. 1990;180:37–47. doi: 10.1016/0014-2999(90)90590-3. [DOI] [PubMed] [Google Scholar]
- Sehgal M, Song C, Ehlers VL, Moyer JR., Jr Learning to learn—intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem. 2013;105:186–199. doi: 10.1016/j.nlm.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenster D, Beckers T, Kindt M. Retrieval per se is not sufficient to trigger reconsolidation of human fear memory. Neurobiol Learn Mem. 2012;97:338–345. doi: 10.1016/j.nlm.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeter M, Kindt M. Dissociating response systems: erasing fear from memory. Neurobiol Learn Mem. 2010;94:30–41. doi: 10.1016/j.nlm.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M. Disrupting reconsolidation: pharmacological and behavioral manipulations. Learn Mem. 2011;18:357–366. doi: 10.1101/lm.2148511. [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M. Erasing fear for an imagined threat event. Psychoneuroendocrinology. 2012;37:1769–1779. doi: 10.1016/j.psyneuen.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M. Stimulation of the noradrenergic system during memory formation impairs extinction learning but not the disruption of reconsolidation. Neuropsychopharmacology. 2012;37:1204–1215. doi: 10.1038/npp.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA. Reconsolidation of drug memories. Neurosci Biobehav Rev. 2012;36:1400–1417. doi: 10.1016/j.neubiorev.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, et al. Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature. 2011;475:377–380. doi: 10.1038/nature10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Tye KM, Stuber GD, de Ridder B, Bonci A, Janak PH. Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature. 2008;453:1253–1257. doi: 10.1038/nature06963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Williams KL, Harding KM. Repeated alcohol extinction sessions in conjunction with MK-801, but not yohimbine or propranolol, reduces subsequent alcohol cue-induced responding in rats. Pharmacol Biochem Behavior. 2014;116:16–24. doi: 10.1016/j.pbb.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouda JA, Diergaarde L, Riga D, van Mourik Y, Schoffelmeer AN, De Vries TJ. Disruption of long-term alcohol-related memory reconsolidation: role of beta-adrenoceptors and NMDA Receptors. Front Behav Neurosci. 2010;4:179. doi: 10.3389/fnbeh.2010.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, et al. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–245. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LY, Sun LL, Shi J, Li P, Zhang Y, Lu L. Effects of beta-adrenergic receptor blockade on drug-related memory reconsolidation in abstinent heroin addicts. Drug Alcohol Depend. 2011;118:224–229. doi: 10.1016/j.drugalcdep.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Takahashi E, Li W, Halt A, Wiltgen B, Ehninger D, et al. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. J Neurosci. 2007;27:13843–13853. doi: 10.1523/JNEUROSCI.4486-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]