

Abstract
Attention-deficit hyperactivity disorder (ADHD) is a neurodevelopmental disorder with high heritability. At least 30% of patients diagnosed in childhood continue to suffer from ADHD during adulthood and genetic risk factors may play an essential role in the persistence of the disorder throughout lifespan. To date, genome-wide association studies (GWAS) of ADHD have been completed in seven independent datasets, six of which were pediatric samples and one on persistent ADHD using a DNA-pooling strategy, but none of them reported genome-wide significant associations. In an attempt to unravel novel genes for the persistence of ADHD into adulthood, we conducted the first two-stage GWAS in adults with ADHD. The discovery sample included 607 ADHD cases and 584 controls. Top signals were subsequently tested for replication in three independent follow-up samples of 2104 ADHD patients and 1901 controls. None of the findings exceeded the genome-wide threshold for significance (PGC<5e−08), but we found evidence for the involvement of the FBXO33 (F-box only protein 33) gene in combined ADHD in the discovery sample (P=9.02e−07) and in the joint analysis of both stages (P=9.7e−03). Additional evidence for a FBXO33 role in ADHD was found through gene-wise and pathway enrichment analyses in our genomic study. Risk alleles were associated with lower FBXO33 expression in lymphoblastoid cell lines and with reduced frontal gray matter volume in a sample of 1300 adult subjects. Our findings point for the first time at the ubiquitination machinery as a new disease mechanism for adult ADHD and establish a rationale for searching for additional risk variants in ubiquitination-related genes.
INTRODUCTION
Attention-deficit hyperactivity disorder (ADHD; OMIM:143465) is a neurodevelopmental disorder characterized by inappropriate levels of hyperactivity, impulsivity, and/or inattention that affects 5–6% of children and persists into adulthood in 30–50% of ADHD patients diagnosed in childhood (Faraone et al, 2000b; Kessler et al, 2005; Kooij et al, 2005). Previous work supports a strong genetic load in ADHD, which places it among the most heritable psychiatric disorders with an estimated heritability of 76% (Biederman, 2005; Biederman and Faraone, 2005; Franke et al, 2012).
The first investigations on genes underlying ADHD focused on genome-wide linkage studies or hypothesis-driven candidate gene association analyses. In most cases, however, these failed to identify consistent and replicable genetic factors and provide modest evidence for the involvement of some specific genes on the basis of meta-analysis (Faraone et al, 2005; Franke et al, 2012; Gizer et al, 2009). This situation, along with advances in high-throughput technologies, stimulated a turn towards genome-wide association studies (GWAS). To date, GWAS of ADHD have been completed in seven independent datasets, six of which used pediatric samples (Franke et al, 2009; Hinney et al, 2011; Lesch et al, 2008; Mick et al, 2010; Neale et al, 2008; Neale et al, 2010a; Stergiakouli et al, 2012; Yang et al, 2013). None of them reported genome-wide significant associations and, apart from the possible involvement of cadherin 13 (CDH13) identified in three reports (Lesch et al, 2008; Neale et al, 2008; Neale et al, 2010a), there is limited overlap between them or with previous linkage or candidate gene association studies. In addition, none of the ‘classic' candidate genes for ADHD were found among the top findings of any GWAS study.
Neale et al (2008), carried out the first GWAS on ADHD in 909 affected triads and identified nominal associations for XKR4 and FAM190A, two genes expressed in the cerebellum. A second family-based multi-site GWAS comprised of 735 affected-offspring trios highlighted the SLC9A9 gene (Mick et al, 2010). Four additional case–control GWAS were also conducted in childhood ADHD and unscreened controls. Neale et al, genotyped 896 cases and 2455 controls and found CDH13 and PRKG1 among the top-ranked genes nominally associated with ADHD, while the other three studies yielded no significant evidence of association neither in the discovery cohort nor in the replication attempts on different datasets (Hinney et al, 2011; Neale et al, 2010a; Stergiakouli et al, 2012; Yang et al, 2013). Finally, a single GWAS analysis has been reported on persistent ADHD. It used a DNA-pooling strategy in a sample of 343 adults with ADHD and 304 controls in conjunction with linkage analysis and pointed to several chromosomal regions likely to contain allelic variants influencing susceptibility to ADHD (Lesch et al, 2008). So far, following the first analysis by Neale et al, secondary analyses considering the same dataset were conducted on ADHD-related phenotypes, such as quantitative measures of DSM-IV ADHD symptoms, age at onset of ADHD symptoms, or conduct problems. None of them identified single-nucleotide polymorphisms (SNPs) achieving genome-wide significance, with the most significant markers located in CDH13 and GFOD1 (Lasky-Su et al, 2008a; Lasky-Su et al, 2008b). All these negative results suggest that an even larger sample may be required, in analogy to approaches in schizophrenia and bipolar disorder where larger sample size did result in significant findings (Ripke et al, 2013a; Ruderfer et al, 2013). However, the first large-scale meta-analysis of ADHD GWAS datasets in 2064 trios, 896 cases, and 2455 controls from four previous studies also failed to identify any genome-wide significant association with childhood ADHD (Neale et al, 2010b).
In an attempt to unravel novel genes and biological pathways related to ADHD, we aimed to improve the design of previous GWAS in ADHD through reducing heterogeneity in the discovery cohort by (1) considering persistent ADHD, which may be influenced by a stronger genetic load than remitting ADHD (Biederman et al, 1996; Chang et al, 2013; Faraone et al, 2000a); (2) recruiting patients from a restricted geographic area by a single clinical group and following a single assessment protocol, which may facilitate the identification of risk variants that might be neglected by a broader recruitment approach used in previous multi-site GWAS; (3) not only considering the overall ADHD sample, but also the combined and inattentive clinical subtypes that may be influenced by shared as well as specific genetic factors; and (4) using screened, non-ADHD controls. Following this strategy we conducted a two-stage case–control GWAS in adulthood ADHD. The first phase included a discovery cohort of 607 adults with ADHD and 584 controls from Spain. Top signals were subsequently tested for replication in three independent follow-up cohorts including 2104 adults with ADHD and 1901 controls from three European countries (Germany, The Netherlands, and Norway) integrated in The International Multicentre persistent ADHD CollaboraTion (IMpACT).
MATERIALS AND METHODS
Participants
Clinical information of the studied cohorts is included in Supplementary Table S1 and Supplementary Material. Briefly, the discovery cohort consisted of 607 adulthood ADHD cases (61% combined, 35% inattentive, 3.5% hyperactive-impulsive and 0.5% with undefined subtype) and 584 healthy controls from Spain. All patients were adults of Caucasian origin and met Diagnostic and Statistical Manual for Mental Disorders-IV (DSM-IV) criteria for ADHD. The diagnosis of ADHD was evaluated with the Structured Clinical Interview for DSM-IV Axis I and II Disorders (SCID-I and SCID-II) and the Conner's Adult ADHD Diagnostic Interview for DSM-IV (CAADID Parts I and II). For the replication study, 2104 adult ADHD patients and 1901 controls of Caucasian origin from three European countries (Germany, The Netherlands and Norway; for more information see Sánchez-Mora et al, 2010) were recruited at three sites of the IMpACT (Supplementary Table S1). Consensus eligibility criteria for the current study across all sites were a diagnosis of ADHD according to the diagnostic criteria of DSM-IV, onset before the age of 7 years via retrospective diagnosis (which was confirmed by a family member, wherever possible), lifelong persistence, and current diagnosis. The study was approved by the ethics committee of each institution and informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Study Design, Genotyping, and Quality Control Assessment
To identify loci conferring susceptibility to ADHD, we carried out a two-stage case–control GWAS. In the initial discovery phase we performed genome-wide genotyping with the Illumina HumanOmni1-Quad BeadChip platform in 607 adults with ADHD and 584 healthy controls. Quality control was implemented at the individual and SNP level using PLINK (Purcell et al, 2007) and included filtering subjects with low call rate (<98%) or gender discrepancy followed by filtering SNPs with minor allele frequency (MAF)<0.01, Hardy–Weinberg equilibrium test P-values, PHWE<1e−06 or call rate<0.99 in either cases or controls. Top signals were further evaluated in three independent follow-up cohorts from Germany, The Netherlands, and Norway consisting of 2104 cases and 1901 controls. The replication cohorts were genotyped using the Kompetitive Allele Specific PCR (KASP) technology (LGC Genomics, UK). Clinical information of the studied cohorts is included in Supplementary Table S1 and Supplementary Material.
Statistical Analysis
Genome-wide association study
Initially, 607 cases and 584 controls were genotyped for 1 138 747 markers. In total, five samples were excluded from the analysis due to low call rate. After stringent quality control assessment, a total of 794 090 SNPs with a mean call rate of 0.9994 for the remaining 603 cases and 583 controls were included in the study. Genome-wide association analysis was performed using the Cochran–Armitage trend test with PLINK. The genomic inflation factor (λ) was calculated and used to correct for the degree of inflations. Quantile–quantile and Manhattan plots were drawn using the SnpStats (Clayton, 2013) and Gap R packages (Zhao, 2013). The genome-wide significance threshold was calculated by considering the three ADHD clinical subtypes and was set at P<1.7e−08 (5e−08/3). The phenotypic variance explained by all autosomal SNPs for ADHD was calculated with GCTA software (Yang et al, 2013). The gene-based association analysis was performed with the VEGAS software following the default settings and considering the HaMap CEU samples to estimate the linkage disequilibrium (LD) structure (Liu et al, 2010). The ALIGATOR software was used to test for over-representation of biological pathways, as indexed by gene ontology (GO) terms, considering genes (RefSeq transcript plus 20 kb up and downstream) with at least one SNP showing P<0.05, 10 000 replicate gene lists and 10 000 replicate studies (Holmans et al, 2009). ALIGATOR corrects for variable gene size, for the presence of LD between SNPs and for overlapping genes through a random selection of SNPs for generation of replication gene lists that are used to calculate the category-specific P-value. The replicate studies are used to assess significance of the numbers of categories reaching various P-values and the study-wide significance levels for individual categories.
Follow-up of top signals from the discovery stage
For the replication analysis, a follow-up threshold of significance was set at PGC<1e−05. A total of 16 independent SNPs reaching this cut-off were considered (Supplementary Table S1 and S2). For the SNP selection we evaluated the LD pattern of markers achieving the follow-up threshold with the LD-select software (r2<0.85; Carlson et al, 2004). Seven markers were selected for replication in the overall ADHD sample, eight in the combined and four in the inattentive clinical subtypes (Supplementary Table S2). Rs1937444 could not be tested due to experimental constraints and therefore 15 SNPs from 10 loci were finally assessed (Supplementary Table S2). We performed a single-marker analysis on the replication samples from the three IMpACT sites separately and then combined the individual study results using both mega-analytic and meta-analytic approaches. We assessed HWE in each control sample and compared genotype frequencies using Cochran–Armitage trend tests with the SNPassoc R package (González et al, 2007). Meta-analyses were conducted using the Meta-R package, under a dominant model of inheritance (Viechtbauer, 2010). To test heterogeneity among studies we used the Q-statistic and heterogeneity, which was considered significant when PQ<0.10. When no heterogeneity was present, the pooled OR was estimated using a fixed-effects model. Otherwise, a random-effects model was applied. Under the more conservative Bonferroni correction taking into account 15 SNPs and three clinical subtypes, the threshold for significance was set at P<1.1e−03 (0.05/15 × 3).
Imputation and gene-based association analysis
Markers at the FBXO33 (F-box only protein 33 gene) locus (ch14:39865674–39902852) were imputed in the discovery sample with the BEAGLE software using data from the 1000 Genomes Project (http://www.1000genomes.org/; Browning and Browning, 2009). The association analysis was performed using the Cochran–Armitage trend test with PLINK (Purcell et al, 2007). The significance threshold under the Bonferroni correction was set at P<4.7e−04, considering the 107 SNPs analyzed in the FBXO33 locus. The prediction of the potential effects of the risk alleles associated with ADHD were tested using CFSSP (Chou & Fasman Secondary Structure Prediction Server), SNPinfo, TFSearch, ESEfinder, ESRSearch, and RESCUE_ESE (Chou and Fasman, 1974; Cartegni et al, 2003; Fairbrother et al, 2002; Heinemeyer et al, 1998; Wang et al, 2004; Xu and Taylor, 2009) as well as the Ensembl database (http://www.ensembl.org/).
Cis Expression Quantitative Trait Loci Analysis
FBXO33 expression levels and genotype data for rs17696574, rs10139591, rs7156962, and rs1056345 from 210 unrelated HapMap individuals, including Utah residents with Northern and Western European ancestry from the CEPH collection (CEU, n=60); Han Chinese from Beijing, China (CHB, n=45); Japanese from Tokyo, Japan (JPT, n=45); and Yoruban from Ibadan, Nigeria (YRI, n=60), were obtained from the whole-genome Illumina lymphoblastoid cell line gene expression data and the HapMap browser (http://www.hapmap.org) (International HapMap Consortium (2003)), respectively. No genotype data were available for marker rs12232110. For the expression data we considered the GSE6536 series dataset at the Gene Expression Omnibus site (http://www.ncbi.nlm.nih.gov/geo; Stranger et al, 2007a; Stranger et al, 2007b) and selected the GI_42558257-S probe corresponding to the FBXO33 transcript NM_203301.3. Association tests between genetic markers and FBXO33 expression were performed using linear regression models with SPSS 17.0 (SPSS, Chicago, USA) and the significant threshold was set at 0.0125 (0.05/4 SNPs) after applying the Bonferroni correction.
Brain Imaging Studies
To further understand the effects of ADHD-related SNPs in FBXO33, we tested rs17696574, rs12232110, rs10139591, rs7156962, and rs1056345 for their effects on brain structure in a pre-existing neuroimaging genetics sample, consisting of 1300 adult subjects (age range 18–40 years, average age 22.9 years) from the Dutch Cognomics Resource Brain Imaging Genetics (BIG; www.cognomics.nl). All participants were self-reportedly healthy subjects of Caucasian descent with no ADHD diagnosis. Genetic data were available from an Affymetrix Genome-Wide Human SNP Array 6.0 and imputed using the 1000 Genomes reference set. The effects of the genetic variation on structural brain changes were tested using voxel-based morphometry (VBM). For this, structural T1 brain scans acquired at 1.5 and 3 Tesla scanners (Siemens, Erlangen, Germany) were used (Supplementary Material). To study the effects of the specific SNPs, a full-factorial ANCOVA was performed using genotype as factor and age, gender, total brain volume, and field strength of the scanner as covariates. Gray and white matter images were analyzed separately. Statistics were corrected for non-stationarity and were applied at Puncorrected<1e−04. Significant clusters were considered at a level of PFWE<0.05. We specified our regions of interest using the WFU pickatlas (Maldjian et al, 2003). These regions included the frontal lobe, caudate nucleus, and the cerebellum selected based on information on FBXO33 gene expression in combination with knowledge of brain regions implicated in ADHD (Cubillo and Rubia, 2010).
RESULTS
Genome-Wide Association Study
In the first stage of the study and after individual and SNP-based standard quality control filtering, we conducted a GWAS in a discovery sample of 603 adult ADHD cases and 583 healthy controls. The quantile–quantile plot showed no departure from the expected P-values distribution with a genomic control inflation factor λ=1.031 used as a final quality control measure (Supplementary Figure S1). None of the association signals exceeded genome-wide thresholds for significance (PGC<1.7e−08). The GWAS of the overall sample, however, highlighted 12 SNPs from five loci that surpassed the follow-up threshold (PGC<1e−05), five of which were located in four genes, PEX19 (rs10594, PGC=3.69e−06), COPA (rs7522166, PGC=1.97e−06 and rs6669995, PGC=6.43e−06), FBXO33 (rs12590172, PGC=7.40e−06), and KCNG4 (rs11646443, PGC=6.51e−07; Table 1; Supplementary Figure S2). When patients were subdivided according to clinical subtype, we identified eight loci in which one or more SNPs showed evidence for association with combined or inattentive ADHD subtypes after considering the cut-off of PGC<1e−05, with the top signal at rs2232429 (PGC=8.88e−08; Tables 2 and 3; Supplementary Figure S2). Six of them were located in five genes: ZSCAN12 (rs2232429, PGC=8.88e−08), FBXO33 (rs3814860, PGC=8.72e−06 and rs12590172, PGC=3.96e−06), KCNG4 (rs11646443, PGC=8.39e−07), PDE4B (rs1937444, PGC=7.12e−06), and ACCN1 (rs1988353, PGC=1.16e−06; Tables 2 and 3; Supplementary Figure S2). The proportion of variance for ADHD explained by the 794 090 autosomal SNPs was estimated at 0.25 (SE=0.01; P=1.2e−08), 0.24 (SE=0.011; P=6.6e−06), and 0.19 (SE=0.07; P=2.9e−05) for overall, combined, and inattentive ADHD, respectively.
Table 1. Case–Control Genome-Wide Association Study in 603 Adult ADHD Patients and 583 Sex-Matched Unrelated Controls: Top Hits.
|
Cases |
Controls |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Base pairsa | Nearest gene | Risk alleleb | 11 | 12 | 22 | Sum | 11 | 12 | 22 | Sum | Adjusted Pc | Adjusted Pc Combined | Adjusted Pc Inattentive |
| rs1557524 | 1 | 160 246 485 | T | 218 (36.2) | 300 (49.8) | 85 (14.1) | 603 | 148 (25.4) | 306 (52.3) | 129 (22.1) | 583 | 3.55e−06 | 2.49e−04 | 3.64e−04 | |
| rs10594d | 1 | 160 247 185 | PEX19e (3′UTR) | G | 218 (36.2) | 300 (49.8) | 85 (14.1) | 603 | 147 (25.3) | 305 (52.6) | 128 (22.1) | 580 | 3.69e−06 | 2.54e−04 | 3.67e−04 |
| rs2795069 | 1 | 160 256 405 | G | 218 (36.2) | 299 (49.6) | 86 (14.3) | 603 | 148 (25.4) | 306 (52.3) | 129 (22.1) | 583 | 4.44e−06 | 3.08e−04 | 3.77e−04 | |
| rs7522166 | 1 | 160 276 709 | COPAe | C | 220 (36.5) | 298 (49.4) | 85 (14.1) | 603 | 147 (25.2) | 307 (52.7) | 129 (22.1) | 583 | 1.97e−06 | 1.71e−04 | 2.33e−04 |
| rs6669995 | 1 | 160 297 924 | COPAe | T | 218 (36.2) | 300 (49.8) | 85 (14.1) | 603 | 150 (25.7) | 305 (52.3) | 128 (22.0) | 583 | 6.43e−06 | 3.83e−04 | 5.18e−04 |
| rs1445594d | 13 | 58 675 299 | PCDH17 | A | 363 (60.2) | 218 (36.2) | 22 (3.6) | 603 | 283 (48.5) | 252 (43.2) | 48 (8.2) | 583 | 5.61e−06 | 4.70e−05 | 0.0017 |
| rs1897792d | 14 | 39 697 568 | A | 311 (51.7) | 244 (40.5) | 47 (7.8) | 602 | 366 (62.9) | 197 (33.8) | 19 (3.3) | 582 | 8.82e−06 | 2.00e−05 | 0.0013 | |
| rs4902569d | 14 | 39 858 941 | G | 275 (45.6) | 254 (42.1) | 74 (12.3) | 603 | 326 (56.0) | 223 (38.3) | 33 (5.7) | 583 | 9.47e−06 | 7.54e−06 | 0.0022 | |
| rs2415545d | 14 | 39 863 802 | A | 165 (27.4) | 293 (48.6) | 145 (24.0) | 603 | 218 (37.4) | 277 (47.5) | 88 (15.1) | 583 | 5.73e−06 | 2.72e−06 | 0.0071 | |
| rs12590172 | 14 | 39 893 535 | FBXO33e | C | 278 (46.1) | 255 (42.3) | 70 (11.6) | 603 | 329 (56.4) | 225 (38.6) | 29 (5.0) | 583 | 7.40e−06 | 3.96e−06 | 0.0031 |
| rs11644983d | 16 | 80 314 901 | DDYNLRB2 | C | 543 (90.0) | 59 (9.8) | 1 (0.2) | 603 | 472 (81.0) | 107 (18.4) | 4 (0.7) | 583 | 9.11e−06 | 6.58e−05 | 0.0057 |
| rs11646443d | 16 | 84 270 476 | KCNG4e (EXON 1) | G | 318 (52.7) | 249 (41.3) | 36 (6.0) | 603 | 228 (39.1) | 291 (49.9) | 64 (11.0) | 583 | 6.51e−07 | 8.39e−07 | 0.0055 |
Base pairs according to hg19—NCBI build 36.
Positive strand.
Cochram–Armitage trend test P-values deflated for a genomic control inflation factor λ=1.031.
SNP selected for follow-up.
SNP within the gene.
Table 2. Case–Control Genome-Wide Association Study in 368 Adult Combined ADHD Patients and 583 Sex-Matched Unrelated Controls: Top Hits.
|
Cases |
Controls |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Base pairsa | Nearest gene | Risk alleleb | 11 | 12 | 22 | Sum | 11 | 12 | 22 | Sum | Adjusted Pc | Adjusted Pc All | Adjusted Pc Inattentive |
| rs2232429d | 6 | 28 359 632 | ZSCAN12e | G | 303 (82.6) | 50 (13.6) | 14 (3.8) | 367 | 540 (92.6) | 42 (7.2) | 1 (0.2) | 583 | 8.88e−08 | 6.13e−05 | 0.66 |
| rs4416909d | 9 | 24 095 294 | ELAVL2 | C | 131 (35.6) | 173 (47.0) | 64 (17.4) | 368 | 137 (23.5) | 289 (49.6) | 157 (26.9) | 583 | 7.65e−06 | 4.64e−05 | 0.084 |
| rs4902569d | 14 | 39 858 941 | G | 159 (43.2) | 164 (44.6) | 45 (12.2) | 368 | 326 (56.0) | 223 (38.3) | 33 (5.7) | 582 | 7.54e−06 | 9.47e−06 | 0.0023 | |
| rs2415543d | 14 | 39 860 216 | A | 113 (30.7) | 191 (51.9) | 64 (17.4) | 368 | 265 (45.5) | 250 (42.9) | 68 (11.7) | 583 | 9.80e−06 | 2.86e−05 | 0.012 | |
| rs2415545d | 14 | 39 863 802 | A | 86 (23.4) | 197 (53.5) | 85 (23.1) | 368 | 218 (37.4) | 277 (47.5) | 88 (15.1) | 583 | 2.72e−06 | 5.73e−06 | 0.0071 | |
| rs3814860d | 14 | 39 868 646 | FBXO33 (3′UTR)e | A | 163 (44.3) | 163 (44.3) | 42 (11.4) | 368 | 331 (56.8) | 223 (38.3) | 29 (5.0) | 583 | 8.72e−06 | 1.16e−05 | 0.0029 |
| rs12590172 | 14 | 39 893 535 | FBXO33e | C | 160 (43.5) | 165 (44.8) | 43 (11.7) | 368 | 329 (56.4) | 225 (38.6) | 29 (5.0) | 583 | 3.96e−06 | 7.40e−06 | 0.0031 |
| rs17696574d | 14 | 39 967 531 | G | 173 (48.1) | 155 (43.1) | 32 (8.9) | 360 | 200 (34.4) | 286 (49.1) | 96 (16.5) | 582 | 4.40e−06 | 1.03e−05 | 0.015 | |
| rs11646443d | 16 | 84 270 476 | KCNG4e (EXON 1) | G | 203 (55.2) | 144 (39.1) | 21 (5.7) | 368 | 228 (39.1) | 291 (49.9) | 64 (11.0) | 583 | 8.39e−07 | 6.51e−07 | 0.0055 |
Base pairs according to hg19—NCBI build 36.
Positive strand.
Cochram–Armitage trend test P-values deflated for a genomic control inflation factor λ=1.0307.
SNP selected for follow-up.
SNP within the gene.
Table 3. Case–Control Association Study in 211 Adult Inattentive ADHD Patients and 583 Sex-Matched Unrelated Controls: Top Hits.
| Cases N (%) | Controls N (%) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
SNP |
Chr |
Base pairsa |
Nearest gene |
Risk alleleb |
11 |
12 |
22 |
Sum |
11 |
12 |
22 |
Sum |
Adjusted Pc |
Adjusted Pc All |
Adjusted Pc Combined |
| rs1937444d | 1 | 66 463 920 | PDE4Be | A | 94 (44.5) | 98 (46.4) | 19 (9.0) | 211 | 359 (61.6) | 200 (34.3) | 24 (4.1) | 583 | 7.12e−06 | 0.0013 | 0.18 |
| rs12333188f | 6 | 143 065 590 | GPR126 | T | 188 (89.1) | 23 (10.9) | — | 211 | 566 (97.1) | 17 (2.9) | — | 583 | 7.63e−06 | 0.0013 | 0.090 |
| rs1962749f | 6 | 153 551 654 | A | 146 (69.2) | 59 (28.0) | 6 (2.8) | 205 | 483 (82.8) | 97 (16.6) | 3 (0.5) | 580 | 8.56e−06 | 0.015 | 0.763 | |
| rs1988353f | 17 | 1 468 941 | ACCN1e | A | 97 (46.0) | 90 (42.7) | 24 (11.4) | 211 | 362 (62.1) | 200 (34.3) | 21 (3.6) | 583 | 1.16e−06 | 0.011 | 0.93 |
Base pairs according to hg19—NCBI build 36.
Positive strand.
Cochram–Armitage trend test P-values deflated for a genomic control inflation factor λ=1.0307.
Not tested in the replication phase due to experimental constraints.
SNP within the gene.
SNP selected for follow-up.
Follow-up of Top Signals from the Discovery Stage
Fifteen SNPs from 10 loci with a significance level of PGC<1e−05 in the discovery sample were taken forward for follow-up in three independent clinic-based adult ADHD samples (2104 cases and 1901 controls). Although nominal associations were detected in some of the populations under study (Supplementary Table S3–S4), no significant differences were observed in the joint analysis of the three replication cohorts neither through a mega-analysis (Supplementary Table S3–S4) nor in a meta-analysis (data not shown).
We subsequently considered the available data from both the discovery and replication stages and, although none of the markers surpassed genome-wide significance, some remained nominally associated with ADHD. When the overall ADHD sample was considered in the pooled analysis, six out of seven SNPs were nominally associated with ADHD: rs10594 (P=8.4e−04), rs11644983 (P=6.6e−04), rs2415545 (P=9.7e−03), rs4902569 (P=0.014), rs11646443 (P=0.049), and rs1897792 (P=0−05; Supplementary Table S3). Rs10594 and rs11644983 showed a significant association with ADHD in the German or Norwegian samples, respectively, with consistent effect size for the same risk allele identified in the discovery sample. When patients were subdivided according to the ADHD subtypes, the five SNPs tested at the FBXO33 locus remained associated with combined ADHD, rs4902569 (P=5.9e−03), rs2415543 (P=6.4e−03), rs2415545 (P=3.0e−03), rs3814860 (P=2.5e−03), and rs17696574 (P=3.1e−03; Supplementary Table S4a). Positive signals were also observed for the three SNPs considered in the inattentive ADHD sample, rs12333188 (P=3.0e−04), rs1962749 (P=2.1e−04), and rs1988353 (P=2.3e−04; Supplementary Table S4b). The mega-analysis, however, showed less significant results for all SNPs than the analysis of the discovery sample.
We subsequently performed a meta-analysis considering the discovery and replication sample sets and, after having detected heterogeneity among populations, a single positive signal was identified at rs17696574, located 5′ from FBXO33, in the combined ADHD clinical subtype (ORrandom=1.34 (1.01–1.78); P=0.041; Table 4 and Supplementary Tables S5–S7). Although the same analysis in the replication cohorts alone showed borderline significant association (combined ADHD: ORrandom=1.20 (0.99–1.46), P=0.056; Table 4b), directionally consistent effects were observed in the populations under study and these differences remained significant when we considered the overall ADHD cohort (ORfixed=1.24 (1.06–1.44; P=5.7e−03).
Table 4. Effect of rs17696574 on combined ADHD considering (a) the discovery and replication cohorts or (b) the replication cohorts alone.
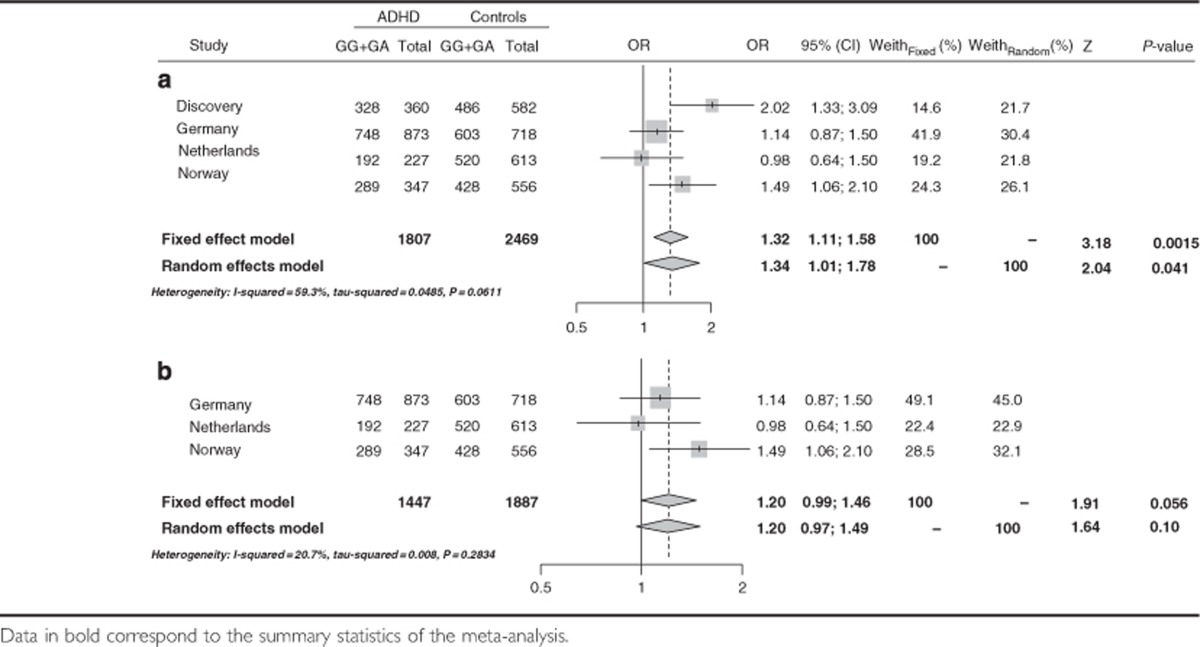
To better delineate the involvement of FBXO33 in combined ADHD and to detect potential functional variants, we imputed markers at this locus in the discovery cohort. Although the locus imputation did not identify any genome-wide significant signal, imputed SNPs within the locus provided stronger evidence for association than genotyped SNPs (Figure 1; Supplementary Table S8). Positive signals were identified along the entire gene, with the top signal, rs12232110 (P=9.02e−07), located in intron 1 (Supplementary Table S8). Although conditioning for the effect of rs12232110 eliminated evidence for association for the majority of SNPs in close proximity, this marker did not explain the entire signal at FBXO33 since a second independent SNP, rs10139591, maintained significance with combined ADHD (P=4.7e−04). This second marker is also located in intron 1, lies 1.5 kb from rs12232110 and resides in a haplotype block that encompasses the entire FBXO33 gene (Figure 1). Other candidate SNPs within the region were also nominally associated with combined ADHD when conditioning for the effect of rs12232110 and include rs7156962 (P=0.05) and rs1056345 (P=3.1e−03) in exons 3 and 4 of the FBXO33 gene, respectively. Given that multiple SNPs mapped to FBX033, we performed a gene-wise association test based on all the SNPs in the gene (n=17) and found additional evidence for its involvement in ADHD (gene-based P-value=1e−06 for overall and combined ADHD). Since FBXO33 acts as a substrate-recognition component of a protein–ubiquitin ligase complex, we also examined the joint effect of genetic variation in biological pathways. After quality control procedures, genotypes on 750 222 SNPs were considered, of which 443 335 lay within genes, covering 22 947 genes and 6188 categories. The pathway analysis showed a total of 293 out of 5264 GO categories reaching nominal significance for over-representation in our GWAS dataset. Among them, significant results were observed for the gene set/pathway referenced as ‘protein ubiquitination' (GO:0016567), which includes the FBXO33 gene (P=0.016; 41 significant genes out of 81 genes in this GO category), and for 31 additional GO categories related to neuronal development, differentiation, and activity (Supplementary Table S9). When the FBXO33 gene was excluded from the pathway analysis, the ‘protein ubiquination' category remained significantly over-represented in our GWAS dataset (P=0.026).
Figure 1.

Regional association plot including the FBXO33 locus on chromosome 14q13.3 from data imputed using the 1000 Genomes Project dataset. The x-axis shows physical distance (kb) and y-axis shows −log (P) values when considering the combined ADHD discovery cohort. In the upper panel genotyped and imputed markers are indicated in black and red dots, respectively. In the lower panel, the color reflects the degree of linkage disequilibrium (r2) between each genotyped (circled) or imputed (squared) marker with the most associated genotyped SNP, rs17696574, shown in diamond.
To provide insight into the molecular mechanisms behind the top FBXO33 SNPs associated with ADHD, we searched for cis eQTLs (expression quantitative trait loci) using a pre-existing dataset (GSE6536; Stranger et al, 2007a; Stranger et al, 2007b) and performed neuroimaging genetics studies using VBM analyses. In total, five SNPs within the FBXO33 locus were considered: rs17696574, identified in the joint meta-analysis of the discovery and the replication samples, rs12232110 and rs10139591, the two independent top signals identified in the discovery sample after genotype imputation, and rs7156962 and rs1056345, two intragenic SNPs nominally associated with ADHD that were located in exons 3 and 4 of the FBXO33 gene, respectively. FBXO33 expression levels in HapMap lymphoblastoid cell lines were significantly associated with genotypes in a dose-dependent manner for rs17696574 and rs7156962 (eQTL P=0.024 and P=8.7e−03, respectively), with lower FBXO33 expression being associated with the rs17696574G and rs7156962C risk alleles (Supplementary Figure S3). After applying the Bonferroni correction, only rs7156962 remained associated with FBXO33 expression levels.
When we investigated the role of FBXO33 SNPs in the brain considering the frontal lobe, caudate nucleus and the cerebellum as regions of interest, we found a nominally significant effect of rs1056345 (AA (n=406)/TA (n=603)/TT (n=211)) on frontal gray matter (peak voxel x=−56, y=−8 and z=49; cluster PFWE=0.02), with FBXO33 risk genotypes being nominally associated with reduced gray matter volume (Supplementary Figure S4). This effect was observed on the border of the frontal lobe and the parietal lobe, covering parts of the left post-central gyrus, an area that is involved in motor processing, which is a deficient cognitive domain in ADHD (Stray et al, 2013). No significant effects were found for the other SNPs or brain regions.
DISCUSSION
We have completed a genome-wide association scan and a follow-up replication study on adult ADHD. To our knowledge, this is the first GWAS on the persistent form of the disorder following individual genotyping, since the only one reported so far used DNA pooling (Lesch et al, 2008). Our results show that common SNPs in total explain 0.25 (SE=0.01) of phenotypic variance for adulthood ADHD, similar to the 0.28 (SE=0.02) described in a previous study of childhood ADHD (Lee et al, 2013). Although none of the findings exceeded genome-wide thresholds for association, neither in the discovery cohort nor in the follow-up meta-analysis, the results of the present study show tentative evidence for the involvement of the FBXO33 gene in the susceptibility to adult ADHD.
FBXO33 is a member of the F-box protein family that acts as a substrate-recognition component of a protein–ubiquitin ligase complex involved in targeting substrates for proteasomal degradation. FBXO33 has embryonic and adulthood expression in a wide range of neural tissues and has previously been associated with autism spectrum disorders in two large cohorts and with the neural response to seizure (Flood et al, 2004; Tai and Schuman, 2008; Wang et al, 2009). In addition, a deletion of chromosome 14 that spans the FBXO33 gene was identified in a patient with signs of attention deficit, hyperactivity, and mild mental retardation (de Bruijn et al, 2010). Furthermore, a balanced t(14;21)(q21.1;p11.2) translocation involving the FBXO33 gene was also reported in a patient with severe progressive autism (de Bruijn et al, 2010). These results are in agreement with previous studies pointing to a shared genetic etiology between ADHD and autism and support the view that common risk factors may be involved in both disorders as well as in their comorbidity (Nijmeijer et al, 2010; Rommelse et al, 2010).
To our knowledge, this is the first study suggesting a role of the ubiquitination machinery in ADHD, which is in line with previous reports showing association between genes from the ubiquitin family and autism, bipolar disorder, or intellectual disability (Glessner et al, 2009; Ryan et al, 2006; Tai and Schuman, 2008). Although the ubiquitin–proteasome pathway modulates synaptic activity, neurotransmitter release, and synaptic vesicle recycling, the exact mechanism by which FBXO33 contributes to ADHD remains to be elucidated. Our expression study suggests that decreased FBXO33 levels may underline the vulnerability to ADHD, which is in agreement with the downregulation of ubiquitin cycle-related genes identified in bipolar post-mortem brains (Ryan et al, 2006). Reduced expression of enzymes involved in this pathway may thus impair targeting of proteins for proteasomal degradation and hence the turnover of synaptic components. The association between the FBXO33 risk variant at the 3′UTR of the gene and reduced gray matter volume in the left post-central gyrus, a region that is part of the attention network of the brain and shows altered activity in patients with combined subtype ADHD (Silk et al, 2005), also supports the role of FBXO33 in the pathogenesis of ADHD. Further studies are needed to confirm whether FBXO33 expression is involved in ADHD and to determine the potential mechanism by which reduced expression of this gene may contribute to the susceptibility to the disorder.
Although the top SNP from this association study, rs17696574, is located 65.8 kb upstream from FBXO33, its involvement in ADHD is supported by multiple highly correlated SNPs within the gene region as well as by the gene-wise association analysis (Figure 1). Interestingly, rs7156962, an evolutionary conserved synonymous change in exon 3 of FBXO33, may affect the regulation of splicing, with the rs7156962C risk allele altering putative exonic splicing enhancers. This sequence variant has a functional significance score (FS) of 0.77, which may reflect its potential deleterious effect (Lee and Shatkay, 2009). Other SNPs within the FBXO33 locus highlighted in the present study may also deserve further attention, since they are highly conserved (rs3814860), affect highly methylated regions (rs12232110), or alter transcription factor binding sites (TFBS; rs2415543). In addition, all these sequence variants, with the exception of rs2415543, are associated with trimethylated histone H3 at lysine 36 (H3K36me3), which is related with transcriptionally active regions. Finally, the correlation between genotypes at rs17696574 or rs7156962, a synonymous change in exon 3, and FBXO33 expression levels in lymphoblastoid cell lines also suggests functional relevance and reinforces the possibility of true causality. In any case, the analysis of gene expression in brain tissue is required to confirm this correlation.
Given that the two top-ranked SNPs associated with ADHD after locus imputation, rs12232110 and rs10139591, are located in intron 1 of FBXO33 and contained in a LD block that spans the entire gene (Figure 1), we cannot rule out that additional coding variants in FBXO33 might exert functional effects. It also remains an open issue whether disease vulnerability is driven by a single variant or by multiple independent SNPs within the region. Further fine mapping and deep sequencing of this genomic region is required to identify causal sequence variants directly involved in the genetic background of ADHD.
Although we have attempted to minimize limitations of previous GWAS, the outcome of the present study should be interpreted in the context of several considerations.
First, to minimize population stratification and genetic heterogeneity we focused on the persistent form of ADHD. Despite the fact that genetic studies have mainly focused on children, there is evidence pointing to the existence of an even stronger genetic component in adult ADHD (Biederman, 2005; Biederman and Faraone, 2005; Chang et al, 2013; Faraone et al, 2005). In addition, different lines of investigation suggest a distinct genetic load between persistent and remitting ADHD, and support the existence of specific genetic factors contributing to the stability of ADHD symptoms across life (Kuntsi et al, 2005; Price et al, 2005; Ribases et al, 2008). Thus, previous GWAS focused on childhood ADHD may have increased heterogeneity by including in the analysis the subset of patients in whom ADHD will remit.
Second, nominal signals were detected both in the discovery cohort and in the joint analysis of the discovery and replication samples, but only borderline significance was observed when the replication cohorts were considered alone. Although this is not a standard replication-based design, the analysis combining the information from both the discovery and replication samples may be more powerful than treating the design as a hypothesis generation followed by independent replication (Skol et al, 2006). Further studies in additional samples and a truly independent replication are warranted both to confirm the association between FBXO33 and ADHD and to estimate the magnitude of its effect.
Third, a part from variation in FBX033, we failed to replicate in our validation cohort any of the top-ranked associations that achieved the follow-up threshold of significance in the discovery stage. Although these results provide little support for association with genes other than FBXO33, several top findings deserve further comments. PEX19 and COPA encode the peroxisomal farnesylated protein and the subunit alpha of the coatomer protein complex, respectively, and they have been associated with different psychiatric disorders. PEX19 was associated with major depression in a meta-analysis carried out by the Psychiatric GWAS Consortium (Ripke et al, 2013b), while COPA expression has been found dysregulated in the orbitofrontal cortex of bipolar disorder patients (Ryan et al, 2006). In addition, KCNG4 codes for a regulatory subunit of voltage-gated potassium channels highly expressed in the brain, involved in the regulation of neurotransmitter release and neuronal excitability, and associated with migraine (Lafreniere and Rouleau, 2012). Interestingly, although rs11646443, the missense change (c.616G>A, p.Arg206Trp) in KCNG4 found associated with ADHD in the present study may not change the secondary structure of the protein or have damaging effects on its function, it might affect the regulation of splicing, since the rs11646443G risk allele lies within a putative exonic splicing repressor. Finally, PCDH17 belongs to the protocadeherin family and its product participates in synaptic function and cell–cell adhesion activity, which has been previously associated with ADHD (Lesch et al, 2008; Neale et al, 2008; Neale et al, 2010b; Rivero et al, 2013; Zhou et al, 2008). These promising candidates may be considered as potential ADHD-related loci worth replicating in larger sample sets.
In addition, the results from our GWAS do not identify hits from previous GWAS or provide genome-wide significant support for any of the previously postulated candidate genes for ADHD (Franke et al, 2009; Hinney et al, 2011; Lesch et al, 2008; Mick et al, 2010; Neale et al, 2008; Stergiakouli et al, 2012). No overlap or LD was observed between our main hits and the top-ranked findings from previous GWAS reports either. Confounding factors, such as comorbidities, IQ, or environmental influences that may modulate the genetic mechanisms predisposing to ADHD could help to explain that positive signals in previous studies do not reach genome-wide significance in our study or were not ranked high enough to be included in our replication stage. It is worth pointing out, however, that rs1937444 in the PDE4B gene showed a borderline significant association with adulthood ADHD in a previous GWAS performed in the persistent form of the disorder (Supplementary Table S10; Lesch et al, 2008). Interestingly, this gene was previously associated with schizophrenia (Millar et al, 2005) and an identically active isoform, the brain-specific phosphodiesterase 4D isoform 6 (PDE4D6), was found duplicated in an ADHD family (Lesch et al, 2011).
Fourth, we aimed to improve our GWAS design by focusing not only on SNPs contributing to the overall risk for ADHD, but also on markers that may participate in the combined or inattentive clinical subtypes. Following this strategy, which allows capturing additional associations that might have been skipped in previous studies that focused on the overall ADHD sample, we found preliminary evidence for the involvement of FBXO33 in combined ADHD but not in the inattentive subgroup. These findings are in agreement with previous reports pointing to the validity of the DSM-IV distinction between the combined and predominantly inattentive types, support previous studies that find differential genetic components across ADHD subtypes and suggest that the combined group may represent a distinctive and more homogeneous phenotype that may facilitate the identification of genetic factors contributing to ADHD (Larsson et al, 2006; Ribases et al, 2008; Ribases et al, 2009; Todd et al, 2001). Thus, the identification of phenotypic characteristics that define subgroups of patients affected by specific sets of genes, such as clinical subtypes, may be an alternative approach to reduce genetic heterogeneity in ADHD. However, we cannot exclude that the limited sample size of the inattentive group may account for the absence of association observed when this ADHD subtype was considered.
Fifth, since heterogeneity may increase in large multi-site designs, cases and controls included in the discovery sample were recruited by a single clinical group in a restricted geographic area (Barcelona) and had the same ethnic background (Spanish Caucasian). Diagnosis of ADHD was based on structured, systematic interviews with no variability in measurements across data. We used one single ascertainment approach with homogeneous semi-structured diagnostic assessments, which may facilitate the identification of risk variants that might be neglected by a broader recruitment strategy used in previous multi-site GWAS. In addition, given that using controls unscreened for the presence of the disorder and genotyping with different platforms may reduce power, the presence of ADHD-related symptoms was excluded in our controls, and a single genotyping platform, Illumina HumanOmni1-Quad, was used for both affected individuals and controls. However, despite our efforts to increase the homogeneity of the discovery cohort, our discovery GWAS did not have sufficient statistical power to unequivocally detect small to medium effects of common SNPs on ADHD at a genome-wide significant level. Our negative results may be explained by the existence of small genetic effects also in the adult form of ADHD, which would require larger and homogeneous sample collections and further replication efforts to be reliably identified.
In conclusion, we performed a two-step GWAS to identify genetic risk factors for persistent ADHD. Our findings show tentative evidence for the involvement of FBXO33 in the susceptibility to adult ADHD, point at the ubiquitination machinery as a new disease mechanism for the disorder and establish a rationale for searching additional risk variants within ubiquitin-related genes through additional collaborative efforts.
FUNDING AND DISCLOSURE
JB has been in the past 3 years a consultant to/member of advisory board of/and/or speaker for Janssen-Cilag BV, Eli Lilly, Bristol-Myers Squibb, Schering-Plough, UCB, Shire, Novartis, and Servier. He is not an employee of any of these companies, and not a stock shareholder of any of these companies. He has no other financial or material support, including expert testimony, patents, and royalties. CCK takes part as an advisor in an Adult ADHD Advisory Workgroup of Eli Lilly and is a lecturer in their educational program ‘Adult-ADHD Academy'. JH has in the past 3 years been a speaker for Janssen-Cilag BV, Eli Lilly, and Novartis. He is not an employee of any of these companies, and not a stock shareholder of any of these companies. AH received funding from the DFG (HE1446/9-1, HI865/2-1) and the BMBF (01GS0820). SVF receives royalties from books published by Guilford Press: Straight Talk about Your Child's Mental Health and Oxford University Press: Schizophrenia: The Facts.
Acknowledgments
We thank all persons who kindly participated in this research. Part of the DNA extractions and genotyping was performed at the Spanish National Genotyping Centre (CEGEN-Barcelona). Principal investigators of the Nijmegen Biomedical Study are L.A.L.M. Kiemeney, J. de Graaf, A.L.M. Verbeek, D.W. Swinkels, and B. Franke. Principal investigators of the Cognomics Initiative are B. Franke, S. Fisher, G. Fernandez, H. Brunner, P. Hagoort, J. Buitelaar, H. van Bokhoven, and D. Norris. We are grateful to Silvia Fochs, Anna Sànchez, Maribel López, Nuria Pey, and Muriel Ferrer for their assistance in contacting the families and administering the questionnaires. Marta Ribasés is a recipient of a Miguel de Servet contract from the ‘Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación', Spain. Financial support was received from ‘Fundació La Marató de TV3' (ref. 092330/31), ‘Instituto de Salud Carlos III-FIS' (PI11/00571, PI11/01629, PI12/01139, PI041436, PI081151, Red INMA G03/176, and CB06/02/0041), ‘Plan Nacional Sobre Drogas' (PNSD#2011-0080), ‘Agència de Gestió d'Ajuts Universitaris i de Recerca-AGAUR, Generalitat de Catalunya ‘(2014SGR1357, 2014SGR0932, and 1999SGR00241), Ministerio de Economía y Competitividad, Spain' (SAF2012-33484), and ‘Departament de Salut', Government of Catalonia, Spain. The Dutch part of the project was supported by the Hersenstichting Nederland and by a Vici grant of The Netherlands Organisation for Scientific Research (NWO) to Barbara Franke. The Dutch controls were derived from the Nijmegen Biomedical Study (www.nijmegenbiomedischestudie.nl); European Commission (ENGAGE project and grant agreement HEALTH-F4-2007-201413). This work made use of the BIG (Brain Imaging Genetics) database, first established in Nijmegen, The Netherlands, in 2007. This resource is now part of Cognomics (www.cognomics.nl), a joint initiative by researchers of the Donders Centre for Cognitive Neuroimaging, the Human Genetics and Cognitive Neuroscience departments of the Radboud University Medical Centre and the Max Planck Institute for Psycholinguistics in Nijmegen. The Cognomics Initiative is supported by the participating departments and centers and by external grants, ie, the Biobanking and Biomolecular Resources Research Infrastructure (The Netherlands; BBMRI-NL), the Hersenstichting Nederland and The Netherlands Organisation for Scientific Research (NWO). Funding for the Norwegian cohort was provided by the K.G. Jebsen Foundation, the Norwegian Research Council, and Regional Health Authority of Western Norway. The sample used in this study is part of the international multicentre persistent ADHD collaboration (IMpACT). IMpACT unites major research centres working on the genetics of ADHD persistence across the lifespan and has participants in the Netherlands, Germany, Spain, Norway, the United Kingdom, the United States, Brazil and Sweden. Principal investigators of IMpACT are: Barbara Franke (chair), Andreas Reif, Stephen V. Faraone, Jan Haavik, Bru Cormand, Antoni Ramos Quiroga, Philip Asherson, Klaus-Peter Lesch, Jonna Kuntsi, Claiton Bau, Jan Buitelaar, Stefan Johansson, Henrik Larsson, Alysa Doyle, and Eugenio Grevet.
Footnotes
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary Material
References
- Biederman J. Attention-deficit/hyperactivity disorder: a selective overview. Biol Psychiatry. 2005;57:1215–1220. doi: 10.1016/j.biopsych.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Biederman J, Faraone S, Milberger S, Curtis S, Chen L, Marrs A, et al. Predictors of persistence and remission of ADHD into adolescence: results from a four-year prospective follow-up study. J Am Acad Child Adolesc Psychiatry. 1996;35:343–351. doi: 10.1097/00004583-199603000-00016. [DOI] [PubMed] [Google Scholar]
- Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248. doi: 10.1016/S0140-6736(05)66915-2. [DOI] [PubMed] [Google Scholar]
- Browning BL, Browning SR. A unified approach to genotype imputation and haplotype phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analysis using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D. snpStats: SnpMatrix and XSnpMatrix classes and methods. R package version 1.14.0 2013.
- Cubillo A, Rubia K. Structural and functional brain imaging in adult attention-deficit/hyperactivity disorder. Expert Rev Neurother. 2010;10:603–620. doi: 10.1586/ern.10.4. [DOI] [PubMed] [Google Scholar]
- Chang Z, Lichtenstein P, Asherson PJ, Larsson H. Developmental twin study of attention problems: high heritabilities throughout development. JAMA Psychiatry. 2013;70:311–318. doi: 10.1001/jamapsychiatry.2013.287. [DOI] [PubMed] [Google Scholar]
- Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–245. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- de Bruijn DR, van Dijk AH, Pfundt R, Hoischen A, Merkx GF, Gradek GA, et al. Severe progressive autism associated with two de novo changes: a 2.6-Mb 2q31.1 deletion and a balanced t(14;21)(q21.1;p11.2) translocation with long-range epigenetic silencing of LRFN5 expression. Mol Syndromol. 2010;1:46–57. doi: 10.1159/000280290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Feighner JA, Monuteaux MC. Assessing symptoms of attention deficit hyperactivity disorder in children and adults: which is more valid. J Consult Clin Psychol. 2000;68:830–842. [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Spencer T, Wilens T, Seidman LJ, Mick E, et al. Attention-deficit/hyperactivity disorder in adults: an overview. Biol Psychiatry. 2000;48:9–20. doi: 10.1016/s0006-3223(00)00889-1. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Flood WD, Moyer RW, Tsykin A, Sutherland GR, Koblar SA. Nxf and Fbxo33: novel seizure-responsive genes in mice. Eur J Neurosci. 2004;20:1819–1826. doi: 10.1111/j.1460-9568.2004.03646.x. [DOI] [PubMed] [Google Scholar]
- Franke B, Faraone SV, Asherson P, Buitelaar J, Bau CH, Ramos-Quiroga JA, et al. The genetics of attention deficit/hyperactivity disorder in adults, a review. Mol Psychiatry. 2012;17:960–987. doi: 10.1038/mp.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke B, Neale BM, Faraone SV. Genome-wide association studies in ADHD. Hum Genet. 2009;126:13–50. doi: 10.1007/s00439-009-0663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. Candidate gene studies of ADHD: a meta-analytic review. Hum Genet. 2009;126:51–90. doi: 10.1007/s00439-009-0694-x. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González JR, Armengol L, Solé X, Guinó E, Mercader JM, Estivill X, et al. SNPassoc: an R package to perform whole genome association studies. Bioinformatics. 2007;23:644–645. doi: 10.1093/bioinformatics/btm025. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinney A, Scherag A, Jarick I, Albayrak O, Putter C, Pechlivanis S, et al. Genome-wide association study in German patients with attention deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:888–897. doi: 10.1002/ajmg.b.31246. [DOI] [PubMed] [Google Scholar]
- Holmans P, Green EK, Pahwa JS, Ferreira MAR, Purcell SM, Sklar P. Gene ontology analysis of GWAS datasets provide insights into the biology of bipolar disorder. Am J Hum Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Adler LA, Barkley R, Biederman J, Conners CK, Faraone SV, et al. Patterns and predictors of attention-deficit/hyperactivity disorder persistence into adulthood: results from the national comorbidity survey replication. Biol Psychiatry. 2005;57:1442–1451. doi: 10.1016/j.biopsych.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooij JJ, Buitelaar JK, van den Oord EJ, Furer JW, Rijnders CA, Hodiamont PP. Internal and external validity of attention-deficit hyperactivity disorder in a population-based sample of adults. Psychol Med. 2005;35:817–827. doi: 10.1017/s003329170400337x. [DOI] [PubMed] [Google Scholar]
- Kuntsi J, Rijsdijk F, Ronald A, Asherson P, Plomin R. Genetic influences on the stability of attention-deficit/hyperactivity disorder symptoms from early to middle childhood. Biol Psychiatry. 2005;57:647–654. doi: 10.1016/j.biopsych.2004.12.032. [DOI] [PubMed] [Google Scholar]
- Lafreniere RG, Rouleau GA. Identification of novel genes involved in migraine. Headache. 2012;52 (Suppl 2:107–110. doi: 10.1111/j.1526-4610.2012.02237.x. [DOI] [PubMed] [Google Scholar]
- Larsson H, Lichtenstein P, Larsson JO. Genetic contributions to the development of ADHD subtypes from childhood to adolescence. J Am Acad Child Adolesc Psychiatry. 2006;45:973–981. doi: 10.1097/01.chi.0000222787.57100.d8. [DOI] [PubMed] [Google Scholar]
- Lasky-Su J, Anney RJ, Neale BM, Franke B, Zhou K, Maller JB, et al. Genome-wide association scan of the time to onset of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1355–1358. doi: 10.1002/ajmg.b.30869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky-Su J, Neale BM, Franke B, Anney RJ, Zhou K, Maller JB, et al. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1345–1354. doi: 10.1002/ajmg.b.30867. [DOI] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PH, Shatkay H. An integrative scoring system for ranking SNPs by their potential deleterious effects. Bioinformatics. 2009;25:1048–1055. doi: 10.1093/bioinformatics/btp103. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Timmesfeld N, Renner TJ, Halperin R, Roser C, Nguyen TT, et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm. 2008;115:1573–1585. doi: 10.1007/s00702-008-0119-3. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T, et al. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry. 2011;16:491–503. doi: 10.1038/mp.2010.29. [DOI] [PubMed] [Google Scholar]
- Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–1239. doi: 10.1016/s1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- Mick E, Todorov A, Smalley S, Hu X, Loo S, Todd RD, et al. Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:898–905.e893. doi: 10.1016/j.jaac.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Neale BM, Lasky-Su J, Anney R, Franke B, Zhou K, Maller JB, et al. Genome-wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1337–1344. doi: 10.1002/ajmg.b.30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Medland S, Ripke S, Anney RJ, Asherson P, Buitelaar J, et al. Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:906–920. doi: 10.1016/j.jaac.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP, et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:884–897. doi: 10.1016/j.jaac.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijmeijer JS, Arias-Vasquez A, Rommelse NN, Altink ME, Anney RJ, Asherson P, et al. Identifying loci for the overlap between attention-deficit/hyperactivity disorder and autism spectrum disorder using a genome-wide QTL linkage approach. J Am Acad Child Adolesc Psychiatry. 2010;49:675–685. doi: 10.1016/j.jaac.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TS, Simonoff E, Asherson P, Curran S, Kuntsi J, Waldman I, et al. Continuity and change in preschool ADHD symptoms: longitudinal genetic analysis with contrast effects. Behav Genet. 2005;35:121–132. doi: 10.1007/s10519-004-1013-x. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribases M, Hervas A, Ramos-Quiroga JA, Bosch R, Bielsa A, Gastaminza X, et al. Association study of 10 genes encoding neurotrophic factors and their receptors in adult and child attention-deficit/hyperactivity disorder. Biol Psychiatry. 2008;63:935–945. doi: 10.1016/j.biopsych.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Ribases M, Ramos-Quiroga JA, Hervas A, Bosch R, Bielsa A, Gastaminza X, et al. Exploration of 19 serotoninergic candidate genes in adults and children with attention-deficit/hyperactivity disorder identifies association for 5HT2A, DDC and MAOB. Mol Psychiatry. 2009;14:71–85. doi: 10.1038/sj.mp.4002100. [DOI] [PubMed] [Google Scholar]
- Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero O, Sich S, Popp S, Schmitt A, Franke B, Lesch KP. Impact of the ADHD-susceptibility gene CDH13 on development and function of brain networks. Eur Neuropsychopharmacol. 2013;23:492–507. doi: 10.1016/j.euroneuro.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Rommelse NN, Franke B, Geurts HM, Hartman CA, Buitelaar JK. Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur Child Adolesc Psychiatry. 2010;19:281–295. doi: 10.1007/s00787-010-0092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Fanous AH, Ripke S, McQuillin A, Amdur RL, Gejman PV, et al. Polygenic dissection of diagnosis and clinical dimensions of bipolar disorder and schizophrenia. Mol Psychiatry. 2013;19:1017–1024. doi: 10.1038/mp.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MM, Lockstone HE, Huffaker SJ, Wayland MT, Webster MJ, Bahn S. Gene expression analysis of bipolar disorder reveals downregulation of the ubiquitin cycle and alterations in synaptic genes. Mol Psychiatry. 2006;11:965–978. doi: 10.1038/sj.mp.4001875. [DOI] [PubMed] [Google Scholar]
- Sánchez-Mora C, Ribasés M, Ramos-Quiroga JA, Casas M, Bosch R, Boreatti-Hümmer A, et al. Meta-analysis of brain-derived neurotrophic factor p.Val66Met in adult ADHD in four European populations. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:512–523. doi: 10.1002/ajmg.b.31008. [DOI] [PubMed] [Google Scholar]
- Silk T, Vance A, Rinehart N, Egan G, O'Boyle M, Bradshaw JL, et al. Fronto-parietal activation in attention-deficit hyperactivity disorder, combined type: functional magnetic resonance imaging study. Br J Psychiatry. 2005;187:282–283. doi: 10.1192/bjp.187.3.282. [DOI] [PubMed] [Google Scholar]
- Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- Stergiakouli E, Hamshere M, Holmans P, Langley K, Zaharieva I, Hawi Z, et al. Investigating the contribution of common genetic variants to the risk and pathogenesis of ADHD. Am J Psychiatry. 2012;169:186–194. doi: 10.1176/appi.ajp.2011.11040551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray LL, Kristensen O, Lomeland M, Skorstad M, Stray T, Tonnessen FE. Motor regulation problems and pain in adults diagnosed with ADHD. Behav Brain Funct. 2013;9:18. doi: 10.1186/1744-9081-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- Todd RD, Rasmussen ER, Neuman RJ, Reich W, Hudziak JJ, Bucholz KK, et al. Familiality and heritability of subtypes of attention deficit hyperactivity disorder in a population sample of adolescent female twins. Am J Psychiatry. 2001;158:1891–1898. doi: 10.1176/appi.ajp.158.11.1891. [DOI] [PubMed] [Google Scholar]
- Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36:1–48. [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–W605. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Neale BM, Liu L, Lee SH, Wray NR, Ji N, et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: genome-wide association study of both common and rare variants. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:419–430. doi: 10.1002/ajmg.b.32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JH. gap: Genetic Analysis Package. R package version 1.1-10 2013.
- Zhou K, Dempfle A, Arcos-Burgos M, Bakker SC, Banaschewski T, Biederman J, et al. Meta-analysis of genome-wide linkage scans of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1392–1398. doi: 10.1002/ajmg.b.30878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.